

ABSTRACT
The two-component control of virulence (Cov) regulator (R)-sensor (S) (CovRS) regulates the virulence of Streptococcus pyogenes (group A Streptococcus [GAS]). Inactivation of CovS during infection switches the pathogenicity of GAS to a more invasive form by regulating transcription of diverse virulence genes via CovR. However, the manner in which CovRS controls virulence through expression of extended gene families has not been fully determined. In the current study, the CovS-regulated gene expression profiles of a hypervirulent emm23 GAS strain (M23ND/CovS negative [M23ND/CovS−]) and a noninvasive isogenic strain (M23ND/CovS+), under different growth conditions, were investigated. RNA sequencing identified altered expression of ∼349 genes (18% of the chromosome). The data demonstrated that M23ND/CovS− achieved hypervirulence by allowing enhanced expression of genes responsible for antiphagocytosis (e.g., hasABC), by abrogating expression of toxin genes (e.g., speB), and by compromising gene products with dispensable functions (e.g., sfb1). Among these genes, several (e.g., parE and parC) were not previously reported to be regulated by CovRS. Furthermore, the study revealed that CovS also modulated the expression of a broad spectrum of metabolic genes that maximized nutrient utilization and energy metabolism during growth and dissemination, where the bacteria encounter large variations in available nutrients, thus restructuring metabolism of GAS for adaption to diverse growth environments. From constructing a genome-scale metabolic model, we identified 16 nonredundant metabolic gene modules that constitute unique nutrient sources. These genes were proposed to be essential for pathogen growth and are likely associated with GAS virulence. The genome-wide prediction of genes associated with virulence identifies new candidate genes that potentially contribute to GAS virulence.
IMPORTANCE The CovRS system modulates transcription of ∼18% of the genes in the Streptococcus pyogenes genome. Mutations that inactivate CovR or CovS enhance the virulence of this bacterium. We determined complete transcriptomes of a naturally CovS-inactivated invasive deep tissue isolate of an emm23 strain of S. pyogenes (M23ND) and its complemented avirulent variant (CovS+). We identified diverse virulence genes whose altered expression revealed a genetic switching of a nonvirulent form of M23ND to a highly virulent strain. Furthermore, we also systematically uncovered for the first time the comparative levels of expression of a broad spectrum of metabolic genes, which reflected different metabolic needs of the bacterium as it invaded deeper tissue of the human host.
INTRODUCTION
Streptococcus pyogenes is a group A Streptococcus (GAS) Gram-positive (Gram+) pathogen that afflicts humans with diseases ranging from mild pharyngitis and impetigo to severe invasive necrotizing fasciitis and toxic shock syndrome. Postinfection sequelae can result in rheumatic fever and glomerulonephritis. The ∼1.8-Mb genome of this species encodes a wide range of virulence determinants with diverse biological activities that involve a complex series of physiological processes. These include gene products that allow GAS adherence and colonization to tissues and cells, resistance to host immune defense systems, and dissemination through barriers presented by the host. These virulence determinants play appropriate temporal roles in specific diseases under conditions of specific challenges from the host. It is well understood that GAS pathogenesis is controlled by finely tuned regulatory systems that have evolved over time.
The control of virulence (Cov) regulator (R)-sensor (S) (CovRS) system is one of the best-known regulatory systems. CovRS is a two-component transcriptional regulator of GAS genes that play roles in its pathogenesis and adaptation to environmental stress. For example, in vivo and in vitro studies have shown that CovRS positively regulates a fibronectin-binding protein (Sfb1), a secreted cysteine protease (SpeB) (1–3), and a streptodornase (Spd3) (3). These proteins allow GAS to adhere to and invade epithelial host cells, disrupt host protection, and establish localized infections. In addition, in order to circumvent host innate immunity, CovRS also influences the expression of bacterial hyaluronic acid (HA) capsule synthesis genes (hasABC) (4), the hemolytic exotoxin streptolysin O gene (slo) (5), a serine endopeptidase gene (prtS) (5), and the gene encoding the human plasminogen (hPg) activator streptokinase (sk) (3). Inactivation of CovS derepressed these virulence determinants and enhanced GAS virulence in murine infection models. A variety of naturally inactivated covRS genotypes with spontaneous inactivating mutations of the CovR and CovS proteins have been identified in GAS isolates of different M serotypes from severe invasive infections, such as M1 (2), M3 (6), M53 (7), and M81 (8). The isolates were phenotypically highly virulent and displayed enhanced resistance to killing by the host immune system. In all cases, the hypervirulent phenotypes can be abrogated in vitro, and in mice in vivo, by complementation of wild-type (WT) covRS alleles. Comparative transcriptomic studies of CovRS mutants and wild-type strains have been performed previously using microarray hybridization in M1-type GAS strains. These investigations revealed that CovRS explicitly regulated diverse virulence factors that are closely associated with pathogenic phenotypes (3, 5, 9, 10). Growth-dependent transcriptome dynamics profiling in M1 strain MGAS5005 (11) demonstrated that the CovRS system also regulated key metabolic genes needed for survival adaption. Notably, a comparative study of GAS gene expression (12) in vitro versus in vivo revealed that an enriched set of genes, including genes involved in metabolic activities, were differentially expressed in response to environmental adaptation between wild-type (WT) GAS M1T1 and its CovS− mutant. These results suggested that CovRS globally regulates virulence determinants by rapidly switching the genotypes of GAS but also appropriately remodels the metabolic system in response to diverse environmental stresses to ensure bacterial survival in hostile host environments (4, 12).
In order to unravel mechanisms of CovRS in regulating GAS pathogenesis and to identify more-comprehensive sets of virulence genes involved in virulence regulation, we performed RNA sequencing (RNA-Seq) on a naturally mutated invasive GAS strain, M23ND/CovS−, and its complemented isogenic strain, M23ND/CovS+. By leveraging the advantages of high-throughput sequencing technology, we were able to quantitatively identify an extended set of virulence-related genes that were regulated by CovS at different growth stages and which likely contributed to GAS pathogenesis. We used S. pyogenes strain M23ND, a serotype M23 strain that was first isolated from a case of severe streptococcal infection (13), for this work because of the documented importance of CovS gene switching in the virulence of M23ND (14) and because the natural isolate caused rapid death in mice. Subsequent genome sequencing of strain M23ND revealed a 5-nucleotide deletion in the covS gene (14) that inactivated the translated protein. However, the strain was less lethal after complementation of the deleted nucleotides in the CovS− mutant, thereby providing a 45% survival rate until at least 10 days postinfection. We hypothesized that the results obtained in this study would not only extend our understanding of the regulatory roles of CovRS in streptococcal pathogenesis but would also provide new perspectives on the dynamics of pathogenesis development in this invasive bacterial strain.
MATERIALS AND METHODS
Strain growth and total RNA isolation.
S. pyogenes serotype M23 strain M23ND (ATCC 21059), containing a natural CovS− mutation, and its isogenic complemented CovS+ derivative, were grown aerobically in Todd-Hewitt (TH) broth supplemented with 0.2% yeast extract (THY medium) at 37°C. The cells were collected at logarithmic phase (LP; A600 = 0.5 to 0.6) and early stationary phase (SP; A600 = 0.9 to 1.0). The collected cells were digested with mutanolysin (500 units/ml) in 300 μl of spheroplasting buffer (20 mM Tris-HCl–10 mM MgCl2–56% raffinose, pH 6.8) and then incubated with 100 mg/ml chloramphenicol at 37°C for 60 min. The cell pellets were harvested by centrifugation at 10,000 rpm for 5 min. Total RNA was isolated and purified from the cell pellets using a DNeasy blood and tissue kit (Qiagen, Valencia, CA). The RNA was treated with DNase I, incubated at room temperature for 20 min, and then washed with RW1 wash buffer. Before elution was performed with nuclease-free water, the purified RNA was treated again with DNase I in order to efficiently remove genomic contaminations. The quality of the RNA was assessed by the A260/A280 ratio and visualized following gel electrophoresis in 1% agarose.
Library construction and sequencing.
The sequencing library was constructed using an Epicentre ScriptSeq v2 Complete kit for bacteria and the protocol companion (Illumina, San Diego, CA). Here, the rRNA was removed from total RNA using a Ribo-Zero rRNA removal kit specifically for Gram+ organisms (Epicenter, Madison, WI). The quality of the preparation was assessed with a model 2100 BioAnalyzer along with an RNA Nano kit (Agilent Technologies, Santa Clara, CA). This analysis provided a RNA integrity number (RIN) of >9.0. The remaining RNA was purified using Agencourt AMPure XP beads (Beckman Coulter, Brea, CA) and then subjected to heat fragmentation at 85°C for 5 min. Only the first strand was reverse transcribed to cDNA in order to obtain strand-specific cDNA sequences. The final library integrity and size distribution were assessed using an Agilent model 2100 Bioanalyzer along with the Agilent DNA 7500 assay. Paired-end 2-by-79-bp sequencing was performed using an Illumina Miseq platform at the Genomics and Bioinformatics Core Facility at the University of Notre Dame.
Read mapping and comparative analysis of differential expression levels of genes.
The sequencing reads were mapped to the previously annotated genome of M23ND (14) using the Burrows-Wheeler Alignment (BWA) tool (15). The reads which were mapped to the noncoding strands or mapped to multiloci were removed. Expression levels were evaluated by counting the reads uniquely falling into the coding regions of the genome and calculated as the number of reads per kilobase per million reads (RPKM) by normalizing to the total number of mappable reads and gene lengths. In order to identify genes that were significantly regulated, the fold change was calculated by comparing the expression levels of the M23ND/CovS+ and M23ND/CovS− strains at both LP and SP. Two biological replicates were used as surrogates for statistical testing based on the DESeq Package in R environments (16). Negative binomial testing was used to assess the P value of the differential levels of expression. The Benjamini-Hochberg multiple-test correction was applied to evaluate the false-discovery rate (FDR) (17, 18). Significantly regulated genes were defined based on the following criteria: (i) a >2.0-fold change with a FDR P value of <0.001 when the maximum RPKM of the compared genes was >200; (ii) a >1.7-fold change with a FDR P value of <10−7 when the maximum RPKM was <200; (iii) a maximum RPKM number of >100 of the genes across different samples.
Identification of differentially regulated gene modules.
In order to comprehensively and systematically capture genes regulated by CovS, we developed modulated methods to identify differentially expressed gene modules instead of individual genes. We searched for consecutively located genes in the genome, within which >2 satisfied the significance criteria for individual genes. If the gene members in the locus shared similar expression patterns and were involved in a common physiological process, we defined this locus as a significantly regulated gene island. When the gene locus contained only one gene member, we then termed it an islet. The pairwise gene distance was not treated as a prerequisite. Although the gene distance cannot be treated as a precise determinant of gene islands, we restricted the sense-strand distance to <500 bp. The expression pattern for each gene was represented as a four-component vector from a four-way comparison (the CovS+ strain versus the CovS− mutant and LP versus SP). The patterns were considered to be similar when the Pearson distance was <0.2.
Construction of the metabolic model.
The transporter genes and enzymes in the same loci were usually self-organized gene clusters involved in the same subsystem or metabolic pathway. Therefore, we assigned gene islands or islets to specific subsystems or pathways on the basis of the SEED subsystem (19) or KEGG pathway database (20) and applied hypergeometric tests for subsystem/pathway enrichment assessments. Manual curation was subsequently performed. The genome-wide metabolic network was constructed based on the BioCyc database (21, 22) using SRI Pathway Tools and manual editing (23, 24).
q-RT-PCR.
RNA-Seq data were confirmed for a subset of genes for both the CovS+ and CovS− strains under all growth conditions. Two independent extractions of total RNA from each strain were used. Total RNA (100 ng) was reverse transcribed to cDNA. Forward and reverse primers (see Table S1 in the supplemental material) were specifically designed for the selected genes and produced amplicons of 100 to 400 bp. Quantitative real-time PCR (q-RT-PCR) was performed for 40 cycles with 12 μl of 2.5× SYBR green PCR master mix (Applied Biosystems) and 10 pM forward and reverse primers, each in a final volume of 30 μl. For each primer pair, H2O was used as a negative control and a RNA sample without reverse transcriptase was used as a DNA control. The transcription level of each gene was analyzed by the 2−ΔΔCT (threshold cycle) method (25). The statistical means of triplicate CT values were calculated for the target and reference gene plr (or the GAPDH [glyceraldehyde-3-phosphate dehydrogenase] gene). All q-RT-PCR products were run on agarose gels to verify that only a single amplicon was produced.
Mouse survival studies.
The C57BL/6 mouse model containing the human plasminogen (hPg) transgene (from D. Ginsburg, Ann Arbor, MI) was employed for survival studies with WT mice as controls. The mice were anesthetized with isoflurane and subcutaneously injected with 1.2 × 107 to 1.8 × 107 CFU/mouse of GAS. Mice were monitored for 10 days for survival status.
HA capsule assays.
The GAS cultures were grown to LP and SP in THY medium. The cells were centrifuged and resuspended in 0.5 ml water. The capsule was liberated by shaking with 1 ml CHCl3. The HA capsule level in the aqueous phase was determined by the A640 after addition of an equal volume of Stains-All solution {20 mg of 1-ethyl-2-[3-(1-ethylnaphtho-[1,2-d]thiazolin-2-ylidene)-2-methylpropenyl]naptho-[1,2-d]thiazolium bromide (Sigma-Aldrich)–60 ml acetic acid–100 ml of 50% formamide}, as previously detailed (26). Standard curves were constructed using concentrations of commercial HA (Sigma).
Fluoroquinolone resistance assay.
The M23ND strain was tested for ciprofloxacin susceptibility using the Kirby-Bauer disk diffusion assay (27). An overnight culture of M23ND was resuspended in 0.7% saline solution to an A600 of 0.5. The solution was streaked on a sheep blood agar plate (Teknova, Hollister, CA) prior to adding a disk (Sensi-Disc; BD Biosciences, San Jose, CA) previously incubated with 5 μg/ml ciprofloxacin. Plates were incubated overnight (16 to 18 h) at 37°C and 5% CO2. The diameter of the diffusion zones was measured by hand using a ruler.
Nucleotide sequence accession numbers.
RNA-Seq data have been submitted to the NCBI Gene Expression Omnibus database with GEO platform accession no. GPL19982, series accession no. GSE67533, and sample accession no. GSM1649095 to GSM1649102. Supplemental data sets are available at https://sites.google.com/a/nd.edu/transgene/Transcriptome.
RESULTS
RNA-Seq provided sufficient coverage for the covRS-regulated transcriptome.
The strand-specific cDNA libraries were constructed and subsequently sequenced using an Illumina Miseq platform. The sequencing generated an average of 3,167,505 paired-end reads, yielding ∼500 million bp/library (see Table S2 in the supplemental material). Averages of 87% of the reads were uniquely mapped to the genome of M23ND (14). The expression level for each gene was evaluated by counting the number of reads uniquely mapped to the gene and normalized to RPKM. At least 93% of the genes in the genome were transcribed under one or more of the experimental growth conditions, with RPKM values of >30. This suggested that the sequencing reached sufficient coverage for the CovRS-regulated transcriptome. While the sequence coverage had a broad dynamic range of 104, the coverage profile was remarkably consistent between biological replicates, with Pearson correlations of 0.97 to 0.99.
Global differential expression profile of M23ND.
It is well established that genes with similar expression patterns tend to localize in the same gene clusters or gene islands, where their gene members facilitate coordinated functions, or are involved in a common physiological process. This phenomenon was also observed in the current transcriptome data. Therefore, in order to comprehensively and systematically capture genes regulated by CovS, we undertook a modular approach for identification of differentially expressed genes, by searching for consecutively located gene islands in the genome and characterizing them as parts of islands or as islets by the criteria outlined in Materials and Methods. The advantages of this approach are that more differentially regulated genes can be identified and their functions can often be inferred based on association in a genetic locus.
By applying this modular approach of identifying genes differentially expressed between M23ND/CovS+ and M23ND/CovS−, we detected 349 genes (18% of the total genes) regulated by CovS at either LP or SP, of which 309 (89%) fell into 86 gene islands and 40 (11%) were considered islets. Among the 349 CovS-regulated genes, a total of 188 were differentially expressed at LP (80 positive and 108 negative), whereas 264 were expressed at SP (183 positive and 81 negative). These results suggest that more genes were regulated by CovS at SP, likely due to the higher accumulation of CovS at the initial growth phase. It was observed that the transcription levels of covR and covS were higher at LP than SP, with 1.5-fold to 2.0-fold differences. The naturally mutated covS gene was transcribed with high abundances of ∼400 RPKM at LP and ∼200 RPKM at SP.
Based on the results of the identification of gene islands, we propose that the total number of genes directly or indirectly regulated by CovS could increase to ∼450 (25%) by considering those with an expression level near the significance threshold that are located in highly regulated gene islands. For example, in the regulated ATP synthase operon, three of the central genes (atpH, atpA, and atpG) did not qualify as significant, based on the statistical criteria that we employed. However, their expression changes were very close to the threshold at 1.4-fold to 1.7-fold and their FDR P values ranged from 1.1 × 10−6 to 1.1 × 10−3. We expect that these gene differences could reach significance with more sequencing capacity and with ideal experimental controls.
The differentially regulated genes include a set of known virulence factors previously demonstrated to be controlled by the CovRS system in GAS. These include positively regulated genes, e.g., speB, speC, spd3, spd1, and sfb1, and negatively regulated genes, e.g., slo, nga, and the hasABC and mga regulons (Fig. 1). In addition to virulence factors, functional classifications showed that the CovS-regulated genes were also involved in several other functional categories, i.e., nutrient uptake, energy metabolism, cell wall and capsule generation, membrane transport, fatty acid biosynthesis, and regulation and cell signaling (Fig. 2A). These genes play pivotal roles in bacterial growth and survival in challenging host environments. The reliability of the quantification of differentially regulated genes from the RNA-Seq expression data was further assessed by quantitative real time-PCR (q-RT-PCR). A subset of 15 genes were selected for q-RT-PCR assays at LP and SP growth. The results showed a high (0.79) Pearson correlation of these genes with the RNA-Seq data (Fig. 2B).
FIG 1.
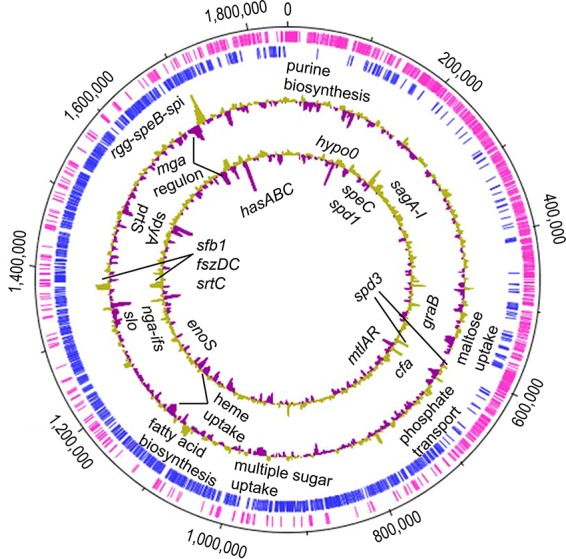
Representation of differential expression profiles of all genes regulated by CovS at LP and SP growth. From the outermost circle to the innermost circle, circle 1 and circle 2 display annotated coding sequences for the forward (pink) and reverse (blue) DNA strands, respectively, and circle 3 and circle 4 depict the differential expression of genes between CovS+ and CovS− at LP and SP growth, respectively. Significantly regulated genes at LP or SP growth are marked. hypo0, hypothetical gene 0.
FIG 2.

Functional classification of genes regulated by CovS at LP or SP growth. (A) The numbers of genes in each category are shown in parentheses for LP and SP, respectively. There is overlap of CovS-regulated genes at LP and SP. (B) Validation of a subset of regulated genes using q-RT-PCR. The data showed a Pearson correlation of 0.79 between RNA-Seq and q-RT-PCR. hypo0, hypothetical gene 0.
The expression profile of phage-carried virulence genes regulated by CovS.
In the genome of M23ND, we previously identified six phage-carried virulence factors. Of these, three, including spd3, speC, and spd1, were significantly upregulated by CovS at both LP and SP whereas only the superantigen, ssa, was suppressed ∼2.2-fold by CovR at both LP and SP (Fig. 3A). A gene of unknown function (hypothetical gene 14) and a paratox (prx) in the same loci as spd3 and ssa, respectively, were also found to be regulated by CovS (Fig. 3A). This indicates that the M23ND strain expressed more superantigen (ssa) in M23ND/CovS−, while another set of toxins, represented by spd3, speC, and spd1, had higher expression in M23ND/CovS+. Previous studies showed that, when present, ssa in the GAS genome may be associated with the pathogenesis of toxic shock syndrome (28) and scarlet fever (29). Thus, we propose that the increased transcriptional expression of ssa in M23ND/CovS− may constitute one of the factors contributing to the enhanced lethality of M23ND/CovS−. The virulence genes, speH and speI, were not remarkably regulated by CovS. However, the pseudogene, speH, was transcribed at moderate abundance, ∼200 RPKM. Recent studies suggested that pseudogenes may be transcribed and even translated to fragmented peptides but may lack properly folded structures (30, 31), thus unnecessarily consuming energy from the bacteria. We did not find a pattern of growth-dependent regulation of the phage-carried virulence genes.
FIG 3.

RNA-Seq changes of levels of expression of virulence gene islands differentially regulated by CovS and q-RT-PCR validation of a subset of regulated genes. (A) Fold changes of phage-carried virulence factors. (B) Fold changes of chromosome-inherited virulence factors. The fold changes determined for each gene between CovS+ (S+) and CovS− (S−) at LP and SP and between SP and LP for both phenotypes (CovS+ and CovS−) are represented with gradient colors. The genes associated with the same gene islands are clustered. Functional categories are classified based on the processes of virulence development. (C) The disk diffusion susceptibility assay was performed using ciprofloxacin, and the zone of growth inhibition surrounding a ciprofloxacin-coated (5-μg-diameter) disk was measured. Both CovS+ and CovS− isolates exhibited a zone of inhibition of >18 mm in diameter, indicating the absence of significant resistance to ciprofloxacin. (D) Validation of RNA-Seq expression data using q-RT-PCR for a subset of genes at LP and SP growth. The expression values were normalized to the plr gene and represented as relative quantities (RQ) in relation to those seen with M23ND/CovS− at LP. The error bar indicates the standard errors of the means (SEM). The overall expression profiles are consistent with the RNA-Seq data. hypo, hypothetical gene.
The majority of other phage-carried genes were inactivated by both M23ND genotypes. However, we did detect several transcribed phage-carried genes with RPKM values in the range of ∼200 to ∼10,000, encoding integrase, tail, phage capsid, endopeptidase, hyaluronidase, lysis, and toxin-antitoxin systems (see Data Set S1 in the supplemental material). These genes seem to function in phage integration, structure, stabilization, and/or adaptation to host genes. Examination of the expression profile of these phage genes showed that they are not regulated by CovRS but more definitively by the LP and SP growth phases.
The expression profile of chromosome-inherited virulence genes regulated by CovS.
In a previous study, we examined the chromosome-inherited virulence genes that are carried by the genome of M23ND. By integrating those previous data and results of other studies, we established the virulome of S. pyogenes M23ND and quantified the expression profiles of the genes therein (14). The virulome is composed of 43 virulence-related genes, 34 of which were differentially regulated by CovS at either LP or SP and 9 of which were not affected (Table 1). The 34 CovS-regulated genes classify into nine gene islands and three islets, while 8 of the 9 unaffected genes were islets. Most of these virulence factors were expressed at SP growth, and their expression abundances differed broadly, depending on the genotypes or growth phase. Notably, it was observed that CovS suppressed the expression of one set of genes, viz., the hasABC operon, slo, nga, the mga regulon, spyA, and prtS, while simultaneously enhancing expression of another set of genes, viz., speB, sfb1, and graB, at both LP and SP (Fig. 3B and Table 1). Meanwhile, expression of endoS was suppressed at LP but mildly enhanced at SP and expression of the sagA operon was upregulated at LP but weakly suppressed at SP in the CovS+ strain. Expression of the dltABCD operon was upregulated only mildly at SP. Expression of the remaining well-established virulence factors, smeZ, speG, spd, ska, sclA, plr, hylA, ideS, and eno, was not regulated by CovRS at either LP or SP (5, 32). The expression patterns of several of the key regulated virulence genes were previously confirmed by q-RT-PCR experiments in M23ND (14).
TABLE 1.
Fold changes of expression of chromosomal virulence genes significantly regulated by covS at LP or SPa
| Gene name | CovS+ vs CovS− |
SP vs LP |
Gene product or function | ||||||
|---|---|---|---|---|---|---|---|---|---|
| LP |
SP |
CovS− |
CovS+ |
||||||
| Fold | FDR P value | Fold | FDR P value | Fold | FDR P value | Fold | FDR P value | ||
| hasC | −53 | 0 | −7.7 | 0 | −7.8 | 0 | −1.1 | 0.47 | UTP-glucose-1-phosphate uridylyltransferase |
| Hypothetical gene 1 | −50 | 0 | −7.5 | 0 | −8.0 | 0 | −1.2 | 0.57 | Hypothetical protein |
| hasB | −55 | 0 | −8.1 | 0 | −11 | 0 | −1.6 | 0.03 | UDP-glucose dehydrogenase |
| hasA | −65 | 0 | −6.0 | 0 | −13 | 0 | −1.2 | 0.41 | Hyaluronan synthase |
| Hypothetical gene 2 | −16 | 0 | −15 | 0 | −1.3 | 0.03 | −1.2 | 0.72 | Hypothetical protein |
| Hypothetical gene 3 | −25 | 0 | −14 | 0 | 1.6 | 0.01 | 2.9 | 0.01 | Hypothetical protein |
| slo | −7.0 | 0 | −18 | 0 | −1.8 | 0 | −4.6 | 0 | Thiol-activated cytolysin |
| ifs | −8.6 | 0 | −15 | 0 | −2.0 | 0 | −3.5 | 0 | Streptococcal NAD glycohydrolase inhibitor |
| nga | −8.7 | 0 | −12 | 0 | −2.2 | 0 | −3.0 | 0 | Nicotine adenine dinucleotide glycohydrolase |
| htpA | −6.0 | 0 | −4.5 | 0 | −1.3 | 0.01 | 1.0 | 0.82 | Streptococcal histidine triad protein |
| lmb | −4.1 | 0 | −4.0 | 0 | −1.4 | 0.01 | −1.3 | 0.22 | Laminin-binding surface protein |
| scpA | −6.3 | 0 | −9.0 | 0 | −2.1 | 0 | −2.9 | 0 | C5a peptidase |
| emm23 | −1.4 | 0 | −1.9 | 0 | −1.7 | 0 | −2.2 | 0 | Antiphagocytic M protein |
| mga | −1.9 | 0 | −2.6 | 0 | −1.1 | 0.10 | −1.6 | 0 | M protein trans-acting positive regulator |
| Hypothetical gene 4 | −21 | 0 | −22 | 0 | −1.0 | 0.71 | −1.1 | 0.90 | Hypothetical protein |
| spyA | −4.1 | 0 | −4.6 | 0 | 1.2 | 0.02 | 1.1 | 0.71 | C3 family ADP-ribosyltransferase |
| prtS | −13 | 0 | −1.3 | 0.01 | 2.3 | 0 | 24 | 0 | Serine endopeptidase ScpC |
| Hypothetical gene 5 | −1.1 | 0.46 | −1.5 | 0.01 | 8.73 | 0 | 6.6 | 0 | Hypothetical protein |
| inlA | −2.2 | 0 | −2.7 | 0 | 1.1 | 0.5 | −1.1 | 0.68 | Internalin, putative |
| endoS | −3.1 | 0 | −1.4 | 0 | 2.1 | 0 | 9.3 | 0 | Secreted endo-beta-N-acetylglucosaminidase |
| eno | 1.2 | 0.03 | −1.0 | 0.92 | −1.1 | 0.09 | −1.4 | 0.01 | Enolase |
| sagA | 1.9 | 0 | −1.3 | 0.03 | 8.5 | 0 | 3.6 | 0 | Streptolysin S precursor |
| sagB | 2.3 | 0 | −1.2 | 0.12 | 8.9 | 0 | 3.3 | 0 | Streptolysin S biosynthesis protein B |
| sagC | 2.0 | 0 | −1.3 | 0.01 | 9.2 | 0 | 3.6 | 0 | Streptolysin S biosynthesis protein C |
| sagD | 2.2 | 0 | −1.4 | 0.01 | 8.8 | 0 | 2.8 | 0 | Streptolysin S biosynthesis protein D |
| sagE | 2.2 | 0 | −1.7 | 0 | 8.0 | 0 | 2.2 | 0 | Streptolysin S self-immunity protein |
| sagF | 2.0 | 0 | −1.5 | 0.01 | 5.9 | 0 | 2.0 | 0 | Streptolysin S biosynthesis protein |
| sagG | 2.4 | 0 | −1.7 | 0 | 6.9 | 0 | 1.7 | 0 | Streptolysin S export protein |
| sagH | 2.3 | 0 | −1.7 | 0 | 7.1 | 0 | 1.5 | 0.01 | Streptolysin S export permease |
| sagI | 2.0 | 0 | −1.5 | 0.01 | 5.2 | 0 | 1.8 | 0 | Streptolysin S export permease |
| prsA | 3.5 | 0 | 26 | 0 | 3.2 | 0 | 24 | 0 | Peptidylproline cis-trans isomerase |
| spi | 3.7 | 0 | 118 | 0 | 5.7 | 0 | 184 | 0 | Streptopain inhibitor |
| speB | 2.8 | 0 | 91 | 0 | 5.0 | 0 | 163 | 0 | Cysteine protease |
| Hypothetical gene 8 | 1.3 | 0.56 | 90 | 0 | 5.3 | 0 | 363 | 0 | Hypothetical protein |
| Hypothetical gene 9 | 1.9 | 0.09 | 52 | 0 | 8.5 | 0 | 231 | 0 | Hypothetical protein |
| ropB | 3.9 | 0 | 3 | 0 | 1.7 | 0.001 | 1.5 | 0.01 | Rgg-like transcription regulator |
| cfa | 8.3 | 0 | 5.7 | 0 | 1.6 | 0.01 | 1.1 | 0.54 | CAMP factor |
| graB | 6.6 | 0 | 4.5 | 0 | 1.66 | 0 | 1.1 | 0.26 | G-related α 2 macroglobulin binding |
| srtB | 3.2 | 0 | 2.7 | 0 | −1.4 | 0.03 | −17 | 0.01 | Sortase B, LPXTG specific |
| fszC | 4.8 | 0 | 6.8 | 0 | −2.6 | 0 | −1.9 | 0 | Cell wall surface anchor family protein |
| fszD | 51 | 0 | 7.7 | 0 | −2.6 | 0 | −1.6 | 0 | Hypothetical protein |
| sfb1 | 2.6 | 0 | 8.6 | 0 | −6.5 | 0 | −1.9 | 0 | Fibronectin-binding protein |
| rofA | 2.4 | 0 | 2.3 | 0 | 1.2 | 0.33 | 1.1 | 0.35 | Transcriptional regulator, rofA |
| parE | 1.3 | 0.02 | 2.0 | 0 | −1.6 | 0 | 1.0 | 0.79 | Topoisomerase IV subunit B |
| Hypothetical gene 10 | 1.4 | 0.01 | 1.7 | 0 | −1.3 | 0.01 | −1.0 | 0.92 | Hypothetical protein |
| parC | 1.3 | 0.01 | 1.7 | 0 | −1.8 | 0 | −13 | 0.02 | Topoisomerase IV subunit A |
| dltA | 1.0 | 1.0 | 1.5 | 0 | −1.2 | 0.02 | 1.2 | 0.11 | d-Alanine polyligase subunit 1 |
| dltB | −1.2 | 0.04 | 1.6 | 0 | −2.0 | 0 | −11 | 0.72 | d-Alanyl transfer protein DltB |
| dltC | −1.1 | 0.29 | 1.4 | 0.01 | −1.9 | 0 | −1.2 | 0.25 | d-Alanine polyligase subunit 2 |
| dltD | −1.1 | 0.29 | 1.5 | 0 | −2 | 0 | −1.2 | 0.10 | Poly-d-alanine transfer protein |
| ska | 1.0 | 1.0 | 1.21 | 0.07 | 2.8 | 0 | 3.3 | 0 | Streptokinase |
| sclA | 4.0 | 0 | −2.5 | 0 | 3.2 | 0 | 5.0 | 0 | Collagen-like surface protein |
| smeZ | 1.3 | 0.12 | −1.3 | 0.15 | 2.1 | 0 | 1.3 | 0.05 | Streptococcal mitogenic exotoxin Z |
| spd | −15 | 0 | 1.1 | 0.22 | 4.5 | 0 | 7.4 | 0 | Streptodornase B; mitogenic factor 1 |
| plr | −1.0 | 0.94 | −1.3 | 0.01 | 1.2 | 0.05 | −1.1 | 0.30 | NADGH |
| hylA | 1.3 | 0.01 | −1.1 | 0.55 | 1.3 | 0.01 | −1.1 | 0.61 | Hyaluronate lyase precursor |
| ideS/mac | −1.3 | 0.16 | −1.6 | 0.02 | −1.5 | 0.01 | −1.9 | 0.01 | Immunoglobulin G-endopeptidase |
| speG | −1.5 | 0.02 | −2.3 | 0 | −1.0 | 1.0 | −1.5 | 0.06 | Exotoxin G |
The FDR P values refer to Benjamini-Hochberg-adjusted P values. LPXTG, Leu-Pro-any amino acid-Thr-Gly.
The expression of the cysteine protease, encoded by speB, and the expression of the fibronectin-binding protein, encoded by sfb1, are among the most widely studied virulence determinants in GAS. These transcripts were robustly upregulated by CovS, consistent with many previous reports. Regarding the present RNA-Seq data, speB was expressed with a high abundance of ∼70,000 RPKM at SP in M23ND/CovS+, corresponding to a 91-fold increase in expression of M23ND/CovS+ versus M23ND/CovS− at SP and a 163-fold increase in expression at SP compared to LP for M23ND/CovS+. The expression of speB was abrogated in CovRS mutants and was correlated with the hypervirulent phenotype of GAS (5, 33). However, the levels of growth-phase dependence of speB expression were divergent in various studies, suggesting multifaceted regulation and complex functions of speB (34, 35). The expression of the sfb1 transcript, along with that of the macroglobulin-binding protein, encoded by graB, as well as phage-carried extracellular nucleases, encoded by spd1 and spd3, was upregulated by CovS through the growth phases. This demonstrated their key roles in the persistence of noninvasive infection.
Several genes that were examined exhibited regulation trends different from those previously reported. graB and sagA (upregulated only at LP) were shown to exhibit elevated expression in a CovR− strain (36) and in animal-passaged invasive strains (10, 37). The inconsistency was proposed to be due to the nonreciprocal regulation of CovR and CovS. This may also partly explain the intact expression of several other genes in the present study, i.e., ska, ideS, and spd, which were suppressed by CovR in other studies (36).
Identification of chromosome-carried virulence genes not previously known to be regulated by CovS.
In this investigation, we discovered several known virulence-related genes that were not previously reported to be regulated by CovS in S. pyogenes. Expression of the cotranscribed gene pair parE and parC, expressing subunits of topoisomerase IV that are important for cell division and replication by relaxing DNA supercoils, was upregulated by CovS+ at SP by 1.7-fold to 2.0-fold. Mutations in this gene pair in another strain of GAS were previously shown to be associated with increased resistance to fluoroquinolones (FQ) and may assist in enhancing the infection of epithelial host cells in superficial diseases of skin and throat (38–40). We performed GAS M23ND antibiotic susceptibility assays using ciprofloxacin-coated disks in blood-agar plates, and the zone of growth inhibition was measured as a function of time. Neither M23ND/CovS+ nor M23ND/CovS− showed significant resistance to ciprofloxacin, displaying a GAS killing zone of >18 mm in diameter (Fig. 3C), a value associated with full ciprofloxacin susceptibility (41). Further examination of the sequences of protein encoded by gene pair parE and parC in M23ND did not reveal mutations previously associated with quinolone resistance, viz., Ser79 to Ala/Phe/Tyr in ParC and Asp437 to Asn in ParE (42).
We also found two inhibitor genes, viz., those encoding a streptococcal NAD glycohydrolase inhibitor (ifs) and a streptopain inhibitor (spi). These genes, situated immediately downstream of nga and speB (see Fig. S1A and B in the supplemental material), respectively, were significantly downregulated (∼15-fold) and upregulated (∼118-fold) by CovS (see Fig. S1A and B). The expression of these two inhibitor genes was highly coordinated with that of their corresponding inhibition targets, nga (∼12-fold) and speB (∼91-fold) (see Fig. S1C and D). These findings are manifestations of self-protection mechanisms of GAS, in addition to its pathogenesis, considering that nga and speB are also toxic to GAS, in addition to their toxicities to host cells.
Two genes encoding surface proteins, viz., internalin (inlA) and histidine triad protein (htpA), were both downregulated by CovS, by 2.2-fold and 2.7-fold at LP and SP and by 6.0-fold and 4.5-fold at LP and SP, respectively. The proteins expressed from these genes were shown to contribute to antiphagocytosis and dissemination in invasive infections in previous studies via attaching to host cell surface (43) or to inhibiting complement deposition through recruitment of complement factor H (44).
Differentially regulated genes with unknown functions.
Through the use of RNA-Seq, we also found 26 differentially regulated genes with unknown functions (see Table S3 in the supplemental material). Eight of these genes are located in the gene islands identified above containing one or more known virulence factors. Sequence homology comparisons showed that 21 of the 26 hypothetical genes are conserved across strains of S. pyogenes with similarities of >90% at both the nucleotide and amino acid levels. Although they are significantly regulated by CovS, information concerning the functions of the hypothetical genes was not accessible by homology searching and hidden Markov model (HMM) profile searching (45) of public databases, i.e., NCBI-nr and Pfam (46). The functions of the hypothetical genes in the virulence are still unclear, but the data indicate that a reservoir of functional genes regulated by CovS has yet to be discovered.
Validation of regulated virulence genes by q-RT-PCR.
We used q-RT-PCR to quantitatively verify several regulated virulence genes (cfa, parE, inlA, htpA, ifs, and hypothetical gene 0) for both strains at LP and SP growth with ideS as a negative control. The expression patterns of the selected genes determined by q-RT-PCR are consistent with that obtained by RNA-Seq (Fig. 3D). The high level of consistency between the two quantification methods suggests the reliability of the current methods for identifying genes differentially regulated by CovS.
The regulatory profile of CovS confers invasiveness and lethality to M23ND.
Infection studies showed that M23ND/CovS− had higher lethality in mice than M23ND/CovS+ (Fig. 4A). In order to correlate the gene regulation profile of CovS with the enhanced virulence of S. pyogenes M23ND/CovS−, we assigned the virulence-related functional classes to the differentially regulated gene islands categorized in the previous section. From the data presented in Fig. 3B, it is seen that genes derepressed in M23ND/CovS−, viz., hasABC, slo and nga, the mga regulon, spyA, and prtS, are highly associated with invasiveness and antiphagocytosis, which are important for deeper colonization and resistance to neutrophil killing of GAS. Previous studies also showed that these genes have higher expression in invasive GAS infections than in noninvasive infections (3, 5, 47), and expression of the genes is considered essential for evasion, persistence, and/or dissemination (48–50). Remarkably, the highly elevated expression of the hasABC operon played a key role in resistance to host killing in invasive infections of GAS. In the present study, the expression of the hasABC operon was greatly increased in M23ND/CovS− at LP (an average of 50-fold) and was confirmed by measuring the HA capsule levels for M23ND/CovS− and M23ND/CovS+ (Fig. 4B). Meanwhile, M23ND/CovS− induced suppression of a set of virulence factors encoding the functions of extracellular protease (speB), exotoxic cytolysis (cfa), immunoglobulin binding (graB), resistance to antibiotics (parE and parC), and adhesion (sfb1).
FIG 4.
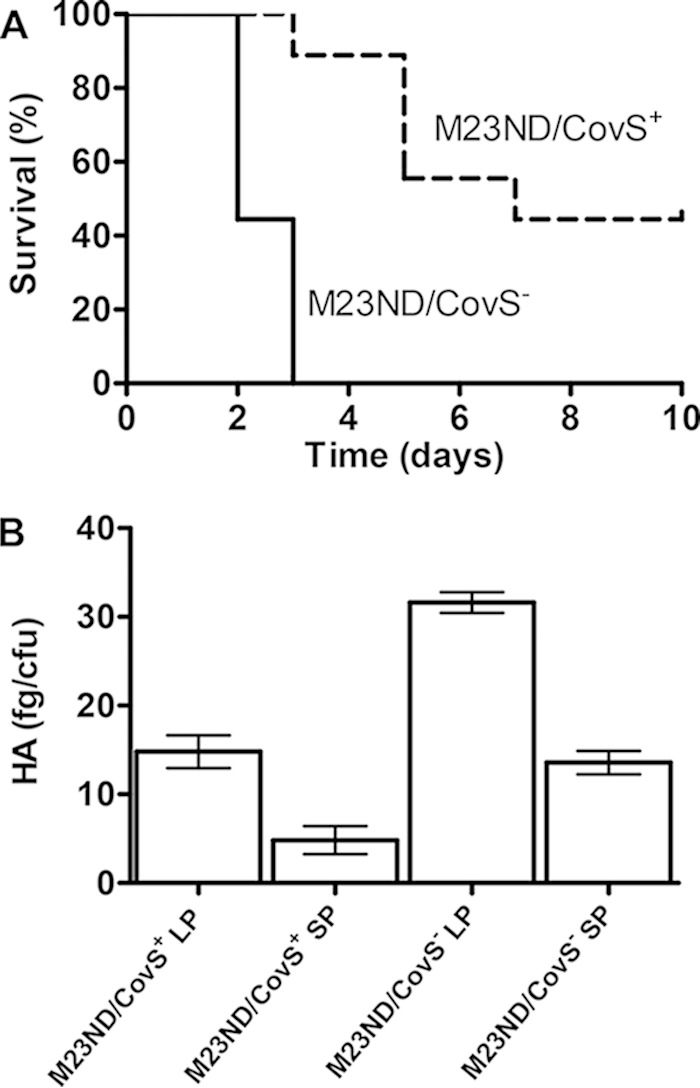
Enhanced lethality and increased capsule expression in M23ND/CovS− compared to M23ND/CovS+ isolates. (A) Kaplan-Meier survival curves of mice (n = 9) after subcutaneous infection with 1.2 × 107 to 1.8 × 107 CFU of strains M23ND/CovS+ and M23ND/CovS−. The mice were monitored for 10 days. The differences between the curves were evaluated by the log-rank test. M23ND/CovS− showed significantly higher lethality than M23ND/CovS+ (P = 0.0003). (B) Measurement of hyaluronic acid capsule levels at LP and SP growth. M23ND/CovS+ showed capsule deficiency during the growth. The error bars indicate the SEM with three repeats.
The opposing regulation characteristics of two sets of virulence factors corresponding to CovS resulted in the switching of two distinct phenotypes in M23ND, viz., lower pathogenicity from M23ND/CovS+ and higher lethality from M23ND/CovS−. Fatal infections were achieved in mice with M23ND/CovS− through enhanced expression of genes necessary for survival (e.g., hasABC), attenuated expression of a gene expressing an inherent degrading protease (e.g., speB), and compromised expression of genes with dispensable functions (e.g., sfb1). Previous studies demonstrated defects in epithelial cell adherence or cell growth of the M1/CovS− clones as a consequence of increased capsule expression (4). Since M1 strains do not express sfb1, it was proposed that, under in vivo conditions, mutant strains are naturally selected for better adaptation and survival in host invasive infections and do not require surface adhesion (4). Natural selection of M23ND/CovS− is also manifested by the intact expression of another set of genes required for survival, e.g., ska, plr, eno, ideS, and spd. They are protected from SpeB-catalyzed degradation in M23ND/CovS−, since this strain does not produce active SpeB, and thus do not require higher expression. Therefore, the current data reflect CovS-mediated reshaping of the regulation profile of virulence genes in response to the original host environments. In addition, CovS was also found to modulate the adaptation of GAS in response to growth conditions.
Nutrient utilization and energy metabolism are finely tuned by CovS.
Transcriptional profiling demonstrated an extensive range of genes involved in nutrient acquisition and energy metabolism that were differentially regulated by CovS. We detected 28 differentially regulated gene islands related to nutrient uptake and utilization and 6 islands related to energy metabolism that covered 114 individual genes (see Data Set S2 in the supplemental material). The utilized nutrients included a variety of components, including multiple sugars, amino acids, nucleotides, glycerol, cofactors, heme iron, and inorganic components, e.g., Mn2+, K+, and phosphate. Their acquisition was primarily mediated by the ATP-binding cassette (ABC) transport system or the phosphotransferase (PTS) system, with the transporter genes located in the same loci as the corresponding enzymes. The expression levels of most of the genes were altered by 2-fold to 3-fold, with several exceptions, e.g., the expression levels of the PTS components for sucrose transport were altered by >5.8-fold and of ABC transporters htsABC for heme uptake by >4.3-fold. The overall regulation of nutrient utilization demonstrated the versatile nutrient utilization capacity and robustly sustainable cellular metabolism for both virulence phenotypes. Although the cells were grown in a rich media, many of the expressed metabolic pathways for nutrient utilization were consistent with those from in vivo infection models.
The wild-type and hypervirulent phenotypes exhibited distinct patterns of nutrient utilization which were explicitly tuned by CovS (Fig. 5). Here, it is illustrated that both the CovS+ and CovS− phenotypes preferentially utilized specific sets of nutrients through the growth phase. The CovS− phenotype preferred polysaccharides, amino sugars, cofactors, heme iron, dipeptides, and glutamates, while the CovS+ phenotype had more inorganic molecules (e.g., Mn2+, K+, and phosphate), glycerol, oligopeptides, methionine, cysteine, and nucleotides (Fig. 5A to C). Generally, the CovS− phenotype revealed higher competence with respect to utilization of large molecules that cannot be synthesized by the pathogen. On the other hand, it was observed that the CovS−-regulated utilization of several carbon sources was growth dependent. As examples, the CovS−-regulated utilization of lactose, galactose, glucose, sucrose, mannose, and deoxyribose was suppressed at LP growth and derepressed at SP growth in M23ND/CovS+, reflecting the high rate of consumption during growth (Fig. 6A and B). The accelerated nutrient acquisition at SP for the CovS+ variant was most likely due to the intense competition for diminishing nutrients in the late growth phase. In contrast, the CovS− phenotype obtained sufficient nutrient sources in LP growth and maintained the lower consumption rate until the SP stage. This is consistent with the pattern of energy metabolism and production, where the genes were more abundantly expressed at SP growth for the highly active CovS+ phenotype (Fig. 5D; see also Data Set S2 in the supplemental material). The higher rate of nutrient uptake and the active energy metabolism are consistent with the higher growth rate of the CovS+ phenotype (Fig. 6C). Only two gene islands encoding the ABC transporter-mediated maltose/maltotriose utilization exhibited the opposite, growth-dependent CovS regulation. These islands were upregulated at LP growth and significantly suppressed at SP growth in the CovS+ phenotype. Compared with the expression of virulence genes, which is relatively independent of the growth phase (except for the sagA and speB regulon), the expression of nutritional genes is highly dependent on growth conditions, indicating their active response to the growth environments.
FIG 5.

Heat map representation of RNA-Seq expression changes of genes involved primarily in extracellular nutrient uptake and energy metabolism in gene islands differentially regulated by CovS. The fold changes in expression of each gene between CovS+ and CovS− at LP and SP and between SP and LP for both the CovS+ and CovS− phenotypes are represented with gradient colors. The genes falling into the same gene islands are clustered. The functional classifications of the gene islands are grouped into four broad categories of nutrient utilization: sugars and glycerol (A); biotin, heme iron, and inorganic molecules (B); peptides, amino acids, and nucleosides (C); and energy metabolism and production (D). An asterisk indicates that the classification is putative.
FIG 6.

Accelerated nutrient consumption rate and higher growth rate of M23ND/CovS+ compared to M23ND/CovS−. (A and B) The temporal dynamics of CovS-regulated genes involved in nutrient utilization. The gene expression levels are shown for CovS+ and CovS− (vertical direction) at the two growth points of LP and SP (horizontal direction). The levels of uptake of lactose, galactose, and sucrose (A) and of β-glucose, mannose, and deoxyribose (B) are shown to be generally suppressed at LP growth and derepressed at SP growth in the CovS+ phenotype, reflecting the accelerated nutrient consumption rate during growth. The expression values are shown in RPKM. (C) Growth curve of the two phenotypes, CovS+ and CovS−, in THY medium measured by A600. The error bar indicates the SEM. M23ND/CovS+ showed more-rapid growth than M23ND/CovS− after 2 h postinfection. The growth trend is consistent with the consumption rate for carbohydrates demonstrated in the expression data.
CovS-dependent regulation of de novo biosynthesis of nutrient molecules.
In addition to the extracellular acquisition of nutrients, the de novo biosynthesis of diverse molecules, primarily nucleotides, amino acids, and fatty acids, was also regulated by CovS (see Data Set S3 in the supplemental material). A total of 49 genes, classified into 9 gene islands and 10 gene islets, were regulated by CovS. The gene islands encoding purine biosynthesis and glutamine biosynthesis were more highly activated in M23ND/CovS− than in M23ND/CovS+. The higher requirements of purine biosynthesis by CovS− may reflect the limited concentration of purines in the THY medium or may represent clones naturally selected by the hypervirulent CovS− strain during host infection. An active purine biosynthesis pathway was also identified in other intracellular pathogens, such as Listeria monocytogenes (51) and Bacillus anthracis (52), due to limited concentration of purines in host cells. Two genes involved in glutamine biosynthesis, glnA and glnR, were also highly activated in M23ND/CovS− at LP growth. Previous studies showed that these genes are related to the virulence of pathogens, likely due to their nitrogen-containing properties. glnA, the homologous gene from S. pneumoniae, was shown to play a role in colonization in pharyngeal cell infections (53), and the glnA mutant strain from S. suis exhibited reduced mortality in murine infection (54).
Prediction of candidate genes associated with virulence among CovS-regulated metabolic genes.
In order to globally represent the regulatory landscape of CovS for nutritional derivation during the growth of GAS, we reconstructed a whole-genome metabolic network for M23ND based on the previously published genome annotation (14) and mapped the nutritional genes to this network (see Fig. S2 in the supplemental material). The network revealed a wide-ranging distribution of CovS-regulated genes in pathways relevant to nutritional derivation. By examining nutritional genes at the genome-wide scale, we found that the nutritional derivation of pathogenic GAS is highly redundant and that many nutrients can be supplied by multiple genetic loci or physical sources with regard to extracellular nutrient uptake or de novo biosynthesis (55). As examples, two loci for β-glucoside transport were simultaneously downregulated by CovS+ at LP and upregulated at SP and de novo biosynthesis of cysteine and the cysteine transporter system were both upregulated at SP by CovS+. Mannose, lactose, galactose, sucrose, maltose, and mannitol can be imported by more than two sets of transporters differentially regulated by CovS. The CovRS regulatory system not only flexibly modulates diverse kinds of virulence factors but also may finely tune versatile metabolic systems, which may substantially contribute to overall virulence. Previous studies demonstrated that bacterial metabolism during growth may be related to the virulence of pathogens in infection (51, 56, 57). Thus, we attempted to identify potential candidates among metabolic genes that may contribute to or be associated with the GAS virulence.
To computationally predict the potential virulence association of the metabolic genes, we defined the regulated gene module as redundant or nonredundant by determining whether the gene module constituted a unique source for obtaining specific nutrients. Those nonredundant gene modules are likely functionally indispensable and may also contribute to virulence. A genome-wide search yielded 16 nonredundant gene islands or islets which are potentially essential for the intracellular growth or virulence development of GAS (Table 2). These genes are involved in derivation of amino acids, fatty acids, peptides, Mn2+, biotin, and heme iron. The genes for energy metabolism and production are unique and hence are also nonredundant.
TABLE 2.
Nonredundant gene modules involved in nutrient derivation from extracellular uptake or de novo biosynthesis
| Nutrient/energy factor(s) | Coding gene(s)a | Functional classification | KEGG pathway/module |
|---|---|---|---|
| Biotin | 1341 | Biotin utilization | Biotin transport system |
| Heme iron | 1168:1177 | Heme iron uptake | Heme iron utilization |
| Mn2+ | 1517:1519 | Mn2+ transport | Mn2+/iron transport system |
| Dipeptide | 1707:1711 | Dipeptide transport | Dipeptide transport system |
| Oligopeptide | 1266:1270 | Oligopeptide transport | Oligopeptide transport system |
| Amino acids (Met) | 1616:1619 | Amino acid transport (Met) | Met transport system |
| Amino acids (Gln) | 1240:1241 | Gln biosynthesis/nitrogen metabolism | Gln biosynthesis |
| Fatty acids | 1132:1144 | Fatty acid biosynthesis | Fatty acid biosynthesis FASII |
| Isoprenoid | 0361:0366; 1042; 1387; 1461 | Fatty acid biosynthesis (isoprenoid) | Terpenoid biosynthesis |
| Aminoacyl-tRNA (Pro) | 1679 | tRNA charging (Pro) | tRNA charging (Pro) |
| Aminoacyl-tRNA (Tyr) | 0077 | tRNA charging (Tyr) | tRNA charging (Tyr) |
| Aminoacyl-tRNA (Arg) | 1813:1814 | tRNA charging (Arg) | tRNA charging (Arg) |
| Aminoacyl-tRNA (Trp) | 1764 | tRNA charging (Trp) | tRNA charging (Trp) |
| Aminoacyl-tRNA (Leu) | 1359 | tRNA charging (Leu) | tRNA charging (Leu) |
| Energy metabolism | 0804:0808; 1547; 0767; 0525; 0656:0657 | Energy metabolism | Pyruvate metabolism |
| Energy production | 0253:0260 | Energy production | Oxidative phosphorylation |
Numbers correspond to the gene identifier(s) (ID) annotated in the genome of S. pyogenes M23ND.
Among the nonredundant gene modules, two, i.e., those encoding a biotin transporter and heme iron uptake, were upregulated in the hypervirulent CovS− phenotype, indicating their close relationships with the development of hypervirulence of the CovS− mutant. These modules were also reported to be essential for infective growth and were important elements in virulence in Staphylococcus aureus (58) and Bordetella pertussis (59).
The heme iron uptake operon is involved in coordinated regulation with the sagA operon.
The heme iron acquisition operon (sia) is one of the nonredundant gene modules and is significantly upregulated in M23ND/CovS− throughout the growth phases. The sequences and architecture of this 10-gene operon (60, 61) are highly conserved across diverse M-type strains of S. pyogenes, and its gene members exhibit robust (∼2-fold to 4-fold) upregulation in the CovS− strain, except cbiOQMN, which shows a smaller (∼1.5-fold) change (see Fig. S3A in the supplemental material). Located in the upstream region of the sia operon, two genes, shp and shr, encoding surface proteins, have been identified as playing a role in utilization of hemoglobin and related iron sources through capture of heme-containing proteins from the host and subsequently in delivering heme iron to the immediate downstream heme-specific ABC transporter, HtsABC (62). This process allows iron from the host to be utilized by GAS for growth and survival in vivo and in vitro (63, 64).
Interestingly, our RNA-Seq data suggest a coordinated requirement of heme iron for GAS by the observation that the sagA operon, responsible for producing streptolysin S (SLS), is significantly upregulated in SP growth, compared to LP growth, by 8.5-fold and 3.6-fold for M23ND/CovS− and M23ND/CovS+, respectively (see Fig. S3B in the supplemental material). The sagA operon was recently demonstrated to be closely tied to the acquisition of heme iron by GAS by responding to the availability of hemoglobin during an invasive infection (65). As iron sources become more limiting, the expression of sagA leads to an increase in the pools of the hemolysin, SLS, that serves to lyse red blood cells in extracellular environments. The upregulation of the sagA operon revealed by our data suggests that as GAS strains enter SP growth, the lower level of iron sources at SP triggers an increase in the production of SLS as a programmed response in order to acquire heme iron for survival needs in the context of invasive infection.
The observation that M23ND/CovS− at SP has higher expression of both sia and the sagA operon suggests a potential coordination between the production of bacterial toxins utilized for invasive infection and heme iron requirements. Indeed, an increased requirement for iron was demonstrated to closely align with enhanced production of virulence factors by invasive GAS strains and to facilitate the progression of severe disease (66).
DISCUSSION
The CovRS two-component regulatory system has been extensively studied due to its key role in regulating pathogenic virulence and its close association with disease phenotypes. Various studies showed that CovRS was mutated in in vivo isolates from subjects with severe invasive infection or animal-passaged derivations, while this two-component regulator/sensor remained intact in isolates from subjects with superficial diseases. Such studies revealed that CovRS functioned as a genetic switch by explicitly altering pathogenic phenotypes. A variety of gene expression studies have been performed to attempt to understand the molecular mechanism of CovRS in regulating pathogenic processes at the posttranscriptional and posttranslational levels. Further studies showed that CovRS altered the in vivo and in vitro expression of a broad spectrum of virulence factors, including exotoxins speA, speC, and speB; surface adhesins sfb1, lmb, and the gene encoding M protein; and antiphagocytic genes graB, prtS, and hasA. Thus, CovRS precisely and flexibly regulated the expression of these virulence factors in response to host environments or other physiochemical stress conditions. However, the manner in which CovRS controls expression of so many other genes in S. pyogenes is not generally understood. In order to address these issues, we performed a RNA-Seq study using a hypervirulent serotype M23 strain of S. pyogenes (M23ND).
The natural isolate of M23ND was found to contain a 5-nucleotide deletion in covS and resulted in rapid death in a murine model, while the isogenic engineered strain with an intact covS gene showed a 45% survival rate in mice. Thus, we studied transcriptomic differences between the two phenotypes to evaluate the manner of their dynamic regulation by the key virulence regulatory gene covS. Both the CovS− isolate and the isogenic CovS+ strain were cultured to LP and SP. The mRNAs were isolated and subjected to transcript sequencing using RNA-Seq. The high resolution and sensitivity of RNA-Seq allowed us to detect 349 (18%) differentially regulated genes.
We established the virulome of M23ND and showed that it contained 43 virulence genes, 34 of which were regulated by CovS. The expression profile of the virulence genes revealed the finely tuned virulence regulation patterns of CovS by simultaneously suppressing and enhancing the expression of distinct sets of virulence factors. The genes derepressed by the CovS− strain, viz., hasABC, slo and nga, the mga regulon, spyA, and prtS, possess invasive and antiphagocytotic functions, which are important for bacterial persistence in the host, as well as allowing assimilation and conferring resistance to neutrophil killing. Meanwhile, the CovS− strain suppressed the expression of a set of genes, i.e., speB, sfb1, cfa, and graB, which are required by the less virulent CovS+ superficial infection phenotype. The overall expression profiles of these genes were consistent with previous reports and hypotheses suggesting that CovS switching was induced by in vivo selection of CovS− mutants in host environments and that the dispensable functions were abandoned by the invasive phenotype, perhaps for energy conservation.
In addition to the known regulated virulence factors, we also uncovered in this study several genes that were not previously reported to be regulated by CovRS, i.e., those encoding topoisomerase IV, associated with increased antibiotic resistance (parE and parC), internalin (inlA), histidine triad protein (htpA), nga inhibitor (ifs), and speB inhibitor (spi). Their differential levels of expression were confirmed by q-RT-PCR. While we did not detect the accumulated mutations responsible for fluoroquinolone resistance in ParE and ParC and we did not observe significant resistance to fluoroquinolones of two phenotypes of M23ND, the upregulation of parE and parC in M23ND/CovS+ may be associated with the rapid replication and higher growth rate of this strain. Both inlA and htpA encode surface-exposed lipoproteins conserved in S. pyogenes strains or other streptococcal species. They were shown to contribute to antiphagocytosis and colonization in invasive infections by eliciting a host immune response (43, 44). Meanwhile, whether transcriptional regulation of the two surface proteins is universal in other streptococcal strains is worthy of study. The remarkable differential levels of expression of two inhibitors of nga and speB (ifs and spi, respectively) may extend the current knowledge of GAS pathogenesis mechanisms. Such information not only should involve the active roles of a wide spectrum of virulence factors but also should include self-protective machineries by inhibiting overexpressed harmful activities, such as those exhibited by proteases and DNases.
By leveraging the advantages of RNA-Seq, we also systematically characterized for the first time the CovS-regulated genes that are involved in nutrient utilization and metabolism in S. pyogenes. Compared to the previous studies of S. pyogenes transcripts, we were able to detect metabolic genes with higher resolution and perform a systematic functional characterization. A total of 114 genes were identified as regulated by CovS− that are involved in diverse nutrient utilization systems, i.e., multiple carbohydrates, glycerol, cofactors, heme iron, amino acids, nucleotides, and inorganic molecules, and de novo biosynthesis of basic materials, i.e., fatty acids, amino acids, and nucleotides, as well as involved in energy metabolism. The overall regulation profile of metabolic genes demonstrated the versatile nutrient utilization capacity and complex metabolic requirements for intracellular growth for both phenotypes. In these cases, CovS-mediated regulation again revealed the two distinct virulence phenotypes. On the one hand, the hypervirulent phenotype, CovS−, exhibited more competence than the noninvasive CovS+ phenotype in utilizing high-molecular-weight molecules (e.g., polysaccharides, amino sugars, cofactors, heme iron) which cannot be synthesized by the pathogens. On the other hand, the CovS+ strain displayed a higher rate of consumption of a variety of sugars (e.g., lactose, galactose, sucrose, β-glucose, mannose, and deoxyribose) and a higher energy production rate. The hypervirulent strain maintained an energy-efficient mode of nutrient consumption during growth. It is not clear whether this mode is related to the morphology of high encapsulation and persistence in response to stress in the host immune system. The expression profile of genes for nutrient acquisition could be dependent to various extents on local nutrient availability in the infection niches or in culture media.
Surprisingly highly versatile and redundant utilization of nutrients is commonly found in other mammalian pathogens, indicating that this should be considered part of pathogenic virulence. Thus, we searched for metabolic genes potentially associated with virulence among the CovS-regulated metabolic genes. By constructing a genome-scale metabolic model of S. pyogenes M23ND, we identified 16 nonredundant metabolic gene modules, including 1 for biotin transport and 1 for heme iron uptake, which constitute the unique source for specific nutrient derivation. The nutrients from these sources can be obtained only by de novo biosynthesis due to their deficiency in host environments or can be derived from the host. Therefore, the computationally predicted nonredundant genes are proposed to be essential for cell growth and may contribute to the development of virulence of pathogenic S. pyogenes.
Observing that the CovS-dependent changes in expression of metabolic genes are generally smaller than those seen with virulence genes, we suggest that CovRS regulation is predominantly responsible for pathogenic virulence. On the other hand, the growth-phase-dependent changes in the expression of metabolic genes were much higher than those seen with virulence genes, indicating that expression of metabolic genes is sensitive to nutrient status and may involve other regulatory elements or processes which interact with the CovRS system (67). The multifaceted regulation of metabolic genes is also manifested by the lower frequency of identification of promoter elements and the CovR-binding motif (ATTARA) (68) within the upstream coding regions of metabolic genes in comparison with levels seen with the virulence genes. We detected the promoter elements and CovR-binding motif for only 4 of the 114 regulated metabolic genes, viz., dppA, araR, glpR, and glnR, compared to 8 of the 34 virulence genes, including hasA, sagA, prtS, ska, spd3, graB, ropB, and covR itself. Among these genes, hasA, sagA, prtS, ska, covR, and dppA have been previously examined for their interactions with CovR (69–73).
In conclusion, the current RNA-Seq study provided quantitative transcriptomic signatures of the CovS regulation in a hypervirulent S. pyogenes strain and addressed aspects of the CovS-modulated virulome. Our studies revealed that CovS finely modulates the pathogenic phenotypes of S. pyogenes M23ND by regulating a broad spectrum of genes, including previously established virulence factors and newly identified nutrient metabolic modules. An extensive investigation of the regulated genes in host infection will help understanding of the virulence mechanisms of S. pyogenes and provide new perspectives into therapeutic strategies.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by NIH grant HL013423 (to F.J.C.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00511-15.
REFERENCES
- 1.Aziz RK, Pabst MJ, Jeng A, Kansal R, Low DE, Nizet V, Kotb M. 2004. Invasive M1T1 group A Streptococcus undergoes a phase-shift in vivo to prevent proteolytic degradation of multiple virulence factors by SpeB. Mol Microbiol 51:123–134. [DOI] [PubMed] [Google Scholar]
- 2.Cole JN, McArthur JD, McKay FC, Sanderson-Smith ML, Cork AJ, Ranson M, Rohde M, Itzek A, Sun H, Ginsburg D, Kotb M, Nizet V, Chhatwal GS, Walker MJ. 2006. Trigger for group A streptococcal M1T1 invasive disease. FASEB J 20:1745–1747. doi: 10.1096/fj.06-5804fje. [DOI] [PubMed] [Google Scholar]
- 3.Treviño J, Perez N, Ramirez-Pena E, Liu Z, Shelburne SA, Musser JM, Sumby P. 2009. CovS simultaneously activates and inhibits the CovR-mediated repression of distinct subsets of group A Streptococcus virulence factor-encoding genes. Infect Immun 77:3141–3149. doi: 10.1128/IAI.01560-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hollands A, Pence MA, Timmer AM, Osvath SR, Turnbull L, Whitchurch CB, Walker MJ, Nizet V. 2010. Genetic switch to hypervirulence reduces colonization phenotypes of the globally disseminated group A streptococcus M1T1 clone. J Infect Dis 202:11–19. doi: 10.1086/653124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kansal RG, Datta V, Aziz RK, Abdeltawab NF, Rowe S, Kotb M. 2010. Dissection of the molecular basis for hypervirulence of an in vivo-selected phenotype of the widely disseminated M1T1 strain of group A Streptococcus bacteria. J Infect Dis 201:855–865. doi: 10.1086/651019. [DOI] [PubMed] [Google Scholar]
- 6.Miyoshi-Akiyama T, Ikebe T, Watanabe H, Uchiyama T, Kirikae T, Kawamura Y. 2006. Use of DNA arrays to identify a mutation in the negative regulator, csrR, responsible for the high virulence of a naturally occurring type M3 group A streptococcus clinical isolate. J Infect Dis 193:1677–1684. doi: 10.1086/504263. [DOI] [PubMed] [Google Scholar]
- 7.Mayfield JA, Liang Z, Agrahari G, Lee SW, Donahue DL, Ploplis VA, Castellino FJ. 2014. Mutations in the control of virulence sensor gene from Streptococcus pyogenes after infection in mice lead to clonal bacterial variants with altered gene regulatory activity and virulence. PLoS One 9:e100698. doi: 10.1371/journal.pone.0100698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia AF, Abe LM, Erdem G, Cortez CL, Kurahara D, Yamaga K. 2010. An insert in the covS gene distinguishes a pharyngeal and a blood isolate of Streptococcus pyogenes found in the same individual. Microbiology 156:3085–3095. doi: 10.1099/mic.0.042614-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graham MR, Smoot LM, Migliaccio CA, Virtaneva K, Sturdevant DE, Porcella SF, Federle MJ, Adams GJ, Scott JR, Musser JM. 2002. Virulence control in group A Streptococcus by a two-component gene regulatory system: global expression profiling and in vivo infection modeling. Proc Natl Acad Sci U S A 99:13855–13860. doi: 10.1073/pnas.202353699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sumby P, Whitney AR, Graviss EA, DeLeo FR, Musser JM. 2006. Genome-wide analysis of group a streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog 2:e5. doi: 10.1371/journal.ppat.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graham MR, Virtaneva K, Porcella SF, Barry WT, Gowen BB, Johnson CR, Wright FA, Musser JM. 2005. Group A Streptococcus transcriptome dynamics during growth in human blood reveals bacterial adaptive and survival strategies. Am J Pathol 166:455–465. doi: 10.1016/S0002-9440(10)62268-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aziz RK, Kansal R, Aronow BJ, Taylor WL, Rowe SL, Kubal M, Chhatwal GS, Walker MJ, Kotb M. 2010. Microevolution of group A streptococci in vivo: capturing regulatory networks engaged in sociomicrobiology, niche adaptation, and hypervirulence. PLoS One 5:e9798. doi: 10.1371/journal.pone.0009798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okamoto H, Minami M, Shoin S, Koshimura S, Shimizu R. 1966. Experimental anticancer studies. XXXI. On the streptococcal preparation having potent anticancer activity. Jpn J Exp Med 36:175–186. [PubMed] [Google Scholar]
- 14.Bao Y, Liang Z, Booyjzsen C, Mayfield JA, Li Y, Lee SW, Ploplis VA, Song H, Castellino FJ. 2014. Unique genomic arrangements in an invasive serotype M23 strain of Streptococcus pyogenes identify genes that induce hypervirulence. J Bacteriol 196:4089–4102. doi: 10.1128/JB.02131-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B 57:289–300. [Google Scholar]
- 18.Ghosh D. 26 July 2012, posting date Incorporating the empirical null hypothesis into the Benjamini-Hochberg procedure. Stat Appl Genet Mol Biol doi: 10.1515/1544-6115.1735. [DOI] [PubMed] [Google Scholar]
- 19.Henry CS, DeJongh M, Best AA, Frybarger PM, Linsay B, Stevens RL. 2010. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat Biotechnol 28:977–982. doi: 10.1038/nbt.1672. [DOI] [PubMed] [Google Scholar]
- 20.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. 2012. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40:D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caspi R, Dreher K, Karp PD. 2013. The challenge of constructing, classifying, and representing metabolic pathways. FEMS Microbiol Lett 345:85–93. doi: 10.1111/1574-6968.12194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caspi R, Altman T, Billington R, Dreher K, Foerster H, Fulcher CA, Holland TA, Keseler IM, Kothari A, Kubo A, Krummenacker M, Latendresse M, Mueller LA, Ong Q, Paley S, Subhraveti P, Weaver DS, Weerasinghe D, Zhang P, Karp PD. 2014. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res 42:D459–D471. doi: 10.1093/nar/gkt1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karp PD. 2001. Pathway databases: a case study in computational symbolic theories. Science 293:2040–2044. doi: 10.1126/science.1064621. [DOI] [PubMed] [Google Scholar]
- 24.Karp PD, Paley SM, Krummenacker M, Latendresse M, Dale JM, Lee TJ, Kaipa P, Gilham F, Spaulding A, Popescu L, Altman T, Paulsen I, Keseler IM, Caspi R. 2010. Pathway Tools version 13.0: integrated software for pathway/genome informatics and systems biology. Brief Bioinform 11:40–79. doi: 10.1093/bib/bbp043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang Z, Zhang Y, Agrahari G, Chandrahas V, Glinton K, Donahue DL, Balsara RD, Ploplis VA, Castellino FJ. 2013. A natural inactivating mutation in the CovS component of the CovRS regulatory operon in a pattern D streptococcal pyogenes strain influences virulence-associated genes. J Biol Chem 288:6561–6573. doi: 10.1074/jbc.M112.442657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schrager HM, Rheinwald JG, Wessels MR. 1996. Hyaluronic acid capsule and the role of streptococcal entry into keratinocytes in invasive skin infection. J Clin Invest 98:1954–1958. doi: 10.1172/JCI118998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bauer AW, Kirby WM, Sherris JC, Turck M. 1966. Antibiotic susceptibility testing by a standardized single disk method. Am J Clin Pathol 45:493–496. [PubMed] [Google Scholar]
- 28.Cunningham MA, Rondeau E, Chen X, Coughlin SR, Holdsworth SR, Tipping PG. 2000. Protease-activated receptor 1 mediates thrombin-dependent, cell-mediated renal inflammation in crescentic glomerulonephritis. J Exp Med 191:455–461. doi: 10.1084/jem.191.3.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davies MR, Holden MT, Coupland P, Chen JH, Venturini C, Barnett TC, Zakour NL, Tse H, Dougan G, Yuen KY, Walker MJ. 2015. Emergence of scarlet fever Streptococcus pyogenes emm12 clones in Hong Kong is associated with toxin acquisition and multidrug resistance. Nat Genet 47:84–87. [DOI] [PubMed] [Google Scholar]
- 30.Williams DL, Slayden RA, Amin A, Martinez AN, Pittman TL, Mira A, Mitra A, Nagaraja V, Morrison NE, Moraes M, Gillis TP. 2009. Implications of high level pseudogene transcription in Mycobacterium leprae. BMC Genomics 10:397. doi: 10.1186/1471-2164-10-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brosch M, Saunders GI, Frankish A, Collins MO, Yu L, Wright J, Verstraten R, Adams DJ, Harrow J, Choudhary JS, Hubbard T. 2011. Shotgun proteomics aids discovery of novel protein-coding genes, alternative splicing, and “resurrected” pseudogenes in the mouse genome. Genome Res 21:756–767. doi: 10.1101/gr.114272.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cole JN, Barnett TC, Nizet V, Walker MJ. 2011. Molecular insight into invasive group A streptococcal disease. Nat Rev Microbiol 9:724–736. doi: 10.1038/nrmicro2648. [DOI] [PubMed] [Google Scholar]
- 33.Kansal RG, McGeer A, Low DE, Norrby-Teglund A, Kotb M. 2000. Inverse relation between disease severity and expression of the streptococcal cysteine protease, SpeB, among clonal M1T1 isolates recovered from invasive group A streptococcal infection cases. Infect Immun 68:6362–6369. doi: 10.1128/IAI.68.11.6362-6369.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Graham MR, Virtaneva K, Porcella SF, Gardner DJ, Long RD, Welty DM, Barry WT, Johnson CA, Parkins LD, Wright FA, Musser JM. 2006. Analysis of the transcriptome of group A Streptococcus in mouse soft tissue infection. Am J Pathol 169:927–942. doi: 10.2353/ajpath.2006.060112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Z, Itzek A, Malke H, Ferretti JJ, Kreth J. 2012. Dynamics of speB mRNA transcripts in Streptococcus pyogenes. J Bacteriol 194:1417–1426. doi: 10.1128/JB.06612-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dalton TL, Collins JT, Barnett TC, Scott JR. 2006. RscA, a member of the MDR1 family of transporters, is repressed by CovR and required for growth of Streptococcus pyogenes under heat stress. J Bacteriol 188:77–85. doi: 10.1128/JB.188.1.77-85.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ravins M, Jaffe J, Hanski E, Shetzigovski I, Natanson-Yaron S, Moses AE. 2000. Characterization of a mouse-passaged, highly encapsulated variant of group A streptococcus in in vitro and in vivo studies. J Infect Dis 182:1702–1711. doi: 10.1086/317635. [DOI] [PubMed] [Google Scholar]
- 38.Drlica K, Zhao X. 1997. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev 61:377–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alonso R, Galimand M, Courvalin P. 2002. parC mutation conferring ciprofloxacin resistance in Streptococcus pyogenes BM4513. Antimicrob Agents Chemother 46:3686–3687. doi: 10.1128/AAC.46.11.3686-3687.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malhotra-Kumar S, Van Heirstraeten L, Lammens C, Chapelle S, Goossens H. 2009. Emergence of high-level fluoroquinolone resistance in emm6 Streptococcus pyogenes and in vitro resistance selection with ciprofloxacin, levofloxacin and moxifloxacin. J Antimicrob Chemother 63:886–894. doi: 10.1093/jac/dkp057. [DOI] [PubMed] [Google Scholar]
- 41.Orscheln RC, Johnson DR, Olson SM, Presti RM, Martin JM, Kaplan EL, Storch GA. 2005. Intrinsic reduced susceptibility of serotype 6 Streptococcus pyogenes to fluoroquinolone antibiotics. J Infect Dis 191:1272–1279. doi: 10.1086/428856. [DOI] [PubMed] [Google Scholar]
- 42.González I, Georgiou M, Alcaide F, Balas D, Linares J, de la Campa AG. 1998. Fluoroquinolone resistance mutations in the parC, parE, and gyrA genes of clinical isolates of viridans group streptococci. Antimicrob Agents Chemother 42:2792–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reid SD, Montgomery AG, Voyich JM, DeLeo FR, Lei B, Ireland RM, Green NM, Liu M, Lukomski S, Musser JM. 2003. Characterization of an extracellular virulence factor made by group A Streptococcus with homology to the Listeria monocytogenes internalin family of proteins. Infect Immun 71:7043–7052. doi: 10.1128/IAI.71.12.7043-7052.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kunitomo E, Terao Y, Okamoto S, Rikimaru T, Hamada S, Kawabata S. 2008. Molecular and biological characterization of histidine triad protein in group A streptococci. Microbes Infect 10:414–423. doi: 10.1016/j.micinf.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 45.Eddy SR. 2011. Accelerated profile HMM searches. PLoS Comput Biol 7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn RD. 2012. The Pfam protein families database. Nucleic Acids Res 40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ato M, Ikebe T, Kawabata H, Takemori T, Watanabe H. 2008. Incompetence of neutrophils to invasive group A streptococcus is attributed to induction of plural virulence factors by dysfunction of a regulator. PLoS One 3:e3455. doi: 10.1371/journal.pone.0003455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cole JN, Pence MA, von Köckritz-Blickwede M, Hollands A, Gallo RL, Walker MJ, Nizet V. 2010. M protein and hyaluronic acid capsule are essential for in vivo selection of covRS mutations characteristic of invasive serotype M1T1 group A Streptococcus. mBio 1:e00191–10. doi: 10.1128/mBio.00191-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kurupati P, Turner CE, Tziona I, Lawrenson RA, Alam FM, Nohadani M, Stamp GW, Zinkernagel AS, Nizet V, Edwards RJ, Sriskandan S. 2010. Chemokine-cleaving Streptococcus pyogenes protease SpyCEP is necessary and sufficient for bacterial dissemination within soft tissues and the respiratory tract. Mol Microbiol 76:1387–1397. doi: 10.1111/j.1365-2958.2010.07065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li J, Zhu H, Feng W, Liu M, Song Y, Zhang X, Zhou Y, Bei W, Lei B. 2013. Regulation of inhibition of neutrophil infiltration by the two-component regulatory system CovRS in subcutaneous murine infection with group A streptococcus. Infect Immun 81:974–983. doi: 10.1128/IAI.01218-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lobel L, Sigal N, Borovok I, Ruppin E, Herskovits AA. 2012. Integrative genomic analysis identifies isoleucine and CodY as regulators of Listeria monocytogenes virulence. PLoS Genet 8:e1002887. doi: 10.1371/journal.pgen.1002887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jenkins A, Cote C, Twenhafel N, Merkel T, Bozue J, Welkos S. 2011. Role of purine biosynthesis in Bacillus anthracis pathogenesis and virulence. Infect Immun 79:153–166. doi: 10.1128/IAI.00925-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kloosterman TG, Hendriksen WT, Bijlsma JJ, Bootsma HJ, van Hijum SA, Kok J, Hermans PW, Kuipers OP. 2006. Regulation of glutamine and glutamate metabolism by GlnR and GlnA in Streptococcus pneumoniae. J Biol Chem 281:25097–25109. doi: 10.1074/jbc.M601661200. [DOI] [PubMed] [Google Scholar]
- 54.Si Y, Yuan F, Chang H, Liu X, Li H, Cai K, Xu Z, Huang Q, Bei W, Chen H. 2009. Contribution of glutamine synthetase to the virulence of Streptococcus suis serotype 2. Vet Microbiol 139:80–88. doi: 10.1016/j.vetmic.2009.04.024. [DOI] [PubMed] [Google Scholar]
- 55.Steeb B, Claudi B, Burton NA, Tienz P, Schmidt A, Farhan H, Maze A, Bumann D. 2013. Parallel exploitation of diverse host nutrients enhances Salmonella virulence. PLoS Pathog 9:e1003301. doi: 10.1371/journal.ppat.1003301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perfect JR. 2004. Genetic requirements for virulence in Cryptococcus neoformans, p 89–111. In Domer JE, Kobayashi GS (ed), The Mycota: human fungal pathogens, vol 12 Springer, New York, NY. [Google Scholar]
- 57.Eisenreich W, Dandekar T, Heesemann J, Goebel W. 2010. Carbon metabolism of intracellular bacterial pathogens and possible links to virulence. Nat Rev Microbiol 8:401–412. doi: 10.1038/nrmicro2351. [DOI] [PubMed] [Google Scholar]
- 58.Torres VJ, Pishchany G, Humayun M, Schneewind O, Skaar EP. 2006. Staphylococcus aureus IsdB is a hemoglobin receptor required for heme iron utilization. J Bacteriol 188:8421–8429. doi: 10.1128/JB.01335-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brickman TJ, Vanderpool CK, Armstrong SK. 2006. Heme transport contributes to in vivo fitness of Bordetella pertussis during primary infection in mice. Infect Immun 74:1741–1744. doi: 10.1128/IAI.74.3.1741-1744.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bates CS, Montanez GE, Woods CR, Vincent RM, Eichenbaum Z. 2003. Identification and characterization of a Streptococcus pyogenes operon involved in binding of hemoproteins and acquisition of iron. Infect Immun 71:1042–1055. doi: 10.1128/IAI.71.3.1042-1055.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lei B, Liu M, Voyich JM, Prater CI, Kala SV, DeLeo FR, Musser JM. 2003. Identification and characterization of HtsA, a second heme-binding protein made by Streptococcus pyogenes. Infect Immun 71:5962–5969. doi: 10.1128/IAI.71.10.5962-5969.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu H, Liu M, Lei B. 2008. The surface protein Shr of Streptococcus pyogenes binds heme and transfers it to the streptococcal heme-binding protein Shp. BMC Microbiol 8:15. doi: 10.1186/1471-2180-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fisher M, Huang YS, Li X, McIver KS, Toukoki C, Eichenbaum Z. 2008. Shr is a broad-spectrum surface receptor that contributes to adherence and virulence in group A streptococcus. Infect Immun 76:5006–5015. doi: 10.1128/IAI.00300-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dahesh S, Nizet V, Cole JN. 2012. Study of streptococcal hemoprotein receptor (Shr) in iron acquisition and virulence of M1T1 group A streptococcus. Virulence 3:566–575. doi: 10.4161/viru.21933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Salim KY, de Azavedo JC, Bast DJ, Cvitkovitch DG. 2007. Role for sagA and siaA in quorum sensing and iron regulation in Streptococcus pyogenes. Infect Immun 75:5011–5017. doi: 10.1128/IAI.01824-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eichenbaum Z, Muller E, Morse SA, Scott JR. 1996. Acquisition of iron from host proteins by the group A streptococcus. Infect Immun 64:5428–5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Horstmann N, Saldana M, Sahasrabhojane P, Yao H, Su X, Thompson E, Koller A, Shelburne SA. 2014. Dual-site phosphorylation of the control of virulence regulator impacts group a streptococcal global gene expression and pathogenesis. PLoS Pathog 10:e1004088. doi: 10.1371/journal.ppat.1004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Federle MJ, Scott JR. 2002. Identification of binding sites for the group A streptococcal global regulator CovR. Mol Microbiol 43:1161–1172. doi: 10.1046/j.1365-2958.2002.02810.x. [DOI] [PubMed] [Google Scholar]
- 69.Gusa AA, Froehlich BJ, Desai D, Stringer V, Scott JR. 2007. CovR activation of the dipeptide permease promoter (PdppA) in group A Streptococcus. J Bacteriol 189:1407–1416. doi: 10.1128/JB.01036-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gao J, Gusa AA, Scott JR, Churchward G. 2005. Binding of the global response regulator protein CovR to the sag promoter of Streptococcus pyogenes reveals a new mode of CovR-DNA interaction. J Biol Chem 280:38948–38956. doi: 10.1074/jbc.M506121200. [DOI] [PubMed] [Google Scholar]
- 71.Sumby P, Zhang S, Whitney AR, Falugi F, Grandi G, Graviss EA, DeLeo FR, Musser JM. 2008. A chemokine-degrading extracellular protease made by group A Streptococcus alters pathogenesis by enhancing evasion of the innate immune response. Infect Immun 76:978–985. doi: 10.1128/IAI.01354-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gusa AA, Scott JR. 2005. The CovR response regulator of group A streptococcus (GAS) acts directly to repress its own promoter. Mol Microbiol 56:1195–1207. doi: 10.1111/j.1365-2958.2005.04623.x. [DOI] [PubMed] [Google Scholar]
- 73.Miller AA, Engleberg NC, DiRita VJ. 2001. Repression of virulence genes by phosphorylation-dependent oligomerization ofCsrR at target promoters in S. pyogenes. Mol Microbiol 40:976–990. doi: 10.1046/j.1365-2958.2001.02441.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.