

Abstract
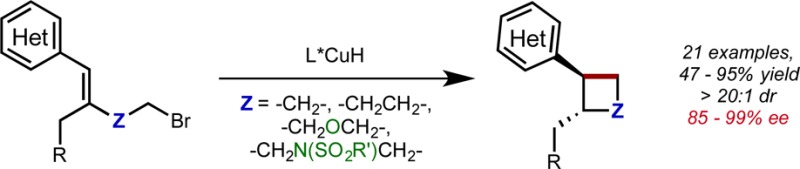
The enantioselective, intramolecular hydroalkylation of halide-tethered styrenes has been achieved through a copper hydride-catalyzed process. This approach allowed for the synthesis of enantioenriched cyclobutanes, cyclopentanes, indanes, and six-membered N- and O-heterocycles. This protocol was applied to the synthesis of the commercial serotonin reuptake inhibitor (−)-paroxetine.
The formation of carbon–carbon bonds has long been recognized as a central process in organic synthesis and remains a fundamental objective in the field. In particular, the construction of C–C bonds between sp3 centers represents a strategically important approach for the introduction of stereochemical information. A diverse array of catalytic and stoichiometric approaches have been developed for the construction of C(sp3)–C(sp3) bonds with high stereoselectivity.1 However, the majority of these methods rely on the presence of nearby reactive functional groups in the target compound. The formation of stereochemically well-defined sp3–sp3 C–C bonds in the absence of such functional groups remains a formidable synthetic challenge.2
The Corey–Posner–Whitesides–House reaction3 between an organocuprate and alkyl halide serves as a prototypical method for the generation of bonds between unfunctionalized saturated carbon atoms. Subsequent work has shown that organocopper species generated under catalytic conditions3c,4 similarly react with alkyl halides to form C–C bonds. Our group’s recent work on copper hydride (CuH)-catalyzed enantioselective alkene hydroamination5 suggested that hydrocupration of an olefin could provide a general approach for the formation of an enantioenriched organocopper species under catalytic conditions (Figure 1). In this context, we posited that an olefin bearing an alkyl (pseudo)halide tether (II, Figure 2) would undergo a formal intramolecular hydroalkylation in the presence of a catalytically generated L*CuH species (I) to furnish enantioenriched, cyclized products.6 Although the analogous borocupration/ring closure sequence has previously been disclosed,4a−4d competititive reduction of alkyl (pseudo)halides by CuH complexes7 renders the proposed hydrocupration/ring closure process nontrivial to execute. Nevertheless, we felt that if suitable conditions could be identified, this strategy would constitute a flexible approach for the synthesis of a variety of 4-, 5-, and 6-membered rings, which are featured prominently in biologically active natural products and pharmaceuticals (Figure 2).8 Herein, we report the implementation of this strategy for the enantioselective synthesis of several classes of compounds, including substituted cyclobutanes, cyclopentanes, indanes, and saturated heterocycles.
Figure 1.
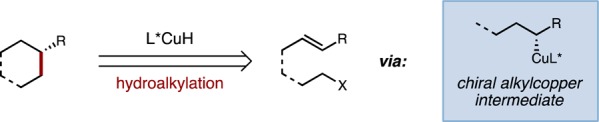
Hydroalkylation as a strategy for the construction of unfunctionalized C(sp3)–C(sp3) bonds.
Figure 2.
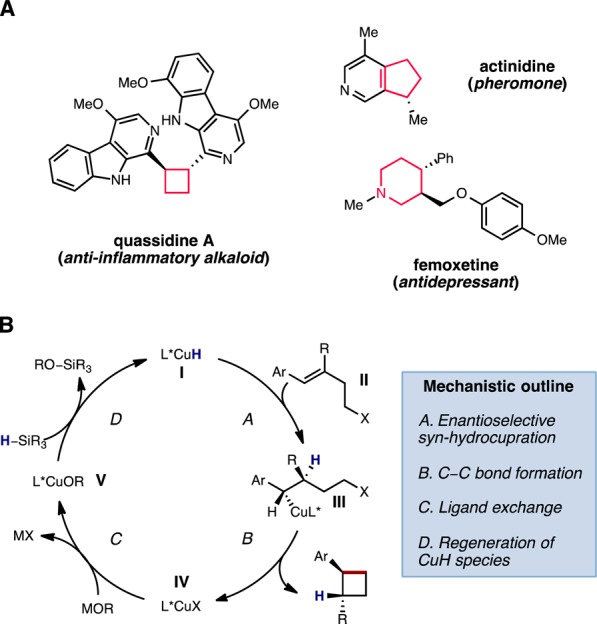
(A) Representative saturated 4-, 5-, and 6-membered rings found in pharmaceuticals and natural products. (B) Proposed catalytic cycle for the CuH-catalyzed enantioselective hydroalkylation.
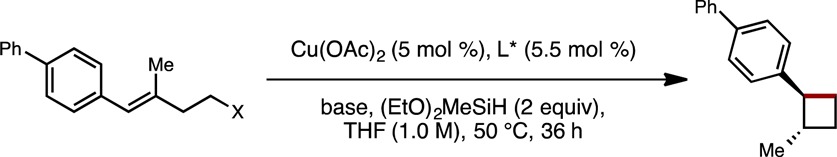
We began our study by examining the reactivity of a homoallylic methanesulfonate in the presence of a DTBM-SEGPHOS-based copper catalyst with diethoxymethylsilane as the hydride source (Table 1). At the outset, we anticipated that the copper (pseudo)halide species (IV, Figure 2) generated upon C–C bond formation would be reluctant to undergo transmetalation with the hydrosilane. We envisioned that the use of an alkoxide base would result in the formation of an intermediate copper alkoxide species (V),9 which would more readily transmetalate with hydrosilane to regenerate copper hydride I. Thus, during preliminary investigations, a range of alkoxide bases were evaluated. Among them, lithium methoxide was found to be uniquely effective in promoting the desired transformation, providing the cyclobutane product in excellent enantioselectivity, albeit in low yield (entry 3). No desired cyclization product was observed in the absence of base, or when other common alkoxide bases were employed (entries 1, 2, and 4).10
Table 1. Optimization of Reaction Conditions.
| entry | X | L* | base (equiv) | % yielda (% eeb) |
|---|---|---|---|---|
| Effect of Base | ||||
| 1 | OMs | L1 | KOt-Bu (2.0) | 0 |
| 2 | OMs | L1 | LiOt-Bu (2.0) | 0 |
| 3 | OMs | L1 | LiOMe (2.0) | 17 (96) |
| 4 | OMs | L1 | NaOMe (2.0) | 0 |
| Effect of Leaving Group | ||||
| 5 | OTs | L1 | LiOMe (2.0) | 9 |
| 6 | Br | L1 | LiOMe (2.0) | 16 |
| 7 | Br | L1 | LiOMe (4.0) | 70 (97) |
| 8 | OMs | L1 | LiOMe (4.0) | 14 |
| Effect of Ligand | ||||
| 9 | Br | L2 | LiOMe (4.0) | 16 |
| 10 | Br | L3 | LiOMe (4.0) | 8 |
| 11 | Br | L4 | LiOMe (4.0) | 5 |
| 12 | Br | L1 | LiOMe (4.0)c | 92d (99) |
NMR yield with 1,3,5-trimethoxybenzene as internal standard, 0.1 mmol scale.
ee determined by chiral HPLC.
4.0 equiv (MeO)2MeSiH at 2.0 M in THF, 55 °C.
83% isolated yield, 0.5 mmol scale.
The effect of the leaving group was then probed in this hydroalkylation protocol. In the presence of additional LiOMe (4.0 equiv), bromide was found to be superior to methanesulfonate as the leaving group, affording the desired cyclobutane product in moderate yield and still excellent enantioselectivity (entry 7 vs entries 3 and 8). Our initial choice of ligand, DTBM-SEGPHOS, was found to be superior to other chiral bisphosphine ligands explored in terms of reactivity (entries 9–11). Finally, the use of dimethoxymethylsilane as the stoichiometric hydride source, as well as further optimization of concentration and temperature, allowed the desired cyclobutane product to be obtained in high yield and excellent enantioselectivity (entry 12).
Under these optimized conditions, we explored the substrate scope of this copper hydride-catalyzed process (see Table 2). Substrates bearing electron-poor aryl substituents reacted efficiently to provide the desired cyclobutane product (2c, 2g, 2h). Replacement of the methyl substituent with a larger ethyl group was also well-tolerated (2b). Substrates containing several functional groups, including the tert-butyl (2g) and methyl (2h) esters, as well as a cis-dialkyl olefin (2e), were also suitable substrates for the transformation. Under these conditions, an alcohol substrate was rapidly transformed to the corresponding silyl ether, which underwent subsequent intramolecular hydroalkylation to furnish the desired cyclization product (2f). Substrates containing heterocycles, including a dibenzofuran (2i), a benzothiophene (2j), and a pyrrole (2k) also proved competent in the cyclization. However, substrates containing an electron-rich aryl substituents tended to provide cyclization product in lower yields, as exemplified by the preparation of 3,4,5-trimethoxyphenyl-substituted cyclobutane 2d.11 Nevertheless, enantioselectivities were uniformly high (≥97% ee) in all cases examined, with only one diastereomer (>20:1 dr) observed by 1H NMR of the crude material. To demonstrate the scalability of the procedure, we performed the synthesis of 2g on a 4.0 mmol scale (1.30 g starting material). The reaction proceeded at this scale without any deleterious effect on yield or enantioselectivity (78% yield, 98% ee).12
Table 2. Substrate Scopea.
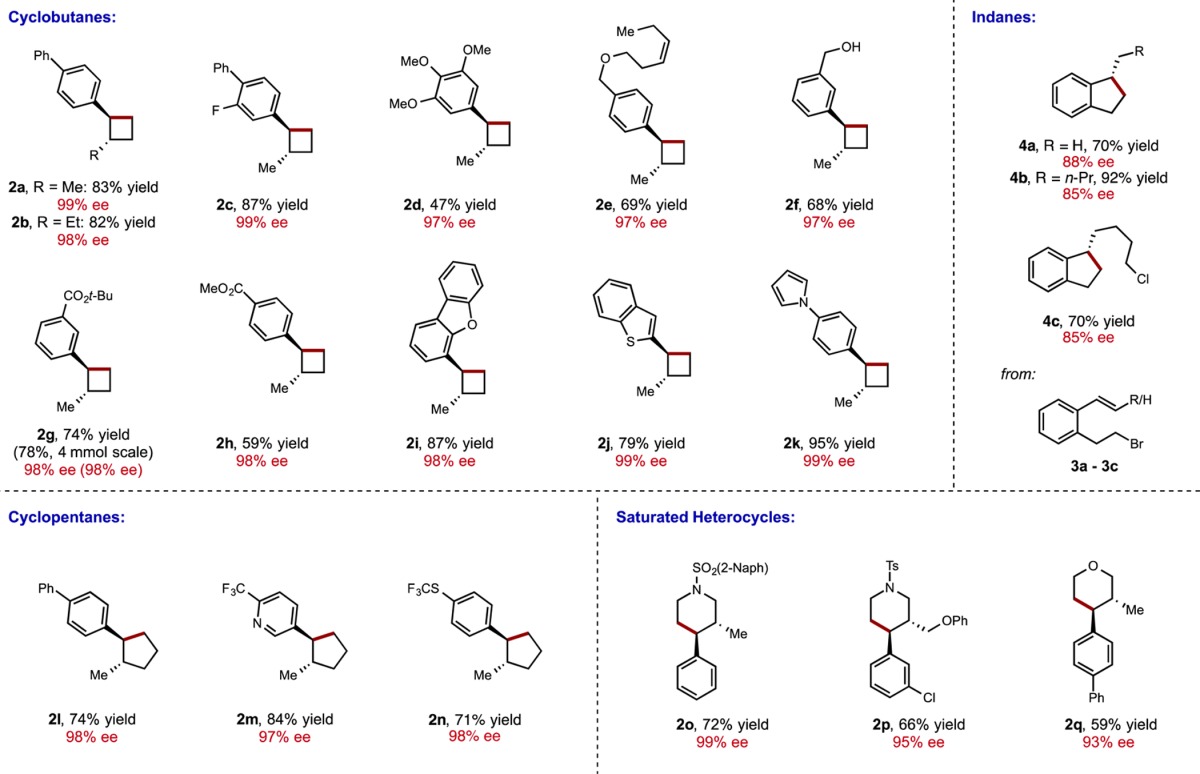
Yields reported are average isolated yields of two runs (0.5 mmol scale). See the Supporting Information for detailed experimental procedures.
Next, we set out to explore the enantioselective synthesis of other ring systems using this approach. Subjection of a homologated substrate to the reaction conditions provided the corresponding cyclopentane product 2l in similarly high yield and enantioselectivity. Notably, a highly electron-poor substrate bearing a 4-trifluoromethyl-3-pyridyl substituent could be used (2m), and a trifluoromethylthio group was also tolerated (2n). As a further extension of this process, we explored the use of substrates with heteroatoms in the tether for the preparation of enantioenriched, saturated heterocycles. Using this approach, 3,4-disubstituted N-sulfonyl piperidines 2o and 2p, as well as tetrahydropyran 2r, could be prepared in moderate to good yield and excellent enantioselectivity. An aryl chloride functional group was tolerated (2p), affording a product that is potentially amenable to subsequent derivatization. The connectivity and stereochemistry of 2o was determined by single crystal X-ray diffraction (Figure 3). As expected, the relative stereochemistry (trans) was consistent with syn-hydrocupration. The absolute stereochemistry was consistent with the sense of stereoinduction observed for the DTBM-SEGPHOS-CuH-catalyzed hydroamination.5a
Figure 3.
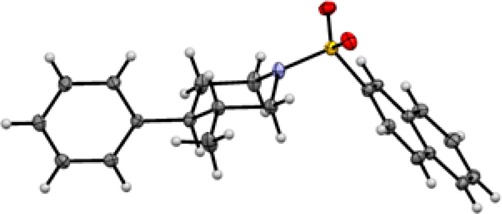
X-ray crystallographic structure of 2o (ellipsoids at 50% probability).
Finally, we sought to expand the hydroalkylation process to the synthesis of 1-alkylindanes through the use of mono- and disubstituted styrenes substrates bearing an alkyl bromide tethered at the ortho position. Indeed, subjection of 3a–3c to hydroalkylation conditions afforded 1-alkylindane products with high synthetic efficiency, though enantioselectivities were somewhat diminished compared to trisubstituted alkene substrates. We note that when a substrate containing both an alkyl chloride and bromide was used, complete selectivity for cyclization at the alkyl bromide was observed while the alkyl chloride remained inert under reaction conditions (4c).
To demonstrate the potential applicability of this process to the synthesis of biologically active molecules, we prepared the commercial selective serotonin reuptake inhibitor (−)-paroxetine in seven steps from known starting materials. The key hydroalkylation step on bromide 1r proceeded smoothly to afford the desired cyclization product in moderate yield and excellent enantioselectivity as one diastereomer. Subsequent deprotection of the sulfonyl group furnished the target compound (Scheme 1). Spectroscopic and optical rotation data matched literature values and further confirmed the stereochemical assignment based on X-ray diffraction (see the Supporting Information).
Scheme 1. Synthesis of (−)-Paroxetine.
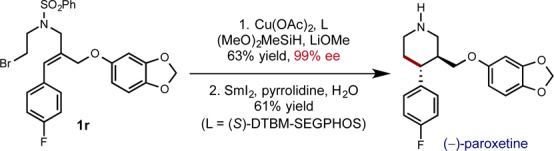
In summary, an intramolecular, enantioselective hydroalkylation of bromide-tethered styrenes was achieved through a copper-catalyzed process. Crucial to the success of this process was the use of lithium methoxide to facilitate regeneration of the proposed copper hydride species and the use of DTBM-SEGPHOS as the supporting ligand. The method presented here was amenable to gram-scale synthesis and could be applied to the synthesis of the pharmaceutical product paroxetine. Importantly, the hydroalkylation process proved general to the synthesis of several scaffolds, including cyclobutanes, cyclopentanes, indanes, and saturated 6-membered heterocycles, all with complete diastereoselectivity and good to excellent levels of enantioselectivity. Efforts toward the development of an intermolecular hydroalkylation reaction are currently under way.
Acknowledgments
Research reported in this publication was supported by the National Institutes of Health under award no. GM46059. Y.-M.W. thanks the National Institutes of Health for a postdoctoral fellowship (GM112218). Á.L.P. gratefully acknowledges the MIT Summer Research Program. We thank Dr. Peter Mueller for X-ray crystallographic data and Drs. Michael Pirnot and Aaron Sather for their advice on the preparation of this manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b07061.
The authors declare no competing financial interest.
Supplementary Material
References
- For selected reviews on the enantioselective catalytic formation of C(sp3)–C(sp3) bonds, see:; a Hoveyda A. H.; Morken J. P. Angew. Chem., Int. Ed. Engl. 1996, 35, 1262. 10.1002/anie.199612621. [DOI] [Google Scholar]; b Kobayashi S.; Ishitani H. Chem. Rev. 1999, 99, 1069. 10.1021/cr980414z. [DOI] [PubMed] [Google Scholar]; c Fürstner A. Chem. Rev. 1999, 99, 991. 10.1021/cr9703360. [DOI] [PubMed] [Google Scholar]; d Johnson J. S.; Evans D. A. Acc. Chem. Res. 2000, 33, 325. 10.1021/ar960062n. [DOI] [PubMed] [Google Scholar]; e Mikami K.; Terada M.; Matsuzawa H. Angew. Chem., Int. Ed. 2002, 41, 3554.. [DOI] [PubMed] [Google Scholar]; f Davies H. M. L.; Beckwith R. E. J. Chem. Rev. 2003, 103, 2861. 10.1021/cr0200217. [DOI] [PubMed] [Google Scholar]; g Ooi T.; Maruoka K. Angew. Chem., Int. Ed. 2007, 46, 4222. 10.1002/anie.200601737. [DOI] [PubMed] [Google Scholar]; h Gaunt M. J.; Johansson C. C. C.; McNally A.; Vo N. T. Drug Discovery Today 2007, 12, 8. 10.1016/j.drudis.2006.11.004. [DOI] [PubMed] [Google Scholar]; i Shibasaki M.; Kanai M. Chem. Rev. 2008, 108, 2853. 10.1021/cr078340r. [DOI] [PubMed] [Google Scholar]; j Trost B. M.; Brindle C. S. Chem. Soc. Rev. 2010, 39, 1600. 10.1039/b923537j. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Terada M. Bull. Chem. Soc. Jpn. 2010, 83, 101. 10.1246/bcsj.20090268. [DOI] [Google Scholar]; l Hassan A.; Krische M. J. Org. Process Res. Dev. 2011, 15, 1236. 10.1021/op200195m. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Negishi E.-i. ARKIVOC 2010, 2011viii34. 10.3998/ark.5550190.0012.803.22514791 [DOI] [Google Scholar]; n Watson I. D. G.; Toste F. D. Chem. Sci. 2012, 3, 2899. 10.1039/c2sc20542d. [DOI] [Google Scholar]; o Yang L.; Huang H. Catal. Sci. Technol. 2012, 2, 1099. 10.1039/c2cy20111a. [DOI] [Google Scholar]; p Scheffler U.; Mahrwald R. Chem. - Eur. J. 2013, 19, 14346. 10.1002/chem.201301996. [DOI] [PubMed] [Google Scholar]; q Tsubogo T.; Ishiwata T.; Kobayashi S. Angew. Chem., Int. Ed. 2013, 52, 6590. 10.1002/anie.201210066. [DOI] [PubMed] [Google Scholar]; r Šebesta R. ChemCatChem 2013, 5, 1069. 10.1002/cctc.201200926. [DOI] [Google Scholar]; s Tasker S. Z.; Standley E. A.; Jamison T. F. Nature 2014, 509, 299. 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]; For selected methods based on stoichiometric auxiliaries, see:; t Meyers A. I. Acc. Chem. Res. 1978, 11, 375. 10.1021/ar50130a002. [DOI] [Google Scholar]; u Evans D. A.; Bartroli J.; Shih T. L. J. Am. Chem. Soc. 1981, 103, 2127. 10.1021/ja00398a058. [DOI] [Google Scholar]; v Myers A. G.; Yang B. H.; Chen H.; McKinstry L.; Kopecky D. J.; Gleason J. L. J. Am. Chem. Soc. 1997, 119, 6496. 10.1021/ja970402f. [DOI] [Google Scholar]; w Ellman J. A.; Owens T. D.; Tang T. P. Acc. Chem. Res. 2002, 35, 984. 10.1021/ar020066u. [DOI] [PubMed] [Google Scholar]; x Job A.; Janeck C. F.; Bettray W.; Peters R.; Enders D. Tetrahedron 2002, 58, 2253. 10.1016/S0040-4020(02)00080-7. [DOI] [Google Scholar]; For an overview of enantioselective C—C bond-forming reactions applied to complex molecule synthesis, see:; y Corey E. J.; Kürti L.. Enantioselective Chemical Synthesis; Academic Press: San Diego, 2013. [Google Scholar]
- For examples of the enantioselective, catalytic generation of C(sp3)–C(sp3) bonds remote from reactive functional groups, see:; a Pino P.; Cioni P.; Wei J. J. Am. Chem. Soc. 1987, 109, 6189. 10.1021/ja00254a052. [DOI] [Google Scholar]; b Long J.; Du H.; Li K.; Shi Y. Tetrahedron Lett. 2005, 46, 2737. 10.1016/j.tetlet.2005.02.161. [DOI] [Google Scholar]; c Arp F. O.; Fu G. C. J. Am. Chem. Soc. 2005, 127, 10482. 10.1021/ja053751f. [DOI] [PubMed] [Google Scholar]; d Saito B.; Fu G. C. J. Am. Chem. Soc. 2008, 130, 6694. 10.1021/ja8013677. [DOI] [PubMed] [Google Scholar]
- a Corey E. J.; Posner G. H. J. Am. Chem. Soc. 1967, 89, 3911. 10.1021/ja00991a049. [DOI] [Google Scholar]; b Whitesides G. M.; Fischer W. P.; San Filippo J.; Bashe R. W.; House H. O. J. Am. Chem. Soc. 1969, 91, 4871. 10.1021/ja01045a049. [DOI] [Google Scholar]; For the copper-catalyzed coupling of alkyl halides and Grignard reagents, see:; c Fouquet G.; Schlosser M. Angew. Chem., Int. Ed. Engl. 1974, 13, 82. 10.1002/anie.197400821. [DOI] [Google Scholar]
- For the construction of C(sp3)–C(sp3) bonds through Cu-catalyzed alkene carboboration, see:; a Ito H.; Kosaka Y.; Nonoyama K.; Sasaki Y.; Sawamura M. Angew. Chem., Int. Ed. 2008, 47, 7424. 10.1002/anie.200802342. [DOI] [PubMed] [Google Scholar]; b Ito H.; Toyoda T.; Sawamura M. J. Am. Chem. Soc. 2010, 132, 5990. 10.1021/ja101793a. [DOI] [PubMed] [Google Scholar]; c Zhong C.; Kunii S.; Kosaka Y.; Sawamura M.; Ito H. J. Am. Chem. Soc. 2010, 132, 11440. 10.1021/ja103783p. [DOI] [PubMed] [Google Scholar]; d Kubota K.; Yamamoto E.; Ito H. J. Am. Chem. Soc. 2013, 135, 2635. 10.1021/ja3104582. [DOI] [PubMed] [Google Scholar]; e Yoshida H.; Kageyuki I.; Takaki K. Org. Lett. 2013, 15, 952. 10.1021/ol4001526. [DOI] [PubMed] [Google Scholar]
- a Zhu S.; Niljianskul N.; Buchwald S. L. J. Am. Chem. Soc. 2013, 135, 15746. 10.1021/ja4092819. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhu S.; Buchwald S. L. J. Am. Chem. Soc. 2014, 136, 15913. 10.1021/ja509786v. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Niljianskul N.; Zhu S.; Buchwald S. L. Angew. Chem., Int. Ed. 2015, 54, 1638. 10.1002/anie.201410326. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Shi S.; Buchwald S. L. Nat. Chem. 2014, 7, 38. 10.1038/nchem.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]; For the independent development of a related system, see:; e Miki Y.; Hirano K.; Satoh T.; Miura M. Angew. Chem., Int. Ed. 2013, 52, 10830. 10.1002/anie.201304365. [DOI] [PubMed] [Google Scholar]; f Miki Y.; Hirano K.; Satoh T.; Miura M. Org. Lett. 2014, 16, 1498. 10.1021/ol5003219. [DOI] [PubMed] [Google Scholar]; For a CuH-catalyzed C—C bond-forming reaction resulting in the enantioselective formation of indolines, see:; g Ascic E.; Buchwald S. L. J. Am. Chem. Soc. 2015, 137, 4666. 10.1021/jacs.5b02316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cu-catalyzed diene hydroalkylation:; a Iwasaki T.; Shimizu R.; Imanishi R.; Kuniyasu H.; Kambe N. Angew. Chem., Int. Ed. 2015, 54, 9347. 10.1002/anie.201503288. [DOI] [PubMed] [Google Scholar]; Cu-catalyzed hydroalkylation of alkynes:; b Uehling M. R.; Suess A. M.; Lalic G. J. Am. Chem. Soc. 2015, 137, 1424. 10.1021/ja5124368. [DOI] [PubMed] [Google Scholar]; c Suess A. M.; Uehling M. R.; Kaminsky W.; Lalic G. J. Am. Chem. Soc. 2015, 137, 7747. 10.1021/jacs.5b03086. [DOI] [PubMed] [Google Scholar]; Pd-catalyzed intermolecular hydroalkylation:; d Urkalan K. B.; Sigman M. S. J. Am. Chem. Soc. 2009, 131, 18042. 10.1021/ja908545b. [DOI] [PMC free article] [PubMed] [Google Scholar]; e DeLuca R. J.; Sigman M. S. Org. Lett. 2013, 15, 92. 10.1021/ol303129p. [DOI] [PMC free article] [PubMed] [Google Scholar]; Co-catalyzed hydroalkylation of [60]fullerene:; f Lin X.; Qing F.-L. Org. Lett. 2013, 15, 4478. 10.1021/ol402032a. [DOI] [PubMed] [Google Scholar]; g Lu S.; Jin T.; Bao M.; Yamamoto Y. J. Am. Chem. Soc. 2011, 133, 12842. 10.1021/ja204982w. [DOI] [PubMed] [Google Scholar]; Sc-catalyzed hydromethylation of olefins:; h Sadow A. D.; Tilley T. D. J. Am. Chem. Soc. 2003, 125, 7971. 10.1021/ja021341a. [DOI] [PubMed] [Google Scholar]; i Fontaine F.-G.; Tilley T. D. Organometallics 2005, 24, 4340. 10.1021/om0505460. [DOI] [Google Scholar]; Lewis acid-catalyzed hydromethylation:; j Parnes Z. N.; Bolestova G. I.; Akhrem I. S.; Vol’pin M. E.; Kursanov D. N. J. Chem. Soc., Chem. Commun. 1980, 748. 10.1039/c39800000748. [DOI] [Google Scholar]; For a two-step, one-pot alkene hydromethylation, see:; k Dao H. T.; Li C.; Michaudel Q.; Maxwell B. D.; Baran P. S. J. Am. Chem. Soc. 2015, 137, 8046. 10.1021/jacs.5b05144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang H.; Cox N.; Lalic G. Angew. Chem., Int. Ed. 2014, 53, 752. 10.1002/anie.201307697. [DOI] [PubMed] [Google Scholar]
- For a review on cyclobutane/cyclopentane natural products and pharmaceuticals, see:; a Sergeiko A.; Poroikov V. V.; Hanus L. O.; Dembitsky V. M. Open Med. Chem. J. 2008, 2, 26. 10.2174/1874104500802010026. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Heasley B. Curr. Org. Chem. 2014, 18, 641. 10.2174/13852728113176660150. [DOI] [Google Scholar]; For a perspective on saturated ring systems in drugs, see:; c Taylor R. D.; MacCoss M.; Lawson A. D. G. J. Med. Chem. 2014, 57, 5845. 10.1021/jm4017625. [DOI] [PubMed] [Google Scholar]
- Tsuda T.; Hashimoto T.; Saegusa T. J. Am. Chem. Soc. 1972, 94, 658. 10.1021/ja00757a069. [DOI] [Google Scholar]
- The use of other bases was found to be ineffective, including LiOEt, LiOPh, NaOPh, NaO(2,6-t-Bu2C6H3), NaOBz, and LiOBz, all of which provided <5% yield of the desired cyclization product.
- We attribute the poorer reactivity of electron-rich substrates to a more difficult hydrocupration step. Reduction of the alkyl bromide to a terminal methyl group was observed as the major side reaction.
- Preliminary experiments indicate that the corresponding (Z)-configured homoallylic bromides were unreactive in the present catalyst system.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.