

ABSTRACT
Bacillus anthracis vegetative forms assemble an S-layer comprised of two S-layer proteins, Sap and EA1. A hallmark of S-layer proteins are their C-terminal crystallization domains, which assemble into a crystalline lattice once these polypeptides are deposited on the bacterial surface via association between their N-terminal S-layer homology domains and the secondary cell wall polysaccharide. Here we show that slaQ, encoding a small cytoplasmic protein conserved among pathogenic bacilli elaborating S-layers, is required for the efficient secretion and assembly of Sap and EA1. S-layer protein precursors cosediment with SlaQ, and SlaQ appears to facilitate Sap assembly. Purified SlaQ polymerizes and when mixed with purified Sap promotes the in vitro formation of tubular S-layer structures. A model is discussed whereby SlaQ, in conjunction with S-layer secretion factors SecA2 and SlaP, promotes localized secretion and S-layer assembly in B. anthracis.
IMPORTANCE S-layer proteins are endowed with the propensity for self-assembly into crystalline arrays. Factors promoting S-layer protein assembly have heretofore not been reported. We identified Bacillus anthracis SlaQ, a small cytoplasmic protein that facilitates S-layer protein assembly in vivo and in vitro.
INTRODUCTION
S-layer proteins (SLPs) are endowed with crystallization domains that, following their secretion and association with envelope polysaccharides, promote self-assembly into a two-dimensional crystalline lattice (1). S-layer proteins are initially synthesized in the bacterial cytoplasm as precursors with N-terminal signal peptides, which are removed during translocation across the plasma membrane (2). Six consecutive immunoglobulin-like domains of Geobacillus stearothermophilus S-layer protein SbsB, organized into ϕ-shaped, disk-like monomeric crystallization units, are stabilized by interdomain calcium ion coordination, which functions as a switch for the formation of a condensed quaternary structure in the mature S-layer (3). The crystallization domains of SbsB and many other S-layer proteins are endowed with the propensity for self-assembly (3–6). Therefore, what mechanisms prevent the crystallization of S-layer proteins in the bacterial cytoplasm, support their secretion, and promote assembly in the bacterial envelope? We sought to address these questions by studying the assembly of S-layer proteins in Bacillus anthracis.
B. anthracis is a Gram-positive, spore-forming bacterium and the causative agent of anthrax (7). Following spore uptake and germination in host tissues, B. anthracis replicates as vegetative forms and disseminates to all organ tissues, eventually triggering the death of infected hosts (8). B. anthracis sporulates in carcass tissues, and contamination of animal products or of the environment promotes spore dissemination to new hosts (8). The pathogenesis of anthrax infections depends on a number of virulence factors that are encoded on two large plasmids, pXO1 and pXO2 (9, 10). Loss of either of these plasmids leads to attenuation of B. anthracis (11, 12), a trait that has been exploited for the generation of whole-cell anthrax vaccines (13). A hallmark of B. anthracis is its propensity to grow as chains of incompletely separated vegetative cells (7), with chain lengths that exceed the capacity for uptake by host phagocytes (14).
Genetic approaches identified determinants of B. anthracis chain length (15, 16). Insertional lesions in bslO, sap, and csaB, as well as patA1 and patA2, increase the chain lengths of B. anthracis vegetative forms without affecting the size of bacterial cells (15–18). bslO encodes a secreted cell wall hydrolase with S-layer homology (SLH) domains that bind to pyruvylated secondary cell wall polysaccharide (SCWP) (15), comprised of the repeating unit [→4)-β-ManNAc-(1→4)-β-GlcNAc-(1→6)-α-GlcNAc-(1→)]n (19, 20). BslO functions as a murein hydrolase when deposited at the division septa of vegetative cells and promotes cell separation, thereby reducing chain length (15). B. anthracis variants lacking the S-layer protein Sap cannot restrict BslO localization to septal rings; similar to bslO mutants, sap variants form elongated chains of vegetative bacilli (16). csaB mutants display the most severe cell separation defect, and the variants are unable to deposit any S-layer or S-layer-associated proteins with SLH domains in the envelope of B. anthracis (17, 21). csaB is thought to encode a pyruvyl transferase that transfers ketal-pyruvyl onto the terminal β-ManNAc residue at the distal end of the SCWP (20). Finally, PatA1 and PatA2 catalyze acetylation of the SCWP, thereby enabling the deposition of EA1 as well as BslO near the septal region of the B. anthracis envelope (18).
B. anthracis mutants with moderate-chain-length phenotypes harbor mutations in the slaP and secA2 genes, which are located upstream of the csaA-csaB-sap-eag gene cluster (22). secA2 encodes a paralogue of SecA, the bacterial secretion ATPase that translocates precursors with N-terminal signal peptides through the SecYEG translocon and across the plasma membrane (23–25). The exacerbated-chain-length phenotype of slaP and secA2 mutants is attributed to the inefficient secretion of the S-layer proteins Sap and EA1 (coded for by eag) (22). Of note, secretion of S-layer-associated proteins (Bsls) or other secretion substrates (e.g., protective antigen [PA]) is not affected in secA2 or slaP mutants (22). SecA2 and SlaP are thought to modify the SecYEG pathway of B. anthracis and provide for the secretion of large amounts of Sap and EA1, which are subsequently deposited into the bacterial S-layer (22).
We entertained the possibility that B. anthracis expresses additional factors involved in the secretion and assembly of S-layer proteins. Here we report on the identification of slaQ, a gene located immediately adjacent to secA2-slaP and the S-layer gene cluster.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
B. anthracis Sterne 34F2 (11) and its mutants (Table 1) were cultured in brain heart infusion (BHI) broth supplemented with 0.8% NaHCO3. Unless otherwise indicated, cultures were incubated at 37°C or at 30°C when harboring the vector pLM4 (26). Escherichia coli strains DH5α (27), K1077 (dcm dam mutant) (28), or BL21(DE3) (29) were cultured in Luria-Bertani broth (LB). Media were supplemented with 20 μg/ml kanamycin or 200 μg/ml spectinomycin to maintain plasmid or mutant selection in B. anthracis and with 50 μg/ml kanamycin or 100 μg/ml ampicillin in E. coli. B. anthracis strains were sporulated in modified G (ModG) medium as described previously (30). Spore preparations were heat treated to kill vegetative bacilli and stored at 4°C. Spores were enumerated by spreading of samples on agar plates and incubation for CFU. Spores were germinated by inoculation into BHI broth and incubation at 37°C.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Descriptiona | Reference or source |
|---|---|---|
| Strains | ||
| B. anthracis | ||
| Sterne 34F2 | pXO1+ pXO2− wild type | 11 |
| SN2 | secA2::aad9 insertion at nt 890561 in 34F2 (Spcr) | 22 |
| SN11 | Deletion of sap (BAS0841 [nt 896758–899063]) in 34F2; Δsap | 22 |
| SN12 | Deletion of eag (BAS0842 [nt 899843–902414]) in 34F2; Δeap | 22 |
| SN13 | Deletion of sap and eag (BAS0841 and BAS0842 [nt 896758–902414]) in 34F2; Δsap eag | 22 |
| SN14 | Deletion of slaP (BAS0837 [nt 889657–890538]) in 34F2; ΔslaP | 22 |
| SN16 | BAS0836::aad9 insertion at nt 889121 in 34F2 | This study |
| SN17 | Deletion of slaQ (BAS0836 [nt 889181–889374]) in 34F2; ΔslaQ | This study |
| SN18 | BAS0835::aphA3 insertion at nt 888714 in 34F2 | This study |
| csaB mutant | Deletion of BAS0840 | 17 |
| E. coli | ||
| BL21(DE3) | Expression of plasmid-borne HisslaQ via T7 polymerase | 29 |
| DH5α | Used for cloning of recombinant plasmids | 27 |
| K1077 | DNA methylation mutant (dcm dam mutant) used for plasmid propagation | 28 |
| Plasmids | ||
| pLM4 | Temperature-sensitive pE194 replicon; Kanr | 26 |
| pJK4 | Pspac; lacI regulator; Kanr | 32 |
| pSN7 | pJK4 expressing slaQ nt 889181–889555 from 34F2 | This study |
| pSN8 | pET15b expressing full-length slaQ with N-terminal 6× His tag; Ampr | This study |
| pSN9 | pJK4 expressing slaQ with N-terminal 6× His tag | This study |
| pSN10 | pJK4 expressing mCherry | This study |
| pSN11 | pJK4 expressing first 100 aa of Sap fused to mCherry | This study |
| pSN12 | pJK4 expressing first 400 aa of Sap fused to mCherry | This study |
| pSN13 | pJK4 expressing full-length Sap fused to mCherry | This study |
| pSN14 | pJK4 expressing first 400 aa of Sap with C-terminal His/STREP tag | This study |
| pSN15 | pJK4 expressing slaQSTREP with C-terminal STREP tag | This study |
| pSY183 | pET24b expressing sapHis with a C-terminal linker and 6× His tag | This study |
| pSY185 | pET24b expressing slaQSTREP | This study |
Kanr, kanamycin resistance; Spcr, spectinomycin resistance; Ampr, ampicillin resistance.
B. anthracis mutants and plasmids.
Plasmid DNA was purified from E. coli K1077 and used to transform B. anthracis (31). Deletion mutants were obtained by allelic replacement using the temperature-sensitive plasmid pLM4 (Table 1) (26). Briefly, 1-kb upstream and downstream DNA sequences flanking the gene of interest were PCR amplified with specific primers (Table 2), cloned by restriction digestion into pLM4, and transformed into B. anthracis. Transformants were grown for 10 h at 42°C in the presence of 20 μg/ml kanamycin; cultures were diluted into fresh medium and grown under the same conditions for four passages. Cultures were then diluted for another four passages into medium lacking antibiotics and grown for 10-h intervals at 30°C. Mutants were screened for growth on BHI agar and no growth on BHI-kanamycin agar. All complementation plasmids were derived from pJK4, and the expression of its Pspac promoter was induced with 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) (32). The primers for the amplification and construction of genes and plasmids are listed in Table 2.
TABLE 2.
Oligonucleotides used in this study
| Primer | Sequence | Gene or plasmid |
|---|---|---|
| smn219 F | TTTTCCCGGGGATGGTTTGGTGATAAGAAAGTTGGG | slaQ |
| smn220 R | TTTTGGTACCTTATTCCCTCCAGCTTAAAGCTCT | slaQ |
| smn221 F | TTTTGGTACCACATCCAGATTTCCATCAAGCG | slaQ |
| smn222 R | TTTTGAATTCCGTGATAACGGTTATGACGTAGCT | slaQ |
| smn223 F | TTTTTCTAGATTGATGTACAAACAGTTACCACACG | pSN7/15 |
| smn224 R | TTTTGGTACCTTATTTCGTCACTAATAAACGTTCAATATGATATTCA | pSN7/9 |
| smn227 F | TTTTCATATGTTGATGTACAAACAGTTACCACACG | pSN8 |
| smn228 R | TTTTGGATCCTTATTTCGTCACTAATAAACGTTCAATATGATATTCA | pSN8 |
| smn242 F | GCGCTCTAGAATGCATCACCATCACCATCACATGTACAAACAGTTACCACACG | pSN9 |
| smn135 F | TTTTTCTAGAATGGTGAGCAAGGGCG | pSN10 |
| smn136 R | TTTTGGTACCTTACTTGTACAGCTCGTCCATG | pSN10 |
| smn98 F | TTTTGCTAGCATGGCAAAGACTAACTCTTACAAAAAAGTAATC | pSN11/14 |
| smn102 R | GCCCTTGCTCACCATCCTTGAGAGTCAGCGAAAGA | pSN11 |
| smn103 F | GCTGACTCTCAAGGCATGGTGAGCAAAGGGCG | pSN11 |
| smn104 R | GCCCTTGCTCACCATAATGAATTGAGCAGCTTCTGCTTTAG | pSN12 |
| smn105 F | GCTGCTCAATTCATTATGGTGAGCAAAGGGCG | pSN12 |
| smn108 R | GCCCTTGCTCACCATTTTATTTTGTTCTGCAACTGTCCAG | pSN13 |
| smn109 F | GCAGAACAAAATAAAATGGTGAGCAAAGGGCG | pSN13 |
| smn188 R | TTTTGGTACCAGATCTTTTTGTTGCAGGTTTTGCTTCTTTAATAG | pSN13 |
| smn189 F | TTTTAGATCTATGGTGAGCAAAGGGCGAG | pSN13 |
| smn287 R | TTACTTCTCAAATTGAGGATGAGACCAGTGATGGTGATGGTGATGAGATCTTTTTGTTGCAGGTTTTGCTTC | pSN14 |
| smn288 F | CATCACCATCACCATCACTGGTCTCATCCTCAATTTGAGAAGTAAGGTACCAGCGCTATCGATCG | pSN14 |
| smn226 R | TTTGGTACCTTACTTCTCAAATTGAGGATGAGACCATTTCGTCACTAATAAACGTTCAATATGATATTCA | pSN15 |
| 411 | TTTTCATATGTACAAACAGTTACCACACGGAGTG | pSY185 |
| 412 | TTTTCTCGAGTTACTTCTCAAATTGAGGATGAGACCATTTC | pSY185 |
| 405 | TTTTCATATGGGTAAAACATTCCCAGACGTTCC | pSY183 |
| 406 | GCGCCTCGAGGTTTTTTTGTTGCAGGTTTTGCTTCTTTAATAG | pSY183 |
| 407 | CACACCGGTGGTCGCAGCAGCCATCACCATCATCACCACTAAGGATCCCACCACCACCACCACCACTGAGATCC | pSY183 |
| 408 | TGATGGCTGCTGCGACCACCGGTGTGATGGATATCTGCGCTATTCGGTTTTTTTGTTGCAGGTTTTGCTTC | pSY183 |
Antibody production.
Recombinant SlaQ was produced in E. coli BL21(DE3) harboring plasmid pSN8. Overnight cultures were diluted 1:50 in LB supplemented with 100 μg ampicillin/ml at 30°C. IPTG (1 mM) was added to the cultures at an A600 of 0.5, and expression of N-terminal His-tagged SlaQ (HisSlaQ) was induced for 3 h. Cells were sedimented by centrifugation, suspended in column buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl), and lysed in a French press. Cell lysates were subjected to affinity purification over an Ni-nitrilotriacetic acid (NTA) column (Qiagen) and eluted with 500 mM imidazole. Eluates were pooled and dialyzed in 1× phosphate-buffered saline (PBS). Purified protein (700 μg) was emulsified in complete Freund's adjuvant (Difco) and injected subcutaneously into a female New Zealand White rabbit. Antibody production was stimulated in 21-day intervals with two booster injections of antigen emulsified in incomplete Freund's adjuvant. Rabbit antibodies were used for immunoblotting experiments.
Microscopy of bacilli.
Vegetative forms were grown as indicated, sedimented by centrifugation, and fixed with 4% buffered formalin. Images were captured with a charge-coupled device (CCD) camera on an Olympus IX81 microscope using 40× or 100× objectives. For immunofluorescence microscopy, samples were incubated with dilute rabbit antiserum raised against purified recombinant Sap, BslO, or EA1 (15, 16) and labeled with secondary antibody conjugated to a fluorophore (Molecular Probes). Cells were counterstained with boron-dipyrromethene (BODIPY)-vancomycin (Invitrogen). A Leica SP5 tandem scanner spectral 2-photon confocal microscope was used to observe cells with a 63.1× objective.
S-layer fractionation.
B. anthracis overnight cultures were diluted 1:100 into fresh media and incubated with rotation until they reached an A600 of 1.0. Cultures were centrifuged at 16,000 × g, and the supernatant (medium [M] fraction) was separated from the sediment. Proteins in the medium were precipitated with (10% vol/vol) trichloroacetic acid (TCA) for 30 min on ice and centrifuged at 16,000 × g for 10 min. The bacterial sediments were washed twice with 1× PBS, and the S-layer was extracted with 3 M urea at 95°C. Cells were sedimented by centrifugation at 16,000 × g, and the supernatant (S-layer [S] fraction) was removed and proteins precipitated with TCA. The bacterial sediment was washed twice with PBS and lysed by physical force using silica bead beating for 3 min at 6.0 m/s (MP Biomedical Fastprep-24). After sedimentation of the beads, proteins in the cell lysates were precipitated with TCA (cell [C] fraction). All TCA precipitates were washed with ice-cold acetone and centrifuged at 16,000 × g for 10 min. Acetone was removed, and protein precipitates were dried. Samples were suspended in 1 M Tris-HCl (pH 8.0)–4% SDS and mixed with an equal volume of 2× sample buffer (4% SDS, 1% β-mercaptoethanol, 10% glycerol, 50 mM Tris-HCl [pH 7.5], 0.2% bromophenol blue). Proteins were separated by SDS-PAGE and analyzed by staining with Coomassie brilliant blue or electrotransferred to polyvinylidene difluoride (PVDF) membrane for immunoblot analysis. Proteins were detected with rabbit antisera raised against purified antigens. Immunoreactive products were revealed by chemiluminescent detection after incubation with horseradish peroxidase (HRP)-conjugated secondary antibody (Cell Signaling Technology). Detection of tagged proteins was assessed using StrepMAB (IBA) or His probe (Promega). Signal intensity was quantified in ImageJ.
Membrane protein extraction.
Diluted B. anthracis cultures were grown to an A600 of 1.0, and vegetative forms were sedimented by centrifugation at 16,000 × g for 10 min. Bacilli were washed and suspended in cytoplasmic buffer (50 mM HEPES, 66 mM potassium acetate, 10 mM magnesium acetate [pH 7.5]). Cells were lysed by bead beating for 10 min at 4.5 m/s. Lysates were subjected to ultracentrifugation at 100,000 × g for 30 min. Soluble (Sol) proteins from the cytoplasm were removed with the supernatant. The plasma membrane sediment (PM fraction) was extracted with 0.1 M sodium carbonate or cytoplasmic buffer and incubated for 30 min. Extracts were again subjected to ultracentrifugation at 100,000 × g for 30 min. Soluble proteins were removed with the supernatant (S fraction). Proteins in the extracted membrane pellet (P fraction) were suspended in cytoplasmic buffer. Proteins in all samples were precipitated with TCA, washed with acetone, and solubilized in sample buffer prior to analysis by SDS-PAGE and immunoblotting.
Affinity chromatography.
Overnight cultures of B. anthracis Sterne(pSN9) were diluted 1:100 in fresh BHI medium supplemented with 20 μg/ml kanamycin and 0.1 mM IPTG and incubated until they reached an A600 of 1.0. Bacilli were sedimented by centrifugation and lysed by bead beating in column buffer at 4.5 m/s for 10 min. Cellular debris was removed by centrifugation at 8,000 × g for 10 min. The cleared lysate was centrifuged at 100,000 × g for 30 min. Soluble proteins (HisSlaQ) in the supernatant were subjected to affinity chromatography using Ni-NTA-Sepharose. HisSlaQ was eluted with 500 mM imidazole. The load (L), wash (W), and eluate (E) fractions were analyzed by immunoblotting and chemiluminescence detection. Signal intensity was quantified using ImageJ analysis.
Purification of SlaQSTREP.
An overnight culture of E. coli BL21(DE3, pSY185) grown at 30°C in LB (50 μg kanamycin/ml) was diluted 1:40 into 2 liters of fresh medium and incubated with 150-rpm rotation at 30°C for 4 h (A600 of 0.65). IPTG (1 mM) was added to induce streptavidin-tagged SlaQ (SlaQSTREP) expression, and the culture was rotated for another 4 h at 30°C (A600 of 1.57). Bacteria were sedimented by centrifugation of the culture at 6,000 × g for 10 min. Wet cell paste (15.8 g) was suspended in 30 ml of 0.05 M Tris-HCl (pH 7.5), 10 mg lysozyme, and 10 mg DNase I and frozen at −80°C. For purification, the sample was thawed on ice, vortexed, and lysed with two passes through a French press at 14,000 lb/in2. The lysate was centrifuged at 32,000 × g for 30 min, and the supernatant was separated from sediment and loaded onto 1.5 ml StrepTactin-Sepharose, preequilibrated with 15 ml column buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl). The column was washed with 15 ml column buffer and 15 ml 20 mM Tris-HCl (pH 8.0) and eluted with 4 ml of 5 mM desthiobiotin in 20 mM Tris-HCl (pH 8.0). The concentration of purified SlaQSTREP was determined by 280-nm absorption, and the sample was diluted to 2 mg/ml and incubated at 4°C. Over time, SlaQSTREP forms visible aggregates that sediment spontaneously.
Purification of SapHis.
An overnight culture of E. coli BL21(DE3, pSY183) grown at 30°C in LB (50 μg/ml kanamycin) was diluted 1:20 into 2 liters fresh medium and incubated with 150-rpm rotation at 30°C for 2 h (A600 of 1.395). IPTG (1 mM) was added to induce Histidine-tagged Sap (SapHis) expression, and the culture was rotated for another 4 h at 30°C (A600 of 1.66). Bacteria were sedimented by centrifugation of the culture at 6,000 × g for 10 min. Cells from 1 liter of culture were suspended in 30 ml 0.05 M Tris-HCl (pH 7.5) and frozen at −80°C. For purification, the sample was thawed on ice, 10 mg/ml lysozyme and 10 mg/ml DNase I were added, the mixture was vortexed, and cells were lysed with two passes through a French press at 14,000 lb/in2. Lysate was centrifuged at 32,000 × g for 30 min, and supernatant was separated from the sediment and loaded onto 1 ml Ni-NTA-agarose, preequilibrated with 20 ml column buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl). The column was washed with 2× 20 ml column buffer and then 2× 20 ml 20 mM Tris-HCl (pH 8.0)–25 mM imidazole and finally eluted with 4 ml of 250 mM imidazole in 20 mM Tris-HCl (pH 8.0). The concentration of purified SapHis was determined by absorption of 280-nm light, and the sample was diluted to 2 mg/ml and incubated at 4°C. Over time, SapHis forms visible aggregates that sediment spontaneously.
Size exclusion chromatography.
Analytical size exclusion chromatography of affinity-purified SlaQSTREP and SapHis at a concentration of 2 mg/ml was performed at 4°C on a Superdex 200 10/300 GL column connected to an Äkta fast-protein liquid chromatography system (GE Healthcare). The running buffer, 20 mM Tris-HCl (pH 8.0), was degassed. Proteins were eluted at a flow rate of 0.5 ml min−1, with the UV absorbance of the eluent monitored at 280 nm. Timed assembly reactions with purified SlaQSTREP and SapHis, either alone or mixed, were set up using identical concentrations of protein in a mixture containing 20 mM Tris-HCl (pH 8.0), 2.5 mM desthiobiotin, and 125 mM imidazole.
Electron microscopy.
Affinity-purified protein or eluates of analytical size exclusion chromatography experiments were spotted onto glow-discharged, carbon-coated (Edwards Auto 306 Evaporator) copper grids (400 mesh) and were subsequently negatively stained using 2% uranyl acetate (Electron Microscopy Services, University of Chicago). Images were recorded using a TeCNAi F30 (Philips/FEI) transmission electron microscope (field emission gun, 300-kV accelerating voltage, with a magnification of 20,000 to 100,000×) and a high-performance CCD camera with a 4,000-by-4,000 resolution. Images were acquired using the Gatan DigitalMicrograph software.
Synteny alignment.
Synteny alignment was constructed using SyntTax (http://archaea.u-psud.fr/synttax/) anchored at the csaA and sap genes with a 10% minimal search threshold.
RESULTS
B. anthracis slaQ is required for S-layer protein secretion.
The secA2-slaP operon is located immediately adjacent to and is divergently transcribed from the S-layer gene cluster (csaA-csaB-sap-eag) (Fig. 1A). Earlier work used transposon mutagenesis in B. anthracis Sterne 34F2(pXO1+, pXO2−) (wild type) and PCR mapping to identify insertional lesions in the vicinity of these operons (16, 33). Here we assessed several of these variants for S-layer protein secretion by fractionating B. anthracis cultures. Briefly, mid-log-phase cultures were centrifuged to separate proteins in the culture medium (M) from the bacteria. S-layer proteins were extracted with 3 M urea and removed with the supernatant after centrifugation (S [S-layer]). The bacterial sediment was lysed to release cellular proteins (lanes C in Fig. 1B). Proteins in each sample were precipitated with trichloroacetic acid (TCA), washed in acetone, and solubilized in hot SDS prior to analysis by Coomassie-stained SDS-PAGE or immunoblotting. The BAS0836::aad9 variant with a transposon insertion 102 nucleotides (nt) upstream of the BAS0836 open reading frame displayed an S-layer protein secretion phenotype (Fig. 1B). BAS0836 and BAS0835 are positioned downstream of and transcribed divergently from secA2-slaP (Fig. 1A). The BAS0836::aad9 variant secreted only small amounts of Sap and EA1 into the medium, whereas S-layer protein secretion was not affected in the BAS0835::aphA3 mutant (Fig. 1B). Secretion of proteins that are not destined for the S-layer, e.g., protective antigen (PA) and sortase A (SrtA), were not perturbed in the BAS0836 and BAS0835 mutants (Fig. 1B).
FIG 1.

The S-layer assembly gene Q (slaQ) is located adjacent to the S-layer gene cluster (csaA-csaB-sap-eag) and is required for efficient S-layer protein secretion. (A) The S-layer cluster includes genes for S-layer proteins Sap (sap) and EA1 (eag) as well as the gene coding for the pyruvyl transferase for modification of the secondary cell wall polysaccharide (csaB) and its associated carbohydrate transport gene (csaA). Genes for the secretion ATPase paralogue (secA2) and S-layer assembly factor P (slaP), together with slaQ, support S-layer protein secretion. Arrowheads identify transposon insertions upstream of slaQ (BAS0836) and within BAS0835, which is not involved in S-layer assembly. (B) Mid-log cultures of B. anthracis Sterne (wild type [WT]) and its BAS0836::aad9 and BAS0835::aphA3 variants were fractionated into medium (M), S-layer (S), and cellular (C) proteins and analyzed by Coomassie-stained SDS-PAGE and immunoblotting with antibodies (α) specific for Sap, EA1, protective antigen (PA [a secreted protein]), or sortase A (SrtA [a membrane protein]). Migratory positions of molecular weight markers are indicated, and the arrow identifies Sap and EA1 by Coomassie-stained SDS-PAGE.
BAS0836 was designated slaQ for S-layer assembly protein Q. The slaQ open reading frame is 375 nucleotides in length and encodes a 124-amino-acid (124-aa) protein with a calculated mass of 14,998.41 Da. To ascertain whether the S-layer phenotype of the BAS0836::aad9 mutant was caused by a defect in slaQ expression, the gene was deleted from the genome of wild-type B. anthracis (Fig. 2). When analyzed for S-layer assembly by fractionation of B. anthracis cultures, the ΔslaQ variant secreted reduced amounts of Sap and EA1 into the culture medium (Fig. 2A and B). Quantification of immune-reactive signals from three independent experimental determinations revealed that the B. anthracis ΔslaQ strain secretes less Sap and EA1 into the culture medium than wild-type bacilli (Table 3).
FIG 2.

Deletion of slaQ (ΔslaQ) diminished S-layer protein secretion in Bacillus anthracis. (A) Mid-log cultures of B. anthracis Sterne (WT) and the ΔslaQ and ΔslaQ(pSN7) (for plasmid-borne expression of wild-type slaQ) variants were fractionated into medium (M), S-layer (S), and cellular (C) proteins and analyzed by Coomassie-stained SDS-PAGE. (B) Fractionated cultures were analyzed by immunoblotting with antibodies (α) specific for PA, BslA, Sap, EA1, BslO, SrtA, SecA, SlaP, and SlaQ. Migratory positions of molecular weight markers are indicated; the arrow identifies Sap and EA1 by Coomassie-stained SDS-PAGE. (C) Then abundance of S-layer proteins (Sap and EA1) and a control, ribosomal protein L6, in B. anthracis cultures was assessed by immunoblotting and signal intensity in three independent experimental determinations quantified with ImageJ and analyzed with the unpaired, two-tailed Student's t test (*, P < 0.01). (D) Vegetative growth of B. anthracis Sterne (WT) and the ΔslaQ and ΔslaQ(pSN7) variants was monitored as increased absorbance at 600 nm (A600).
TABLE 3.
Distribution of S-layer proteins in wild-type Bacillus anthracis and the ΔslaQ mutant
| Strain and protein location | % of protein in medium, S-layer, or cell lysatea |
|||
|---|---|---|---|---|
| Sap | EA1 | BslA | BslO | |
| Wild type | ||||
| Medium | 40.2 ± 0.05 | 50.8 ± 0.09 | 37.4 ± 0.13 | 17.1 ± 0.18 |
| S-layer | 38.4 ± 0.04 | 42.4 ± 0.04 | 40.7 ± 0.06 | 76.9 ± 0.06 |
| Cell lysate | 21.4 ± 0.03 | 6.9 ± 0.05 | 21.9 ± 0.79 | 17.7 ± 0.04 |
| ΔslaQ strain | ||||
| Medium | 20.1 ± 0.03* | 24.0 ± 0.08* | 37.3 ± 0.02 | 26.8 ± 0.05 |
| S-layer | 56.7 ± 0.03* | 65.9 ± 0.08* | 44.8 ± 0.01 | 63.8 ± 0.05 |
| Cell lysate | 23.2 ± 0.06 | 10.1 ± 0.03 | 17.9 ± 0.021 | 9.5 ± 0.01 |
| ΔslaQ(pSN7) strain | ||||
| Medium | 40.4 ± 0.12 | 46.6 ± 0.14 | 40.4 ± 0.05 | 19.1 ± 0.18 |
| S-layer | 45.0 ± 0.05 | 46.9 ± 0.15 | 49.0 ± 0.07 | 76.0 ± 0.17 |
| Cell lysate | 14.6 ± 0.07 | 6.5 ± 0.06 | 10.6 ± 0.02 | 4.8 ± 0.01 |
*, P < 0.05 by Student's t test (n = 3).
When assessed by immunoblotting with specific antibodies, slaQ expression was abolished in the ΔslaQ variant; however, the abundance of SecA2 and SlaP, the known secretion factors for S-layer proteins, was not affected (Fig. 2B). Furthermore, S-layer deposition of BslA and BslO, secretion of PA, or the abundance of SrtA was not affected (Fig. 2B). Immunoblotting revealed that the abundance of Sap and EA1 was reduced in the ΔslaQ mutant compared to wild-type B. anthracis, indicating that S-layer assembly was diminished (Fig. 2C). The defects in S-layer protein secretion and assembly in the ΔslaQ mutant were restored to wild-type levels when the mutant was transformed with pSN7, which provides for expression of wild-type slaQ (Fig. 2A to C). B. anthracis vegetative growth in BHI broth was not affected by the ΔslaQ mutation or by pSN7 (Fig. 2D).
The ΔslaQ mutant mislocalizes Sap and BslO in the bacterial S-layer.
BslO, an SLH domain-containing murein hydrolase, has been implicated in the separation of vegetative bacilli following cell division and in controlling chain length (15). Furthermore, the assembly of Sap on the surface directs proper localization of BslO to the site of activity at the septum (16). To analyze BslO localization in wild-type and ΔslaQ mutant bacilli, vegetative cells that had been sampled 2 or 4 h following germination were incubated with BslO-specific antibodies and analyzed by fluorescence microscopy. BODIPY-vancomycin was used to stain newly synthesized peptidoglycan at the division septum. BslO puncta were observed proximal to the septum in both wild-type and ΔslaQ cells, albeit that BslO staining appeared increased in the ΔslaQ mutant (Fig. 3B).
FIG 3.

Deposit of Sap and BslO into the S-layer of ΔslaQ mutant Bacillus anthracis. Spores derived from B. anthracis Sterne (WT) and its ΔslaQ and ΔslaQ(pSN7) variants were germinated, and vegetative forms were fixed after 2 h (A and B) and 4 h (C and D) of incubation. Vegetative cells were subjected to immunofluorescence using rabbit antibodies specific for Sap (A and C) or BslO (B and D) followed by anti-rabbit secondary fluorophore conjugates. Bacilli were also stained with BODIPY-vancomycin to identify the cell wall envelope and septa of vegetative chains. Scale bar, 2 μm.
When stained with Sap-specific antibodies, the ΔslaQ mutant displayed discontinuous patches of intensity on the cell surface, unlike wild-type bacilli, which distribute the S-layer protein uniformly along their cylindrical envelope (Fig. 3A). Of note, discontinuous distribution of Sap was only observed for cells harvested at early time points during growth—i.e., 2 h postgermination. At the 4-hour time point, a more uniform S-layer distribution of Sap was observed, albeit its abundance, measured by the intensity of its staining, was reduced compared to that of wild-type bacilli (Fig. 3C and D). The abundance and distribution of Sap and BslO were restored to wild-type levels in the B. anthracis ΔslaQ(pSN7) strain (Fig. 3). The distributions of EA1, the late-stage S-layer protein, appeared similar in wild-type and ΔslaQ mutant cells: i.e., EA1 was deposited at the poles and septum (Fig. 4). Thus, inactivation of ΔslaQ results in the transient mislocalization of Sap during early exponential growth and in the reduced deposition of S-layer proteins in the envelope throughout the growth cycle.
FIG 4.

Deposition of EA1 into the S-layer of ΔslaQ mutant Bacillus anthracis. Spores derived from B. anthracis Sterne (WT) and its ΔslaQ and ΔslaQ(pSN7) variants were germinated, and vegetative forms were fixed after 8 h of incubation. Vegetative cells were subjected to immunofluorescence using rabbit antibodies specific for EA1 followed by anti-rabbit secondary antibody–fluorophore conjugates. Bacilli were also stained with BODIPY-vancomycin to identify the cell wall envelope and septa of vegetative chains. Scale bar, 2 μm.
SlaQ sedimentation in B. anthracis lysates requires S-layer protein production.
Data in Fig. 2B indicate that SlaQ is located in bacterial cells and not secreted or deposited in the S-layer. In agreement with this, SlaQ does not encompass a hydrophobic signal peptide or membrane anchor (34). To localize SlaQ in bacterial cells, wild-type B. anthracis and the ΔslaQ variant were grown to the mid-log phase. Bacilli were sedimented by centrifugation and lysed, and cleared lysates were subjected to ultracentrifugation for 30 min at 100,000 × g, thereby separating soluble proteins in the supernatant (Sol) from proteins that sediment with the plasma membrane (PM) (Fig. 5). The experiment was performed in triplicate to quantify immune-reactive species: 54.45% ± 0.12% of SlaQ was found soluble in the cytoplasm (Sol), but 45.55% ± 0.12% sedimented with the plasma membrane (PM). Proteins in the plasma membrane fraction were further subjected to extraction with 0.1 M sodium carbonate (Na2CO3 [pH 11]) (35). Next, samples were subjected to ultracentrifugation for 30 min at 100,000 × g to separate proteins that are solubilized by sodium carbonate (soluble [S]) from integral membrane proteins that are refractory to sodium carbonate extraction and remain in the sediment (pellet [P]) (Fig. 5). Treatment with sodium carbonate extracted 93.02% ± 0.092% of SlaQ from membranes. Thus, although SlaQ cosediments with membrane proteins, this polypeptide does not insert into the lipid bilayer. As a control, sortase A (SrtA), an integral membrane protein that cannot be extracted with sodium carbonate treatment, was identified by immunoblotting (Fig. 5). SlaQ association with the plasma membrane was also examined in Δsap eag mutant bacilli, which cannot synthesize S-layer protein precursors (22). In Δsap eag mutant cells, SlaQ was found to be mostly soluble, and its membrane association was diminished (Fig. 5). These data suggest that SlaQ associates with the plasma membrane of B. anthracis during the secretion of S-layer protein precursors.
FIG 5.

SlaQ sedimentation in lysates of B. anthracis requires S-layer protein precursor expression. Mid-log-phase cultures of B. anthracis Sterne (WT) or its ΔslaQ and Δsap eag variants were lysed, unbroken cells were removed (8,000 × g), and the lysate was subjected to ultracentrifugation (100,000 × g) to separate soluble proteins (Sol) from others that sediment with the plasma membrane (PM). The sediment was then extracted with the buffer control (−) or with 0.1 M sodium carbonate at pH 11 (Na2CO3) and again subjected to ultracentrifugation (100,000 × g), separating soluble proteins in the supernatant (S) from insoluble proteins in the pellet (P). Proteins in all samples were precipitated with trichloroacetic acid, washed in acetone, and analyzed by immunoblotting with antibodies specific for SlaQ and SrtA, a membrane protein control.
Secretion and S-layer assembly of Sap-mCherry hybrids.
Sap is synthesized as an 814-amino-acid precursor protein harboring an N-terminal signal peptide (residues 1 to 30), three SLH domains (residues 34 to 197), and a large C-terminal domain (residues 210 to 814) that promotes the crystallization of the S-layer protein (4, 36) (Fig. 6A). Predictions of secondary sequences suggest a high β-strand content in the C-terminal crystallization domain, with the possible formation of a bacterial Ig-like domain 2-fold between residues 211 and 476 (16) (Fig. 6A). We generated translational hybrids between the 3′ ends of various sap truncation mutants and the mCherry gene, which encodes a 28.8-kDa reporter protein (37). The first 100, 400, or 814 (full-length) codons of sap were fused to the 5′ end of the mCherry open reading frame. All constructs were cloned into pJK4, which provides for their IPTG-inducible expression via the Pspac promoter (16), and recombinant plasmids were transformed into B. anthracis Sterne (WT). Vegetative bacilli were grown to mid-log-phase cultures, fractionated into culture medium (M), S-layer (S), and bacterial cells (C), and analyzed by immunoblotting (Fig. 6B). When expressed in B. anthracis, mCherry remained in the bacterial cytoplasm, whereas Sap1–100-mCherry was secreted into the culture medium (Fig. 6B). Sap1–400-mCherry, which encompasses the signal peptide and the three SLH domains of Sap, was secreted into the culture medium and deposited in the S-layer compartment, similar to the fractionation profile of wild-type Sap and Sap1–814-mCherry (Fig. 6B).
FIG 6.

Secretion and S-layer assembly of Sap-mCherry hybrids. (A) The 3′ end of full-length sap coding sequence was fused to the 5′ end of the mCherry open reading frame to generate Sap1–814-mCherry. Other translational hybrids between sap and mCherry omitted coding sequence for the crystallization domain (Sap1–400-mCherry) with or without the SLH domains (Sap1–100-mCherry) from sap and were compared to expression with mCherry alone. (B) Plasmids expressing mCherry, Sap1–100-mCherry, Sap1–400-mCherry, or Sap1–814-mCherry were transformed into B. anthracis Sterne, and mid-log cultures were fractionated to separate proteins in the medium (M), S-layer (S), and cellular lysate (C). Samples were immunoblotted with antibodies specific for Sap, mCherry, and SrtA. The arrow denotes the migratory position of Sap, whereas the arrowheads identify Sap-mCherry hybrids. Plasmids for the expression of Sap1–400-mCherry or Sap1–814-mCherry were transformed into secA2, slaP, and ΔslaQ mutant strains and analyzed as described in the legend to panel B.
mCherry reporter plasmids were transformed into secA2, slaP, or slaQ mutant B. anthracis, and protein secretion and deposition into the bacterial envelope were examined. Similar to wild-type B. anthracis, mCherry and Sap1–100-mCherry were found in the cytoplasm and in the culture medium, respectively (data not shown). In contrast to wild-type bacilli, the secA2 and slaP mutants secreted reduced amounts of Sap1–400-mCherry and Sap1–814-mCherry into the culture medium, indicating that efficient secretion of the Sap signal peptide and SLH domains requires both SecA2 and SlaP (Fig. 6C). Secretion of Sap1–814-mCherry but not of Sap1–400-mCherry required the expression of slaQ. These data suggest that SlaQ contributes to secretion and assembly of the C-terminal crystallization domain of Sap, which is located between residues 211 and 814, whereas signal peptide-mediated initiation of the precursor or transport of the SLH domains is dependent on SecA2 and SlaP but not SlaQ (Fig. 6C).
Purification of SlaQ from B. anthracis.
To purify SlaQ from bacilli, the coding sequence for the 6-histidine affinity tag was inserted after the start codon of slaQ and the recombinant HisslaQ gene was expressed from the Pspac promoter in pSN9. As assessed by immunoblotting, pSN9 restored the expression of slaQ and the secretion of Sap and EA1 in the B. anthracis ΔslaQ mutant (Fig. 7A). For purification, B. anthracis ΔslaQ(pSN9) vegetative cells were lysed, cellular debris was removed by centrifugation at 8,000 × g, and bacterial lysate was subjected to ultracentrifugation at 100,000 × g, thereby separating soluble and insoluble proteins. The cleared lysate (soluble proteins) was subjected to affinity chromatography on Ni-NTA-Sepharose and eluted with imidazole (Fig. 7B). HisSlaQ was detected in the eluate by silver-stained SDS-PAGE and immunoreactivity to recombinant SlaQ (Fig. 8B and C). Several other proteins were detected in the Ni-NTA affinity chromatography eluate from the B. anthracis ΔslaQ(pSN9) lysate; however, these proteins were also detected in Ni-NTA affinity chromatography eluate from B. anthracis ΔslaQ(pSN7) (for plasmid-borne expression of wild-type SlaQ), suggesting that they do not specifically associate with HisSlaQ (Fig. 7B). Aliquots of the lysate (L), wash (W), and eluate (E) derived from affinity chromatography experiments were analyzed by immunoblotting, which revealed the purification of HisSlaQ but not of SlaQ (Fig. 7D). Sap, EA1, SecA2, and SlaP did not copurify with soluble HisSlaQ, suggesting that soluble SlaQ does not form stable cytoplasmic complexes with other secretion factors or S-layer proteins (Fig. 7D).
FIG 7.

Purification of HisSlaQ from Bacillus anthracis. (A) Mid-log cultures of B. anthracis Sterne (WT), ΔslaQ and ΔslaQ(pSN9) (for plasmid-borne expression of affinity tagged HisSlaQ) were fractionated into medium (M), S-layer (S), and cellular (C) proteins, and the proteins were subjected to Coomassie-stained SDS-PAGE or immunoblotting with antibodies (α) specific for PA, Sap, EA1, SrtA, and SlaQ. Migratory positions of molecular weight markers are indicated; the arrow identifies Sap and EA1 on Coomassie-stained SDS-PAGE. (B and C) Cleared lysates derived from B. anthracis ΔslaQ(pSN7) (SlaQ) and ΔslaQ(pSN9) (HisSlaQ) were subjected to affinity chromatography on Ni-NTA-Sepharose and eluted with imidazole, and the eluate was analyzed by silver-stained SDS-PAGE (B) or His probe staining (C). The arrow denotes the migratory position of HisSlaQ. (D) Load, wash, and eluate fractions of the affinity chromatography samples in panel B were analyzed by immunoblotting with antibodies specific for Sap, EA1, SecA2, SlaP, and SlaQ.
FIG 8.
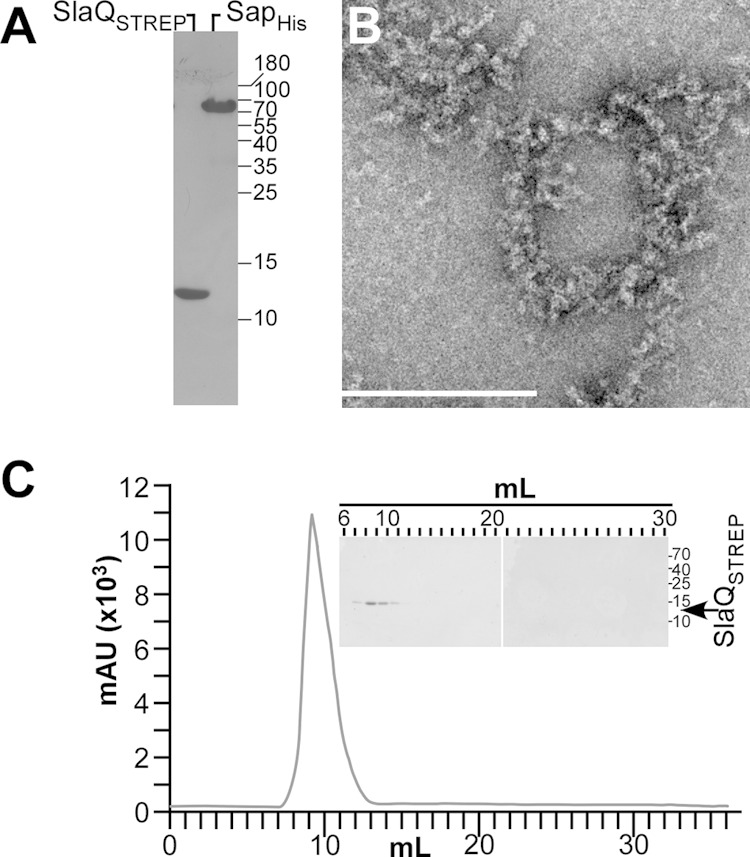
Assembly of SlaQ into high-molecular-weight structures. (A) Recombinant SlaQSTREP was purified by StrepTactin affinity chromatography from cleared lysate of E. coli. Recombinant SapHis, lacking the N-terminal signal peptide of wild-type Sap, was purified by Ni-NTA affinity chromatography from cleared lysate of E. coli. Both proteins were diluted to a concentration of 2 mg/ml and analyzed by Coomassie-stained SDS-PAGE. (B) Affinity-purified SlaQSTREP was stained with uranyl acetate and viewed by transmission electron microscopy. Bar, 200 nm. (C) SlaQSTREP was subjected to analytical size exclusion chromatography, and milliabsorbance units (mAU) at 280 nm were recorded. (Inset) Eluate fractions from panel C were analyzed by Coomassie-stained SDS-PAGE.
SlaQ promotes in vitro S-layer assembly of Sap.
To study the role of SlaQ in in vitro Sap S-layer assembly, we generated affinity-tagged proteins, expressing slaQSTREP with an insertion of eight codons upstream of the stop codon, thereby generating SlaQSTREP with a C-terminal streptavidin tag (WSHPQFEK). Plasmid pSN15, with IPTG-inducible expression of slaQSTREP cloned into pJK4, was used to confirm that SlaQSTREP complements the Sap S-layer secretion and assembly phenotypes of the B. anthracis ΔslaQ mutant (data not shown). pSY185, with IPTG-inducible expression of slaQSTREP from the T7 promoter of pET24b, was used to purify recombinant SlaQSTREP via StrepTactin affinity chromatography from cleared lysates of E. coli BL21(DE3, pSY185) (Fig. 8A). When subjected to size exclusion chromatography on Superdex 200 at a concentration of 2 mg/ml, SlaQSTREP eluted as a peak at Mr 700,000 to 2,000,000, suggesting that the protein forms large structures (Fig. 8C). High-molecular-weight structures formed by SlaQSTREP could indeed be detected by electron microscopy of uranyl acetate-stained affinity-purified protein (Fig. 8B).
The sapHis gene was expressed from the T7 promoter of pSY183 in E. coli BL21(DE3) and lacks coding sequence for the N-terminal signal peptide of wild-type sap. Further, sapHis was modified with coding sequence for C-terminal histidine tag and linker identical to that used for the purification of G. stearothermophilus SbsB (KPNSADIHHTGGRSSHHHHHH) (38). Expression of sapHis in the cytoplasm of E. coli leads to the formation of crystalline assemblies (data not shown). Nevertheless, soluble SapHis could be purified by Ni-NTA affinity chromatography from E. coli lysate (Fig. 8A). When subjected to size exclusion chromatography on Superdex 200 at a concentration of 2 mg/ml, immediately following affinity chromatography (day 0), SapHis eluted over many fractions, indicating random multimer formation (Fig. 9A). However, when SapHis (2 mg/ml) was incubated for 4 days at 4°C prior to chromatography, the protein eluted in a narrow and well-defined absorption peak with an elution volume of 13 to 15 ml (Fig. 9B). Electron microscopy of this sample revealed tubular S-layer structures formed by SapHis (Fig. 9C and D). Thus, similar to Geobacillus SbsB (39), purified SapHis is endowed with the propensity of self-assembly into an S-layer structure. Self-assembly of SapHis at a concentration of 2 mg/ml required time and could be observed after 4 days of incubation at 4°C.
FIG 9.

Self-assembly of Sap into S-layers. Purified SapHis was incubated at a concentration of 2 mg/ml at 4°C. (A) When subjected to analytical size exclusion chromatography immediately after affinity purification and dilution (day 0), eluate was monitored via absorbance at 280 nm, silver-stained SDS-PAGE, and electron microscopy. S-layer assemblies of SapHis were not detectable on day 0. (B) When analyzed by analytical size exclusion chromatography after 4 days of incubation at 4°C, SapHis eluted as a defined peak at 14 ml. (C and D) Electron microscopy of 14 ml eluate revealed tubular S-layer structures.
When SapHis was mixed with SlaQSTREP immediately after affinity chromatography purification and subjected to size exclusion chromatography, the elution profile revealed a defined, narrow peak at 8 to 10 ml that contained both proteins (Fig. 10A). A similar size exclusion elution profile was observed following incubation over 4 days, and large amounts of SlaQSTREP/SapHis eluted together at 8 to 10 ml (Fig. 10B). Electron microcopy revealed abundant and elongated bundles of tubular S-layer structures several micrometers in length immediately after the mixing of affinity-purified SlaQSTREP and SapHis (Fig. 10C). Similar structures were observed in the 8-ml elution peak of SlaQSTREP/SapHis during size exclusion chromatography (Fig. 10D). Thus, in the presence of SlaQSTREP, SapHis rapidly forms elongated tubular S-layer structures. Taken together, our data suggest that SlaQ may facilitate Sap assembly at the membrane and, in conjunction with SecA2/SlaP, promote S-layer protein secretion and assembly in B. anthracis.
FIG 10.

SlaQ accelerates Sap S-layer assembly. Purified SapHis and SlaQSTREP were mixed and incubated at a concentration of 2 mg/ml at 4°C. (A) Mixed SapHis and SlaQSTREP were subjected to analytical size exclusion chromatography immediately after affinity purification and dilution (day 0). The SapHis/SlaQSTREP peak at 8 ml was detected by absorbance and silver-stained SDS-PAGE. (B) Analytical size exclusion chromatography after 4 days of incubation (day 4), revealed a similar pattern. (C) Mixed SapHis and SlaQSTREP were stained with uranyl acetate and viewed by transmission electron microscopy. Bar, 200 nm. (D) Electron microscopy of the 8-ml size exclusion chromatography sample on day 4 (B) detected tubular S-layer structures. Bar, 200 nm.
DISCUSSION
Proteins destined for secretion are synthesized as precursors with N-terminal signal peptides (40). In bacteria, the SecYEG proteins form a translocation channel in the plasma membrane that is engaged when SecA, the secretion ATPase, recruits precursor proteins and moves them through the SecYEG channel (25, 41, 42). The genomes of some Gram-positive bacteria harbor two secA genes: one that encodes the canonical SecA ATPase of the protein secretory pathway (23) and another that specifies a paralogue, SecA2, supporting the secretion of only one or a few select substrates (43). SecA2 was first identified in Mycobacterium tuberculosis, where it promotes the transport of proteins across the mycobacterial envelope that aid in the pathogenesis of tuberculosis (44, 45). Mycobacterial SecA2 recognizes the mature domains of a few secretory precursors to assist in their secretion, suggesting its primary function may be maintenance of export competence for proteins with the propensity for rapid folding (46). Listeria monocytogenes secA2 was identified because mutations in the structural gene affect colony shape and cellular morphology, a phenotype that is attributed to the diminished secretion of cell wall hydrolases (i.e., p60 autolysin, NamA hydrolase, and two dozen other substrates) (47–49). The gene for the Listeria SecA2 p60 substrate, cwhA, is located immediately adjacent to secA2, and this locus is conserved among pathogenic and nonpathogenic Listeria species (50). Thus, unlike M. tuberculosis, the Listeria secA2 pathway does not appear to be specifically involved in disease pathogenesis.
Streptococcus gordonii secA2 was first identified in a screen for diminished binding of bacteria to human platelets (51). This phenotype could be assigned to the loss of secretion of GspB, a large, glycosylated cell-wall-anchored protein (51). In streptococci and related microbes expressing GspB paralogues, secA2 does not act alone, as GspB secretion requires accessory factors, including secY2, a paralogue of secY, and asp1 to -5 (52). The Asp1 to -5 factors are unique to the accessory GspB pathway and interact with mature domains of the secretion precursor (53, 54).
Gram-positive microbes that express SecA paralogues may or may not express a SecY paralogue. For example, the genome of B. anthracis harbors secY2; however, the structural gene is not clustered with secA2, slaP, and slaQ, and secY2 does not contribute to S-layer protein secretion (22). Clostridium difficile, another Gram-positive spore-forming microbe, assembles its S-layer from secreted S-layer proteins (SLPs), and SlpA and CwpV, a cell wall protein, require SecA2 for secretion of their precursors (55, 56). The genome of C. difficile does not encode a SecY paralogue (56). We show here that B. anthracis requires slaP and slaQ, encoding small cytoplasmic polypeptides, along with secA2 for efficient secretion of S-layer protein precursors, Sap and EA1. While SecA2 and SlaP are required for efficient secretion of mCherry hybrids encompassing the signal peptide and SLH domains of Sap, SlaQ functions to promote secretion of mCherry hybrids that include the C-terminal crystallization domain. Work with S-layer proteins from several different bacteria, including B. anthracis Sap, demonstrated the propensity of crystallization domains to rapidly assemble into two-dimensional paracrystalline lattices (4, 5, 57). Factors that promote the assembly of S-layer proteins have not yet been reported.
We observed that B. anthracis SlaQ, a small cytoplasmic protein, sediments with bacterial membranes in a manner requiring expression of S-layer protein precursors (Sap and EA1). Affinity chromatography experiments revealed that soluble HisSlaQ, purified from sedimented B. anthracis cell extracts, did not copurify with Sap or EA1. Thus, soluble SlaQ does not appear to form stable complexes with either of these two proteins. Purified SlaQSTREP, on the other hand, assembles into high-molecular-weight structures that can be purified by size exclusion chromatography and are detectable by electron microscopy. We propose this attribute of SlaQSTREP can assist in the secretion and S-layer assembly of Sap and EA1 precursors at the bacterial membrane. In agreement with this model, self-assembly of purified SapHIS is accelerated in the presence of SlaQSTREP. It is conceivable that SlaQ may constitute a scaffold for the rapid assembly of Sap; however, we cannot exclude other possibilities—for example, that SlaQ functions as a folding chaperone for S-layer assembly. Whatever the molecular underpinnings of SlaQ function, both proteins Sap and SlaQ physically interact with one another, as evidenced by the in vivo cosedimentation data and by the coelution of SapHis and SlaQSTREP during size exclusion chromatography. Future experiments need to distinguish between several different possibilities, including SlaQ-mediated stabilization of S-layer folding intermediates or the chelation of calcium ions that coordinate interdomain interactions of crystallization units for the formation of a condensed quaternary structure (3).
The function of SlaQ toward S-layer assembly may be conserved in pathogenic bacilli, including B. anthracis, Bacillus cereus, Bacillus thuringiensis, and Bacillus cytotoxicus, whose genomes contain S-layer gene clusters (csaA-csaB-sap-eag) with accessory secretion genes (secA2-slaP-slaQ) (see Fig. S1 in the supplemental material). Thus, the accessory secretion factors SlaP and SlaQ appear to have evolved specifically to support secretion and assembly of Bacillus S-layer proteins. If so, S-layer protein precursors in other bacteria (for example, C. difficile) may have coevolved with specific secretion factors that support membrane translocation of their envelope binding domain, designated CWB2 in C. difficile, or of their crystallization domains (1).
Supplementary Material
ACKNOWLEDGMENTS
We thank members of our laboratory and Yimei Chen (Electron Microscopy facility at the University of Chicago) for experimental advice and discussion.
This work was supported by a grant from the National Institute of Allergy and Infectious Diseases, Infectious Diseases Branch (AI069227), to O.S. S.-M.N.-M. was supported by NIH training grant GM007183 (Molecular Cell Biology).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00492-15.
REFERENCES
- 1.Fagan RP, Fairweather NF. 2014. Biogenesis and functions of bacterial S-layers. Nat Rev Microbiol 12:211–222. doi: 10.1038/nrmicro3213. [DOI] [PubMed] [Google Scholar]
- 2.Etienne-Toumelin I, Sirard JC, Duflot E, Mock M, Fouet A. 1995. Characterization of the Bacillus anthracis S-layer: cloning and sequencing of the structural gene. J Bacteriol 177:614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baranova E, Fronzes R, Garcia-Pino A, Van Gerven N, Papapostolou D, Péhau-Arnaudet G, Pardon E, Steyaert J, Howorka S, Remaut H. 2012. SbsB structure and lattice reconstruction unveil Ca2+ triggered S-layer assembly. Nature 487:119–122. doi: 10.1038/nature11155. [DOI] [PubMed] [Google Scholar]
- 4.Candela T, Mignot T, Hagenrelle X, Haustant M, Fouet A. 2005. Genetic analysis of Bacillus anthracis Sap S-layer protein crystallization domain. Microbiology 151:1485–1490. doi: 10.1099/mic.0.27832-0. [DOI] [PubMed] [Google Scholar]
- 5.Pum D, Toca-Herrera JL, Sleytr UB. 2013. S-layer protein self-assembly. Int J Mol Sci 14:2484–2501. doi: 10.3390/ijms14022484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shin SH, Comolli LR, Tscheliessnig R, Wang C, Nam KT, Hexemer A, Siegerist CE, De Yoreo JJ, Bertozzi CR. 2013. Self-assembly of “S-bilayers,” a step toward expanding the dimensionality of S-layer assemblies. ACS Nano 7:4946–4953. doi: 10.1021/nn400263j. [DOI] [PubMed] [Google Scholar]
- 7.Koch R. 1876. Die Ätiologie der Milzbrand-Krankheit, begründet auf die Entwicklungsgeschichte des Bacillus anthracis. Beitr Biol Pflanz 2:277–310. [Google Scholar]
- 8.Mock M, Fouet A. 2001. Anthrax. Annu Rev Microbiol 55:647–671. doi: 10.1146/annurev.micro.55.1.647. [DOI] [PubMed] [Google Scholar]
- 9.Green BD, Battisti L, Koehler TM, Thorne CB, Ivins BE. 1985. Demonstration of a capsule plasmid in Bacillus anthracis. Infect Immun 49:291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okinaka RT, Cloud K, Hampton O, Hoffmaster AR, Hill KK, Keim P, Koehler TM, Lamke G, Kumano S, Mahillon J, Manter D, Martinez Y, Ricke D, Svensson R, Jackson PJ. 1999. Sequence and organization of pXO1, the large Bacillus anthracis plasmid harboring the anthrax toxin genes. J Bacteriol 181:6509–6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sterne M. 1937. Avirulent anthrax vaccine. Onderstepoort J Vet Sci Anim Ind 21:41–43. [PubMed] [Google Scholar]
- 12.Pasteur L. 1881. Le vaccin du charbon. C R Hebd Scéances Acad Sci 92:666–668. [Google Scholar]
- 13.Turnbull PCB. 1991. Anthrax vaccines: past, present and future. Vaccine 9:533–539. doi: 10.1016/0264-410X(91)90237-Z. [DOI] [PubMed] [Google Scholar]
- 14.Ruthel G, Ribot WJ, Bavari S, Hoover T. 2004. Time-lapse confocal imaging of development of Bacillus anthracis in macrophages. J Infect Dis 189:1313–1316. doi: 10.1086/382656. [DOI] [PubMed] [Google Scholar]
- 15.Anderson VJ, Kern JW, McCool JW, Schneewind O, Missiakas DM. 2011. The SLH domain protein BslO is a determinant of Bacillus anthracis chain length. Mol Microbiol 81:192–205. doi: 10.1111/j.1365-2958.2011.07688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kern VJ, Kern JW, Theriot JA, Schneewind O, Missiakas DM. 2012. Surface (S)-layer proteins Sap and EA1 govern the binding of the S-layer associated protein BslO at the cell septa of Bacillus anthracis. J Bacteriol 194:3833–3840. doi: 10.1128/JB.00402-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kern J, Ryan C, Faull K, Schneewind O. 2010. Bacillus anthracis surface-layer proteins assemble by binding to the secondary cell wall polysaccharide in a manner that requires csaB and tagO. J Mol Biol 401:757–775. doi: 10.1016/j.jmb.2010.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lunderberg JM, Nguyen-Mau SM, Richter GS, Wang YT, Dworkin J, Missiakas DM, Schneewind O. 2013. Bacillus anthracis acetyltransferases PatA1 and PatA2 modify the secondary cell wall polysaccharide and affect the assembly of S-layer proteins. J Bacteriol 195:977–989. doi: 10.1128/JB.01274-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choudhury B, Leoff C, Saile E, Wilkins P, Quinn CP, Kannenberg EL, Carlson RW. 2006. The structure of the major cell wall polysaccharide of Bacillus anthracis is species specific. J Biol Chem 281:27932–27941. doi: 10.1074/jbc.M605768200. [DOI] [PubMed] [Google Scholar]
- 20.Forsberg LS, Abshire TG, Friedlander A, Quinn CP, Kannenberg EL, Carlson RW. 2012. Localization and structural analysis of a conserved pyruvylated epitope in Bacillus anthracis secondary cell wall polysaccharides and characterization of the galactose deficient wall polysaccharide from avirulent B. anthracis CDC 684. Glycobiology 22:1103–1117. doi: 10.1093/glycob/cws080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mesnage S, Fontaine T, Mignot T, Delepierre M, Mock M, Fouet A. 2000. Bacterial SLH domain proteins are non-covalently anchored to the cell surface via a conserved mechanism involving wall polysaccharide pyruvylation. EMBO J 19:4473–4484. doi: 10.1093/emboj/19.17.4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen-Mau S-M, Oh SY, Kern V, Missiakas D, Schneewind O. 2012. Secretion genes as determinants of Bacillus anthracis chain length. J Bacteriol 194:3841–3850. doi: 10.1128/JB.00384-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oliver DB, Beckwith J. 1981. E. coli mutant pleiotropically defective in the export of secreted proteins. Cell 25:765–772. doi: 10.1016/0092-8674(81)90184-7. [DOI] [PubMed] [Google Scholar]
- 24.Economou A, Wickner W. 1994. SecA promotes preprotein translocation by undergoing ATP-driven cycles of membrane insertion and deinsertion. Cell 78:835–843. doi: 10.1016/S0092-8674(94)90582-7. [DOI] [PubMed] [Google Scholar]
- 25.Duong F, Wickner W. 1997. The SecDFYajC domain of preprotein translocase controls preprotein movement by regulating SecA membrane cycling. EMBO J 16:4871–4879. doi: 10.1093/emboj/16.16.4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marraffini LA, Schneewind O. 2006. Targeting proteins to the cell wall of sporulating Bacillus anthracis. Mol Microbiol 62:1402–1417. doi: 10.1111/j.1365-2958.2006.05469.x. [DOI] [PubMed] [Google Scholar]
- 27.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–572. doi: 10.1016/S0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 28.Fulford W, Model P. 1984. Specificity of translational regulation by two DNA-binding proteins. J Mol Biol 173:211–226. doi: 10.1016/0022-2836(84)90190-6. [DOI] [PubMed] [Google Scholar]
- 29.Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. 1990. Use of T7 polymerase to direct expression of cloned genes. Methods Enzymol 185:60–89. doi: 10.1016/0076-6879(90)85008-C. [DOI] [PubMed] [Google Scholar]
- 30.Kim HU, Goepfert JM. 1974. A sporulation medium for Bacillus anthracis. J Appl Bacteriol 37:265–267. doi: 10.1111/j.1365-2672.1974.tb00438.x. [DOI] [PubMed] [Google Scholar]
- 31.Gaspar AH, Marraffini LA, Glass EM, DeBord KL, Ton-That H, Schneewind O. 2005. Bacillus anthracis sortase A (SrtA) anchors LPXTG motif-containing surface proteins to the cell wall envelope. J Bacteriol 187:4646–4655. doi: 10.1128/JB.187.13.4646-4655.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kern JW, Schneewind O. 2008. BslA, a pXO1-encoded adhesin of Bacillus anthracis. Mol Microbiol 68:504–515. doi: 10.1111/j.1365-2958.2008.06169.x. [DOI] [PubMed] [Google Scholar]
- 33.Tam C, Glass EM, Anderson DM, Missiakas D. 2006. Transposon mutagenesis of Bacillus anthracis strain Sterne using bursa aurealis. Plasmid 56:74–77. doi: 10.1016/j.plasmid.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 34.Eisenberg D, Schwarz E, Komaromy M, Wall R. 1984. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J Mol Biol 179:125–142. doi: 10.1016/0022-2836(84)90309-7. [DOI] [PubMed] [Google Scholar]
- 35.Steck TL, Yu J. 1973. Selective solubilization of proteins from red blood cell membranes by protein perturbants. J Supramol Struct 1:220–232. doi: 10.1002/jss.400010307. [DOI] [PubMed] [Google Scholar]
- 36.Kern JW, Wilton R, Zhang R, Binkowski A, Joachimiak A, Schneewind O. 2011. Structure of the SLH domains from Bacillus anthracis surface array protein. J Biol Chem 286:26042–26049. doi: 10.1074/jbc.M111.248070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. 2004. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein Nat Biotechnol 22:1567–1572. [DOI] [PubMed] [Google Scholar]
- 38.Howorka S, Sára M, Wang Y, Kuen B, Sleytr U, Lubitz W, Bayley H. 2000. Surface-accessible residues in the monomeric and assembled forms of a bacterial surface layer protein. J Biol Chem 275:37876–37886. doi: 10.1074/jbc.M003838200. [DOI] [PubMed] [Google Scholar]
- 39.Kinns H, Badelt-Lichtblau H, Egelseer EM, Sleytr UB, Howorka S. 2010. Identifying assembly-inhibiting and assembly-tolerant sites in the SbsB S-layer protein from Geobacillus stearothermophilus. J Mol Biol 395:742–753. doi: 10.1016/j.jmb.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 40.Blobel G, Dobberstein B. 1975. Transfer of proteins across membranes. I. Presence of proteolytically processed and unprocessed nascent immunoglobulin light chains on membrane-bound ribosomes of murine myeloma. J Cell Biol 67:835–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Economou A, Pogliano JA, Beckwith J, Oliver DB, Wickner W. 1995. SecA membrane cycling at SecYEG is driven by distinct ATP binding and hydrolysis events and is regulated by SecD and SecF. Cell 83:1171–1181. doi: 10.1016/0092-8674(95)90143-4. [DOI] [PubMed] [Google Scholar]
- 42.Chatzi KE, Sardis MF, Karamanou S, Economou A. 2013. Breaking on through to the other side: protein export through the bacterial Sec system. Biochem J 449:25–37. doi: 10.1042/BJ20121227. [DOI] [PubMed] [Google Scholar]
- 43.Feltcher ME, Braunstein M. 2012. Emerging themes in SecA2-mediated protein export. Nat Rev Microbiol 10:779–789. doi: 10.1038/nrmicro2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Braunstein M, Brown AM, Kurtz S, Jacobs WRJ. 2001. Two nonredundant SecA homologues function in mycobacteria. J Bacteriol 183:6979–6990. doi: 10.1128/JB.183.24.6979-6990.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Braunstein M, Espinosa B, Chan J, Beisle JT, Jacobs WRJ. 2003. SecA2 functions in the secretion of superoxide dismutase A and in the virulence of Mycobacterium tuberculosis. Mol Microbiol 48:453–464. doi: 10.1046/j.1365-2958.2003.03438.x. [DOI] [PubMed] [Google Scholar]
- 46.Feltcher ME, Gibbons HS, Ligon LS, Braunstein M. 2013. Protein export by the mycobacterial SecA2 system is determined by the preprotein mature domain. J Bacteriol 195:672–681. doi: 10.1128/JB.02032-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lenz LL, Portnoy DA. 2002. Identification of a second Listeria secA gene associated with protein secretion and the rough phenotype. Mol Microbiol 45:1043–1056. doi: 10.1046/j.1365-2958.2002.03072.x. [DOI] [PubMed] [Google Scholar]
- 48.Lenz LL, Mohammadi S, Geissler A, Portnoy DA. 2003. SecA2-dependent secretion of autolytic enzymes promotes Listeria monocytogenes pathogenesis. Proc Natl Acad Sci U S A 100:12432–12437. doi: 10.1073/pnas.2133653100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Renier S, Chambon C, Viala D, Chagnot C, Hébraud M, Desvaux M. 2013. Exoproteomic analysis of the SecA2-dependent secretion in Listeria monocytogenes EGD-e. J Proteomics 80:183–195. doi: 10.1016/j.jprot.2012.11.027. [DOI] [PubMed] [Google Scholar]
- 50.Mishra KK, Mendonca M, Aroonnual A, Burkholder KM, Bhunia AK. 2011. Genetic organization and molecular characterization of secA2 locus in Listeria species. Gene 489:76–85. doi: 10.1016/j.gene.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 51.Bensing BA, Sullam PM. 2002. An accessory sec locus of Streptococcus gordonii is required for export of the surface protein GspB and for normal levels of binding to human platelets. Mol Microbiol 44:1081–1094. doi: 10.1046/j.1365-2958.2002.02949.x. [DOI] [PubMed] [Google Scholar]
- 52.Seepersaud R, Bensing BA, Yen YT, Sullam PM. 2010. Asp3 mediates multiple protein-protein interactions within the accessory Sec system of Streptococcus gordonii. Mol Microbiol 78:490–505. doi: 10.1111/j.1365-2958.2010.07346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bensing BA, Sullam PM. 2010. Transport of preproteins by the accessory Sec system requires a specific domain adjacent to the signal peptide. J Bacteriol 192:4223–4232. doi: 10.1128/JB.00373-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Siboo IR, Chaffin DO, Rubens CE, Sullam PM. 2008. Characterization of the accessory Sec system of Staphylococcus aureus. J Bacteriol 190:6188–6196. doi: 10.1128/JB.00300-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fagan RP, Albesa-Jove D, Qazi O, Svergun DI, Brown KA, Fairweather NF. 2009. Structural insights into the molecular organization of the S-layer from Clostridium difficile. Mol Microbiol 71:1308–1322. doi: 10.1111/j.1365-2958.2009.06603.x. [DOI] [PubMed] [Google Scholar]
- 56.Fagan RP, Fairweather NF. 2011. Clostridium difficile has two parallel and essential Sec secretion systems. J Biol Chem 286:27483–27493. doi: 10.1074/jbc.M111.263889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sára M, Sleytr UB. 2000. S-layer proteins. J Bacteriol 182:859–868. doi: 10.1128/JB.182.4.859-868.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.