

Caserta et al. generated and analyzed Pten knock-in mice harboring a C2 domain missense mutation at phenylalanine 341 (PtenFV), found in human cancer. This PTEN noncatalytic missense mutation exposes a core tumor suppressor function distinct from inhibition of canonical AKT signaling that predisposes to organ-selective cancer development in vivo.
Keywords: F341V, breast, cancer, endometrium, pten, uterus
Abstract
Inactivation of phosphatase and tensin homology deleted on chromosome 10 (PTEN) is linked to increased PI3K–AKT signaling, enhanced organismal growth, and cancer development. Here we generated and analyzed Pten knock-in mice harboring a C2 domain missense mutation at phenylalanine 341 (PtenFV), found in human cancer. Despite having reduced levels of PTEN protein, homozygous PtenFV/FV embryos have intact AKT signaling, develop normally, and are carried to term. Heterozygous PtenFV/+ mice develop carcinoma in the thymus, stomach, adrenal medulla, and mammary gland but not in other organs typically sensitive to Pten deficiency, including the thyroid, prostate, and uterus. Progression to carcinoma in sensitive organs ensues in the absence of overt AKT activation. Carcinoma in the uterus, a cancer-resistant organ, requires a second clonal event associated with the spontaneous activation of AKT and downstream signaling. In summary, this PTEN noncatalytic missense mutation exposes a core tumor suppressor function distinct from inhibition of canonical AKT signaling that predisposes to organ-selective cancer development in vivo.
In human cancer, phosphatase and tensin homology deleted on chromosome 10 (PTEN) is subject to large and small chromosomal deletions, missense and nonsense mutations, and silencing by promoter CpG island methylation (for review, see Song et al. 2012). Humans carrying a germline mutation in PTEN may be developmentally delayed and are highly predisposed to thyroid, endometrial, and breast cancer, symptoms collectively known as PTEN hamartoma tumor syndrome (PHTS), of which Cowden syndrome is the best known (Pilarski et al. 2013). Mice carrying a germline-null Pten allele develop tumors in a variety of organs typically affected in PHTS patients, demonstrating a causal relationship between PTEN inactivation and cancer (Di Cristofano et al. 1998; Suzuki et al. 1998; Podsypanina et al. 1999; Trotman et al. 2003; Wang et al. 2010; Papa et al. 2014; Sun et al. 2014).
The PTEN protein is a lipid phosphatase that negatively regulates cellular concentration of PtdIns (3,4,5)P3 (PIP3), a second messenger linked to signaling pathways that control cell survival, proliferation, and motility (Maehama and Dixon 1998). Recent studies, however, suggest additional unexpected roles of PTEN that are unrelated to its phosphatase activity or PIP3 levels (Shi et al. 2012). PTEN encodes two isoforms: a 403-amino-acid protein that localizes to the cytoplasm and nucleus and a recently identified larger protein containing a leader peptide that facilitates its secretion into the extracellular environment (Hopkins et al. 2013). Both isoforms contain three well-characterized functional modules: an N-terminal catalytic domain, a C2 domain, and an extended C-terminal tail. The N-terminal active center catalyzes the dephosphorylation of PIP3 to PIP2 (Lee et al. 1999); however, a number of protein substrates have also been identified (Tamura et al. 1999; Raftopoulou et al. 2004; Zhang et al. 2011; Juric et al. 2014). The C2 well-structured domain mediates interactions with lipids and proteins important for its spatial compartmentalization within the cell (Lee et al. 1999; Meuillet et al. 2004). The C-terminal tail regulates protein stability and access of the C2 domain to lipid substrates (Georgescu et al. 1999; Vazquez et al. 2000, 2001; Wu et al. 2000a,b; Torres and Pulido 2001; Al-Khouri et al. 2005; Okahara et al. 2006; Yim et al. 2007). Several layers of regulation are used to keep overall PTEN protein levels and activity under tight control, including transcriptional, translational, post-translational, and subcellular trafficking mechanisms (for review, see Fata et al. 2012). However, the spatial segregation of PTEN regulation and activity within the cell is poorly understood and is likely critical for its broad impact on physiology, including cognition, metabolism, aging, and cancer.
Genetically engineered mice carrying missense mutations within the N-terminal catalytic domain, mice carrying a nonsense mutation that generates a C-terminal truncated product, or mice with a complete null allele of Pten have been described (Di Cristofano et al. 1998; Suzuki et al. 1998; Wang et al. 2010; Papa et al. 2014; Sun et al. 2014). Here, we used a knock-in approach to generate mice harboring a phenylalanine-to-valine substitution within the C2 β-sheet domain at amino acid position 341 (PtenFV), which has been identified in human glioblastoma (Kato et al. 2000) and colorectal and endometrial cancer as well as Cowden syndrome patients (Ali et al. 1999; Minaguchi et al. 2001; Parsons et al. 2005; Peterson et al. 2012). We show that mutant PTENFV protein has a short half-life and is dramatically depleted from the nucleus, yet cells and tissues retain intact AKT signaling, and homozygous PtenFV/FV embryos are carried to term. Intriguingly, mice with this mutation developed cancer only in a subset of organs typically impacted by Pten deficiency. Whereas cancer in susceptible organs ensued in the absence of overt AKT activation, progression to carcinoma in cancer-resistant organs required an additional focal event that we show biochemically to be associated with the spontaneous activation of AKT signaling. Thus, the in vivo analysis of this noncatalytic mutation exposes a tumor suppressor function of PTEN distinct from its canonical role in AKT signaling that mechanistically defines stage- and organ-specific cancer development.
Results
PtenFV supports embryonic development
In human cancer, the majority of sequence alterations in PTEN result in the abrogation of phosphatase activity by either missense mutations that cluster around the catalytic domain or nonsense mutations that lead to truncated proteins (Bonneau et al. 2000). However, some patients harbor missense mutations in the C2 domain that have as of yet undefined molecular consequences. We used standard homologous recombination approaches to introduce a C2 missense mutation into the mouse genome, where phenylalanine 341 was substituted by valine (Fig. 1A). Southern blot, PCR genotyping, and sequencing of mouse genomic DNA confirmed correct integration of the mutant PtenFV allele into mouse embryonic stem (ES) cells (Fig. 1B,C). Targeted ES cells were then used to generate mice carrying the germline-transmitted PtenFV allele, and positive offspring were bred with EIIA-cre mice to remove the LoxP-flanked neomycin selection cassette.
Figure 1.

Generation, targeting, and embryonic development of PtenFV knock-in mice. (A) Schematic diagram of the Pten locus (top); the Pten targeting vector with a missense mutation in exon 8 (*), containing the selectable phosphoglycerate kinase promoter (PGK)-neo cassette flanked by LoxP sites (triangles; middle); and the targeted mutant PtenFV allele lacking the PGK-neo cassette (after mating with EIIA-cre-expressing mice) (+cre; bottom). (Probe A and probe B) DNA probes used for Southern blot analysis; (prim1–3) primers used for PCR genotyping. (B) Southern blot analyses using probe A (top panel) and probe B (middle panel). (Bottom panel) Genotyping PCR of tail DNA with the indicated genotypes. The expected band sizes are indicated for each allele. (C) Sequence analysis of tail DNA isolated from Pten+/+ and PtenFV/FV newborn pups. Chromatograms depicting the successful targeting of the Pten locus, with translated amino acids shown below the codons (underlined). Red letters and bold numbers denote the targeted amino acid F341V. The black arrows point to the targeted nucleotide. (D) The observed and expected number of pups from heterozygous intercrosses harvested at birth (P0) and 7 d post-birth (P7) are indicated. The expected and observed numbers of embryos were compared using a χ2 test. (*) P = 0.003; (**) P < 0.001; (+) wild-type allele; (FV) F341V mutant allele. (E) Photographs of Pten+/+ and PtenFV/FV littermate newborn offspring derived from the PtenFV/+ heterozygous intercrosses in D. The arrow points to the stomach of the wild-type pup containing milk, which was not observed in homozygous mutant pups.
We next evaluated the developmental consequences of this mutation in vivo. Heterozygous PtenFV/+ mice were viable and fertile (Fig. 1D). Interbreeding heterozygous animals yielded homozygous PtenFV/FV embryos that were born at Mendelian ratios. While mutant newborn pups appeared externally normal with anatomically well-formed organs, they failed to ingest milk from their nursing mothers and died within their first week of life (Fig. 1D,E). We noted that the hippocampus in PtenFV/FV embryonic day 18.5 (E18.5) embryos had focal areas of neuronal disorganization (heterotypia), indicating improper migration of neurons during development (Supplemental Fig. 1a,b). Aberrant development of the nervous system may have contributed to the inability of newborns to suckle properly on nursing mothers. In conclusion, the postnatal lethality of mice was the first salient in vivo phenotype distinguishing the PtenFV mutation from previously characterized Pten-null (Di Cristofano et al. 1998; Suzuki et al. 1998; Wang et al. 2010), missense (Wang et al. 2010; Papa et al. 2014), or nonsense mutations (Sun et al. 2014), which result in unregulated PI3K–AKT signaling and embryonic death before E9.5.
Reduced protein stability of mutant PTENFV
To explore the molecular alterations caused by this mutation, we isolated PtenFV/FV and littermate control Pten+/+ mouse embryo fibroblasts (MEFs) and evaluated PTEN protein levels and downstream canonical AKT signaling. When compared with controls, PTEN protein levels in PtenFV/FV MEFs were markedly reduced (Fig. 2A; Supplemental Fig. 2a,b), whereas Pten mRNA levels were unchanged (Supplemental Fig. 2c). Time-course experiments with cycloheximide treatment demonstrated a significant reduction in the half-life of mutant PTENFV protein [∼t(1/2) = 5.5 h for PTENFV vs. t(1/2) > 24 h for wild-type PTEN] (Fig. 2B,C). Despite significantly reduced protein levels, downstream canonical PI3K signaling in PtenFV/FV cells, as measured by AKT phosphorylation at Thr308 (P-AKTT308) or Ser473 (P-AKTS473), remained intact, whereas in Pten-deleted cells (PtenΔ/Δ), signaling was robustly activated (Fig. 2A; Supplemental Fig. 2a,b). Moreover, phosphorylation levels of GSK3β, a direct target of AKT, was similar in Pten+/+ and PtenFV/FV cells (Supplemental Fig. 2b).
Figure 2.
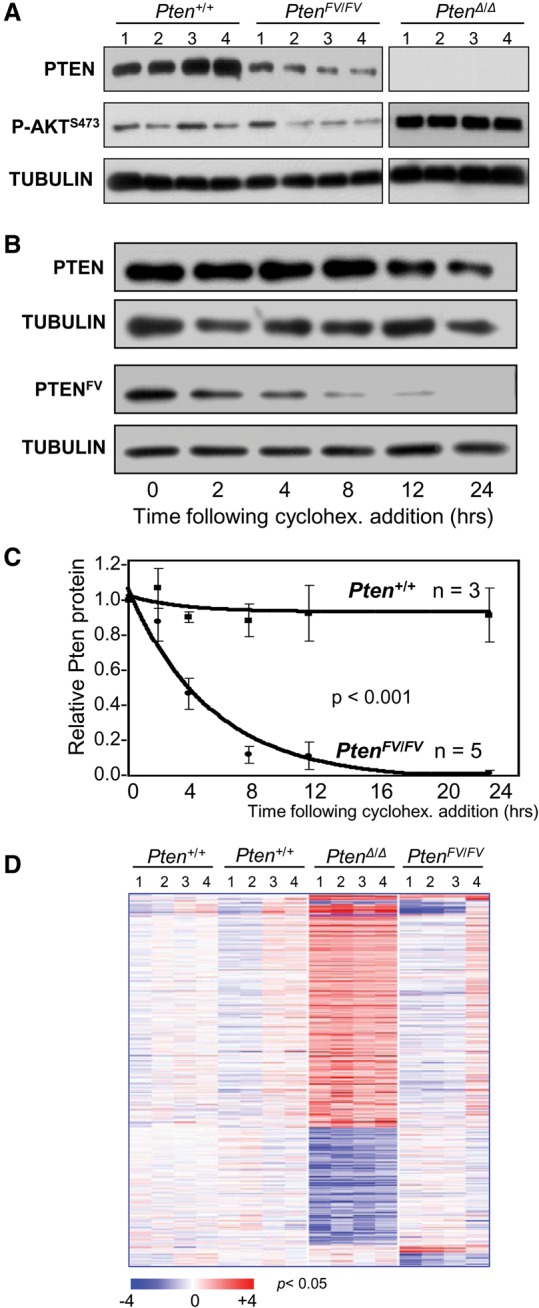
Shorter half-life of mutant PTENFV protein and normal PI3K–AKT signaling in PtenFV/FV MEFs. (A) Immunoblots of whole-cell lysates from MEFs with the indicated genotypes were probed with specific antibodies for PTEN and P-AKTS473; Tubulin was used as the loading control. (B) Cycloheximide-treated (cyclohex.) MEFs with the indicated genotypes were harvested at the indicated times after treatment, and lysates were immunoblotted with antibodies specific for PTEN; Tubulin was used as the loading control. (C) PTEN protein was quantified from immunoblots in B and normalized to Tubulin; wild-type and mutant PTEN protein levels (normalized) were then plotted. (n) Number of independent experiments. Two-way ANOVA with an interaction term (group × time) was used to compare group differences across time. (D) Heat map of differentially expressed genes (greater than twofold; P < 0.05) in MEFs with the indicated genotypes. Two groups of corresponding littermate Pten+/+ MEFs were used as controls for each mutant genotype group.
We also compared the transcriptomes of PtenFV/FV and PtenΔ/Δ MEFs. To this end, RNA isolated from PtenFV/FV, PtenΔ/Δ, and the corresponding wild-type littermate MEFs was used to generate cDNA and probe Affymetrix Mouse Genome 430 2.0 microarrays. Robust Multichip Average (RMA)-normalized gene expression was standardized to the geometric mean of the corresponding Pten+/+ groups, and clustering of differentially expressed genes was visualized by heat maps (greater than twofold; P < 0.005) (Fig. 2D). A subset of differentially expressed genes was confirmed by real-time quantitative PCR (qPCR) assays (Supplemental Fig. 2d). When compared with littermate control Pten+/+ MEFs, profound gene expression changes were observed in PtenΔ/Δ MEFs, whereas no significant changes were observed in PtenFV/FV MEFs that could be confirmed by individual PCRs (Fig. 2D; Supplemental Fig. 2d). Thus, mutant PTENFV protein, while less stable, retains the capacity to effectively inhibit PI3K–AKT signaling and support a relatively normal gene expression program.
Nuclear depletion of mutant PTENFV protein
We next examined the intracellular distribution of mutant PTENFV protein in MEFs and E9.5 embryos. Cellular fractionation and immunoblotting with compartment-specific markers confirmed appropriate enrichment of cytoplasmic, membrane, nuclear, chromatin, and cytoskeletal-associated fractions. As expected, wild-type PTEN protein was mostly distributed between the cytoplasmic and cell membrane compartments, with some protein present in the nucleus (Fig. 3A; Supplemental Fig. 3a). Mutant PTENFV protein was significantly decreased in both the cytoplasmic and membrane compartments and was undetectable in the nuclear fraction.
Figure 3.
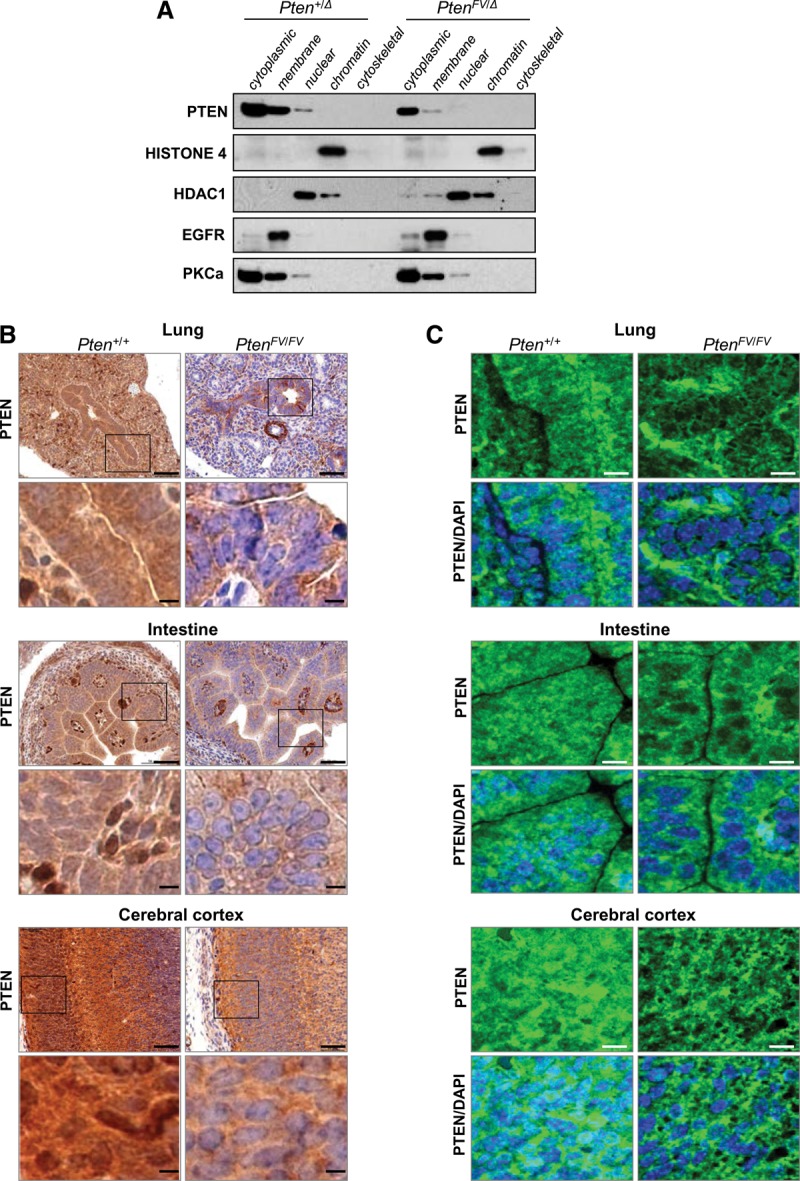
PTENFV is depleted in the nucleus. (A) Cell compartment fractionation of MEFs with the indicated genotypes. Protein lysates from each fraction were immunoblotted and probed with compartment-specific markers (Histone 4, HDAC1, EGFR, and PKCa) and a PTEN-specific antibody. (B) Immunohistochemistry (IHC) of sections from the indicated organs of E15.5 Pten+/+ and PtenFV/FV embryos using a PTEN-specific antibody; IHC stained sections were counterstained with hematoxylin. The boxes in the top panels indicate regions shown at high magnification in the panels directly below. Bars: low magnification, 200 µm; high magnification, 50 µm. (C) Confocal images showing PTEN localization (green) in E15.5 embryonic tissues that were probed with the PTEN-specific antibody. Tissue sections were counterstained with DAPI (blue) to demarcate nuclei. Bar, 50 µm.
PTEN protein was also examined in vivo by immunohistochemistry (IHC) and confocal microscopy. Both cytoplasmic and nuclear PTEN protein could be uniformly detected in most organs of wild-type E15.5 embryos (Fig. 3B,C). The overall levels of PTEN protein were significantly decreased in littermate PtenFV/FV embryos. Intriguingly, nuclear PTEN was markedly depleted in a number of organs, including the budding lungs, intestines, and brains of PtenFV/FV embryos (Fig. 3B,C), even though cytoplasmic protein levels in some of these organs were relatively unaffected. Consistent with the normal growth and appearance of mutant embryos, P-AKT levels were identical in PtenFV/FV and wild-type embryos (Supplemental Fig. 3c). Collectively, these observations suggest that mutant PTENFV protein is unstable and nuclear-depleted and maintains normal canonical AKT signaling, supporting embryonic development.
Organ-specific cancer development in PtenFV/+ knock-in mice
We and others have previously shown that heterozygous animals carrying a germline Pten-null allele (Pten−/+) develop spontaneous tumors in a broad spectrum of organs by 9 mo of age (Wang et al. 2010; Papa et al. 2014; Sun et al. 2014). To determine whether this human cancer-derived C2 mutation results in loss of tumor suppressor function, a cohort of 98 heterozygous PtenFV/+ and 52 wild-type control animals was followed for 18 mo and evaluated histologically for development of neoplastic lesions. Like Pten−/+ animals described previously (Wang et al. 2010), heterozygous PtenFV/+ mice developed cancer in the adrenal medulla, thymus, stomach, and mammary gland (Fig. 4; Supplemental Fig. 4). However, no significant lesions beyond low-grade hyperplasia were observed in the thyroids, prostates, and uteri of PtenFV/+ mice, which are additional organs highly predisposed to cancer development in Pten−/+ animals. Wild-type control mice also had no evidence of carcinoma in these three organs (Supplemental Fig. 4). These findings demonstrate that PTENF341V is a relevant human cancer noncatalytic mutation that, when introduced into mice, is sufficient to incite cancer development with remarkable tissue selectivity.
Figure 4.
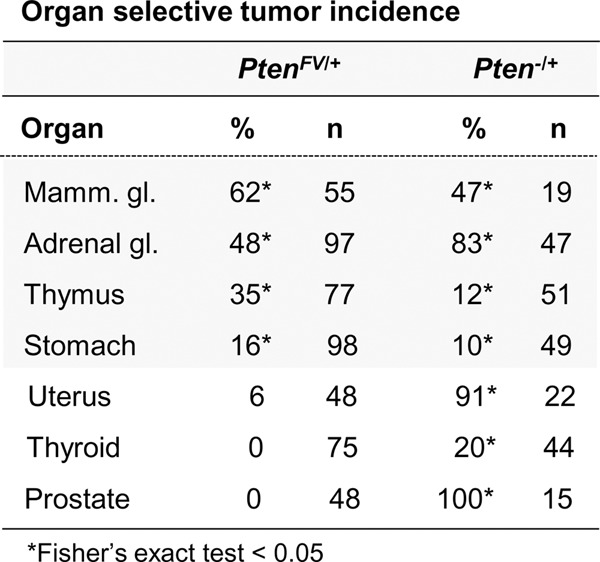
Organ-selective cancer development in PtenFV/+ mice. Histopathological analysis of the indicated tissues from PtenFV/+ males and females was performed and scored as described in the Materials and Methods and Supplemental Figure 4. The table shows the percentage (%) of 18-mo-old PtenFV/+ animals with cancer in the noted organs compared with the percentage of PtenΔ/+ animals with cancer at 9 mo of age described previously (Wang et al. 2010). Carcinoma in control Pten+/+ animals was not observed. (Total n) Number of animals analyzed for each organ site. Fisher's exact tests were used to compare differences between PtenFV/+ and control Pten+/+ animals. (*) P < 0.05.
Progression to uterine carcinoma in Pten+/Δ and PtenFV/Δ mice is associated with activation of P-AKT
To explore the basis for the dichotomous pattern of organ-specific cancer, we first focused on the uterus, as it was one of the three organs in PtenFV/+ mice resistant to cancer. Because PtenFV/FV pups die perinatally, we used a conditional gene ablation approach. To this end, we used a uterine-specific Sprr2f-cre transgene (Contreras et al. 2010) and a conditional allele of PtenL to compare neoplasia development in Pten+/+, PtenΔ/Δ, and PtenFV/Δ females. Whereas wild-type control females had normal uteri, PtenΔ/Δ females developed widespread high-grade endometrial hyperplasia by the time puberty was completed and multifocal carcinoma in situ by 2 mo of age (Fig. 5A). Many of these lesions invaded into the myometrium by 3 mo of age (invasive carcinoma), and, in some mice, there was clear evidence of metastasis to the sublumbar lymph nodes and ovaries (Fig. 5A,B; Supplemental Fig. 5a). In contrast, young PtenFV/Δ females exhibited widespread low-grade neoplasia, with epithelial cells maintaining their normal baso–luminal orientation (data not shown). In 6-mo-old PtenFV/Δ females, however, we noted occasional uterine lesions where epithelial cells lacked normal baso–luminal polarity and proliferated into the luminal spaces, marking a clear transition from low-grade hyperplasia to carcinoma in situ (Fig. 5C).
Figure 5.

Progression of endometrial cancer in PtenFV/Δ females requires a second event associated with activation of P-AKT and downstream signaling. (A) Sections of uteri harvested from Pten+/+, Pten+/Δ, and PtenΔ/Δ females were stained with H&E. The images show examples of normal tissue (Pten+/+), high-grade hyperplasia (Pten+/Δ), carcinoma in situ (PtenΔ/Δ), and invasive carcinoma (PtenΔ/Δ). (B) Full-body necropsy was performed in PtenΔ/Δ females, and sections from tissues with metastatic lesions were stained with H&E. Dotted yellow lines demarcate the borders of metastases. Bar, 50 µm. (C, top two rows) Sections from uteri of 6-mo-old Pten+/+, PtenΔ/Δ, and PtenFV/Δ females were stained with H&E. Boxes in the low-magnification panels indicate regions shown at high magnification in the panels directly below. Consecutively cut sections were also processed for IHC using antibodies specific for PTEN or P-AKTS473 as indicated. Bars: low magnification, 200 µm; high magnification 50 µm. (D) Sections from the uteri of 6-mo-old Pten+/+, PtenΔ/Δ, and PtenFV/Δ females were processed for IHC using antibodies specific for P-AKTS473 or FOXO3A. IHC stained sections were counterstained with hematoxylin. Bar, 20 µm. (E) Sections from paraffin-embedded uteri of 6-mo-old mice with the indicated genotypes were probed with PTEN-specific antibody for confocal microscopy. Images show PTEN localization (green) in the tissues and DAPI staining (blue) demarcating the nuclei in the composite images. Bar, 20 µm.
In PtenFV/Δ females, the spontaneous transition from hyperplasia to carcinoma in situ was associated with the focal loss of PTEN protein expression and a striking accumulation of P-AKT (Fig. 5C; Supplemental Fig. 5b,c). Moreover, the nuclear FOXO3A transcription factor, a well-established AKT target, was shuttled to the cytoplasm (Fig. 5D; Supplemental Fig. 5c). In contrast, the entire uterine glandular epithelial network in PtenΔ/Δ females stained positive for P-AKT and cytoplasmic FOXO3A.
Similar to the PtenFV/FV embryonic tissues described above, there was reduced nuclear PTEN protein in PtenFV/Δ endometrial glands (Fig. 5C,E). Previous work showed an association between reduced nuclear PTEN levels and increased DNA damage and genomic instability (Bassi et al. 2013). Consistent with these earlier observations, the uteri of PtenFV/Δ and PtenΔ/Δ females exhibited widespread γH2AX staining that was not restricted to the neoplastic lesions (Supplemental Fig. 5d). Detection of faint nuclear and strong focal γH2AX staining suggested DNA damage-associated apoptosis, which was confirmed by IHC with cleaved caspase 3 (CC3)-specific antibodies (Supplemental Fig. 5d). Thus, we propose that transition to carcinoma in situ in the uterus is associated with a clonal event that selects for the loss of PTEN expression and the subsequent activation of P-AKT and downstream signaling.
Mammary tumor development in the absence of overt P-AKT signaling
We next focused on the mammary gland, as it was the organ most susceptible to carcinoma development in PtenFV/+ mice. Inspection of whole-mount mammary glands from post-puberty PtenFV/+ females revealed normal ductal development (Supplemental Fig. 6a). However, low-grade mammary intraepithelial neoplasia (Min) was observed by 11 mo of age, high-grade Min with occasional small carcinomas was observed by 17 mo of age, and large adenosquamous carcinoma and adenomyoepitheliomas were observed by 24 mo of age (Fig. 6A; Supplemental Fig. 6b).
Figure 6.
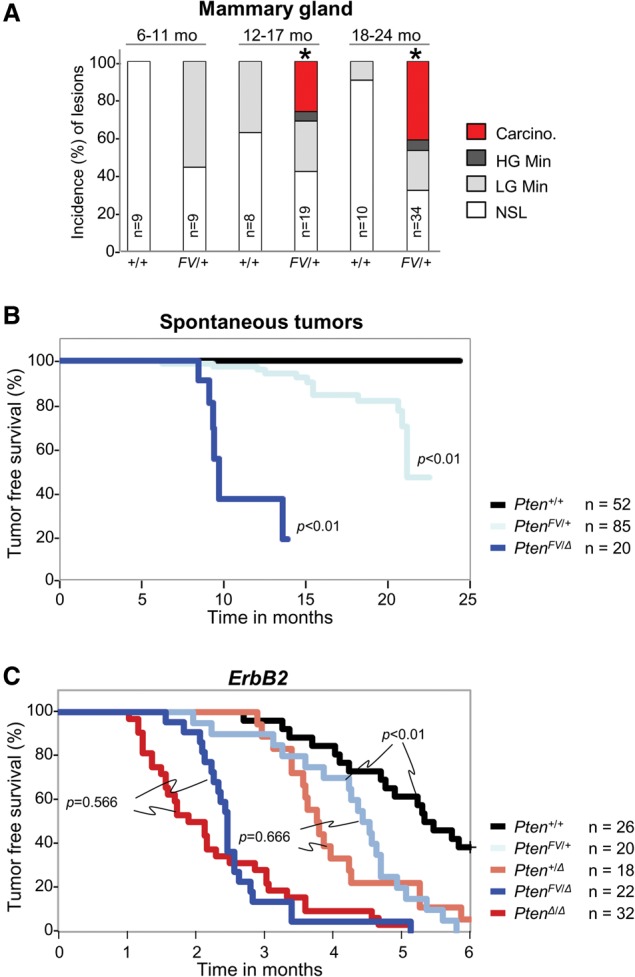
Mammary tumor development in mice harboring the PtenFVmutation. (A) The incidence of neoplastic lesions in mammary glands of Pten+/+ and PtenFV/+ female mice analyzed at the various ages indicated (in months). Histopathological grades are described in detail in the Materials and Methods. (Carcino) Carcinoma; (LG) low grade; (HG) high grade; (NSL) no significant gross or microscopic neoplastic lesions. Two sample tests were used to compare proportions of groups into two categories (carcinoma and/or high-grade Min vs. low-grade Min and/or NSL) within each age group. (*) P < 0.01. No test was performed for the 6- to 11-mo age group, since carcinoma or high-grade Min was not observed. (B,C) Kaplan-Meier curves for tumor-free survival of mice with the indicated Pten genotypes in either the absence (B) or presence (C) of the MMTV-ErbB2 oncogene. Tumors were detected and measured weekly by palpation; tumors were recorded if ≥0.5 cm2. (n) Number of mice. Log-rank test was used to compare Kaplan-Meier curves for each mutant genotype with wild type (B) or between the indicated genotypes (C).
Ablation of the conditional PtenL allele in the mammary ducts of PtenFV/L females using a mammary gland-specific MMTV-cre transgene (Andrechek et al. 2000) led to palpable tumors in 50% of PtenFV/Δ females by 9.5 mo (P < 0.01) (Fig. 6B), which was significantly earlier than in PtenFV/+ females (21 mo). We also examined mammary tumorigenesis in the context of a human-relevant oncogene by introducing the mammary-specific MMTV-ErbB2 transgene (ErbB2) into Pten+/+, PtenΔ/+, PtenΔ/Δ, PtenFV/+, and PtenFV/Δ mice. In this setting, tumors developed in 50% of control ErbB2;Pten+/+ females by 5.5 mo of age, 50% of ErbB2;PtenΔ/+ and ErbB2;PtenFV/+ females by 4–5 mo of age, and 50% of ErbB2;PtenΔ/Δ and ErbB2;PtenFV/Δ females by 2.5 mo of age (Fig. 6C). Tumor sections stained with H&E displayed a uniform, solid, and well-differentiated appearance in all genetic cohorts (Supplemental Fig. 6c). Thus, in mice expressing a human-relevant oncogene, tumorigenesis was accelerated similarly by the PtenFV and PtenΔ alleles.
The levels of PTEN protein and P-AKT activation were then examined in mammary glands and mammary tumors. IHC showed a marked reduction of PTEN protein in epithelial ducts of PtenFV/Δ mammary glands when compared with ducts of Pten+/+ and Pten+/Δ glands, yet P-AKT levels were below detection in preneoplastic mammary ducts of all of these three genotypes (Fig. 7A). Confocal microscopy confirmed the overall reduction in mutant PTENFV protein, with a particular depletion from the nucleus (Fig. 7B). As expected, P-AKT levels were markedly increased in PtenΔ/Δ mammary glands (Fig. 7A). In mammary tumors from ErbB2;PtenFV/Δ mice, we observed low levels of PTEN protein and almost undetectable levels of P-AKT and nuclear FOXO3A (Supplemental Fig. 7a). In tumors from ErbB2;PtenΔ/Δ mice, there was robust accumulation of P-AKT and effective shuttling of FOXO3A to the cytoplasm (Supplemental Fig. 7a,b).
Figure 7.
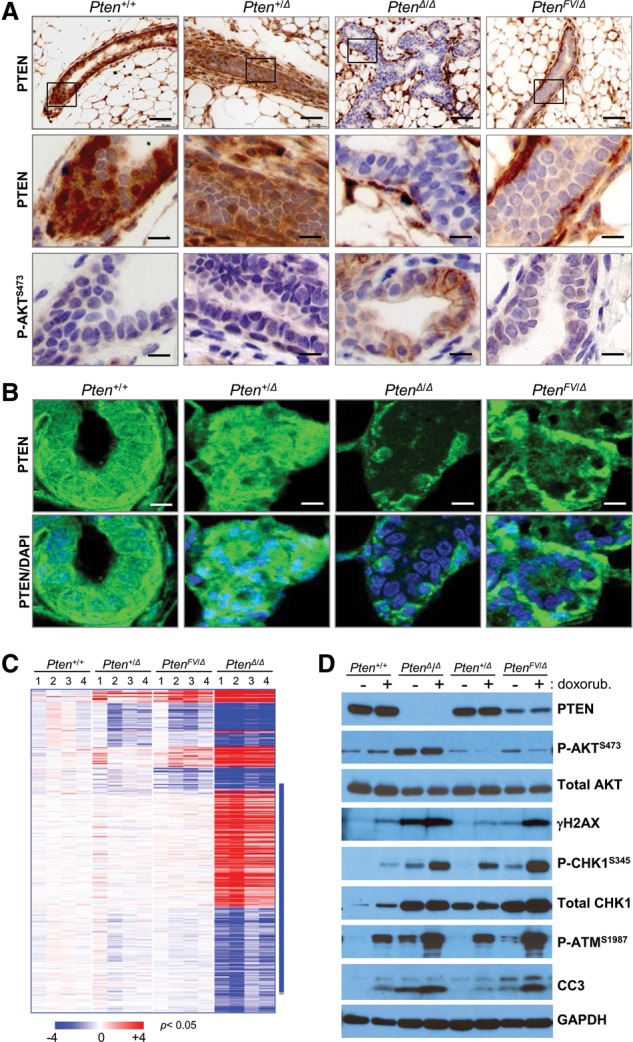
Normal P-AKT signaling and genomic instability in mammary glands, tumors, and MEFs expressing mutant PTENFV protein. (A) IHC staining of sections from pretumoral mammary glands in females with the indicated genotypes using antibodies specific for PTEN (top and middle panels) or P-AKTS473 (bottom panels); IHC stained sections in A and B were counterstained with hematoxylin. Bars: low magnification, 200 µm; high magnification, 50 µm. Boxes in the low-magnification panels indicate regions shown at high magnification in the panels directly below. (B) Confocal microscopy images showing PTEN localization (green) and DAPI-stained nuclei (blue) in the mammary glands harvested from 4-mo-old mice with the indicated genotypes. Bar, 50 µm. (C) Heat map of differentially expressed genes (434 genes; P < 0.05; greater than twofold) between Pten+/+ and Pten+/Δ, PtenΔ/Δ, and PtenFV/Δ MECs. (Blue bar) Genes differentially expressed only in PtenΔ/Δ cells. (D) Immunoblots of whole-cell lysates derived from MEFs with the indicated genotypes that were cultured in the absence (−) or presence (+) of doxorubicin. Blots were probed with antibodies specific for γH2AX, P-AKTS473, total AKT, P-CHK1S345, and total CHK1, P-ATMS1987; GAPDH was used as the loading control.
The transcriptome of Pten+/+, Pten+/Δ, PtenFV/Δ, and PtenΔlΔ mammary epithelial cells (MECs) was interrogated by Affymetrix microarrays. Normalized gene expression was standardized to the geometric mean of the Pten+/+ group, and clustering of differentially expressed genes was visualized by heat maps (greater than twofold; P < 0.05) (Fig. 7C). The expression of a subset of differentially regulated mRNAs was confirmed by real-time qPCR assays (Supplemental Fig. 7c). This analysis showed a profound dysregulation of gene expression in PtenΔlΔ MECs when compared with control Pten+/+ MECs. In contrast, profiles from PtenFV/Δ and Pten+/Δ cells were remarkably similar, suggesting that downstream signaling was not severely impacted by the PtenFV mutation. Collectively, these findings demonstrate that mutant PTENFV protein, while less stable, is able to effectively suppress the persistent activation of P-AKT and global transcription changes.
DNA damage and downstream response mechanisms were also evaluated in MEFs, mammary glands, and mammary tumors using antibodies against γH2AX, P-CHK1Ser345, and P-ATMSer1987. Little or no γH2AX, P-CHK1Ser345, and P-ATMSer1987 was detected in Pten+/+ and Pten+/Δ cells, whereas baseline levels of these three markers was significantly higher in both PtenΔ/Δ and PtenFV/Δ samples (Fig. 7D). Cells of all four genotypes responded to doxorubicin (Fig. 7D), a therapeutic cytotoxic drug used in the clinic that causes double-stranded breaks. However, PtenΔ/Δ and PtenFV/Δ cells incurred significantly more DNA damage in response to doxorubicin than Pten+/+ and Pten+/Δ cells, which resulted in the robust activation of P-CHK1Ser345 and P-ATMSer1987. Similarly, tumors from ErbB2;Pten+/+ and ErbB2;Pten+/Δ mice lacked any measurable γH2AX staining, whereas tumors from ErbB2;PtenFV/Δ and ErbB2;PtenΔ/Δ mice stained strongly positive for γH2AX (Supplemental Fig.7e,f). Interestingly, Pten+/+, Pten+/Δ, PtenFV/Δ, and PtenΔlΔ mammary glands did not exhibit any detectable γH2AX staining (Supplemental Fig. 7g). Together, these results suggest that nuclear depletion of mutant PTENFV protein under certain contexts (immortalized cells and tumors) is associated with increased DNA damage and persistent activation of a CHK1–ATM signaling response.
Discussion
PTEN is genetically and epigenetically disrupted in a broad range of solid and hematologic malignancies with exceptional frequency. Comprehensive evidence suggests that inactivation of its lipid phosphatase function by missense and nonsense mutations or by complete gene deletion is strongly linked to the accumulation of the PIP3 second messenger and persistent engagement of canonical PI3K–AKT signaling (Maehama and Dixon 1998; Vivanco and Sawyers 2002; Maroulakou et al. 2007; Wang et al. 2010). Here, we generated a Pten knock-in mouse model with a missense mutation in a β-sheet stretch of the C2 domain (PTENFV) that was originally identified in human cancer patients. The first salient observation distinguishing the PtenFV allele from previously Pten mutant alleles analyzed was that PtenFV/FV embryos could be carried to full term. Molecular analysis of cells and tissues revealed that mutant PTENFV protein is less stable than wild-type protein, and, consequently, total PTEN was decreased, and nuclear PTEN was acutely depleted. However, by a number of criteria, canonical AKT signaling remained intact in these cells. This is in stark contrast to embryos lacking Pten or expressing a lipid phosphatase-dead product of Pten (Wang et al. 2010; Papa et al. 2014), where AKT-signaling is persistently activated, and embryos die in utero well before E9.5. Moreover, the observation that mouse embryos expressing a constitutively active mutant form of Pik3ca, which is frequently observed in human cancer (Pik3caH1047R), also die in utero before E9.5 suggests that unregulated PI3K–AKT signaling through either the inactivation of PTEN or the activation of PI3K is detrimental to embryonic development (Hare et al. 2015). Thus, the analysis of PtenFV/FV embryos, which can be carried to term in the absence of overt AKT activation, suggests that the F341V missense mutation (and possibly other missense mutations within the C2 domain) disrupts a function that is distinct from the well-established lipid phosphatase function of PTEN.
The analysis of aging PtenFV/+ mice establishes PTENF341V as a relevant human cancer driver mutation. Intriguingly, these heterozygous mice developed cancer in the adrenal medulla, thymus, stomach, and mammary glands but not in other organs typically susceptible to Pten deficiency, including the thyroid, prostate, and uterus. This remarkable selectivity in cancer development appears to be based on the differential requirement of tissues for persistent AKT activation. Indeed, progression to carcinoma in the mammary gland ensued in the absence of overt P-AKT activation, whereas cancer development in the uterus required additional alterations. The transition from hyperplasia to carcinoma in the uterus was directly associated with, but not necessarily caused by, the focal and persistent activation of P-AKT. The view that overt AKT signaling may be required for carcinogenesis in an organ-selective manner is consistent with previous work showing a profound inhibition of carcinogenesis in the uterus and prostate of Pten+/− mice lacking Akt1 (Pten+/−;Akt1−/−) and only a modest inhibition in the mammary gland or adrenal medulla of these animals (Chen et al. 2006). Our finding that a single noncatalytic missense Pten mutation recapitulates and magnifies this organ-selective cancer susceptibility is remarkable and underscores the importance of the noncanonical tumor suppressor function of PTEN.
Others have proposed that the diverse roles of PTEN in the control of cell proliferation, survival, and motility and genomic integrity may reflect the spatial subcompartmentalization of PTEN protein in cells (Shi et al. 2012). The challenge has been to identify and link the many molecular functions of PTEN at each distinct subcellular location to the processes and physiological systems under its control, such as cognition, metabolism, aging, and cancer. Our analysis of the PtenFV missense mutation provides a clear in vivo example of spatial segregation of PTEN's functions relevant to cancer. While the precise mechanism remains to be deciphered biochemically, the fundamental function disrupted by the PtenFV mutation appears to be related to nuclear depletion and increased DNA damage, which leads to an attempt by cells to activate ATM-mediated DNA repair processes. Nuclear PTEN has been proposed to have noncanonical functions (Freeman et al. 2003; Chen et al. 2005; Shen et al. 2007; Gu et al. 2011; Song et al. 2011; Zhang et al. 2011; Shi et al. 2014), and its depletion correlated with loss of genomic integrity and cancer progression (Trotman et al. 2007; Song et al. 2008; Bassi et al. 2013). However, definitive experiments have yet to fully test this hypothesis. Our studies provide in vivo evidence in support of a nuclear AKT-independent role for PTEN in cancer. This interpretation is in agreement with the sequencing of human endometrial cancer genomes, which shows that PTEN inactivation coexists with either activating PIK3CA mutations or loss-of-function PIK3R mutations (The Cancer Genome Atlas Network 2013), strongly advocating that PTEN and PI3K are not simply signaling through the same linear pathway but rather through parallel pathways that cooperate in carcinogenesis. Furthermore, the recent genomic analysis of a small cohort of breast cancer patients with an activating PIK3CA mutation shows that the subsequent loss of PTEN mediates the acquired resistance of tumor cells to the PI3Kα inhibitor BYL719, suggesting the involvement of a PTEN non-PI3K/AKT-associated pathway in this process (Juric et al. 2014). Thus, the discovery of a novel noncanonical tumor suppressor function of PTEN, exposed by the analysis of PtenFV/+ mice here, provides an explanation for the PTEN-mediated resistance to PI3Kα inhibition and may suggest a reason for the failure of recent clinical trials targeting PTEN-deficient tumors with PI3K inhibitors. We propose that PTEN possesses a noncanonical function distinct from its well-defined role in inhibiting AKT signaling, which is critical for suppression of human cancer.
Materials and methods
Generation of PtenFV knock-in mice
Standard cloning techniques were used to construct targeting vectors. The targeting vector contained a 1690-base-pair (bp)-long short arm, a floxed phosphoglycerate kinase promoter (PGK)-neo cassette, and a 5577-bp-long long arm (Fig. 1A). The short (starting 330 bp upstream of exon 8) and long arms were PCR-amplified (see Supplemental Fig. 9 for primers). Site-directed mutagenesis was used to mutate AACCGATACTTCTCTCC AAATTTT to AATCGATACTTCTCTCCAAATGTT in exon 8 (TTT > GTT) to replace phenylalanine at position 341 with valine (F341V). This was done to generate a PtenFV knock-in mutation. An additional silent mutation was induced using the same primers to generate a ClaI site. The final targeting vector was confirmed by direct DNA sequencing. Next, TC1 129Sv/Ev ES cells were electroporated with 50 μg of NotI-linearized targeting vectors. Homologous recombination was selected with G418 and FIAU (ganciclovir), confirmed by Southern blot analysis of genomic DNA digested using XbaI, and probed using probe 1 and probe 2 external probes (see Fig. 1A; Supplemental Fig. 9 for probes). Probe 1 is a 427-bp-long PCR product, and probe 2 is a 482-bp-long PCR product. Both probes were confirmed by sequencing. Probe 1 gave 7.3-kb (targeted) and 21.3-kb (wild-type) bands, confirming the short arm targeting, and probe 2 gave 16-kb (targeted) and 21.3-kb (wild-type) bands, confirming the long arm targeting. ES cell clones carrying the expected PtenFV mutation were injected into E3.5 C57BL/6 blastocysts that were subsequently transferred into foster mothers. The resulting chimeras were bred with National Institutes of Health Black Swiss female mice, and agouti offspring were genotyped by PCR and confirmed by Southern blot analysis. Offspring with germline transmission of PtenFV knock-in targeted alleles were bred with EIIA-cre mice to excise the PGK-neo. The knock-in mutation was confirmed by Southern blot and sequencing of tail DNA samples from agouti mice.
The knock-in mice with the PGK-Neo cassette were genotyped using primer 2 (spanning the Neo cassette) and primer 3 (spanning the LoxP site left of intron 7) to generate a 320-bp fragment.
The knock-in mice after excision of floxed PGK-Neo were genotyped using forward primer 1 and reverse primer 3, which generated 180-bp and 220-bp PCR products from the wild-type and the knock-in alleles, respectively (see Fig. 1A).
Generation of MEFs, retroviral infections, and cell culture conditions
Primary MEFs were isolated from E13.5 embryos using standard methods. MEFs were cultured in DMEM containing 15% fetal bovine serum (FBS). For the production of retrovirus, the cre cDNA was cloned into pBabe-puromycin (Morgenstern and Land 1990). High-tittered retroviruses were produced by transient transfection of retroviral constructs into the Phoenix-Eco packaging cell line as described previously (Pear et al. 1993). MEFs were infected by incubating the cells for 5 h with the viral supernatants from the transfected cells containing 4 μg/mL polybrene (Sigma-Aldrich). Subsequent to infection, cells were grown in selection medium containing 2.5 μg/mL puromycin (Sigma) for 3–5 d. Introduction of cre in the MEFs resulted in deletion of the floxed wild-type allele, which was confirmed by genotyping of DNA extracted from the MEFs and Western blot analysis using PTEN antibody.
Protein and RNA analysis
Cells were scraped from culture dishes in chilled phosphate buffer solution (PBS), centrifuged, and washed once with ice-cold PBS. Total protein extracts were prepared by incubating cells in radioimmunoprecipitation assay (RIPA) buffer for 30 min on ice, and protein content was measured by Bradford assay. For PTEN protein half-life analysis, stably expressing wild-type or PtenFV/Δ MEFs were treated with 30 μg/mL cycloheximide in DMEM and 15% FBS. Whole-cell protein lysate was extracted at 0, 2, 4, 8, 12, and 24 h after addition of cycloheximide. In total, 30 μg of protein was then separated by 10% SDS-PAGE and transferred to PVDF membranes. Blots were probed with antibodies specific for PTEN (1:1000 dilution; Cell Signaling Technology, Inc., catalog no. 9559), P-AKTT308 (1:1000 dilution; Cell Signaling Technology, Inc., catalog no. 4056) P-AKTS473 (1:1000 dilution; Cell Signaling Technology, Inc., catalog no. 4060), AKT (1:1000 dilution; Cell Signaling Technology, Inc., catalog no. 4685), P-GSK3βS9 (Cell Signaling Technology, Inc., catalog no. 9223), GSK3β (1:1000 dilution; Cell Signaling Technology, Inc., catalog no. 9315), γH2AXS139 (1:1000 dilution; Cell Signaling Technology, Inc., catalog no. 9718), and Tubulin (1:5000 dilution; Sigma, T6199). GAPDH (1:5000; Santa Cruz Biotechnology, sc-25778) was used to determine protein loading. Endogenous sumoylated PTEN was detected following published protocols (Ho et al. 2015) with some modifications. Briefly, cells were scraped from culture dishes in chilled PBS, centrifuged, and washed once with ice-cold PBS. Total protein extracts were prepared by incubating cells in Laemmli sample buffer without bromophenol blue (60 mM Tris-HCl at pH 6.8, 2% SDS, 10% glycerol, 5% β-mercaptoethanol) and were sonicated by two 10-sec pulses (lowest setting, Branson sonicator) followed by 10 min of incubation on ice and a 5-sec sonication pulse. Lysates were centrifuged at 15,000g for 10 min at 4°C. The clarified lysate was then diluted 13-fold in Tris-buffered saline (50 mM Tris at pH 7.5, 150 mM NaCl) containing 1% Triton-X-100 (TBS-T). The diluted lysate was precleared with protein A/G agarose beads (Calbiochem) for 30 min at 4°C. The precleared lysate was incubated with anti-Sumo2/3 18H8 antibody (Cell Signaling Technology, Inc., catalog no. 4971) overnight at 4°C. Protein A/G agarose beads were then added to capture the immune complex for 1 h, and the beads were washed four times with TBS-T. The immune complex was resolved by SDS-PAGE and immunoblotted with anti-PTEN 138G6 antibody (Cell Signaling Technology, Inc., catalog no. 9559) to visualize the sumoylated PTEN protein.
Total RNA was extracted from MEFs using Trizol (Invitrogen) following the manufacturer's protocol. Reverse transcription of 5 μg of total RNA was performed by combining 1 μL of SuperScript III reverse transcriptase (Invitrogen, Life Technologies Corp.), 4 μL of 5× buffer, 0.5 μL of 100 μM oligo(dT) primer, 0.5 μL of 25 mM deoxyribonucleotide (dNTP), 1.0 μL of 0.1 M DTT, 1.0 μL of RNase inhibitor (Roche), and water up to a volume of 20 μL. Reactions were incubated for 60 min at 50°C and then diluted fivefold with 80 μL of water. Pten qRT–PCR was performed using a Bio-Rad iCycler PCR machine. Each PCR contained 0.5 μL of cDNA template and primers at a concentration of 100 nM in a final volume of 25 μL of SYBR Green reaction mix (Bio-Rad). Each PCR yielded only the expected amplicon, as shown by the melting temperature profiles of the final products and by gel electrophoresis. Standard curves were generated using cDNA to determine the linear range and PCR efficiency of each primer pair. Reactions were performed in triplicates, and relative amounts of cDNA were normalized to GAPDH. Primer sequences are listed in Supplemental Figure 10.
Transgenic mouse lines
Pten conditional knockout mice (PtenL) were generated with loxP sites flanking exons 4 and 5, which contain the lipid phosphatase domain (Trimboli et al. 2009); MMTV-Cre mice were a gift from Dr. Willam J. Muller (McGill University Canada) (Andrechek et al. 2000), Sprr2F-Cre mice were a gift from Dr. Diego H. Castrillon (Contreras et al. 2010), and MMTV-ErbB2 mice were purchased from Jackson Laboratories. The initial embryonic and cellular studies described in Figures 1–3 used first-generation (50%) FVB background mice; the perinatal lethality described in Figure 1D was also confirmed with eighth-generation FVB background mice. Tumor studies described in Figures 4 and 6A used second-generation FVB mice, allowing these studies to be compared with previous tumor studies (Wang et al. 2010). Tumor studies described in Figure 6, B and C, used fourth-generation FVB background mice. Littermate mice were used as controls.
Whole-mount staining of mammary glands
Inguinal mammary glands were isolated from 6-wk-old and 9-wk-old female mice (third FVB/N) and mounted on glass slides using a Q-tip to spread the tissue. Tissue was fixed using Carnoy's fixative (60% ethanol, 30% chloroform, 10% acetic acid) overnight at 4°C and then stained with carmine alum stain (Sigma) for 12 h. After thorough rinsing in water, mammary glands were dehydrated with alcohol and preserved in xylene.
Histopathology and IHC
For the analysis of neuronal migration in the newborn mice, the P0 (postnatal day 0) pups were decapitated, and the heads were fixed in modified Davidson's solution. The heads were then cross-sectioned longitudinally at the midpoint and stained with H&E by standard methods.
Mammary glands from third FVB/N females were collected and fixed in 10% formalin in neutral phosphate buffer and then stained with H&E by standard methods. The IHC was performed using a Leica BondRX autostainer (see Supplemental Fig. 11 for details of the programs used). Paraffin-embedded tissues (4-μm sections) were baked offline for 15 min at 65°C and autostained on the machine (dewax, rehydration, retrieval, block, 1°C incubation, after 1°C and detection). Dehydration was done using EtOH, and tissues were mounted in xylene. The solutions used were Bond Dewax Soln (catalog no. AR922), Bond Wash Soln (catalog no. AR9590), Bond Epitope Retrieval Soln 1 (pH 6; catalog no. AR9961), Bond Epitope Retrieval Soln2 (pH 9; catalog no. AR9640), PowerVisionIHC/ISH Super Block (catalog no. PV6122), Bond Primary Antibody Diluent (catalog no. AR9352), Bond Polymer Refine Detection (HRP-DAB, catalog no. DS9390), Bond Polymer Refine Red Detection (AP-RED, catalog no. DS9800), and Post primary anti-rabbit rat antibody (catalog no. Al4001, Vector Laboratories). Paraffin-embedded sections were stained with PTEN antibody (Cell Signaling Technology, Inc., catalog no. 9559L), P-AKTS473-specific antibody (Cell Signaling Technology, Inc., catalog no. 3787), γH2AXS139 antibody (Cell Signaling Technology, Inc., catalog no. 9718), and FOXO3A antibody (Cell Signaling Technology, Inc., catalog no. 12829).
Mammary gland tumors
Proliferative lesions of the mammary gland from PtenFV/+ mice (second FVB/N) were characterized according to the published grading scheme, which is based on the published histopathologic criteria (Cardiff et al. 2000). Diagnostic categories included no significant lesions (NSL), low-grade and high-grade Min (LG Min and HG Min, respectively), and “neoplasia.” Tumors in the “neoplasia” category included adenomyoepithelioma (moderate to well-differentiated duct-like cytokeratin-8-positive epithelial structures within tracts of spindle-shaped, smooth muscle actin-positive myoepithelial cells with mucinous extracellular matrix) and adenosquamous carcinoma (solid sheets of atypical mammary acinar cells embedded within desmoplastic stroma with areas of squamous differentiation and keratin formation). Adenomyoepitheliomas demonstrated absent to moderate degrees of local invasion, while all adenosquamous carcinomas were locally invasive. Regional lymph nodes, lungs, and livers were examined, and no metastases were observed.
MMTV-ErbB2-initiated (fourth FVB/N) mammary tumors and lungs were harvested from mice that were tumor-bearing for 4 wk. They presented as solid carcinomas composed of moderate to poorly differentiated epithelial cells with numerous mitotic figures and frequent areas of necrosis. Tumor stroma was usually sparse, and local invasion was common; however, occasional tumors had more pronounced fibrosis. A few mice demonstrated pulmonary metastases.
Endometrial tumors
Endometrial proliferative lesions were classified as follows: Simple hyperplasia (SH) of the endometrium was characterized by increased density of endometrial glands lined by hyperplastic epithelium. Some hyperplastic glands were dilated (cystic type). Carcinoma in situ (CIS) featured an enlarged, irregularly shaped gland, often displaying a prominent cribiform pattern, and was confined within the endometrium. The glandular epithelium displayed prominent features of cellular atypia and dysplasia, including loss of cellular polarity, anisocytosis, anisokarosis, prominent nucleoli, increased mitotic figures, and single-cell necrosis. CIS lesions were focal in Sprr2f-Cre;Pten+/Δ and Sprr2f-Cre;PtenFV/Δ mice but affected nearly every gland in Sprr2f-Cre;PtenΔ/Δ mice. Invasive carcinoma (IC), seen only Sprr2f-Cre;PtenΔ/Δ mice, was characterized by invasion of well-differentiated endometrial epithelium into the myometrium. These cells formed small nests and cords that separated bundles of smooth muscle and were occasionally observed within lymphatic vessels between the inner and outer muscle layers. Microscopic metastatic foci were observed in the ovary (two mice) and a sublumbar lymph node (one mouse).
Isolation of MECs using CDH1 antibodies
MMTV-Cre;Pten+/L (Pten+/Δ) mice were crossed with PtenL/L or PtenFV/L to generate the MMTV-Cre;PtenL/L, MMTV-Cre;PtenFV/L, and MMTV-Cre;Pten+/L test mice. To prepare tissues for fluorescent-activated cell sorting (FACS), mammary glands were isolated from ∼8-wk-old mice and minced into 1- to 2-mm fragments using microdissection scissors. Tissue was placed in a round-bottom tube (BD Falcon) with prewarmed PBS containing 5 mg/mL collagenase (Worthington Biochemical Corporation), 120 µ/mL DNase I (Roche), and 1 mM MgCl2 in 5 mL. Samples were placed in a 37°C shaker incubator and pipetted at 10- to 15-min intervals (with a 1-mL pipette) to obtain a single-cell suspension. After 30 min, 8 mL of complete medium was added to the cell suspension, and cells were pelleted by centrifugation (1200 rpm for 5 min). The cell pellet was resuspended in 10 mL of sterile PBS and filtered through a 40-µm cell strainer (BD Falcon), and the cells were counted after filtering. One microliter of fluorescently labeled CDH1 (e-cadherin) antibody (R&D Systems, catalog no. FAB7481P) was added per 1 × 106 cells. Samples were placed on a rotor in the cold room for 30 min, and cells were collected by centrifugation (2500 rpm for 5 min). Cells were then resuspended in 500 µL to 1 mL of flow solution (1 mM EDTA at pH 8.0, 1 mM MgCl2, 60 U/mL DNase I [Roche], 3% heat-inactivated FBS) and subjected to FACS analysis. Cells isolated using FACS were collected by centrifugation (1200 rpm for 5 min) and used for RNA isolation using 1 mL of Trizol reagent.
RNA analysis and microarrays
RNA extraction
RNA was extracted using Trizol reagent according to the manufacturer's instructions (Invitrogen), precipitated using glycogen (Roche), and quantified using nanodrop (RNA 6000 Nano-assays or 2100-Bioanalyzer).
cDNA preparation
Using qScript cDNA Supermix (Quanta Biosciences), 1 µg or less of total RNA was mixed with 5× qScript cDNA supermix (containing optimized concentrations of MgCl2, dNTPs, recombinant RNase inhibitor protein, qScript reverse transcriptase, random primers, oligo9dT primer, and stabilizers) and brought to a final volume of 20 µL with nuclease-free water. After gentle mixing and brief centrifugation, samples were incubated in a thermocycler for 5 min at 25°C, 30 min at 42°C, and 5 min at 85°C. Following the RT reaction, samples were diluted with nuclease-free water to make an approximate final concentration of 10 ng/µL. Typically, 5 µL (50 ng) of cDNA was used for each qRT–PCR reaction.
qRT–PCR
Quantitative gene expression was performed using 50 ng of complementary DNA per reaction. TaqMan Roche Universal Probe Library system probes and primers (Roche) were used following the manufacturer's instructions. Reactions were performed on the StepOne Plus (Applied Biosystems) real-time machines. Primers used for qRT–PCR were designed using the Web-based ProbeFinder software provided by Roche (see Supplemental Fig. 12) The reference gene used for all qRT–PCR assays was Rpl4, and melt curve analysis and agarose gels were used to confirm the specificity of the reaction.
Culturing MECs
Primary MECs were isolated as follows: Dissected mammary glands from 8- to 9-wk-old female mice were minced in an Eppendorf tube using microdissection scissors and digested with collagenase solution (20% mammary epithelial growth medium, 0.15% collagenase I, 0.2% NaHCO3, 0.75% Media199, 160 U/mL hyaluronidase, 1 µg/mL hydrocortisone, 10 µg/mL insulin with penicillin and streptomycin) overnight in a 5% CO2 incubator at 37°C. The following day, the tissue was passed through a 1-mL pipette tip ∼30 times, until large tissue chunks were broken up, and the solution had a cloudy appearance. The collagenase was neutralized with DMEM containing 10% FBS and then removed by centrifugation and aspiration. Cells were resuspended in fresh complete medium and subjected to 12 min of gravity separation. Supernatants were removed, and fresh medium was added for four consecutive rounds of gravity separation for 10 min each. After the final sedimentation, the cells remaining in the supernatant were plated in epithelial selective medium HuMEC (Gibco) containing 10% FBS, bovine pituitary extract, and HuMEC supplement (as provided by the manufacturer) in a 5% CO2 incubator at 37°C in 100-mm dishes with feeder cells (Bryja et al. 2006) generated by treating MEFs with 10 µg/mL mitomycin C (Roche) for 2 h. Treated cells were split and allowed to attach for 24 h prior to the addition of MECs.
Mouse embryonic fibroblasts for DNA damage analysis
The cell isolation was the same as above for primary culture. To promote cell immortalization, 300,000 cells after second passage were plated in 60-mm dishes and replated at this same concentration every 3 d. Cells were cultured with 10% FBS DMEM with 100 µg/mL penicillin–streptomycin throughout this process of immortalization. Total cell lysates were extracted by RIPA buffer. Fifteen micrograms of total protein was resolved by 10% SDS-PAGE and transferred onto PVDF membrane to detect PTEN, P-AKTS473, AKT, P-CHK1S345, CHK1, P-ATMS1987, and γH2AX by immunoblotting. For DNA damage response, exponentially growing MEFs were treated with 10 μg/mL doxorubicin for 4 h. Total protein was extracted, and immunoblotting was performed. Antibodies against PTEN (catalog no. 9559), AKT (catalog no. 4691S), P-AKTS473 (catalog no. 4060S), P-CHK1S345 (catalog no. 2348), and CHK1 were from Cell Signaling Technology, Inc. (catalog no. 2345). Antibody against human P-ATMS1981 was from Novus Biologicals (catalog no. NB100-306) and was used to detect mouse P-ATMS1987. P-γH2AXS139 antibody was from Millipore (catalog no. 05-636); GAPDH antibody was from Santa Cruz Biotechnology (catalog no. SC25778).
Affymetrix global gene expression profiling analyses
RNA samples were submitted to the Ohio State University Comprehensive Cancer Center Microarray Shared Resource facility for generation of biotinylated cRNA and hybridization to mouse genome 430 2.0 (MOE4302.0) Affymetrix chips. GeneChip Scanner 3000 (Affymetrix) was used for data acquisition. For each sample, preprocessing of raw signal intensities from .CEL files was performed using the RMA method and consisted of background adjustment, quantile normalization, and median polish. R was used to generate class comparisons to determine differentially expressed genes at a significance level of P-value < 0.05 under the Student's t-test. For construction of heat maps, the geometric mean of genes after RMA normalization from control samples was set as reference, with the color (blue or red) corresponding to the ratio between expression of genes in mutant samples and the reference value. Functional annotation of genes was performed using DAVID.
Image acquisition and analysis
Samples were imaged using the PerkinElmer Vectra multispectral slide analysis system to automatically acquire 40 fields of interest per slide. The automatic workflow consisted of the following: (1) monochrome imaging of the entire slide, (2) RGB low-power imaging of the tumor tissue using an inForm tissue-finding algorithm, and (3) multispectral high-power imaging (400×) of the fields containing at least 90% tumor epithelium by means of an inForm HPF-finding algorithm. The multispectral images were reviewed and analyzed using inForm tissue-finder software. A pattern recognition algorithm was used for batch processing as follows: (1) cell segmentation to locate the subcellular compartments and (2) scoring to bin the spectral nuclear signals into two categories, negative and positive, providing data in percentage. The average number of nuclei analyzed per slide was 40,000.
Supplementary Material
Acknowledgments
We thank Jason Bice and Daphne Bryant for excellent assistance with histology. This work was supported by the Microarray, Nucleic Acid and Mouse Transgenic Shared Resources at Ohio State University. This work was funded by National Institutes of Health grants to G.L. (R01CA121275 and P01 CA97189). C.K., E.C, C.M., and T.P. were recipients of Pelotonia Fellowships, and H.W. was a recipient of a post-doctoral fellowship from the American Cancer Society.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.262568.115.
References
- Ali IU, Schriml LM, Dean M. 1999. Mutational spectra of PTEN/MMAC1 gene: a tumor suppressor with lipid phosphatase activity. Natl Cancer Inst 91: 1922–1932. [DOI] [PubMed] [Google Scholar]
- Al-Khouri AM, Ma Y, Togo SH, Williams S, Mustelin T. 2005. Cooperative phosphorylation of the tumor suppressor phosphatase and tensin homologue (PTEN) by casein kinases and glycogen synthase kinase 3β. J Biol Chem 280: 35195–35202. [DOI] [PubMed] [Google Scholar]
- Andrechek ER, Hardy WR, Siegel PM, Rudnicki MA, Cardiff RD, Muller WJ. 2000. Amplification of the neu/erbB-2 oncogene in a mouse model of mammary tumorigenesis. Proc Natl Acad Sci 97: 3444–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassi C, Ho J, Srikumar T, Dowling RJ, Gorrini C, Miller SJ, Mak TW, Neel BG, Raught B, Stambolic V. 2013. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science 341: 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonneau D, Longy M. 2000. Mutations of the human PTEN gene. Hum Mutat 16: 109–122. [DOI] [PubMed] [Google Scholar]
- Bryja V, Bonilla S, Cajanek L, Parish CL, Schartz CM, Luo Y. 2006. An efficient method for derivation of mouse embryonic stem cells. Stem Cells 24: 844–849. [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network 2013. Integrated genomic characterization of endometrial carcinoma. Nature 497: 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardiff RD, Anver MR, Gusterson BA, Henninghousen L, Jensen RA, Merino MJ, Rehm S, Russo J, Tavassoli FA, Wakefield LM, et al. 2000. The mammary papthology of genetically engineered mice: the consensus report and recommendations from the Annapolis meeting. Oncogene 19: 968–988. [DOI] [PubMed] [Google Scholar]
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al. 2005. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 436: 725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ML, Xu PZ, Peng XD, Chen WS, Guzman G, Yang X, Di Cristofano A, Pandolfi PP, Hay N. 2006. The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/− mice. Genes Dev 20: 1569–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras CM, Akbay EA, Gallardo TD, Haynie JM, Sharma S, Tagao O, Bardeesy N, Takahashi M, Settleman J, Wong KK, et al. 2010. Lkb1 inactivation is sufficient to drive endometrial cancers that are aggressive yet highly responsive to mTOR inhibitor monotherapy. Dis Model Mech 3: 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. 1998. Pten is essential for embryonic development and tumour suppression. Nat Genet 19: 348–355. [DOI] [PubMed] [Google Scholar]
- Fata JE, Debnath S, Jenkins EC Jr, Fournier MV. 2012. Nongenomic mechanisms of PTEN regulation. Int J Cell Biol 2012: e379685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R, Whale AD, Martinez-Diaz H, Rozengurt N, Cardiff RD, et al. 2003. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell 3: 117–130. [DOI] [PubMed] [Google Scholar]
- Georgescu MM, Kirsch KH, Akagi T, Shishido T, Hanafusa H. 1999. The tumor-suppressor activity of PTEN is regulated by its carboxyl-terminal region. Proc Natl Acad Sci 96: 10182–10187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu T, Zhang Z, Wang J, Guo J, Shen WH, Yin Y. 2011. CREB is a novel nuclear target of PTEN phosphatase. Cancer Res 71: 2821–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare LM, Scharz Q, Wiszniak S, Gurung R, Montgomery KG, Mitchell CA, Phillips WA. 2015. Heterozygous expression of the oncogenic Pik3ca(H1047R) mutation during murine development results in fatal embryonic and extraembryonic defects. Dev Biol 404: 14–26. [DOI] [PubMed] [Google Scholar]
- Ho J, Bassi C, Stambolic V. 2015. Characterization of nuclear PTEN and its post translational modifications. Methods 77-78: 104–111. [DOI] [PubMed] [Google Scholar]
- Hopkins BD, Fine B, Steinbach N, Dendy M, Rapp Z, Shaw J, Pappas K, Yu JS, Hodakoski C, Mense S, et al. 2013. A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science 341: 399–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juric D, Castel P, Griffith M, Griffith OL, Won HH, Ellis H, Ebbesen SH, Ainscough BJ, Ramu A, Iyer G, et al. 2014. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nature 518: 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Kato S, Kumabe T, Sonoda Y, Yoshimoto T, Kato S, Han SY, Suzuki T, Shibata H, Kanamaru R, et al. 2000. Functional evaluation of p53 and PTEN gene mutations in gliomas. Clin Cancer Res 6: 3937–3943. [PubMed] [Google Scholar]
- Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P, Pavletich NP. 1999. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 99: 323–334. [DOI] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. 1998. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 273: 13375–13378. [DOI] [PubMed] [Google Scholar]
- Maroulakou IG, Oemler W, Naber SP, Tsichlis PN. 2007. Akt1 ablation inhibits, whereas Akt2 ablation accelerates, the development of mammary adenocarcinomas in mouse mammary tumor virus (MMTV)-ErbB2/neu and MMTV-polyoma middle T transgenic mice. Cancer Res 67: 167–177. [DOI] [PubMed] [Google Scholar]
- Meuillet EJ, Mahadevan D, Berggren M, Coon A, Powis G. 2004. Thioredoxin-1 binds to the C2 domain of PTEN inhibiting PTEN's lipid phosphatase activity and membrane binding: a mechanism for the functional loss of PTEN's tumor suppressor activity. Arch Biochem Biophys 429: 123–133. [DOI] [PubMed] [Google Scholar]
- Minaguchi T, Yoshikawa H, Oda K, Ishino T, Yasugi T, Onda T, Nakagawa S, Matsumoto K, Kawana K, Taketani Y. 2001. PTEN mutation located only outside exons 5, 6, and 7 is an independent predictor of favorable survival in endometrial carcinomas. Clin Cancer Res 7: 2636–2642. [PubMed] [Google Scholar]
- Morgenstern JP, Land H. 1990. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res 18: 3587–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okahara F, Itoh K, Nakagawara A, Murakami M, Kanaho Y, Maehama T. 2006. Critical role of PICT-1, a tumor suppressor candidate, in phosphatidylinositol 3,4,5-trisphosphate signals and tumorigenic transformation. Mol Biol Cell 17: 4888–4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa A, Wan L, Bonora M, Salmena L, Song MS, Hobbs RM, Lunardi A, Webster K, Ng C, Newton RH, et al. 2014. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell. 157: 595–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Wang TL, Samuels Y, Bardelli A, Cummins JM, DeLong L, Silliman N, Ptak J, Szabo S, Willson JK, et al. 2005. Colorectal cancer: mutations in a signalling pathway. Nature 436: 792. [DOI] [PubMed] [Google Scholar]
- Pear WS, Nolan GP, Scott ML, Baltimore D. 1993. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci 90: 8392–8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson LM, Kipp BR, Halling KC, Kerr SE, Smith DI, Distad TJ, Clayton AC, Medeiros F. 2012. Molecular characterization of endometrial cancer: a correlative study assessing microsatellite instability, MLH1 hypermethylation, DNA mismatch repair protein expression, and PTEN, PIK3CA, KRAS, and BRAF mutation analysis. Int J Gynecol Pathol 31: 195–205. [DOI] [PubMed] [Google Scholar]
- Pilarski R, Burt R, Kohlman W, Pho L, Shannon KM, Swisher E. 2013. Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J Natl Cancer Inst 105: 1607–1616. [DOI] [PubMed] [Google Scholar]
- Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, Cordon-Cardo C, Catoretti G, Fisher PE, Parsons R. 1999. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci 96: 1563–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftopoulou M, Etienne-Manneville S, Self A, Nicholls S, Hall A. 2004. Regulation of cell migration by the C2 domain of the tumor suppressor PTEN. Science 303: 1179–1181. [DOI] [PubMed] [Google Scholar]
- Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, Yin Y. 2007. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 128: 157–170. [DOI] [PubMed] [Google Scholar]
- Shi Y, Paluch BE, Wang X, Jiang X. 2012. PTEN at a glance. J Cell Sci 125: 4687–4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Wang J, Chandarlapaty S, Cross J, Thompson C, Rosen N, Jiang X. 2014. PTEN is a protein tyrosine phosphatase for IRS1. Nat Struct Mol Biol 21: 522–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MS, Salmena L, Carracedo A, Egia A, Lo-Coco F, Teruya-Feldstein J, Pandolfi PP. 2008. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature 455: 813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MS, Carracedo A, Salmena L, Song SJ, Egia A, Malumbres M, Pandolfi PP. 2011. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell 144: 187–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MS, Salmena L, Pandolfi PP. 2012. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 13: 283–296. [DOI] [PubMed] [Google Scholar]
- Sun Z, Huang C, He J, Lamb KL, Kang X, Gu T, Shen WH, Yin Y. 2014. PTEN C-terminal deletion causes genomic instability and tumor development. Cell Rep 6: 844–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, de la Pompa JL, Stambolic V, Elia AJ, Sasaki T, del Barco Barrantes I, Ho A, Wakeham A, Itie A, Khoo W, et al. 1998. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol 8: 1169–1178. [DOI] [PubMed] [Google Scholar]
- Tamura M, Gu J, Takino T, Yamada KM. 1999. Tumor suppressor PTEN inhibition of cell invasion, migration, and growth: differential involvement of focal adhesion kinase and p130Cas. Cancer Res 59: 442–449. [PubMed] [Google Scholar]
- Torres J, Pulido R. 2001. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem 276: 993–998. [DOI] [PubMed] [Google Scholar]
- Trimboli AJ, Cantemir-Stone CZ, Li F, Wallace JA, Merchant A, Creasao N, Thompson JC, Caserta E, Wang H, Chong JL, et al. 2009. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature 461: 1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, Khoo AS, Roy-Burman P, Greenberg NM, Van Dyke T, et al. 2003. Pten dose dictates cancer progression in the prostate. PLoS Biol 1: e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, Pavletich NP, Carver BS, Cordon-Cardo C, Erdjument-Bromage H, et al. 2007. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 128: 141–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. 2000. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol 20: 5010–5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez F, Grossman SR, Takahashi Y, Rokas MV, Nakamura N, Sellers WR. 2001. Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J Biol Chem 276: 48627–48630. [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. 2002. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer 2: 489–501. [DOI] [PubMed] [Google Scholar]
- Wang H, Karikomi M, Naidu S, Rajmohan R, Caserta E, Chen HZ, Rawahneh M, Moffitt J, Stephens JA, Fernandez SA, et al. 2010. Allele-specific tumor spectrum in pten knockin mice. Proc Natl Acad Sci 107: 5142–5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Hepner K, Castelino-Prabhu S, Do D, Kaye MB, Yuan XJ, Wood J, Ross C, Sawyers CL, Whang YE. 2000a. Evidence for regulation of the PTEN tumor suppressor by a membrane-localized multi-PDZ domain containing scaffold protein MAGI-2. Proc Natl Acad Sci 97: 4233–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Dowbenko D, Spencer S, Laura R, Lee J, Gu Q, Lasky LA. 2000b. Interaction of the tumor suppressor PTEN/MMAC with a PDZ domain of MAGI3, a novel membrane-associated guanylate kinase. J Biol Chem 275: 21477–21485. [DOI] [PubMed] [Google Scholar]
- Yim JH, Kim YJ, Ko JH, Cho YE, Kim SM, Kim JY, Lee S, Park JH. 2007. The putative tumor suppressor gene GLTSCR2 induces PTEN-modulated cell death. Cell Death Differ 14: 1872–1879. [DOI] [PubMed] [Google Scholar]
- Zhang S, Huang WC, Li P, Guo H, Poh SB, Brady SW, Xiong Y, Tseng LM, Li SH, Ding Z, et al. 2011. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat Med 17: 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.