

RNA profiling of maize kernel compartments revealed coexpression modules for each major cell type in the endosperm, including a module regulating differentiation of the basal endosperm transfer layer.
Abstract
Endosperm is an absorptive structure that supports embryo development or seedling germination in angiosperms. The endosperm of cereals is a main source of food, feed, and industrial raw materials worldwide. However, the genetic networks that regulate endosperm cell differentiation remain largely unclear. As a first step toward characterizing these networks, we profiled the mRNAs in five major cell types of the differentiating endosperm and in the embryo and four maternal compartments of the maize (Zea mays) kernel. Comparisons of these mRNA populations revealed the diverged gene expression programs between filial and maternal compartments and an unexpected close correlation between embryo and the aleurone layer of endosperm. Gene coexpression network analysis identified coexpression modules associated with single or multiple kernel compartments including modules for the endosperm cell types, some of which showed enrichment of previously identified temporally activated and/or imprinted genes. Detailed analyses of a coexpression module highly correlated with the basal endosperm transfer layer (BETL) identified a regulatory module activated by MRP-1, a regulator of BETL differentiation and function. These results provide a high-resolution atlas of gene activity in the compartments of the maize kernel and help to uncover the regulatory modules associated with the differentiation of the major endosperm cell types.
INTRODUCTION
Seed development is initiated by double fertilization of the haploid egg cell and the dikaryotic central cell to produce two filial structures, a diploid embryo and a triploid endosperm, respectively (Faure, 2001; Hamamura et al., 2012). Endosperm functions as an absorptive structure that supports embryo development or seedling germination in angiosperms (Lopes and Larkins, 1993). Recent evidence also indicates that endosperm plays a critical role in regulation of seed development through interaction with the embryo and the seed coat (Berger et al., 2006; Lafon-Placette and Köhler, 2014). The endosperm of cereal grains occupies a large portion of the mature seed, holds large amounts of proteins and carbohydrates required for seedling development, and is an important source of food, feed, and renewable industrial raw materials (Lopes and Larkins, 1993; Olsen, 2001, 2004; Sabelli and Larkins, 2009; FAO, 2012).
In most flowering plants, endosperm development begins with the formation of a coenocyte, as the fertilized central cell undergoes multiple rounds of nuclear divisions without cytokinesis. The multinucleated coenocyte then undergoes cellularization and cell differentiation (Olsen, 2004; Sabelli and Larkins, 2009). In dicots, the endosperm is mostly absorbed by the developing embryo shortly after cellularization. By contrast, in monocots, and particularly in cereals, the endosperm enlarges significantly after cellularization through many rounds of cell division accompanied by cell enlargement and organelle proliferation. Consequently, the cereal endosperm acquires a high storage capacity of carbohydrates and proteins prepared for mobilization upon seedling germination (Lopes and Larkins, 1993; Sreenivasulu and Wobus, 2013). The acquisition of endosperm storage capacity is enabled in part through the activity of specialized cell types or compartments that mediate uptake of nutrients from the maternal structures and their storage in the inner compartments of the endosperm. Therefore, elucidating how cell differentiation is regulated during endosperm development is central to understanding endosperm structure and function.
Because of its relatively large size and economic importance, the maize (Zea mays) endosperm represents an excellent model system to study early regulatory processes that regulate regional and cellular differentiation events. The initial coenocytic phase of endosperm growth in maize occurs during the first 2 d after pollination (DAP), and this is followed by a period of cellularization during 3 to 4 DAP. Following cellularization, the endosperm cells undergo two major phases of mitotic proliferation, an early phase that lasts until 8 to 12 DAP in the central region, and a late phase that continues until 20 to 25 DAP in the outer endosperm layers. Starting at ∼8 to 10 DAP, the central portion of endosperm cells gradually switches from mitosis to endoreduplication and becomes filled with starch and storage proteins (Brink and Cooper, 1947; Olsen, 2001, 2004; Sabelli and Larkins, 2009; Becraft and Gutierrez-Marcos, 2012; Olsen and Becraft, 2013; Leroux et al., 2014).
Differentiation of maize endosperm cells occurs primarily at 4 to 6 DAP (following endosperm cellularization and before the initiation of mitotic proliferation), resulting in four main cell types, including the starchy endosperm (SE), the aleurone (AL), the embryo-surrounding region (ESR), and the basal endosperm transfer layer (BETL) (Olsen, 2001; Becraft and Gutierrez-Marcos, 2012; Leroux et al., 2014). The SE is the cell type that accumulates starch and storage proteins. The SE itself contains at least three subregions, including the central starchy endosperm (CSE), the conducting zone (CZ), and the subaleurone (Becraft, 2001; Olsen, 2001, 2004; Sabelli and Larkins, 2009). The AL is a single peripheral layer of cells that produces hydrolytic enzymes to mobilize the storage products in the SE when activated during seed germination. The ESR is believed to act as a physical barrier and messenger between endosperm and embryo (Olsen, 2004). The BETL is a transfer cell layer that transports nutrients from the maternal tissue into the inner endosperm cells, including the developing SE, in order to enable starch and protein synthesis (Sabelli and Larkins, 2009; Becraft and Gutierrez-Marcos, 2012). Recent studies have identified many genes expressed specifically in the BETL, including multiple genes encoding cysteine-rich proteins that are thought to act as antimicrobial or intercellular signal molecules (Tailor et al., 1997; Marshall et al., 2011), and MRP-1 (Myb-Related Protein-1), a MYB-related (MYBR) transcription factor previously shown to activate a number of these genes in the BETL (Gómez et al., 2002, 2009; Gutiérrez-Marcos et al., 2004). Moreover, ectopic expression of MRP-1 in the AL has been shown to produce a transient BETL-like structure (Gómez et al., 2009). Additional recent efforts have enabled genome-wide identification of gene expression during nearly all stages of endosperm development (Sekhon et al., 2013; Chen et al., 2014; Li et al., 2014). However, little is known about the gene regulatory networks (GRNs) that regulate the differentiation and determine the function of the individual cell types or compartments of the endosperm in maize.
Here, we used a coupled laser-capture microdissection (LCM) and RNA sequencing (RNA-Seq) strategy to comprehensively profile the mRNA populations present in each of the main cell types of the maize endosperm, as well as the embryo and four maternal compartments of the kernel at 8 DAP. We identified mRNAs that specifically accumulate in each of the captured compartments. Also, using an unbiased network analysis tool, we detected modules of coexpressed genes that are either predominantly expressed in a single compartment or expressed in multiple compartments, including several endosperm-correlated modules that are enriched for temporally upregulated genes and/or imprinted genes that we previously identified. By focusing on the analysis of genes in a BETL-correlated coexpression module, we identified and experimentally validated a regulatory module of the BETL GRN that is activated by MRP-1.
RESULTS
Capture and Analysis of mRNA Populations of Filial and Maternal Compartments of 8-DAP Kernel
To identify the genes active in each of the endosperm cell types, we used LCM to isolate and profile mRNA populations of five endosperm compartments (cell types) of maize inbred line B73 at 8 DAP. The compartments analyzed included AL, BETL, ESR, and two subregions of SE, the CSE and the CZ. To compare endosperm gene expression programs with embryonic and maternal programs, we also captured the embryo (EMB), nucellus (NU), placento-chalazal region (PC), pericarp (PE), and the vascular region of the pedicel (PED) at 8 DAP (Figure 1A; Supplemental Figures 1 to 3 and Supplemental Table 1). We selected this time point because it follows differentiation of the main cell types of the endosperm, which occurs at 4 to 6 DAP, and precedes developmental programs associated with endosperm function, including the activation of storage product synthesis and deposition program, which initiates at ∼8 to 10 DAP (Becraft, 2001; Olsen, 2001, 2004; Sabelli and Larkins, 2009; Becraft and Gutierrez-Marcos, 2012; Olsen and Becraft, 2013; Leroux et al., 2014). Total RNA extracted from biological triplicates for the endosperm compartments and embryo, and single replicates of maternal compartments (22 samples) were reverse-transcribed to cDNA using oligo(dT) and random primers, amplified, and paired-end sequenced using an Illumina HiSequation 2000 platform.
Figure 1.

Profiles of Sequenced RNAs from the Captured Filial and Maternal Compartments of 8-DAP Maize Kernel.
(A) Graphic representation of an 8-DAP maize kernel showing the relative position of the 10 captured filial and maternal compartments used for RNA sequencing.
(B) Numbers of TF genes, non-TF protein-coding genes, microRNA genes, transposable elements, and pseudogenes expressed in the 10 captured compartments.
(C) Proportions of genes expressed at different levels (based on FPKM) in the 10 kernel compartments.
The resulting reads were quality checked and mapped to the maize reference genome (B73 RefGen_v3). Of the resulting mapped reads (8.9 to 34.7 million, 52.3 to 89.7% of total reads), 3.2 to 11.3 million (22.1 to 39.5%) were mapped to exonic sequences (Supplemental Table 2). The exonic reads were normalized using Cufflinks (Trapnell et al., 2012) and reported as fragments per kilobase of transcript per million mapped reads (FPKM). A gene was considered expressed in a given sample if the lower boundary of its FPKM 95% confidence interval (FPKM_conf_lo) was greater than zero (Hansey et al., 2012). Based on this criterion, 29,369 genes were identified as expressed in at least one of the 22 samples (Supplemental Data Set 1). Using pairwise Spearman correlation coefficient (SCC) analysis, the triplicate FPKM values from each of the endosperm compartments and the embryo were shown to be highly correlated (ρ = 0.87 to 0.91; Supplemental Figure 4). Accordingly, we pooled each triplicate set of exonic reads and renormalized the data using Cufflinks. Using the same cutoff as indicated above, we detected 30,665 genes expressed in at least one of the 10 compartments (Supplemental Data Set 2 and Supplemental Figure 5), 10,725 genes expressed in all 10 compartments (Supplemental Figure 6A), and between 15,910 and 23,853 (NU and ESR, respectively) expressed in individual compartments (Figure 1B; Supplemental Table 3). In all cases, the proportion of transcription factor (TF) genes detected as expressed tracked closely with the total number of expressed genes (between ∼5.3 and 6.0% for CZ and PED, respectively; Figure 1B; Supplemental Table 3). The proportions of high-expressing (FPKM ≥ 10), medium-expressing (2 ≤ FPKM < 10), and low-expressing (FPKM < 2) genes were relatively similar in all compartments (Figure 1C; Supplemental Table 4). Collectively, 22,703 genes were detected as expressed in EMB, and 28,078 and 22,989 genes expressed in at least one captured endosperm and maternal compartment, respectively. The three sources of captured tissues shared 19,009 expressed genes in total (Supplemental Figure 6B). Taken together, our analysis of LCM-derived RNA-Seq data indicates that we have obtained sufficient coverage of the transcriptome of the filial and maternal compartments of the kernel for subsequent analysis of gene networks.
Filial and Maternal Compartments of the Kernel Exhibit Distinct mRNA Populations
To understand the relationships between the mRNA populations isolated from the individual compartments, we performed a principal component analysis (PCA) (Figure 2A; Supplemental Table 5) and a hierarchical clustering of data from the SCC analysis (Figure 2B) of normalized expression levels for the 30,665 genes expressed in at least one compartment. Those compartments with highest overlap in mRNA populations are expected to be more closely associated in such analyses and are likely to share functions. Both analyses showed high correlation among endosperm cell types and a distinct clustering of these cell types in comparison to the maternal compartments (Figures 2A and 2B). As expected, the filial EMB showed closer correlation with endosperm cell types as compared with the maternal compartments. However, the endosperm AL showed a closer correlation with EMB (ρ = 0.80) than with any other endosperm cell type (Figure 2B). An analysis of our data for expression of two AL marker genes, namely, VPP1 (VACUOLAR H+-TRANSLOCATING INORGANIC PYROPHOSPHATASE1) (Wisniewski and Rogowsky, 2004) and AL-9 (Gómez et al., 2009), indicated that the captured EMB RNA sample was not contaminated by the AL RNAs (Supplemental Data Set 2). Therefore, our observation suggests that the AL is distinct in some zygotic functions typically not found in the other endosperm cell types. Together, our data indicate that maternal and filial gene expression programs are divergent as they arise from distinct genetic origins and that the captured compartments show sufficient diversity at the mRNA level to allow identification of unique gene sets for each compartment.
Figure 2.

Relationship between the RNA Populations Obtained from the Filial and Maternal Compartments of 8-DAP Kernel.
(A) PCA of genes expressed in the captured kernel compartments. Principal components one through three (PC1 to PC3) collectively explained 61.9% of the variance in the mRNAs obtained from the 10 compartments.
(B) SCC analysis of the mRNA data for the 10 kernel compartments using log2-transformed FPKM values of the 30,665 expressed genes. The hierarchical clustering dendrogram was inferred by applying (1 − SCC) as distance function.
Identification of Gene Sets Specifically Expressed in Each of the 8-DAP Kernel Compartments
To discover the gene expression programs that characterize each kernel compartment, we identified mRNAs that specifically accumulate in each compartment at 8 DAP by applying a compartment specificity (CS) scoring algorithm (see Methods) to the genes with FPKM ≥ 2 in at least one compartment. In this analysis, we defined the corresponding genes with CS score > 0.3 as being expressed in a compartment-specific pattern. Using this cutoff, 13,009 compartment-specific genes were identified in total for all captured compartments (Supplemental Data Set 3; Figure 3). In contrast to the similarity of the overall mRNA profiles detected among the compartments as described above (Figure 1C), the numbers of detected compartment-specific genes showed dramatic differences among the 10 compartments (Figure 3B). The endosperm cell types showed the lowest number of compartment-specific genes, ranging from a low of 331 in the CSE (1.6% of all CSE-expressed genes) to a high of 912 in the BETL (4.4% of all BETL-expressed genes) compared with the maternal compartments that ranged from 1390 in the PC (8.6% of all PC-expressed genes) to 2432 in the PE (13.2% of all PE-expressed genes) (Figure 3B). For the EMB, 2235 genes were identified as compartment specific, which corresponds to 9.8% of all EMB-expressed genes (Figure 3B). The proportion of TF genes among the compartment-specific genes did not track uniformly across all compartments, varying from a low of 3.9% (CZ) to a high of 9.9% (EMB) (Figure 3B). The variable number and proportion of compartment-specific genes and the associated variation in the proportion of TF genes suggest that most of the endosperm cell types captured express less complex gene sets compared with the maternal compartments or the embryo. Alternatively, the complexity of expression may simply reflect the complexity of the captured compartments, as the captured EMB and maternal compartments likely contain more than one cell type.
Figure 3.

Compartment-Specific Gene Sets Identified Using the CS Scoring Method.
(A) Heat map of scaled FPKM values of the 13,009 compartment-specific genes identified in all 10 kernel compartments.
(B) Numbers of TF and non-TF genes in each compartment-specific gene set.
We used three sets of expression localization data to validate the cell type-specific patterns of mRNA accumulation in the endosperm. First, we performed a series of mRNA in situ hybridizations for genes that were shown to be highly specific to a single compartment based on CS scores ranging from 0.74 to 0.99, including 20 genes expressed in the CSE (2), ESR (1), AL (3), and BETL (14) (Supplemental Figure 7). Second, we previously performed in situ hybridization for 10 genes specifically expressed in CSE (3), CZ (1), ESR (3), AL (1), and BETL (2) (Li et al., 2014). These genes showed CS scores in the given compartments ranging from 0.47 to 0.99. Third, we summarized previously reported in situ hybridization or promoter activity data for cell-specific genes from the literature (Hueros et al., 1995, 1999; Magnard et al., 2000, 2003; Serna et al., 2001; Woo et al., 2001; Gómez et al., 2002, 2009; Gutiérrez-Marcos et al., 2004; Wisniewski and Rogowsky, 2004; Balandín et al., 2005; Massonneau et al., 2005; Muñiz et al., 2006, 2010; Royo et al., 2014) and also found these genes to have a relatively high range of CS scores (0.51 to 0.99). Altogether, these comprise 44 genes showing cell-specific expression in the endosperm (Supplemental Table 6). All of these genes showed highly specific mRNA localization patterns within the cell types with high CS scores, indicating that our RNA-Seq data accurately reflect the accumulation of endogenous mRNAs in the endosperm.
Identification of Gene Coexpression Modules of 8-DAP Maize Kernel
To begin to understand the nature of the GRNs in each of the captured compartments or cell types of the 8-DAP kernel, we identified coexpressed gene sets by applying weighted gene coexpression network analysis (WGCNA) (Zhang and Horvath, 2005; Langfelder and Horvath, 2008) to the expressed genes after excluding the ones with low FPKM (average FPKM < 1) and/or low coefficient of variation (<1) across all 10 compartments. The 9361 genes that fulfilled these stringent criteria fell into 18 coexpression modules (M1 to M18), containing from 83 (M1) to 1209 (M4) genes, including 3 (M1) to 111 (M8) coexpressed TF genes (Figure 4B; Supplemental Data Set 4). Trend-plot analysis of Z-scores of genes in each module showed that these gene sets were expressed in a highly coordinated manner (Supplemental Figure 8). Significantly, a permutation test showed that the average topological overlap of the 18 observed modules was greater than randomly sampled modules of the same size (P value < 10−5; Supplemental Table 7), suggesting that the assignment of gene sets to each of the modules was highly robust. Association of each coexpression module with each compartment or cell type was quantified by Pearson correlation coefficient (PCC) analysis and visualized using a hierarchically clustered heat map (Figure 4A). Interestingly, the 18 coexpression modules fell into two distinct categories showing a relatively high correlation (r ≥ 0.35) with either the filial (i.e., M1, M2, M8, M9, M10, M12, M15, M17, and M18) or the maternal (i.e., M3-M6, M11, M13, M14, and M16) compartments, except for only one module (M7) that was highly correlated with both EMB (r = 0.68) and PE (r = 0.64). Furthermore, 10 of the 18 modules, including M3, M4, M6, M8, M10, M11, M12, M15, M17, and M18, specifically correlated with individual compartments (r ≥ 0.85 for one compartment and r < 0.35 for other compartments), indicating that the expression of genes in these modules are highly compartment or cell type specific. The other eight modules showed relatively high correlation to at least two compartments (r ≥ 0.35). Consistent with the high correlation of mRNAs detected between EMB and AL described above (Figure 2B), three modules including M1, M2, and M9 showed high correlation with both EMB and AL (r ≥ 0.35).
Figure 4.

Gene Coexpression Modules Detected Using WGCNA.
(A) Heat map of the correlations between detected modules (M1 to M18) and kernel compartments hierarchically clustered based on Euclidean distance. The PCC values are quantitative indicators of relative expression levels of all genes in each module.
(B) Numbers of TF and non-TF genes in each coexpression module.
(C) Relationships of the compartment-specific gene sets with the corresponding compartment-correlated coexpression modules obtained using WGCNA. The heat map indicates P values of hypergeometric tests of overrepresentation of genes in a given tested pair of gene sets. Compartment-specific gene sets are noted on the x axis and the corresponding WGCNA coexpression modules (those with a specific pattern for a single compartment) on the y axis. Boxes contain the numbers of overlapping genes and proportions (in parentheses) of these genes in the WGCNA-identified modules.
(D) Relationships of the temporal gene sets (Li et al., 2014) with the coexpression modules obtained using WGCNA. The heat map indicates P values (-log10) of hypergeometric tests of overrepresentation of genes in a given tested pair of gene sets. Temporal gene sets are noted on the x axis and all WGCNA coexpression modules on the y axis. Boxes contain the numbers of overlapping genes. Numbers of genes in each temporal gene set: up@2DAP, 54; up@3DAP, 68; up@4DAP, 92; up@6DAP, 523; up@8DAP, 1,402; up@10DAP, 552; and up@12DAP, 241.
(E) Relationships of the imprinted gene sets (Xin et al., 2013) with the coexpression modules obtained using WGCNA. The heat map indicates P values (-log10) of hypergeometric tests of overrepresentation of genes in a given tested pair of gene sets. Imprinted gene sets are noted on the x axis and all WGCNA coexpression modules on the y axis. Boxes contain the numbers of overlapping genes. Numbers of genes in each imprinted gene set: 7-DAP MEG, 37; 10-DAP MEG, 185; 15-DAP MEG, 15; 7-DAP PEG, 80; 10-DAP PEG, 50; and 15-DAP PEG, 48.
To confirm that the assignment of genes to these coexpression modules using WGCNA reflected valid compartment-based patterns of mRNA accumulation, we examined the number of overlapping genes between these modules and the compartment-specific gene sets identified above (Supplemental Data Set 3). This analysis showed that from ∼52 to 94% of genes in the compartment-correlated coexpression modules were in fact detected within the corresponding compartment-specific gene sets and that the overlap between each compartment-correlated module and the corresponding compartment-specific gene set was greater than expected by chance (hypergeometric test, P value < 10−5) (Figure 4C). These data indicate that a large proportion of coexpressed genes in each module detected through WGCNA is related to compartment- or cell type-specific functions. Taken together, these results indicate that each of the filial and maternal compartments of the maize kernel is associated with one or more coexpression modules that reflect the gene regulatory processes specific to each compartment and are indicators of the differentiation programs functioning within each compartment.
The Endosperm-Associated Coexpression Modules Are Associated with Distinct Temporal Programs of Expression
We previously described a set of temporal programs of gene activity during early kernel and endosperm development in maize and suggested that some of these programs correlated with cell differentiation (Li et al., 2014). The identified temporal programs included gene sets exhibiting temporal upregulation at (“up@”) specific stages (e.g., up@6DAP refers to a gene set exhibiting low expression at 0 to 4 DAP and high expression at 6 to 12 DAP). Comparison of the spatial coexpression modules described here with the temporally upregulated gene sets showed that all of the five endosperm compartment-correlated modules significantly overlapped with the up@6DAP gene set (P value < 10−5), while four of them significantly overlapped with the up@8DAP gene set (Figure 4D). Consistent with this, an analysis of the overall expression levels for each coexpression module using the available developmental RNA-Seq data generated by us and others (Chen et al., 2014; Li et al., 2014) from whole kernel and whole endosperm material showed that nearly all endosperm-correlated modules (M10, M12, M15, M17, and M18) showed upregulation at 6 to 8 DAP (Supplemental Figures 9 and 10). However, the extent of the upregulated patterns varied among the endosperm-correlated modules with the expression of genes in the AL-, ESR-, and BETL-correlated modules (M12, M15, and M18, respectively), showing a more rapid decline by 10 DAP, whereas the expression of CSE- and CZ-correlated modules (M10 and M17, respectively) exhibited a more gradual decline beyond 22 DAP (Supplemental Figures 9 and 10). Interestingly, genes in modules M1, M2, and M9 with high correlations with both EMB and AL also showed a similar temporal expression pattern as those of the endosperm-correlated modules (Supplemental Figure 11). Together, these data indicate that the coexpression modules associated with the major endosperm compartments at 8 DAP are regulated in a highly coordinated manner in both space and time.
The Endosperm-Associated Modules Are Enriched for Endosperm-Imprinted Genes
Gene imprinting has been suggested to be involved in the regulation of nutrient allocation from the maternal tissues to endosperm (Haig and Westoby, 1989; Moore and Haig, 1991; Costa et al., 2012). To investigate the relationship between the spatial programs of gene expression and gene imprinting, we examined the overlap between the WGCNA-generated coexpression modules with the imprinted genes that we previously identified in developing endosperm (Xin et al., 2013). This analysis showed that a subset of genes in the CZ- and BETL-correlated coexpression modules (M17 and M18, respectively) significantly overlapped with a subset of the previously described paternally expressed gene sets (PEGs); the CSE-correlated module M10 and the modules associated with the maternal compartments (M3, M5, M6, M11, M13, M14, and M16) showed significant overlap with the maternally expressed gene sets (MEGs); and a subset of the ESR-correlated coexpression module exhibited significant overlap with both the previously described PEGs and MEGs (hypergeometric P value < 10−5; Figure 4E). In support of these data, a similar pattern of overlaps was observed when we applied less stringent criteria to identify genes with allele-biased expression patterns using the same set of normalized RNA-Seq data (Supplemental Figure 12). Interestingly, many of the imprinted/allele-biased genes assigned by WGCNA to the endosperm-associated coexpression modules were TF genes from multiple families (Supplemental Table 8). These results indicate an extensive interplay between epigenetic programs that regulate allelic expression and the transcriptional regulatory programs involved in the cellular differentiation and function of endosperm.
Biological Processes Enriched in Coexpression Modules of the Filial Compartments of the Kernel
The identified coexpression modules (Figure 4A) are likely associated with specific biological processes or pathways involved in the development or function of each compartment. To identify the major biological processes associated with the filial coexpression modules, we used Blast2GO (Conesa et al., 2005; Conesa and Götz, 2008; Götz et al., 2008) to identify the processes that were significantly enriched (false discovery rate < 0.05) in the modules that showed high correlation with endosperm compartments and/or the EMB. These included modules M1, M2, M8, M9, M10, M17, M12, M15, and M18. As expected, the CSE-correlated M10 was shown to be enriched for “starch biosynthetic process” and “glycogen biosynthetic process” (Supplemental Figure 13), with the former Gene Ontology (GO) category including the Shrunken-2 (Sh2), Brittle-2 (Bt2), STARCH-BRANCHING ENZYME1 (SBE1), and Waxy1 genes, which all have well characterized functions in starch biosynthesis (Shure et al., 1983; Giroux et al., 1994; Blauth et al., 2002). Additionally, close inspection of genes in M10 revealed that this module also contained other starch synthesis-related genes without any current GO annotation, including the Sh1, Sugary1 (Su1), and STARCH SYNTHASE1 (SS1) genes (Chourey and Nelson, 1979; James et al., 1995; Commuri and Keeling, 2001).
In the case of zein-related genes, the mRNAs for only four genes encoding the 15-kD β-zein, the 16-kD γ-zein, the 27-kD γ-zein, and the 18-kD δ-zein were detected in our analysis (FPKM_conf_lo > 0) in at least one cell type (Supplemental Data Set 2). Interestingly, all four zein genes, as well as the Floury-1 gene, which encodes an endoplasmic reticulum membrane protein involved in the targeted localization of an 22-kD α-zein in protein body formation (Holding et al., 2007), were contained within the M10 module (Supplemental Data Set 4). This observation correlates well with previous reports that the formation of zein-containing protein bodies start as small accretions consisting primarily of β- and γ-zeins (Woo et al., 2001), suggesting that the M10 module contains the key early genes necessary for storage protein body biogenesis. The M10 module also included the TF genes Opaque-2 (O2) and PBF (PROLAMIN-BOX BINDING FACTOR), with the relatively high M10 module membership (MM) scores of 0.93 and 0.97, respectively (Supplemental Data Set 4). Furthermore, visualization of M10 using VisANT (Hu et al., 2004) showed that these two TFs are among the most highly connected intramodular hubs of this module (Supplemental Figure 14A). O2 and PBF have previously been shown to regulate storage program gene expression in maize endosperm (Schmidt et al., 1990, 1992; Marzábal et al., 2008). For example, the 15-kD β-zein and the 27-kD γ-zein have been shown to be regulated by O2 and PBF, respectively (Cord Neto et al., 1995; Marzábal et al., 2008). Therefore, the M10 coexpression module likely includes a number of direct gene targets of both TFs.
For the CZ-correlated M17 module, key functional overrepresentations included “glycolysis,” “response to hydrogen peroxide,” “response to cadmium ion,” and “response to heat” (Supplemental Figure 13). This module showed a modest correlation with the CSE (r = 0.27; Figure 4A), suggesting that the CZ may express an overlapping set of genes that function similarly to those detected in the CSE. Conversely, the captured CSE cells are expected to have contained a portion of the CZ cells (Supplemental Figure 1) as the latter likely extend basally and centrally into the captured CSE region (Cooper, 1951; Charlton et al., 1995; Becraft, 2001). This may explain the modest level of correlation of M10 with CZ (r = 0.29; Figure 4A).
The AL-correlated module M12 was enriched for “single-organism process” (Supplemental Figure 13), and the two previously described markers of the aleurone, VPP1 (Wisniewski and Rogowsky, 2004) and AL-9 (Gómez et al., 2009), were assigned within this module with high MM values (0.91 and 0.85, respectively).
Likely due to the greater structural complexity of the EMB compared with the captured endosperm compartments, the EMB-specific module M8 was enriched for more diverse GO categories in comparison to the endosperm-specific modules. The four biological processes that were most significantly enriched for this module included “regulation of transcription, DNA-templated,” “floral organ development,” “phyllome development,” and “response to hormone.” Modules M1, M2, and M9 showed relatively high correlation with both the AL and the EMB (r ≥ 0.35). Among these, M9 also showed a slightly lower correlation with CSE (r = 0.33). Among these modules, M2 contained the largest number (477) of genes. Similar to M8, this module was also enriched for many GO categories, most of which were biological processes related to DNA replication and mitotic cell division (e.g., “DNA replication,” “DNA repair,” “mitotic spindle assembly checkpoint,” and “microtubule-based movement,” etc.). Similarly, M9 (containing 194 genes) was enriched for “cell cycle process,” “chromosome segregation,” “regulation of DNA replication,” and “single-organism organelle organization,” etc. Furthermore, M1, M2, and M9 shared enrichment for “nucleosome assembly” and “DNA duplex unwinding,” which are also biological processes involved in DNA replication and cell division (Supplemental Figure 13). These GO enrichments suggest that the EMB and AL, and to some degree the CSE, are programmed to undergo extensive mitotic cell proliferation at 8 DAP via the coordinated expression of a relatively extensive gene network.
Consistent with the presumptive function of the ESR in mediating endosperm-embryo interaction and expressing antimicrobial products (Sabelli and Larkins, 2009), the ESR-specific module M15 was enriched for “cell-cell signaling involved in cell fate commitment” (Supplemental Figure 13). This module also included many genes isolated previously by virtue of their highly specific ESR expression pattern, including ESR-1, ESR-2, ESR-3, ESR-6, ESR-6B, and AE-3 (Schel et al., 1984; Opsahl-Ferstad et al., 1997; Bonello et al., 2000; Balandín et al., 2005; Sosso et al., 2010). In our analysis, many of the same genes, including ESR-1, ESR-2, ESR-3, and ESR-6, were positioned within the intramodular hubs with high MM (e.g., 0.99) to M15 (Supplemental Figure 14C and Supplemental Data Set 4).
As expected from the BETL’s reported role as mediator of sugar and metabolite uptake into the endosperm through its interaction with the underlying maternal placenta-chalazal region (Sabelli and Larkins, 2009), the BETL-correlated module M18 was found to be enriched for “transmembrane transport,” “ion transport,” and “sucrose transport” functions (Supplemental Figure 13). The M18 module contained nearly all of the previously identified BETL-expressed genes (Hueros et al., 1995, 1999; Cheng et al., 1996; Doan et al., 1996; Serna et al., 2001; Magnard et al., 2003; Gutiérrez-Marcos et al., 2004; Massonneau et al., 2005; Gruis et al., 2006; Muñiz et al., 2006, 2010; Brugière et al., 2008; Gómez et al., 2009), including BETL-1, 3, 4, 9, and 10; BAP-1A, 1B, 2, 3A, and 3B; TCRR-1 and 2; and INCW2 (CELL WALL INVERTASE2), EBE-2 (EMBRYO SAC/BASAL ENDOSPERM-LAYER/EMBRYO-SURROUNDING REGION-2), IPT-2 (ISOPENTENYL TRANSFERASE-2), and CC-8 (CORN CYSTATIN-8) (Supplemental Data Set 4). In addition, this module contained 11 of the 13 MEG genes (with MEG-4 and MEG-14 being the two exceptions) that have been identified in the B73 genome (Supplemental Data Set 4), including MEG-1, which is a BETL-specific gene that has been shown to be important for the development and differentiation of the BETL (Costa et al., 2012). The BETL-, BAP-, and MEG-type genes encode small, secreted, cysteine-rich proteins that have been suggested to protect the embryo from maternally transmitted pathogens (Tailor et al., 1997) and to serve as signaling molecules that coordinate the supply of nutrients to the embryo during kernel development (Marshall et al., 2011), while the TCRR genes encode type-A response regulators (Muñiz et al., 2006, 2010). Significantly, all of these genes showed high MM (>0.90) to module M18, and many of them were among the top-scoring hubs (Supplemental Figure 14E and Supplemental Data Set 4). Furthermore, the BETL-specific TF gene MRP-1, previously shown to be involved in regulation of BETL differentiation (Gómez et al., 2002, 2009), was also detected in M18 with a high MM score (1.00; Supplemental Data Set 4). This is consistent with its role as an activator of many BETL-specific genes, including BETL-1, BETL-9, BETL-10, BAP-2, MEG-1, TCRR-1, and TCRR-2 (Gómez et al., 2002, 2009; Gutiérrez-Marcos et al., 2004; Muñiz et al., 2006, 2010). Therefore, these data suggest that MRP-1 acts as a major regulator of a subset of genes in the M18 coexpression module. Together, our results suggest that the WGCNA-identified coexpression modules can be used as starting points for identification of GRNs functioning in each endosperm compartment.
De Novo Identification of cis-Motifs Associated with the Endosperm Coexpression Modules
As a first step toward identification of the endosperm GRNs, we used MEME software (Bailey and Elkan, 1994) to detect putative cis-regulatory elements in upstream gene sequences from each of the five endosperm coexpression modules identified using WGCNA, including M10, M12, M15, M17, and M18 (Figure 4A). We searched for 10- to 12-bp sequence motifs overrepresented within −1 to +0.5 kb (relative to transcription start site) of genes in each module and further identified motifs that were significantly similar (q-value < 0.05) to the known plant cis-motifs available in the JASPAR CORE database using TOMTOM program (Gupta et al., 2007). We found that most of the detected motifs were shared among the majority of the modules as exemplified by the motifs that contained exclusively CG- or AT-rich sequences (Supplemental Table 9) with significant similarity to the reported binding sites of ABI4 (a maize AP2-EREBP protein) and SOC1 (an Arabidopsis thaliana MIKC-type MADS box protein), respectively (Riechmann et al., 1996; Niu et al., 2002). Coincidently, four of the five endosperm modules (M10, M12, M15, and M17) included at least one MIKC-type MADS box gene, while all five modules included at least one AP2-EREBP gene based on the current maize genome annotation (Supplemental Data Set 4), indicating that the MIKC-type MADS box and AP2-EREBP families may play broad regulatory roles in maize endosperm development.
In contrast, a small number of motifs were detected specifically in single modules. In one instance, the Motif 10 that was enriched in the CSE-correlated module M10 showed significant similarity to a cis-motif that has been shown to bind a bZIP TF in snapdragon (Antirrhinum majus) (Supplemental Table 9) (Martínez-García et al., 1998). Accordingly, two bZIP TF genes, bZIP46 and the storage program regulator O2, were detected within M10 (Supplemental Data Set 4). These results suggest that a subset of genes in module M10 may be regulated by the bZIP genes in the same module. In a second case, Motifs 3, 6, and 8 that were detected in upstream sequences of M18 genes each contained a repeated GATA sequence (Figure 5A) similar to a sequence previously shown to be involved in binding and activation of target genes by MRP-1 (Baranowskij et al., 1994; Barrero et al., 2006), whereas no GATA-containing motifs were detected in any of the other endosperm coexpression modules. In support of a transcriptional regulatory role for the motifs, our analysis indicated a biased distribution of Motifs 3, 6, and 8 upstream to the transcription start site (Figure 5B). A close inspection of the submotifs, namely, the variant sequences of each motif, of Motifs 3, 6, and 8 revealed that at least 148 genes in the M18 module contained one or more submotifs within their upstream sequences. Collectively, our data suggest that a large subset of genes in the M18 coexpression module is likely regulated by MRP-1 through binding at GATA-rich sequences.
Figure 5.

De Novo GATA-Rich Sequence Motifs Overrepresented in the Upstream Sequences of the Coexpression Module M18 Genes Identified by the MEME Program.
(A) Motifs 3, 6, and 8 enriched among the M18 genes.
(B) Position frequencies of the same motifs determined as number of motifs per 50-bp bins in the upstream gene regions.
Identification of a Gene Regulatory Module Associated with BETL Cell Differentiation
We focused on the BETL-associated coexpression module M18 to decipher a portion of the BETL GRN. The BETL transports nutrients from the maternal tissue into the endosperm and is important for the proper development of the endosperm and the endosperm’s capacity as a storage organ (Thompson et al., 2001; Costa et al., 2012). Based on the available data (Baranowskij et al., 1994; Gómez et al., 2002, 2009; Gutiérrez-Marcos et al., 2004; Barrero et al., 2006; Muñiz et al., 2006, 2010) including our identification of the coexpression module M18 and its association with GATA-rich sequence motifs (discussed above), we hypothesized that a GRN for BETL differentiation is minimally composed of MRP-1 and a large set of target genes that are activated upon binding of MRP-1 to Motifs 3, 6, and 8. A close examination of all the submotifs of the 10 motifs enriched for M18 showed that each of the motifs is represented by two to 21 submotifs that appeared one to 90 times within the promoters of the M18 genes (Supplemental Table 10).
We tested for binding of MRP-1 to Motifs 3, 6, and 8 using directed yeast one-hybrid (Y1H) assays. We introduced two constructs into yeast cells, one that resulted in expression of MRP-1 and a second that comprised the test sequence fused upstream of the yeast AUR1-C gene. AUR1-C confers resistance to aureobasidin A (AbA) (Heidler and Radding, 1995). In this assay, MRP-1 binding to the test sequence results in AUR1-C activation and, thus, growth of cells on plates containing AbA, whereas absence of MRP-1 binding results in absence of growth on plates containing AbA. Testing for MRP-1 binding to all of the 24 submotifs comprising Motifs 3, 6, and 8 indicated strong binding to four of the submotifs. These submotifs included Motifs 3a/8a, 6a/8b, 8c, and 8f, with Motifs 3a and 6a identical to 8a and 8b, respectively (Table 1; Supplemental Figure 15). Comparison of these submotifs with the previously reported MRP-1 binding sites showed that Motifs 6a/8b and 8c were identical to the reported MRP-1 binding site in the promoters of BETL-1 (Motif IV) and BETL-2, respectively (Barrero et al., 2006), while Motifs 3a/8a and 8f represent newly identified MRP-1 binding sites.
Table 1. Results of Y1H Assays for Binding of MRP-1 to the MEME-Identified Sequence Motifs of M18.
| Submotif | Sequencea | Y1H Resultsb |
|---|---|---|
| 3a/8a | TAGATAGATAGA | + |
| 3b | TAGATATATAGA | – |
| 3c | TAGATAAATAGA | – |
| 6a/8b | TAGATATAGATA | + |
| 6b | TAGATATATATA | – |
| 6c | TAAATATAAATA | – |
| 6d | TAGATATAGAAA | – |
| 6e | TAGATATAAATA | – |
| 6f | TAAATATAAAAA | – |
| 6g | TAAATATATAAA | – |
| 6h | TATATATATATA | – |
| 6i | TAAATATATATA | – |
| 6j | TAGATAAAAAAA | – |
| 6k | TAAATATAGAAA | – |
| 6l | TAGATATATAAA | – |
| 6m | TAGATATAAAAA | – |
| 8c | TAGATAGAGATA | + |
| 8d | TAGAGAGAGAGA | – |
| 8e | TAGATATAGAGA | – |
| 8f | TAGATAGATATA | + |
| 8g | AAGATAGAGAGA | – |
| 8h | TAGATAGAGAGA | – |
Flanking sequence information is provided in Supplemental Table 14.
Scoring: + indicates strong positive; – indicates negative. Images of the Y1H growth assays are shown in Supplemental Figure 15.
We identified 93 genes that contain at least one of the four submotifs that were shown to be bound by MRP-1 in the Y1H assays (Supplemental Table 11). All the 93 genes showed relatively high MM values to M18, ranging from 0.62 to 1.00, with 78 of them higher than 0.95 (Supplemental Table 11). These results suggest that these 93 genes constitute a regulatory module of the MRP-1-regulated GRN. Significantly, 7 of the 14 BETL-expressed genes described previously, including MEG-1, BETL-1, BETL-10, BAP-1A, BAP-2, BAP-3A, and BAP-3B, were present in this regulatory module (Supplemental Table 11). Based on the available functional annotation of maize genes, this regulatory module contained at least 17 cysteine-rich protein-coding genes (including 3 BETL-, 4 BAP-, and 10 MEG-type genes; Supplemental Table 11), among which BETL-1, BETL-2, BETL-10, and MEG-1 have previously been shown to be regulated by MRP-1. Notably, six TF genes were detected within this regulatory module, including three MYBR-, two C2C2-GATA, one AP2-EREBP, and one DBB family genes, suggesting strongly that MRP-1 indirectly regulates a subset of the BETL-expressed genes by regulating these TFs. In addition, this regulatory module also included a gene (GRMZM2G406552) that encodes a putative nonspecific lipid transfer protein (Supplemental Table 11). Furthermore, comparison of the genes in the regulatory module to the GenBank nonredundant (nr) and Swissprot protein databases (BLASTX, E-value < 10−6) revealed that a number of genes encoding putative transporters of peptide (GRMZM2G156794), calcium (GRMZM5G836886), phosphate (GRMZM2G466545), and magnesium (GRMZM2G054632) were also included in this regulatory module (Supplemental Data Sets 5 and 6).
To validate this putative network further, we used Y1H assays to test for binding of MRP-1 to 22 gene promoters from the M18 module containing motifs positive for MRP-1 binding. We also tested 19 gene promoters lacking these motifs. Of the 41 promoters tested, 31 were shown to bind MRP-1 and 10 did not. Significantly, all promoters containing the MRP-1 binding submotifs were shown to bind MRP-1 (Supplemental Figure 16 and Supplemental Table 12). Interestingly, 7 of the 19 promoters lacking the MRP-1 binding submotifs were found to bind MRP-1 in our assays, indicating that MRP-1 binds to additional sequences not identified in the MEME analysis.
These results suggest strongly that the 93 genes containing the MRP-1 binding submotifs, as well as many additional genes (at least seven), are directly regulated by MRP-1 and that this gene set constitutes a regulatory module within the BETL GRN (Figure 6A). If so, these genes should exhibit a similar temporal pattern of expression to that of MRP-1. An analysis of the expression of the 93 putative MRP-1 target genes throughout development using the RNA-Seq data generated and/or processed by Chen et al. (2014) showed that a large subset of these genes (including most of the cysteine-rich protein-coding genes) were primarily expressed in the endosperm peaking at 6 to 8 DAP, mirroring the pattern of MRP-1 expression (Figure 6B). Interestingly, this set included genes that displayed restricted patterns of mRNA accumulation throughout development, with most showing low mRNA prevalence in the vegetative organs, but showing a wide range of mRNA levels in the 6- to 8-DAP endosperm/kernel (Supplemental Figure 17). In comparison to the rest of the M18 genes, the MRP-1 regulatory module genes showed a significantly higher level of expression in the 8-DAP BETL (Figure 6C; P value = 6.4e-15 based on unpaired t test). As the M18 genes showed significant overlap with the genes in the two temporal programs up@6DAP and up@8DAP (Figure 4D), we examined the overlap between the 93 genes containing the MRP-1 binding submotifs and the two temporal clusters. The result showed that 49 of the 93 genes lie within the up@6DAP gene set (Figure 6D; hypergeometric P value = 8.1e-89), while only three genes were found to overlap with the up@8DAP gene set, indicating that a significant portion of the MRP-1 regulatory module is coordinately upregulated by MRP-1 at 6 DAP in BETL. Taken together, these results suggest that a large subset of M18 genes constitutes a portion of the MRP-1-regulated gene network and that these genes are activated by MRP-1 through binding to specific upstream cis-regulatory sequences.
Figure 6.

A Regulatory Module of the MRP-1-Regulated GRN Based on the Analysis of MRP-1-Bound Submotifs.
(A) A network of the 93 M18 genes associated with at least one MRP-1-bound submotifs visualized using Cytoscape (Shannon et al., 2003). Thickness of the arrows indicates the number of motifs associated with each gene.
(B) Expression pattern of the MRP-1 target genes in seed tissues compared with vegetative and reproductive tissues based on the log2-transformed RPKM data from Chen et al. (2014). The fraction of MRP-1 target genes that have available expression data is indicated in parentheses. Blue line indicates the expression of MRP-1 itself. The selected data include vegetative (Veg) tissues shoots (Sh), roots (R), leaf (L), shoot apical meristem (SAM; replicate 1); reproductive (Rep) tissues ear (E), tassel (T; replicate 1), preemergence cob (C), silk (Si), anther (A), ovule (O), and pollen (P); and whole kernels, endosperm, and embryos of different developmental stages (in DAP).
(C) Expression level of genes within the MRP-1-regulated regulatory module in BETL compared with all other genes in M18. Asterisk indicates P value < 10−5 (unpaired t test).
(D) Venn diagram showing overlaps between the genes that are upregulated at 6 DAP (Li et al., 2014) with those of the M18 coexpression module and those with detected MRP-1 binding sites. Asterisk indicates hypergeometric P value < 10−5.
The boxes in (B) and (C) represent the interquantile range, green lines the median, red dots the mean, and whiskers 1.5 times the interquantile range.
DISCUSSION
We used an LCM RNA-Seq profiling approach to comprehensively detect mRNA populations for 10 filial and maternal compartments of an 8-DAP maize kernel, and subsequently identified highly correlated gene expression programs associated with each compartment using WGCNA. The endosperm coexpression modules are expected to reflect the state of cellular differentiation within individual endosperm compartments or cell types. Our data indicate that the timing and extent of these differentiation processes are unique to each compartment as suggested previously (Olsen, 2001, 2004; Sabelli and Larkins, 2009; Becraft and Gutierrez-Marcos, 2012; Leroux et al., 2014; Li et al., 2014). As a test case, we deciphered an MRP-1 regulatory module containing 93 genes that are likely involved in BETL cellular differentiation.
The high-quality RNAs isolated from the laser-captured cells (Supplemental Figures 2 and 3 and Supplemental Table 1) and the resulting highly reproducible RNA-Seq data (Supplemental Figure 4) enabled us to detect mRNAs of 30,666 genes accumulated in at least one captured compartment (Supplemental Data Set 2) and 28,078 genes expressed in at least one endosperm compartment (Supplemental Figure 6B). The latter is similar to the 33,084 genes that we previously detected as expressed in the 8-DAP whole endosperm (Li et al., 2014). The difference is likely due to the use of different cutoff criteria for defining genes as expressed in the two studies and to the fact that we likely did not collect every portion of the 8-DAP endosperm in our LCM analysis.
Application of a CS scoring method to the 30,666 expressed genes identified 13,009 genes that were predominantly expressed in single compartments (Supplemental Data Set 3). These cell-specific patterns were validated with 20 genes using in situ hybridization and by comparisons with the expression patterns of previously reported endosperm-expressed genes (Supplemental Figure 7 and Supplemental Table 6). In all 44 cases tested, the CS patterns closely matched the experimentally observed patterns, indicating that our RNA-Seq data accurately reflect the accumulation of endogenous mRNAs in the endosperm.
As expected, the PCA and SCC analysis showed that each captured compartment exhibited a distinct mRNA population, with the maternal and filial compartments forming two separate groups. Consistent with this, WGCNA identified both coexpression modules that were correlated with multiple compartments (r ≥ 0.35) and modules that were specifically correlated with each of the captured compartments (r ≥ 0.85 for one compartment and r < 0.35 for other compartments), with the filial and maternal compartment-correlated coexpression modules falling into two nearly distinct groups (Figure 4A). Notably, the SCC analysis also showed that the AL was more closely related to the EMB than to any of the other endosperm compartments (ρ = 0.80; Figure 2B). Accordingly, WGCNA identified three coexpression modules (M1, M2, and M9) that showed relatively high correlation (r ≥ 0.35) to both AL and EMB, with M9 also exhibiting a modest correlation to CSE (r = 0.27) (Figure 4A). GO analysis indicated that these modules were enriched for genes involved in mitotic cell proliferation (Supplemental Figure 13). This suggests that partially overlapping sets of cell proliferative programs distinguish the AL and EMB from the rest of the filial compartments of an 8-DAP kernel.
GO enrichment of the coexpression modules correlated with single endosperm compartments (M10, M12, M15, M17, and M18) were generally consistent with the presumptive functions of each compartment as described previously (Olsen, 2001, 2004; Sabelli and Larkins, 2009; Becraft and Gutierrez-Marcos, 2012). This indicates that these coexpression modules can be used to decipher the key regulatory programs associated with cellular differentiation and related functions of each compartment. In support of this notion, these modules were shown to contain many cell type-specific genes that had been described previously and also identified by us using the CS scoring method (Figure 4C; Supplemental Data Set 4). The latter, in effect, constitutes a high-resolution atlas of spatially specific gene expression programs in the differentiating maize endosperm.
We previously performed RNA-Seq with whole kernels or isolated endosperm at eight temporal stages and identified gene sets exhibiting temporal patterns of gene expression (Li et al., 2014). Among these were temporally upregulated genes exhibiting relatively low expression at early stages and relatively high expression at later stages. Analysis of the temporally upregulated genes among the WGCNA coexpression modules detected significant enrichment of the up@6DAP and/or up@8DAP temporal programs in the endosperm compartment-specific modules including the BETL-correlated M18 (Figure 4D). Similarly, a broad survey of the endosperm-correlated modules for expression during development indicated that these modules follow distinct dynamics of expression with an onset of activation at ∼6 DAP, yet the ESR, BETL, and AL (M15, M18, and M12, respectively) show a more rapid downregulation pattern compared with the CZ and CSE (M17 and M10, respectively) (Supplemental Figures 9 to 11). These data suggest that although the endosperm compartments captured for this study already exhibit characteristics of the differentiated state, the associated coexpression modules nonetheless reflect active regulatory processes that may underlie continuous differentiation and specialization of the relevant cell types. However, the fact that the previously identified temporal programs lack spatial resolution within the endosperm limits our ability to accurately correlate the spatial programs with the developmental dynamics of individual cell types at this time. Therefore, further capture and analysis of endosperm compartments from multiple early stages of endosperm will enable a more comprehensive understanding of these regulatory processes.
Analysis of endosperm-imprinted genes (Xin et al., 2013) in the WGCNA-identified coexpression modules (Figure 4E) revealed that the BETL- and CZ-associated modules (M17 and M18, respectively) were enriched for PEGs, the CSE-correlated module M10 was enriched for MEGs, while the ESR-associated module M15 was enriched for both PEGs and MEGs. These observations indicate that the five endosperm compartment-correlated modules identified in this study can be differentiated in part by memberships of imprinted genes that are likely a reflection of compartment’s function. For example, BETL and CZ presumably function in the nutrient transport from the maternal tissue to the inner endosperm cells including the developing SE (Becraft, 2001; Sabelli and Larkins, 2009). The association of these compartments with the expression of PEGs is consistent with the parental conflict model, which predicts opposite roles for MEGs and PEGs in regulating nutrient allocation from the mother to offspring (Haig and Westoby, 1989; Moore and Haig, 1991). On the other hand, the association of CSE with some MEGs and the dual association of ESR with PEGs and MEGs suggest a more complex relationship between gene imprinting and endosperm cell function.
The CSE, as a major subregion within the SE, is responsible for the storage of most starch and storage proteins in the endosperm (Olsen, 2001, 2004; Sabelli and Larkins, 2009). Correspondingly, as revealed by the GO enrichment analysis, the CSE-correlated module M10 included many starch biosynthetic genes (Supplemental Figure 13). In addition, a number of early zein genes, as well as the storage program regulators O2 and PBF, were also detected in this module (Supplemental Data Set 4). Because the expression of a large subset of the storage protein genes, considered to be regulated by O2 and/or PBF, is not fully activated by 8 DAP, only a few known targets of O2 and PBF were detected in our data set. These included the 15-kD β-zein gene and the cyPPDK1 gene (encoding a cytoplasmic pyruvate orthophosphate dikinase) known to be regulated by O2, and the 27-kD γ-zein gene regulated by PBF (Cord Neto et al., 1995; Gallusci et al., 1996; Maddaloni et al., 1996; Marzábal et al., 2008). Therefore, although our data may not allow us to fully decipher the GRNs regulating the storage function of the SE, they provide an insight into the early phase of the storage program activation.
The BETL-correlated module M18 contained numerous previously described BETL-specific genes (Supplemental Data Set 4), including MRP-1 and seven genes regulated by MRP-1 (Gómez et al., 2002, 2009; Gutiérrez-Marcos et al., 2004; Muñiz et al., 2006, 2010). The identification of GATA-containing motifs among a subset of M18 genes (Figure 5) and confirmation of binding of MRP-1 to these sequences using Y1H assays (Supplemental Table 11) allowed us to propose a 93-gene, direct-target regulatory module for MRP-1 (Figure 6). Functional annotation of the 93-gene regulatory module indicates a diverse array of gene functions activated by MRP-1, including putative signaling and nutrient transport (Supplemental Table 11). The larger M18 gene set also exhibits a wide range of putative functions including those expected for a transfer cell layer (Supplemental Figure 13). These gene functions are likely sufficient to support BETL differentiation, as a recent study showed that the ectopic expression of MRP-1 via an aleurone-specific gene promoter was capable of inducing differentiation of an ectopic BETL in the aleurone albeit in a transient manner (Gómez et al., 2009). On the other hand, ectopic activation of these genes may not produce viable cells outside the endosperm context, as attempts with overexpressing MRP-1 in maize using ubiquitously expressing promoters produced no transformants (Gómez et al., 2009). Further understanding of the MRP-1-regulated network and the associated gene functions will likely require characterization of complete or partial loss-of-function mutants.
The gene set activated by MRP-1 is likely significantly larger than the 93-gene set discussed above for two reasons. First, our Y1H assays showed activation of seven genes that lack motifs identified in our MEME analysis (Supplemental Figure 16 and Supplemental Table 12), indicating that MRP-1 binds to additional sequences not identified in our MEME analysis and that M18 contains additional MRP-1-regulated genes. Second, MRP-1 likely directly regulates six TFs, including MYBR24, MYBR33, GATA7, GATA33, and EREB137, and a DBB family TF (Supplemental Table 11). Each of these TFs may activate a gene set within M18. Additional protein-DNA interaction studies, such as electrophoretic mobility shift assays, and transient directed-expression assays would be necessary to further characterize the interaction between MRP-1 and the full spectrum of its direct targets. Thus, the entire regulatory module activated by MRP-1 (i.e., both directly and indirectly) probably encompasses a much larger proportion of the M18 module than what we report here. Studies devoted to identifying the full spectrum of MRP-1 binding sites and to the identification of the target genes activated by the TFs activated by MRP-1 are in progress to fully characterize this module.
Furthermore, it is notable that in addition to MRP-1 itself, M18 contains at least 48 coexpressed TF genes including six MYBR genes (Supplemental Data Set 4). The latter may regulate overlapping gene sets with MRP-1, possibly through binding to the identified GATA-containing motifs. Limitations of the de novo cis-motif detection approaches utilized here and an absence of an extensive cis-motif-TF database for plants have precluded identification of additional TF targets in M18. Therefore, further approaches including directed Y1H assays for specific TF-target interactions in combination with transient expression studies of the TFs and analysis of any available TF gene mutants will be required to determine the full extent of the BETL GRN.
In summary, our data set provides a high-resolution atlas of gene expression in differentiating endosperm compartments and maternal compartments of an early maize kernel. This data set provides insights into the functions of the endosperm cell types and into the coexpressed gene sets that establish the differentiated states and functions of these cell types. Furthermore, as exemplified by our initial analysis of the MRP-1 regulatory module, this data set can be used as a starting point to dissect the modules regulating endosperm cell differentiation. The analyses provided here constitutes a significant step toward the identification of GRNs that regulate maize endosperm cell differentiation and determine its function.
METHODS
Plant Materials and Growth
Plants of the reference maize (Zea mays) genotype, B73, were grown under greenhouse conditions (16-h day) at the University of Arizona during April to July 2012 and self-pollinated to obtain 8-DAP kernels (Li et al., 2014) for LCM. The kernels for morphological analysis shown in Supplemental Figure 1 were collected during October and November 2013. Kernel compartments were delineated for capture using tissue sections obtained from Farmer’s fixed (see below) or from paraformaldehyde-fixed material that was further stained with Toluidine Blue as described previously (Drews, 1998). The tissue sections for each biological replicate of a given compartment were captured from multiple kernels of a single ear (Supplemental Table 1).
LCM, RNA Isolation, and cDNA Synthesis and Amplification
Kernels were harvested from plants mid-day and cut at the pedicel, punctured through the pericarp, vacuum infiltrated with cold Farmer’s fixative (ethanol:glacial acetic acid, 3:1) (Kerk et al., 2003), and stored in cold fixative overnight. Fixed kernels were then dehydrated in a graded ethanol series, cleared in n-butanol, embedded in Paraplast X-tra (McCormick Scientific Leica) using microwave (Takahashi et al., 2010), cut to 10-μm sections, and mounted on PEN-coated slides. Shortly before capture, sections were deparaffinized in Xylenes and air-dried (Takahashi et al., 2010). Individual cell types or kernel compartments were captured directly into an aliquot of Arcturus PicoPure RNA extraction buffer (Applied Biosystems) using a Leica LMD6500 laser microdissection system (Leica Microsystems). RNA was extracted following the manufacturer’s suggested protocol (Arcturus PicoPure kit), checked for quality, DNase treated using TURBO DNase (Life Technologies), and further purified using the Arcturus PicoPure columns (Applied Biosystems). For each of the 22 samples (Supplemental Table 1), 10 ng purified RNA was used for cDNA synthesis and amplification. cDNA synthesis with oligo(dT) and random primers and cDNA amplifications were performed using an Ovation RNA-Seq System V2 kit (Nugen Technologies) following the manufacturer’s protocols with minor modifications. The quality and profile of the RNA and amplified cDNA samples were checked on an Agilent 2100 Bioanalyzer (Agilent Technologies) using an Agilent RNA 6000 Pico Kit (Agilent Technologies) and an Agilent High Sensitivity DNA Kit (Agilent Technologies), respectively.
RNA-Seq Library Construction and Sequencing
Construction and sequencing of RNA-Seq libraries were performed at the University of Arizona Genetics Core. Using an Illumina TruSeq DNA sample preparation kit (v2; Illumina), nearly 1 µg amplified cDNA for each of the 22 samples was used to generate multiplexed RNA-Seq libraries (mean size 350 to 380 bp, including 120-bp adapters) by following the manufacturer’s suggested procedures. The samples were sequenced in batches on four flow cell lanes of an Illumina HiSequation 2000 platform using a TruSeq SBS kit (v3) to produce 2 × 100-nucleotide paired-end reads. Two of the four lanes each contained libraries for a single replicate of the captured endosperm compartments (AL, BETL, ESR, CSE, and CZ); the third lane contained libraries for a single replicate of the endosperm compartments plus the library for a single replicate of the maternal compartment PC; and the fourth lane contained libraries for EMB (three replicates) and the other three maternal compartments (NU, PE, and PED, one replicate each). After the raw reads were generated, adapter sequences were trimmed using the Trimmomatic program (Bolger et al., 2014). Quality of the trimmed reads was checked using the FastQC program (Andrews, 2010).
Reads Mapping and Analysis
RNA-Seq reads were aligned to the maize reference genome version 3 (B73 RefGen_v3) (Hubbard et al., 2002) using TopHat v2.0.9 (Trapnell et al., 2009). Intron length was set to 30 to 8000 nucleotides, while maximum number of mismatches per read was set to 3. To eliminate the effect of reads mapping in intergenic and/or repeated genomic regions on the estimation of effective library size that may be caused by the cDNA amplification method, reads mapped to exonic regions were extracted using the intersect function of BEDTools v2.17.0 (Quinlan and Hall, 2010) and were provided as input to Cufflinks v2.1.1 (Trapnell et al., 2010) for normalization and estimation of gene expression level. The multi-mapped reads correction and fragment bias correction options of Cufflinks were used. Gene expression levels were reported as FPKM (Mortazavi et al., 2008). The upper and lower bound FPKM values (FPKM_conf_hi and FPKM_conf_lo, respectively) for the 95% confidence interval of each gene were also provided by Cufflinks. A gene was defined as expressed in a sample if the FPKM_conf_lo was greater than zero. Information regarding maize genome annotation used in these analyses was obtained from Ensembl Plants (plants.ensembl.org, release 19).
SCC analysis (Zar, 1972; Hollander et al., 2013) was used to quantify the reproducibility of data between the triplicates of endosperm compartments and EMB. SCCs was calculated from log2-transformed FPKM values [i.e., log2 (FPKM + 1)] of the expressed genes. Based on the high correlation of gene expression profiles among the replicated samples, exonic reads were merged to create a union of each triplicate, and FPKMs were recalculated for the six merged samples. With the resulting FPKM data (log2-transformed) of the expressed genes (FPKM_conf_lo > 0 after renormalization), PCA and SCC analyses were used to compare gene expression profiles among all 10 compartments. The prcomp and cor.test functions in R were used for PCA and SCC analysis, respectively.
Identification of Compartment-Specific Gene Sets
The genes specifically expressed in each kernel compartment were identified using a CS scoring algorithm that compares the expression level of a gene in a given compartment with its maximal expression level in the other nine compartments (Ma and Wang, 2012; Ma et al., 2014). For a given gene i, its expression values in 10 compartments are denoted as 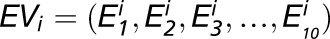 , and the CS score of this gene in compartment j is defined as:
, and the CS score of this gene in compartment j is defined as: 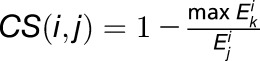 , where 1 ≤ k ≤10, k ≠ j. Thus, CS scores range from 0 to 1, and the higher the CS score of a gene for a compartment, the more likely the gene is specifically expressed in that compartment.
, where 1 ≤ k ≤10, k ≠ j. Thus, CS scores range from 0 to 1, and the higher the CS score of a gene for a compartment, the more likely the gene is specifically expressed in that compartment.
In Situ Hybridization Localization of mRNAs
Staged kernels were obtained from B73 plants grown at the University of Utah. Kernels were harvested, fixed, processed, and hybridized to probes as previously described (Li et al., 2014). Primers used to generate the probe clones are listed in Supplemental Table 13.
Identification of Coexpression Modules
The R package WGCNA (Zhang and Horvath, 2005; Langfelder and Horvath, 2008) was used to identify modules of highly correlated genes based on the FPKM data. Genes with low FPKM (mean FPKM < 1 for 10 compartments) or low coefficient of variation of FPKM (CV < 1 among 10 compartments) were filtered out. Using the FPKM values of the remaining 9361 genes, a matrix of pairwise SCCs between all pairs of genes was created and transformed into a matrix of connection strengths (an adjacency matrix) by raising the correlation matrix to the power 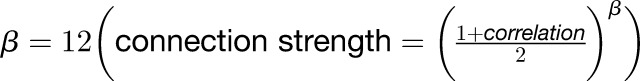 . The power β was interpreted as a soft threshold of the correlation matrix. The resulting adjacency matrix was then converted to a topological overlap (TO) matrix by the TOMsimilarity algorithm. Genes were hierarchically clustered based on TO similarity. The Dynamic Tree Cut algorithm was used to cut the hierarchal clustering tree, and modules were defined as branches from the tree cutting. Modules with fewer than 30 genes were merged into their closest larger neighbor module. Each module was summarized by the first principal component of the scaled module expression profiles (referred to as module eigengene [ME]). MM (also known as module eigengene-based connectivity kME) of a gene to a given module was calculated as PCC between the expression levels (FPKMs, in 10 compartments) of the gene and the ME of the module using the signedKME algorithm. Finally, genes were reassigned using the moduleMergeUsingKME algorithm to ensure each gene possesses the highest MM in its own assigned module. Module-compartment associations were quantified by PCC analysis where each module was represented by its ME, and each compartment was represented with a numeric vector with “1” for the compartment of interest, and “0” for all other compartments.
. The power β was interpreted as a soft threshold of the correlation matrix. The resulting adjacency matrix was then converted to a topological overlap (TO) matrix by the TOMsimilarity algorithm. Genes were hierarchically clustered based on TO similarity. The Dynamic Tree Cut algorithm was used to cut the hierarchal clustering tree, and modules were defined as branches from the tree cutting. Modules with fewer than 30 genes were merged into their closest larger neighbor module. Each module was summarized by the first principal component of the scaled module expression profiles (referred to as module eigengene [ME]). MM (also known as module eigengene-based connectivity kME) of a gene to a given module was calculated as PCC between the expression levels (FPKMs, in 10 compartments) of the gene and the ME of the module using the signedKME algorithm. Finally, genes were reassigned using the moduleMergeUsingKME algorithm to ensure each gene possesses the highest MM in its own assigned module. Module-compartment associations were quantified by PCC analysis where each module was represented by its ME, and each compartment was represented with a numeric vector with “1” for the compartment of interest, and “0” for all other compartments.
Computational Validation of Module Robustness
Average TO for each identified coexpression module was calculated and compared with the average TO of modules of the same size generated by randomly assigning the 9361 tested genes to 18 modules; 100,000 permutations of randomly sampled modules were tested. The observed modules were considered robust if the average TOs were significantly higher than the randomly generated modules (P value < 10−5).
Visualization of Hub Genes
Genes with highest degree of connectivity within a module are referred to as intramodular hub genes (Langfelder and Horvath, 2008). The top 200 connections (based on topological overlap) among the top 100 genes in each module ranked by kME was visualized by VisANT (Hu et al., 2004).
Gene Annotation and Functional Enrichment Analysis
Locus names and functional annotation of maize genes were obtained from Ensembl Plants. The recently annotated MEG family members (Xiong et al., 2014) were also incorporated. Annotation of TF family members were based on information from Plant Transcription Factor Database v3.0 (Jin et al., 2014) and GrassTFDB of GRASSIUS (Gray et al., 2009; Yilmaz et al., 2009). Annotation of zein genes in the B73 genome was based on the information as summarized by Chen et al. (2014). Putative functions of genes of interest were identified and inspected manually. cDNA sequences of the longest isoform of each gene were obtained from Ensembl Plants and used for homology searches against the NCBI nr and Swissprot protein databases, respectively, using the BLASTX program. Only the top five hits for each gene (E-value < 10−6) were considered in our analysis of putative gene functions.
GO term enrichment analyses of the WGCNA-identified coexpression modules were performed using a modified Fisher's exact test in Blast2GO software (false discovery rate < 0.05). GO annotations for maize genes were obtained from Gramene (gramene.org, release 40). For each module, only protein-coding genes (i.e., transposable elements, microRNA genes, and pseudogenes excluded) were subject to GO enrichment analysis, with the most specific biological processes reported.
Identification of cis-Motifs
MEME program was used to identify de novo motifs in the promoter regions of genes in each coexpression module. We defined the promoter regions as 1 kb upstream and 500 bp downstream of transcription start sites and obtained the genomic sequences using a customized Perl script. For each module, 10 motifs (Motifs 1 through 10) were reported by MEME, and the ones with E-value higher than 10−6 were excluded manually. Using the TOMTOM motif comparison tool, the resulting motifs were aligned with motifs in the JASPAR CORE Plantae database (Mathelier et al., 2014) to identify significantly similar known cis-motifs (q-value < 0.05). A customized Perl script was used to identify the genes that contain at least one of the four submotifs shown to be bound by MRP-1 in the Y1H assays.
Y1H Assays
The Matchmaker Gold kit from Clontech for Y1H assays was used to validate MRP-1-target sequence interactions following the manufacturer’s procedures with modifications, including the use of the yeast CUP1 promoter (Etcheverry, 1990) in place of the yeast ADH promoter to drive MRP-1 expression in yeast cells. The plasmid containing the MRP-1 coding region driven by the CUP1 promoter was generated as follows. The CUP1 promoter was provided in plasmid pMB465 by Marcus Babst. A NotI/SacI fragment containing the CUP1 promoter fragment was subcloned into yeast vector pRS425 to make pCUP425. The MRP-1 open reading frame was generated by PCR using primers MRP1-FEco (5′-GGCCGAATTCAATCCCAACTTCAACAGTGTG-3′) and MRP1-RBam (5′-GGCCGGATCCTCGGTTATATATCTGGCTCTCC-3′). The resulting PCR product was cloned into pGADT7 (Clontech) using the EcoRI and BamHI restriction sites introduced during PCR. The resulting plasmid was called MRP1/pGADT7. Using primers NoAD-FNot (5′-GATCGCGGCCGCATGGAGTACCCATACGACG-3′) and PGAD-R2Sal (5′-GATCGTCGACGGAATATGTTCATAGGGTAG-3′), fragments containing an HA tag, the MRP-1 open reading frame, and the ADH1 terminator were amplified. The resulting PCR product was cloned into pCUP425 through the use of the NotI and SalI restriction sites introduced during PCR. The final plasmid was called MRP1/pCUP425. The identical plasmid lacking the MRP-1 coding region was called pCUP425.
The motif:AUR1-C constructs included 147 bp of sequence upstream of the translational start codon of TCRR-1. The motif-promoter fragments were generated by PCR amplification using the primers listed in Supplemental Table 14. The promoter:AUR1-C constructs included 257 to 1187 bp of sequence upstream of the translational start codon. The promoter fragments were generated by PCR amplification using the primers listed in Supplemental Table 15. The resulting PCR products were cloned into pAbAi (Clontech) through the use of unique 5′ and 3′ restriction sites introduced during PCR (Supplemental Tables 14 and 15). The resulting plasmids then were then integrated into the yeast genome of strain Y1HGold following the manufacturer’s recommended protocol (Clontech).
We introduced the MRP1/pCUP425 plasmid into each of the promoter:AUR1-C and motif:AUR1-C yeast strains; these were called +MRP-1 strains in the Y1H figures. To generate control strains, we also introduced pCUP425 into each of the promoter:AUR1-C yeast strains and motif:AUR1-C; these were called -MRP-1 (or empty vector) strains in the Y1H figures. Yeast transformations were performed using the lithium acetate procedure.
Yeast growth assays were performed as follows. Equal numbers of cells from single colonies were spotted in a 1:5 dilution series (∼625 cells/spot, ∼125 cells/spot, ∼25 cells/spot, and ∼5 cells/spot) onto plates. The plate media was deficient in leucine and contained 750 to 2000 ng/mL AbA (variation in the concentration of AbA used was due to manufacturer batch variability) and 0 or 0.1 mM CuSO4. As a growth control, each cell suspension was also spotted onto plates lacking AbA. This procedure was followed for both the +MRP1 and −MRP strains. The plates containing the spotted yeast were incubated at 30°C for ∼48 h and then images of the yeast cells were captured. This procedure was performed with two colonies per strain for each experiment, and each experiment was performed twice on separate days with independently transformed strains and independently prepared plates (i.e., four independent colonies tested per strain). In all cases, all four colonies exhibited very similar growth patterns.
Yeast growth was scored as follows. Cell growth fell into three general categories. In the first category, the +MRP-1 and −MRP-1 strains both grew well on plates lacking AbA but failed to grow on plates containing AbA. This growth pattern indicated no binding of MRP-1 to the test sequence and these strains were scored as negative. In the second category, the −MRP-1 strains grew well on plates lacking AbA but failed to grow on plates containing AbA, and the +MRP-1 strains grew equally well (or nearly so) on plates lacking and containing AbA. This growth pattern indicated strong binding of MRP-1 to the test sequence and these strains were scored as positive. In the third category, growth of the +MRP-1 strains was reduced on plates containing AbA relative to plates lacking AbA but this growth was significantly greater than that of the –MRP-1 strains on plates containing AbA. This growth pattern suggested weak binding of MRP-1 to the test sequence and these strains were scored as weak positives.
Accession Numbers
Sequence data for the genes cited in this article can be found in the Gramene database or GenBank/EMBL libraries under the following accession numbers: 15-kD β-zein, GRMZM2G086294; 16-kD γ-zein, GRMZM2G060429; 18-kD δ-zein, GRMZM2G100018; 27-kD γ-zein, GRMZM2G138727; AL-9, GRMZM2G091054; BAP-1A, GRMZM2G008271; BAP-1B, GRMZM2G008403; BAP-2, GRMZM2G152655; BAP-3A, GRMZM2G133382; BAP-3B, GRMZM2G133370; BETL-1, GRMZM2G082785; BETL-3, GRMZM2G175976; BETL-4, GRMZM2G073290; BETL-9, GRMZM2G087413; BETL-10, GRMZM2G091445; Bt2, GRMZM2G068506; cyPPDK1, GRMZM2G306345; EBE-2, GRMZM2G167733; ESR-1, GRMZM2G046086; ESR-2, GRMZM2G315601; ESR-3, GRMZM2G140302; ESR-6, GRMZM2G048353; ESR-6B, GRMZM2G437040; Floury-1, GRMZM2G094532; INCW2, GRMZM2G119689; IPT-2, GRMZM2G084462; MEG-1, GRMZM2G354335; MEG-2, GRMZM2G502035; MEG-3, GRMZM2G344323; MEG-4, GRMZM2G137959; MEG-6, GRMZM2G094054; MEG-7, GRMZM2G116212; MEG-8, GRMZM2G123153; MEG-9, GRMZM2G088896; MEG-10, GRMZM2G086827; MEG-11, GRMZM2G181051; MEG-12, GRMZM2G175896; MEG-13, GRMZM2G175912; MEG-14, GRMZM2G145466; MRP-1, GRMZM2G111306; O2, GRMZM2G015534; PBF, GRMZM2G146283; Sh1, GRMZM2G089713; Sh2, GRMZM2G429899; SS1, GRMZM2G129451; Su1, GRMZM2G138060; TCRR-1, GRMZM2G016145; TCRR-2, GRMZM2G090264; VPP1, GRMZM2G069095; Waxy1, GRMZM2G024993; bZIP46, GRMZM2G037910; EREB137, GRMZM2G028386; GATA33, GRMZM2G048850; GATA7, GRMZM2G118214; MYBR24, GRMZM2G049695; and MYBR33, GRMZM2G422083. The data reported in this article have been deposited in the Gene Expression Omnibus database (www.ncbi.nlm.nih.gov/geo; accession number GSE62778).
Supplemental Data
Supplemental Figure 1. Structure of an 8-DAP B73 maize kernel.
Supplemental Figure 2. Representative images of kernel compartments that were marked and collected for the LCM RNA-Seq analyses reported in this study.
Supplemental Figure 3. Representative quality assessments of LCM-derived RNAs used in sequencing.
Supplemental Figure 4. Reproducibility of RNA-Seq reads for each triplicate of the filial compartments.
Supplemental Figure 5. Distribution profile of RNA-Seq reads along the length of the gene models.
Supplemental Figure 6. Profiles of sequenced RNAs from the captured kernel compartments.
Supplemental Figure 7. Validation of the compartment-specific expression patterns using in situ hybridization localization of the mRNAs.
Supplemental Figure 8. Z-score plots showing expression profiles of all genes in the WGCNA-generated coexpression modules M1-M18.
Supplemental Figure 9. Expression pattern of genes in the endosperm-associated modules identified by WGCNA based on previously reported RNA-Seq data.
Supplemental Figure 10. Expression pattern of genes in the endosperm-associated modules identified by WGCNA based on RNA-Seq data from Chen et al. (2014).
Supplemental Figure 11. Expression pattern of genes in the WGCNA coexpression modules (M1, M2, and M9) associated with both AL and EMB (in comparison to the other two EMB-associated modules M7 and M8) based on RNA-Seq data from Chen et al. (2014).
Supplemental Figure 12. Relationships of new sets of allele-biased genes with the coexpression modules obtained using WGCNA.
Supplemental Figure 13. Enrichment of biological processes in the filial compartment-correlated modules.
Supplemental Figure 14. Visualization of the five endosperm compartment-correlated coexpression modules using the VisANT program.
Supplemental Figure 15. Yeast one-hybrid assays for binding of MRP-1 to the sequence motifs listed in Table 1.
Supplemental Figure 16. Yeast one-hybrid assays for binding of MRP-1 to the promoters of the genes listed in Supplemental Table 12.
Supplemental Figure 17. Expression pattern of the 93 genes containing MRP-1 binding submotifs based on RNA-Seq data from Chen et al. (2014).
Supplemental Table 1. The origins, quality, and quantities of RNAs isolated using laser-capture microdissection.
Supplemental Table 2. Summary statistics of RNA-Seq reads and mapping.
Supplemental Table 3. Number of genes expressed in each of the ten compartments.
Supplemental Table 4. Number of genes expressed at different FPKM levels in the ten compartments.
Supplemental Table 5. Summary of principal component analysis of the ten compartments.
Supplemental Table 6. Summary of the available in situ hybridization and promoter analysis data for endosperm cell-specific genes.
Supplemental Table 7. Robustness of WGCNA-generated coexpression modules based on a permutation test of average topological overlap.
Supplemental Table 8. Allele-biased genes enriched in the endosperm-associated coexpression modules M10, M15, M17, and M18.
Supplemental Table 9. Motif analysis of the endosperm compartment-correlated modules M10, M12, M15, M17, and M18.
Supplemental Table 10. Occurrences of submotifs of each motif enriched in upstream sequences of M18 genes as identified by MEME.
Supplemental Table 11. Putative functions of the 93 M18 genes that contain at least one MRP-1 binding submotifs (Motifs 3a/8a, 6a/8b, 8c, and 8f).
Supplemental Table 12. Results of Y1H assays for binding of MRP-1 to the promoters of genes within M18.
Supplemental Table 13. Sequences of the primers used to generate clones for the in situ hybridization probes.
Supplemental Table 14. Sequences of the primers used to generate the constructs to test the submotifs in Y1H assays.
Supplemental Table 15. Sequences of the primers used to generate the constructs to test the promoters in Y1H assays.
Supplemental Data Set 1. Normalized expression levels in FPKM for the 29,369 expressed genes expressed in at least one of the 22 sequenced samples.
Supplemental Data Set 2. Normalized expression levels in FPKM for the 30,665 genes expressed in at least one of the ten compartments after merge of replicates.
Supplemental Data Set 3. Compartment specificity scores for the 13,009 compartment-specific genes.
Supplemental Data Set 4. Module assignment and module memberships for the 9361 genes selected for WGCNA.
Supplemental Data Set 5. BLASTX search of the 93 M18 genes that contain at least one MRP-1 binding submotifs against the NCBI nr database (E-value < 10−6).
Supplemental Data Set 6. BLASTX search of the 93 M18 genes that contain at least one MRP-1 binding submotifs against the Swissprot database (E-value < 10−6).
Supplementary Material
Acknowledgments
We thank John Harada, Julie Pelletier, and Bob Goldberg for suggestions and advice on improving the protocols for LCM, RNA isolation, and cDNA amplification. J.Z. was supported by a graduate fellowship from the China Scholarship Council. This work was supported by National Science Foundation Grant IOS-0923880 (to G.N.D. and R.Y.).
AUTHOR CONTRIBUTIONS
D.T., D.W., G.N.D., and R.Y. designed the project. D.T., A.L., N.M.N., A.M.A., W.J.B., and K.O.L. performed research. J.Z., C.M., and X.W. conducted bioinformatics analysis. J.Z., D.T., C.M., X.W., G.N.D., and R.Y. analyzed the data. J.Z., D.T., G.N.D., and R.Y. wrote the article.
Glossary
- DAP
days after pollination
- SE
starchy endosperm
- AL
aleurone
- ESR
embryo-surrounding region
- BETL
basal endosperm transfer layer
- CSE
central starchy endosperm
- CZ
conducting zone
- GRN
gene regulatory network
- LCM
laser-capture microdissection
- RNA-Seq
RNA sequencing
- EMB
embryo
- NU
nucellus
- PC
placento-chalazal region
- PE
pericarp
- PED
pedicel
- FPKM
fragments per kilobase of transcript per million mapped reads
- SCC
Spearman correlation coefficient
- TF
transcription factor
- PCA
principal component analysis
- CS
compartment specificity
- WGCNA
weighted gene coexpression network analysis
- PCC
Pearson correlation coefficient
- PEG
paternally expressed gene
- MEG
maternally expressed gene
- GO
Gene Ontology
- MM
module membership
- Y1H
yeast one-hybrid
- AbA
aureobasidin A
- TO
topological overlap
- ME
module eigengene
Footnotes
Articles can be viewed without a subscription.
References
- Andrews S. (2010). FastQC: A quality control tool for high-throughput sequence data. (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). [Google Scholar]
- Bailey T.L., Elkan C. (1994). Fitting a Mixture Model by Expectation Maximization to Discover Motifs in Bipolymers. (San Diego, CA: University of California; ). [PubMed] [Google Scholar]
- Balandín M., Royo J., Gómez E., Muniz L.M., Molina A., Hueros G. (2005). A protective role for the embryo surrounding region of the maize endosperm, as evidenced by the characterisation of ZmESR-6, a defensin gene specifically expressed in this region. Plant Mol. Biol. 58: 269–282. [DOI] [PubMed] [Google Scholar]
- Baranowskij N., Frohberg C., Prat S., Willmitzer L. (1994). A novel DNA binding protein with homology to Myb oncoproteins containing only one repeat can function as a transcriptional activator. EMBO J. 13: 5383–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrero C., Muñiz L.M., Gómez E., Hueros G., Royo J. (2006). Molecular dissection of the interaction between the transcriptional activator ZmMRP-1 and the promoter of BETL-1. Plant Mol. Biol. 62: 655–668. [DOI] [PubMed] [Google Scholar]
- Becraft P.W. (2001). Cell fate specification in the cereal endosperm. Semin. Cell Dev. Biol. 12: 387–394. [DOI] [PubMed] [Google Scholar]
- Becraft P.W., Gutierrez-Marcos J. (2012). Endosperm development: dynamic processes and cellular innovations underlying sibling altruism. Dev. Biol. 1: 579–593. [DOI] [PubMed] [Google Scholar]
- Berger F., Grini P.E., Schnittger A. (2006). Endosperm: an integrator of seed growth and development. Curr. Opin. Plant Biol. 9: 664–670. [DOI] [PubMed] [Google Scholar]
- Blauth S.L., Kim K.-N., Klucinec J., Shannon J.C., Thompson D., Guiltinan M. (2002). Identification of Mutator insertional mutants of starch-branching enzyme 1 (sbe1) in Zea mays L. Plant Mol. Biol. 48: 287–297. [DOI] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonello J.F., Opsahl-Ferstad H.G., Perez P., Dumas C., Rogowsky P.M. (2000). Esr genes show different levels of expression in the same region of maize endosperm. Gene 246: 219–227. [DOI] [PubMed] [Google Scholar]
- Brink R.A., Cooper D.C. (1947). The endosperm in seed development. Bot. Rev. 13: 479–541. [Google Scholar]
- Brugière N., Humbert S., Rizzo N., Bohn J., Habben J.E. (2008). A member of the maize isopentenyl transferase gene family, Zea mays isopentenyl transferase 2 (ZmIPT2), encodes a cytokinin biosynthetic enzyme expressed during kernel development. Cytokinin biosynthesis in maize. Plant Mol. Biol. 67: 215–229. [DOI] [PubMed] [Google Scholar]
- Charlton W.L., Keen C.L., Merriman C., Lynch P., Greenland A.J., Dickinson H.G. (1995). Endosperm development in Zea mays; implication of gametic imprinting and paternal excess in regulation of transfer layer development. Development 121: 3089–3097. [Google Scholar]
- Chen J., Zeng B., Zhang M., Xie S., Wang G., Hauck A., Lai J. (2014). Dynamic transcriptome landscape of maize embryo and endosperm development. Plant Physiol. 166: 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W.-H., Taliercio E.W., Chourey P.S. (1996). The Miniature1 seed locus of maize encodes a cell wall invertase required for normal development of endosperm and maternal cells in the pedicel. Plant Cell 8: 971–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chourey P.S., Nelson O.E. (1979). Interallelic complementation at the sh locus in maize at the enzyme level. Genetics 91: 317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commuri P.D., Keeling P.L. (2001). Chain-length specificities of maize starch synthase I enzyme: studies of glucan affinity and catalytic properties. Plant J. 25: 475–486. [DOI] [PubMed] [Google Scholar]
- Conesa A., Götz S. (2008). Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics 2008: 619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A., Götz S., García-Gómez J.M., Terol J., Talón M., Robles M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21: 3674–3676. [DOI] [PubMed] [Google Scholar]
- Cooper D.C. (1951). Caryopsis development following matings between diploid and tetraploid strains of Zea mays. Am. J. Bot. 38: 702–708. [Google Scholar]
- Cord Neto G., Yunes J.A., da Silva M.J., Vettore A.L., Arruda P., Leite A. (1995). The involvement of Opaque 2 on beta-prolamin gene regulation in maize and Coix suggests a more general role for this transcriptional activator. Plant Mol. Biol. 27: 1015–1029. [DOI] [PubMed] [Google Scholar]
- Costa L.M., Yuan J., Rouster J., Paul W., Dickinson H., Gutierrez-Marcos J.F. (2012). Maternal control of nutrient allocation in plant seeds by genomic imprinting. Curr. Biol. 22: 160–165. [DOI] [PubMed] [Google Scholar]
- Doan D.N., Linnestad C., Olsen O.-A. (1996). Isolation of molecular markers from the barley endosperm coenocyte and the surrounding nucellus cell layers. Plant Mol. Biol. 31: 877–886. [DOI] [PubMed] [Google Scholar]
- Drews G.N. (1998). In situ hybridization. Methods Mol. Biol. 82: 353–371. [DOI] [PubMed] [Google Scholar]
- Etcheverry T. (1990). Induced expression using yeast copper metallothionein promoter. Methods Enzymol. 185: 319–329. [DOI] [PubMed] [Google Scholar]
- FAO (2012). FAO Statistical Yearbook 2012. (http://www.fao.org/docrep/015/i2490e/i2490e00.htm).
- Faure J.-E. (2001). Double fertilization in flowering plants: discovery, study methods and mechanisms. C. R. Acad. Sci. III 324: 551–558. [DOI] [PubMed] [Google Scholar]
- Gallusci P., Varotto S., Matsuoko M., Maddaloni M., Thompson R.D. (1996). Regulation of cytosolic pyruvate, orthophosphate dikinase expression in developing maize endosperm. Plant Mol. Biol. 31: 45–55. [DOI] [PubMed] [Google Scholar]
- Giroux M.J., Boyer C., Feix G., Hannah L.C. (1994). Coordinated transcriptional regulation of storage product genes in the maize endosperm. Plant Physiol. 106: 713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez E., Royo J., Guo Y., Thompson R., Hueros G. (2002). Establishment of cereal endosperm expression domains: identification and properties of a maize transfer cell-specific transcription factor, ZmMRP-1. Plant Cell 14: 599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez E., Royo J., Muñiz L.M., Sellam O., Paul W., Gerentes D., Barrero C., López M., Perez P., Hueros G. (2009). The maize transcription factor myb-related protein-1 is a key regulator of the differentiation of transfer cells. Plant Cell 21: 2022–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz S., García-Gómez J.M., Terol J., Williams T.D., Nagaraj S.H., Nueda M.J., Robles M., Talón M., Dopazo J., Conesa A. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36: 3420–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J., et al. (2009). A recommendation for naming transcription factor proteins in the grasses. Plant Physiol. 149: 4–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruis D.F., Guo H., Selinger D., Tian Q., Olsen O.-A. (2006). Surface position, not signaling from surrounding maternal tissues, specifies aleurone epidermal cell fate in maize. Plant Physiol. 141: 898–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S., Stamatoyannopoulos J.A., Bailey T.L., Noble W.S. (2007). Quantifying similarity between motifs. Genome Biol. 8: R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez-Marcos J.F., Costa L.M., Biderre-Petit C., Khbaya B., O’Sullivan D.M., Wormald M., Perez P., Dickinson H.G. (2004). maternally expressed gene1 Is a novel maize endosperm transfer cell-specific gene with a maternal parent-of-origin pattern of expression. Plant Cell 16: 1288–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haig D., Westoby M. (1989). Parent-specific gene expression and the triploid endosperm. Am. Nat. 134: 147–155. [Google Scholar]
- Hamamura Y., Nagahara S., Higashiyama T. (2012). Double fertilization on the move. Curr. Opin. Plant Biol. 15: 70–77. [DOI] [PubMed] [Google Scholar]
- Hansey C.N., Vaillancourt B., Sekhon R.S., de Leon N., Kaeppler S.M., Buell C.R. (2012). Maize (Zea mays L.) genome diversity as revealed by RNA-sequencing. PLoS ONE 7: e33071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidler S.A., Radding J.A. (1995). The AUR1 gene in Saccharomyces cerevisiae encodes dominant resistance to the antifungal agent aureobasidin A (LY295337). Antimicrob. Agents Chemother. 39: 2765–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holding D.R., Otegui M.S., Li B., Meeley R.B., Dam T., Hunter B.G., Jung R., Larkins B.A. (2007). The maize floury1 gene encodes a novel endoplasmic reticulum protein involved in zein protein body formation. Plant Cell 19: 2569–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander M., Wolfe D.A.. Chicken E. (2013). Nonparametric Statistical Methods. (New York: John Wiley & Sons; ). [Google Scholar]
- Hu, Z., Mellor, J., DeLisi, C. (2004). Analyzing networks with VisANT. In Current Protocols in Bioinformatics, Andreas D. Baxevanis, ed (New York: John Wiley & Sons), pp. 8.8.1–8.8.24. [DOI] [PubMed] [Google Scholar]
- Hubbard T., et al. (2002). The Ensembl genome database project. Nucleic Acids Res. 30: 38–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hueros G., Royo J., Maitz M., Salamini F., Thompson R.D. (1999). Evidence for factors regulating transfer cell-specific expression in maize endosperm. Plant Mol. Biol. 41: 403–414. [DOI] [PubMed] [Google Scholar]
- Hueros G., Varotto S., Salamini F., Thompson R.D. (1995). Molecular characterization of BET1, a gene expressed in the endosperm transfer cells of maize. Plant Cell 7: 747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James M.G., Robertson D.S., Myers A.M. (1995). Characterization of the maize gene sugary1, a determinant of starch composition in kernels. Plant Cell 7: 417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J., Zhang H., Kong L., Gao G., Luo J. (2014). PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 42: D1182–D1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerk N.M., Ceserani T., Tausta S.L., Sussex I.M., Nelson T.M. (2003). Laser capture microdissection of cells from plant tissues. Plant Physiol. 132: 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafon-Placette C., Köhler C. (2014). Embryo and endosperm, partners in seed development. Curr. Opin. Plant Biol. 17: 64–69. [DOI] [PubMed] [Google Scholar]
- Langfelder P., Horvath S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9: 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroux B.M., Goodyke A.J., Schumacher K.I., Abbott C.P., Clore A.M., Yadegari R., Larkins B.A., Dannenhoffer J.M. (2014). Maize early endosperm growth and development: from fertilization through cell type differentiation. Am. J. Bot. 101: 1259–1274. [DOI] [PubMed] [Google Scholar]
- Li G., et al. (2014). Temporal patterns of gene expression in developing maize endosperm identified through transcriptome sequencing. Proc. Natl. Acad. Sci. USA 111: 7582–7587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes M.A., Larkins B.A. (1993). Endosperm origin, development, and function. Plant Cell 5: 1383–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C., Wang X. (2012). Application of the Gini correlation coefficient to infer regulatory relationships in transcriptome analysis. Plant Physiol. 160: 192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C., Xin M., Feldmann K.A., Wang X. (2014). Machine learning-based differential network analysis: a study of stress-responsive transcriptomes in Arabidopsis. Plant Cell 26: 520–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddaloni M., Donini G., Balconi C., Rizzi E., Gallusci P., Forlani F., Lohmer S., Thompson R., Salamini F., Motto M. (1996). The transcriptional activator Opaque-2 controls the expression of a cytosolic form of pyruvate orthophosphate dikinase-1 in maize endosperms. Mol. Gen. Genet. 250: 647–654. [DOI] [PubMed] [Google Scholar]
- Magnard J.-L., Le Deunff E., Domenech J., Rogowsky P.M., Testillano P.S., Rougier M., Risueño M.C., Vergne P., Dumas C. (2000). Genes normally expressed in the endosperm are expressed at early stages of microspore embryogenesis in maize. Plant Mol. Biol. 44: 559–574. [DOI] [PubMed] [Google Scholar]
- Magnard J.L., Lehouque G., Massonneau A., Frangne N., Heckel T., Gutierrez-Marcos J.F., Perez P., Dumas C., Rogowsky P.M. (2003). ZmEBE genes show a novel, continuous expression pattern in the central cell before fertilization and in specific domains of the resulting endosperm after fertilization. Plant Mol. Biol. 53: 821–836. [DOI] [PubMed] [Google Scholar]
- Marshall E., Costa L.M., Gutierrez-Marcos J. (2011). Cysteine-rich peptides (CRPs) mediate diverse aspects of cell-cell communication in plant reproduction and development. J. Exp. Bot. 62: 1677–1686. [DOI] [PubMed] [Google Scholar]
- Martínez-García J.F., Moyano E., Alcocer M.J., Martin C. (1998). Two bZIP proteins from Antirrhinum flowers preferentially bind a hybrid C-box/G-box motif and help to define a new sub-family of bZIP transcription factors. Plant J. 13: 489–505. [DOI] [PubMed] [Google Scholar]
- Marzábal P., Gas E., Fontanet P., Vicente-Carbajosa J., Torrent M., Ludevid M.D. (2008). The maize Dof protein PBF activates transcription of gamma-zein during maize seed development. Plant Mol. Biol. 67: 441–454. [DOI] [PubMed] [Google Scholar]
- Massonneau A., Condamine P., Wisniewski J.-P., Zivy M., Rogowsky P.M. (2005). Maize cystatins respond to developmental cues, cold stress and drought. Biochim. Biophys. Acta 1729: 186–199. [DOI] [PubMed] [Google Scholar]
- Mathelier A., et al. (2014). JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 42: D142–D147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore T., Haig D. (1991). Genomic imprinting in mammalian development: a parental tug-of-war. Trends Genet. 7: 45–49. [DOI] [PubMed] [Google Scholar]
- Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5: 621–628. [DOI] [PubMed] [Google Scholar]
- Muñiz L.M., Royo J., Gómez E., Baudot G., Paul W., Hueros G. (2010). Atypical response regulators expressed in the maize endosperm transfer cells link canonical two component systems and seed biology. BMC Plant Biol. 10: 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñiz L.M., Royo J., Gómez E., Barrero C., Bergareche D., Hueros G. (2006). The maize transfer cell-specific type-A response regulator ZmTCRR-1 appears to be involved in intercellular signalling. Plant J. 48: 17–27. [DOI] [PubMed] [Google Scholar]
- Niu X., Helentjaris T., Bate N.J. (2002). Maize ABI4 binds coupling element1 in abscisic acid and sugar response genes. Plant Cell 14: 2565–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen O.A. (2001). Endosperm development: cellularization and cell fate specification. Annu. Rev. Plant Physiol. Plant Mol. Biol. 52: 233–267. [DOI] [PubMed] [Google Scholar]
- Olsen O.-A. (2004). Nuclear endosperm development in cereals and Arabidopsis thaliana. Plant Cell 16 (Suppl): S214–S227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen, O.-A., and Becraft, P.W. (2013). Endosperm development. In Seed Genomics, P.W. Becraft, ed (New York: John Wiley & Sons), pp. 43–62.
- Opsahl-Ferstad H.G., Le Deunff E., Dumas C., Rogowsky P.M. (1997). ZmEsr, a novel endosperm-specific gene expressed in a restricted region around the maize embryo. Plant J. 12: 235–246. [DOI] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riechmann J.L., Wang M., Meyerowitz E.M. (1996). DNA-binding properties of Arabidopsis MADS domain homeotic proteins APETALA1, APETALA3, PISTILLATA and AGAMOUS. Nucleic Acids Res. 24: 3134–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royo J., Gómez E., Sellam O., Gerentes D., Paul W., Hueros G. (2014). Two maize END-1 orthologs, BETL9 and BETL9like, are transcribed in a non-overlapping spatial pattern on the outer surface of the developing endosperm. Front. Plant Sci. 5: 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabelli P.A., Larkins B.A. (2009). The development of endosperm in grasses. Plant Physiol. 149: 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schel J., Kieft H., Lammeren A.v. (1984). Interactions between embryo and endosperm during early developmental stages of maize caryopses (Zea mays). Can. J. Bot. 62: 2842–2853. [Google Scholar]
- Schmidt R.J., Burr F.A., Aukerman M.J., Burr B. (1990). Maize regulatory gene opaque-2 encodes a protein with a “leucine-zipper” motif that binds to zein DNA. Proc. Natl. Acad. Sci. USA 87: 46–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt R.J., Ketudat M., Aukerman M.J., Hoschek G. (1992). Opaque-2 is a transcriptional activator that recognizes a specific target site in 22-kD zein genes. Plant Cell 4: 689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekhon R.S., Briskine R., Hirsch C.N., Myers C.L., Springer N.M., Buell C.R., de Leon N., Kaeppler S.M. (2013). Maize gene atlas developed by RNA sequencing and comparative evaluation of transcriptomes based on RNA sequencing and microarrays. PLoS ONE 8: e61005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serna A., Maitz M., O’Connell T., Santandrea G., Thevissen K., Tienens K., Hueros G., Faleri C., Cai G., Lottspeich F., Thompson R.D. (2001). Maize endosperm secretes a novel antifungal protein into adjacent maternal tissue. Plant J. 25: 687–698. [DOI] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13: 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shure M., Wessler S., Fedoroff N. (1983). Molecular identification and isolation of the Waxy locus in maize. Cell 35: 225–233. [DOI] [PubMed] [Google Scholar]
- Sosso D., Wisniewski J.-P., Khaled A.-S., Hueros G., Gerentes D., Paul W., Rogowsky P.M. (2010). The Vpp1, Esr6a, Esr6b and OCL4 promoters are active in distinct domains of maize endosperm. Plant Sci. 179: 86–96. [Google Scholar]
- Sreenivasulu N., Wobus U. (2013). Seed-development programs: a systems biology-based comparison between dicots and monocots. Annu. Rev. Plant Biol. 64: 189–217. [DOI] [PubMed] [Google Scholar]
- Tailor R.H., Acland D.P., Attenborough S., Cammue B.P., Evans I.J., Osborn R.W., Ray J.A., Rees S.B., Broekaert W.F. (1997). A novel family of small cysteine-rich antimicrobial peptides from seed of Impatiens balsamina is derived from a single precursor protein. J. Biol. Chem. 272: 24480–24487. [DOI] [PubMed] [Google Scholar]
- Takahashi H., Kamakura H., Sato Y., Shiono K., Abiko T., Tsutsumi N., Nagamura Y., Nishizawa N.K., Nakazono M. (2010). A method for obtaining high quality RNA from paraffin sections of plant tissues by laser microdissection. J. Plant Res. 123: 807–813. [DOI] [PubMed] [Google Scholar]
- Thompson R.D., Hueros G., Becker H., Maitz M. (2001). Development and functions of seed transfer cells. Plant Sci. 160: 775–783. [DOI] [PubMed] [Google Scholar]
- Trapnell C., Pachter L., Salzberg S.L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7: 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski J.-P., Rogowsky P.M. (2004). Vacuolar H+-translocating inorganic pyrophosphatase (Vpp1) marks partial aleurone cell fate in cereal endosperm development. Plant Mol. Biol. 56: 325–337. [DOI] [PubMed] [Google Scholar]
- Woo Y.M., Hu D.W.N., Larkins B.A., Jung R. (2001). Genomics analysis of genes expressed in maize endosperm identifies novel seed proteins and clarifies patterns of zein gene expression. Plant Cell 13: 2297–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin M., et al. (2013). Dynamic expression of imprinted genes associates with maternally controlled nutrient allocation during maize endosperm development. Plant Cell 25: 3212–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y., Mei W., Kim E.D., Mukherjee K., Hassanein H., Barbazuk W.B., Sung S., Kolaczkowski B., Kang B.H. (2014). Adaptive expansion of the maize maternally expressed gene (MEG) family involves changes in expression patterns and protein secondary structures of its members. BMC Plant Biol. 14: 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz A., Nishiyama M.Y. Jr., Fuentes B.G., Souza G.M., Janies D., Gray J., Grotewold E. (2009). GRASSIUS: a platform for comparative regulatory genomics across the grasses. Plant Physiol. 149: 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zar J.H. (1972). Significance testing of the Spearman rank correlation coefficient. J. Am. Stat. Assoc. 67: 578–580. [Google Scholar]
- Zhang B., Horvath S. (2005). A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 4: e17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.