

Abstract
Background and Purpose
Diabetic patients are at an increased risk of cardiovascular disease, in part due to inflammation and oxidative stress. These two pathological mechanisms also affect other organs and cells including the kidneys and progenitor cells. Angiotensin-(1–7) [Ang-(1–7)] has previously been shown to counterbalance pathological effects of angiotensin II, including inflammation and oxidative stress. The aim of this study was to investigate the effects of short-term (2 weeks) Ang-(1–7) treatment on cardiovascular and renal function in a mouse model of type 2 diabetes (db/db).
Experimental Approach
Eight- to nine-week-old db/db mice were administered either vehicle, Ang-(1–7) alone, or Ang-(1–7) combined with an inhibitor (losartan, PD123319, A-779, L-NAME or icatibant) daily for 14 days.
Key Results
An improvement in physiological heart function was observed in Ang-(1–7)-treated mice. Ang-(1–7) also reduced cardiomyocyte hypertrophy, fibrosis and inflammatory cell infiltration of the heart tissue and increased blood vessel number. These changes were blocked by antagonists of the MAS1, AT2 and bradykinin receptors and inhibition of NO formation. Treatment with Ang-(1–7) reduced glomerular damage and oxidative stress in kidney tissue. Bone marrow and circulating endothelial progenitors, as well as bone marrow mesenchymal stem cells, were increased in mice treated with Ang-(1–7).
Conclusions and Implications
Short-term Ang-(1–7) treatment of young db/db mice improved heart function and reduced kidney damage. Treatment also improved bone marrow and circulating levels of endothelial and mesenchymal stem cells. All of this may contribute to improved cardiovascular and renal function.
Tables of Links
| LIGANDS | |
|---|---|
| A-779 | L-NAME |
| Angiotensin II | Losartan |
| Angiotensin-(1-7) | NO |
| Icatibant | PD123319 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14
Alexander et al., 2013a,b,).
Introduction
Type 2 diabetes mellitus (T2DM) affects over 8% of the US population (Rabelo et al., 2011), and is associated with a number of long-term complications including nephropathy, retinopathy, stroke and cardiovascular disease, ultimately leading to a decreased quality of life and reduced life expectancy. It is estimated that the risk of death of diabetic patients is twice as high as that of age-matched non-diabetic individuals (Centers for Disease Control2014).
The renin–angiotensin system (RAS) has been implicated in the pathophysiology of multiple diabetes-related complications, including hypertension and subsequent cardiovascular disease (Weekers et al., 2005). Patients with T2DM have two to four times the risk of developing heart disease as compared with healthy individuals (Centers for Disease Control2014). Uptake of fatty acids in cardiac cells exceeds their oxidation rate, which leads to the accumulation of fat and promotes lipotoxicity. This, as a consequence, might induce apoptosis and abnormal remodelling of cardiac tissue (McGavock et al., 2006). Hyperglycaemia leads to overproduction of advanced glycation end products that have pro-inflammatory, pro-fibrogenic and mitogenic properties in the heart (Marwick, 2006). The increased levels of angiotensin II (Ang-II) observed in diabetic patients, through binding to AT1 receptors, activate various intracellular pathways causing cardiomyocyte hypertrophy (Mehta and Griendling, 2006). This can lead to cellular apoptosis, decreased contractility, cardiac remodelling and heart failure. Elevated Ang-II promotes chronic inflammation and the formation of oxygen radicals, which also contribute to tissue fibrosis and remodelling (Mehta and Griendling, 2006; Zablocki and Sadoshima, 2013).
Diabetic heart disease is strongly associated with renal complications. Clinical data show that patients with kidney disease have a 20- to 30-fold higher risk for developing a heart condition (Sarnak et al., 2003). One of the major factors causing chronic kidney disease is high BP. Hypertension is not only seen as the cause but also as a consequence of kidney disorders and it may contribute to multi-organ dysfunction including heart failure. Initially, increased BP causes hyperfiltration and mechanical stress leading to increased permeability and proteinuria. Shear stress then activates signalling pathways involved in the overproduction of extracellular matrix and mesangial expansion. In addition, high blood glucose levels stimulate cells to produce increased amounts of growth factors, cytokines and other cellular mediators that lead to glomerular dysfunction (Schena, 2005). Ang-II has been shown to play an important role in diabetic kidney pathogenesis. Inflammation and oxidative stress (OS) due to elevated Ang-II induce tissue fibrosis, increased endothelial permeability and glomerular damage (Navarro and Mora, 2006; Kashihara et al., 2010).
Heart and kidney functions are also strongly dependent on vascular health. Endothelial progenitor cells (EPCs) are critical in the formation of new vessels and have been suggested to be a primary source of repair in cardiovascular disorders (Kawamoto et al., 2002; Capobianco, 2010). Increased levels of glucose seen in diabetic patients increase the production of reactive oxygen species in bone marrow (BM) and result in a reduction of NO availability, which in turn leads to a decrease in the number of EPCs (Chen et al., 2007; Thum et al., 2007). This may cause impaired endothelial repair mechanisms in diabetic patients due to decreased levels of progenitor cells (Tsai et al., 2012; Westerweel et al., 2013). Micro- and macrovascular complications, resulting from a decreased number of progenitor cells and decreased density of vessels in the heart tissue, may lead to a three- to fivefold increased risk of death in diabetic patients (Benter et al., 2006).
Studies have shown that blockade of the RAS, specifically via inhibition of Ang-II synthesis or the AT1 receptor, with the use of pharmacological agents such as ACE inhibitors and angiotensin receptor blockers (ARBs), may decrease the risk or development of cardiovascular and renal complications in diabetic patients (Wolf and Ritz, 2005; Burnier and Zanchi, 2006). Some of the beneficial effects of ARBs and ACE inhibitors may be in part due to their ability to increase the levels of angiotensin-(1–7) [Ang-(1–7)], a heptapeptide of the RAS which has been shown to oppose many of the actions of Ang-II (Iyer et al., 1998; 2000,). Ang-(1–7) actions are thought to be mediated primarily through binding to a GPCR called MAS1. In addition, several studies have shown that Ang-(1–7) can also activate other receptors such as the AT2 receptor (Castro et al., 2005; Walters et al., 2005; Bosnyak et al., 2011). In contrast to Ang-II, Ang-(1–7) acts through anti-inflammatory, antioxidant and vasodilator mechanisms. Some of the effects of Ang-(1–7) are ascribed to the release of NO and bradykinin, following activation of MAS1 receptor.
More recently, evidence has surfaced demonstrating the cardioprotective effects of Ang-(1–7) in various animal models, for example, streptozotocin-induced type 1 diabetes, spontaneously hypertensive rats and fructose-fed rats (a model of metabolic syndrome) (Ebermann et al., 2008; Giani et al., 2010; Yousif et al., 2012). In addition to its effects on the heart, Ang-(1–7) has been also shown to be renoprotective in streptozotocin-diabetic rats, Zucker diabetic fatty rats and in spontaneously hypertensive rats (Benter et al., 2007; Giani et al., 2010; 2012,).
Due to the counter-regulatory role of Ang-(1–7) in opposition of Ang-II in diabetes, this study was designed to investigate the extent of cardiorenal dysfunction in young (10- to 11-week-old) type 2 diabetic mice (db/db), as well as the effects of Ang-(1–7) treatment on diabetes-related renal and cardiovascular dysfunction. The potential mechanisms of action were studied using various inhibitors of RAS and Ang-(1–7) signalling mediators (NO, bradykinin). The inhibitors included: A-779 – a MAS1 receptor antagonist, losartan – an AT1 receptor inhibitor, PD123319 – an AT2 receptor antagonist, Nω-nitro-L-arginine methyl ester hydrochloride (L-NAME) – an inhibitor of NOS, and icatibant – an inhibitor of bradykinin B2 receptors.
Methods
The animal welfare and ethical statement
The NIH Principles of Laboratory Animal Care were followed, and the Department of Animal Resources at the University of Southern California approved this study. Procedures used were as humane as possible. The animal use in this study complies with recommendations of ARRIVE (Kilkenny et al., 2010; McGrath et al., 2010).
Animal procedures
Eight- to nine-week-old male BKS.Cg-Dock7m +/+ Leprdb/J (db/db) mice and age-matched heterozygous controls (non-diabetic) were purchased from Jackson Laboratories (n = 7/group) (Bar Harbor, ME, USA). Food and water were available ad libitum. Mice were kept on a 12 h light/dark cycle.
db/db mice and the heterozygous controls were administered either saline (vehicle), Ang-(1–7) alone (500 μg·kg−1·day−1) or Ang-(1–7) (500 μg·kg−1·day−1) combined with an inhibitor (losartan, PD123319, A-779 or L-NAME at 10 mg·kg−1·day−1 or icatibant at 0.4 mg·kg−1·day−1) via s.c. bolus injection, daily for 14 days. BM studies included groups that were treated with inhibitors alone.
Pharmacological agents and inhibitors
Ang-(1–7) and A-779 were purchased from Bachem (Torrance, CA, USA). Losartan, PD123319 and L-NAME were purchased from Sigma-Aldrich (Saint Louis, MO, USA). Icatibant was purchased from Tocris Bioscience (Ellisville, MO, USA).
Echocardiographic analysis
In vivo murine cardiac function was assessed non-invasively using an echocardiography system consisting of a Philips SONOS 5500 ultrasound machine equipped with a 6–15 MHz linear transducer (Philips Healthcare, Andover, MA, USA). Anaesthesia was induced and maintained using isoflurane (Abbot Laboratories, Abbott Park, IL, USA) to maintain a heart rate between 400 and 450 beats min−1. The left ventricle ejection fraction, fractional shortening and left ventricle internal dimensions at both systole and diastole were measured and calculated in accordance with the American Society of Echocardiography guidelines (Lang et al., 2006). Cardiac output (L·min−1) and cardiac index (L·min−1·mg−1) were determined using the modified Simpson’s rule.
Thermodilution methodology
To assess maximum (dP/dTmax) and minimum (dP/dTmin) changes in left ventricle pressure, mice were anaesthetized with i.p. injection of ketamine (Clipper Distributing Company, St. Joseph, MO, USA) and xylazine (Phoenix Pharmaceutical, St. Joseph, MD, USA) (100 and 10 mg/kg body weight respectively). A midline incision was made in the ventral neck area and a thermocouple probe was introduced into the carotid artery to monitor the temperature of the test fluid. A PE10 (Braintree Scientific, Braintree, MA, USA) catheter was filled with Lactated Ringer’s (Baxter, Deerfield, IL, USA) solution and inserted into the jugular vein. Changes in blood temperature were measured using a Cardiomax-II Thermodilution Cardiac Output Computer (Columbus Instruments, Columbus, OH, USA) connected to the thermocouple probe. The analogue signal was digitized and processed with a BIOPAC MP150 Data Acquisition and Analysis System using AcqKnowledge software version 4.2 (BIOPAC Systems, Incorporated, Goleta, CA, USA).
Histology and immunohistochemistry
Hearts and kidneys were fixed in formalin, embedded in paraffin and cut into 5 μm sections.
Cardiomyocyte hypertrophy
Heart samples were sectioned longitudinally and stained with haematoxylin and eosin. Ten random images of each heart section were taken at 10× magnification along the left ventricular wall, avoiding the septal wall for consistency. Cardiomyocyte area and width was determined using ImageJ version 1.47v (National Institutes of Health, USA) at positions without visible nuclei exposing substantial lengths of uniform width to avoid a large standard error (Tracy and Sander, 2011).
Heart fibrosis
Heart samples were sectioned longitudinally and stained using picrosirius red method. Twenty pictures of the left ventricle were taken at 40× and evaluated using a macro developed by Hadi et al. (2011). The septal wall was avoided for consistency. The fibrosis was expressed as percentage of cardiomyocyte area stained red.
Immunohistochemistry
An avidin–biotin complex method of detection was used for each staining.
CD31
Goat anti-mouse monoclonal antibody directed against CD31 (BD Biosciences, San Jose, CA, USA) was used at a 1:250 dilution. After the samples had been mounted on slides, enumeration of CD31-positive vessels in the heart sections was performed using light microscopy.
Nitrotyrosine
Rabbit anti-mouse polyclonal antibody directed against nitrated tyrosine residues (EMD Millipore, Billerica, MA, USA) was used at 1:500 dilution. Ten random images of kidney cortex at 10× were evaluated for extent of nitrotyrosine staining using ImageJ version 1.47v and expressed as percentage of area positively stained.
Phospho-eNOS Ser1177
Rabbit anti-mouse polyclonal antibody to eNOS (Phospho Ser1177) (GeneTex Inc., Irvine, CA, USA) was used at 1:100 dilution. Twenty random images of kidney cortex at 40× were evaluated for extent of staining using ImageJ version 1.47v and expressed as percentage of area positively stained.
Phospho-eNOS Thr495
Rabbit anti-mouse polyclonal antibody to eNOS (Phospho Thr495) (Bioss Inc., Woburn, MA, USA) was used at 1:100 dilution. Twenty random images of kidney cortex at 40× were evaluated for extent of staining using ImageJ version 1.47v and expressed as percentage of area positively stained.
Glomerular area and mesangial expansion measurements
The kidney sections were stained using the periodic acid-Schiff (PAS) staining method. Twenty images of random cortical glomeruli were obtained at 40× magnification. The images were analysed using ImageJ version 1.47v. Mesangial expansion was expressed as percentage of glomerular area stained with PAS.
Apoptosis
Apoptosis was evaluated using the Click-iT® TUNEL Assay (Life Technologies, Grand Island, NY, USA).
Gene expression in kidney tissue
qRT-PCR was performed as described previously (Mordwinkin et al., 2012). Briefly, RNA was reverse transcribed and real-time PCR was performed using SYBR green PCR Master Mix (Applied Biosystems by Life Technologies, Thermo Fisher Scientific, Waltham, MA, USA). Relative expression of each of the genes of interest was evaluated using an ABI 7300 instrument (Applied Biosystems by Life Technologies, Thermo Fisher Scientific). Abundance of targeted mRNA was normalized against 18S mRNA.
EPC counts
Blood was collected by cardiac puncture and the bone marrow was harvested by flushing both femurs with PBS containing 2% fetal calf serum. Cells were fixed in 2% paraformaldehyde (bone marrow – before; blood cells – after the staining). Cells were labelled with isotype control or fluorescently labelled Flk-1 and Sca-1 antibodies (eBioscience, San Diego, CA, USA) and samples were stored at 4°C in the dark until analysis on a flow cytometer.
Mesenchymal stem cell (MSC) cultures
Following isolation of bone marrow (BM), cells were incubated at 2.5 × 105 cell/mL in Complete MesenCult® Mouse Medium (STEMCELL Technologies, Incorporated, Vancouver, British Columbia, Canada). Cells were cultured for 8 days at 37°C in 5% CO2 and BM-derived colony-forming unit – fibroblast (CFU-F) colonies were enumerated using phase contrast microscopy.
Statistical analysis
GraphPad Prism version 5.0d for Mac OS X (GraphPad Software, San Diego, CA, USA) was used to analyse the data. One-way anova followed by Tukey’s test or Kruskal–Wallis test was used to compare data from more than two groups. The level of statistical significance was set at 5%. Data are expressed as mean value ± SEM.
Nomenclature
Drug and target nomenclature used in this paper conforms to BJP’s Concise Guide to Pharmacology (Alexander et al., 2013a,b,).
Results
Ang-(1–7) treatment improves heart function in db/db mice
Young db/db mice had significant reductions in both cardiac output (43% decrease) and cardiac index (65% decrease) compared with non-diabetic controls as measured by echocardiography (Figure 1A–B). Administration of Ang-(1–7) in db/db mice resulted in a significant improvement in both parameters. Ang-(1–7) had no effect in non-diabetic mice (data not shown). Echocardiography was also used to assess fractional shortening – a measurement of the difference between end-diastolic and end-systolic left ventricle dimensions. Fractional shortening was significantly decreased by 21% in db/db mice compared with non-diabetic controls (Figure 1E). Mice treated with Ang-(1–7) had a significant 48% increase in fractional shortening compared with vehicle-treated diabetic mice. Using thermodilution methodology, dP/dTmax and dP/dTmin were found significantly decreased in db/db mice compared with non-diabetic controls (Figure 1C–D). Treatment with Ang-(1–7) resulted in 37% increase in dP/dTmax and a 41% increase in dP/dTmin.
Figure 1.
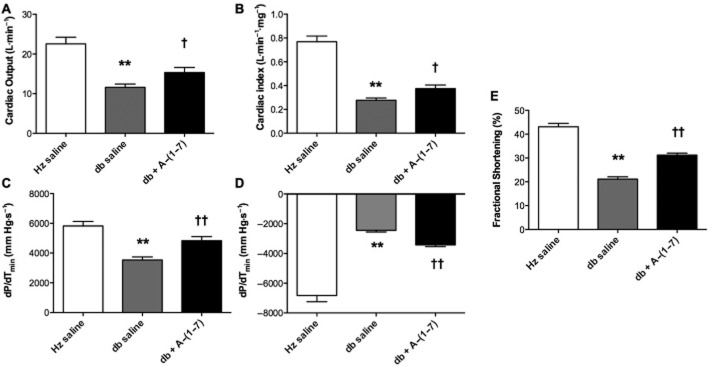
Cardiac function. Cardiac function was decreased in db/db mice and increased by Ang-(1–7) treatment as measured by echocardiography and thermodilution method. The measures of cardiac function included (A) cardiac output, (B) cardiac index, (C) dP/dTmin, (D) dP/dTmax and (E) fractional shortening. (Hz – heterozygous; db – diabetic). **Significantly reduced compared with heterozygous mice (P < 0.01). Significantly increased compared with db saline, †P < 0.05; ††P < 0.01, calculated using one-way anova.
Cardiomyocyte hypertrophy, but not heart size, is reduced following Ang-(1–7) treatment
There were no significant differences in heart weight, a measure of cardiac hypertrophy, between any of the groups (data not shown). In order to investigate the cardiomyocyte hypertrophy, cell dimensions were measured in haematoxylin and eosin stained sections. In diabetic mice, mean cardiomyocyte area and width were significantly increased compared with non-diabetic controls (Figure 2, Supporting Information Fig. S1). Following Ang-(1–7) administration, both measurements were decreased to sizes similar to those observed in non-diabetic mice. One of the mechanisms leading to cardiac remodelling and hypertrophy is uncontrolled apoptosis. However, in this study, no difference in the number of apoptotic cells in the heart tissue was found between any of the groups as measured by TUNEL assay (data not shown).
Figure 2.
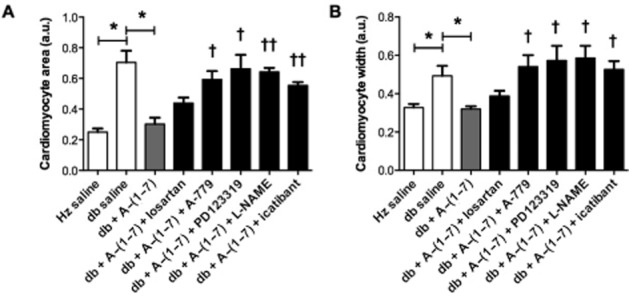
Cardiomyocyte hypertrophy. Cardiomyocyte area (A) and width (B) were assessed. Cardiomyocyte size was increased in db/db mice. Treatment with Ang-(1–7) reversed this change. Co-administration of Ang-(1–7) with either PD123319, A-779, icatibant or L-NAME, but not losartan, blocked the effects of treatment (Hz – heterozygous; db – diabetic; a.u. – arbitrary units). *P < 0.05. Significantly different compared with db + Ang-(1–7), †P < 0.05; ††P < 0.01; or †††P < 0.001; calculated using one-way anova.
Ang-(1–7) decreases cardiac damage by improving tissue vascularization and reducing fibrosis and inflammatory cell infiltration
Cardiac blood vessel density was significantly decreased in db/db mice compared with non-diabetic controls (Figure 3A). Treatment with Ang-(1–7) increased cardiac CD31-positive blood vessel density in db/db mice to levels comparable to those seen in heterozygous controls. Co-administration of Ang-(1–7) with A-779, PD123319, L-NAME and icatibant blocked the effects of Ang-(1–7), while co-administration with losartan had no effect on Ang-(1–7) action. Treatment with Ang-(1–7) also reduced fibrosis in diabetic animals as measured by picrosirius red staining (Figure 3B). Co-administration of Ang-(1–7) and losartan further reduced fibrosis in db/db mice whereas A-779 blocked the effects on Ang-(1–7). Co-administration of either PD123319, L-NAME or icatibant did not have an effect on Ang-(1–7) action. Furthermore, an increase in the number of granulocytes (PMN) in the cardiac muscle was found in db/db mice compared with heterozygous controls (Figure 3C). This increase was not observed in the cardiac tissue of db/db mice administered Ang-(1–7). Co-administration with A-779, PD123319, L-NAME and icatibant, but not losartan, blocked the anti-inflammatory effects of Ang-(1–7).
Figure 3.
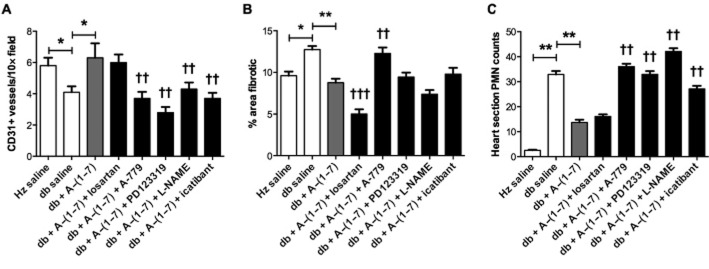
Vessel density and inflammatory cell infiltration in cardiac tissue. Density of CD31-positive blood vessels was assessed as a measurement of tissue vascularization. Fibrosis is expressed as percentage of cell area stained with picrosirius red. Inflammatory cell infiltration was measured by enumerating PMNs. A decrease in blood vessel density (A), increase in tissue fibrosis (B) and increase in PMN number (C) were observed in the heart tissue of db/db mice compared with heterozygous controls (Hz). Ang-(1–7) treatment reversed all of these changes. Co-administration of Ang-(1–7) with either PD123319, A-779, icatibant or L-NAME, but not losartan, blocked the effects of Ang-(1–7) on blood vessel density and PMN count. Co-administration of losartan enhanced anti-fibrotic effects of Ang-(1–7) whereas A-779 blocked the action of Ang-(1–7) (Hz – heterozygous; db – diabetic; PMN – granulocytes). *P < 0.05; **P < 0.01.Significantly different compared with db + Ang-(1–7), †P < 0.05; ††P < 0.01; or †††P < 0.001; calculated using one-way anova.
Administration of Ang-(1–7) reduces glomerular area and mesangial expansion in diabetic mice
Heart disease can be accelerated by kidney dysfunction. To assess glomerular function, glomerular size and mesangial expansion were measured. A significant increase in glomerular area and mesangial expansion was seen in vehicle-treated diabetic animals compared with non-diabetic controls (Figure 4, Supporting Information Fig. S2). Treatment with Ang-(1–7) significantly reduced both of these parameters.
Figure 4.
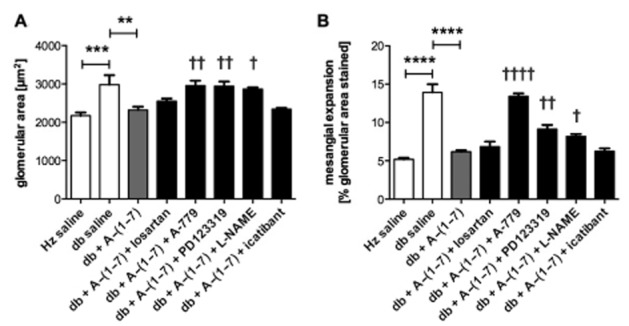
Glomerular area and mesangial expansion. Glomerular area (A) and mesangial expansion (B) were increased in diabetic mice treated with vehicle. Treatment with Ang-(1–7) significantly reduced both of these parameters. Addition of A-779, PD123319 and L-NAME, but not losartan or icatibant, resulted in inhibition of the effects of Ang-(1–7) (Hz – heterozygous; db – diabetic). **P < 0.01; ***P < 0.001; ****P < 0.0001. Significantly increased compared with db + Ang-(1–7), †P < 0.05; ††P < 0.01; or ††††P < 0.0001; calculated using one-way anova.
Ang-(1–7) reduces inflammation and OS in kidneys of db/db mice
OS can lead to severe kidney damage by activating hypertrophic, fibrotic and remodelling signals. To assess the extent of damage in the kidneys due to OS, expression of eNOS, p22-phox (a subunit of NADPH oxidase) and superoxide dismutase (SOD) was measured. In the kidneys of diabetic animals treated with vehicle, expression of eNOS was highly increased (Figure 5A). Ang-(1–7) administration significantly decreased eNOS expression in diabetic mice. This effect was blocked by co-administration of A-779, PD123319, L-NAME and icatibant but not losartan. No significant changes in expression of p22-phox or SOD were observed between heterozygous or diabetic control animals and any other treatment groups (data not shown). The abundance of phosphorylated forms of eNOS was assessed using immunohistochemistry (Figure 5B–C, Supporting Information Fig. S3). eNOS phosphorylation on Ser1177 (activated form) was more abundant in kidneys from diabetic animals. Staining was further increased by administration of Ang-(1–7). This action of Ang-(1–7) was mediated specifically through MAS1 receptor. Administration of A-779 resulted in increased phosphorylation of eNOS on Ser1177. None of the remaining inhibitors had an effect on this marker. Deactivated form of eNOS (phosphorylation on Thr495) was more abundant in db/db mice. Treatment with Ang-(1–7) decreased levels of this form of eNOS in kidneys from diabetic mice. This effect was reversed by co-administration of A-779. Diabetic kidneys also had increased extent of nitrotyrosine staining, which was reversed by Ang-(1–7) treatment (Figure 5F, Supporting Information Fig. S3). A-779, PD123319 and L-NAME blocked the effects of Ang-(1–7) on this marker.
Figure 5.
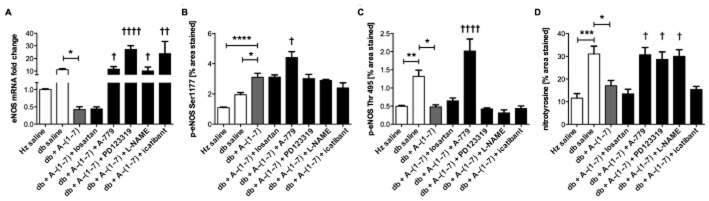
OS in kidneys. Expression of eNOS was highly increased in diabetic animals, suggesting eNOS uncoupling (A). Treatment with Ang-(1–7) reduced levels of eNOS mRNA. Co-administration with A-779, PD123319, L-NAME and icatibant inhibited effects of Ang-(1–7). Level of eNOS phosphorylated on both Ser1177 (active form) (B) and Thr495 (inactive form) (C) was increased in diabetic animals. Treatment further elevated levels of eNOS phosphorylation at Ser1177. Co-administration of A-779 increased levels of this marker. Ang-(1–7) reduced abundance of the deactivated form of eNOS. This effect was reversed after treatment with A-779. Ang-(1–7) decreased nitrotyrosine staining in kidney sections from diabetic animals (D). Co-administration of A-779, PD123319 or L-NAME reversed the effects of Ang-(1–7) on nitrotyrosine formation (Hz – heterozygous; db – diabetic). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Significantly different compared with db + Ang-(1–7), †P < 0.05; ††P < 0.01; or ††††P < 0.0001; calculated using Kruskal–Wallis (A) or one-way anova (B–D).
Ang-(1–7) treatment increases BM-derived MSC and EPC numbers, as well as the number of circulating EPCs
BM-derived MSC numbers were decreased in diabetic mice compared with heterozygous mice (Figure 6C). Ang-(1–7) significantly increased MSC numbers residing in BM. Co-administration of Ang-(1–7) with either PD123319, A-779, icatibant or L-NAME blocked the effects of Ang-(1–7) on this parameter.
Figure 6.

Bone marrow (BM)-derived and circulating EPC counts and BM-derived MSC counts. EPC from the BM (A) and circulation (B), as well as BM-derived MSC (C), were significantly decreased in db/db mice. Ang-(1–7) administration significantly increased these outcomes. Ang-(1–7) effects were blocked by PD123319, A-779, L-NAME and icatibant, but not losartan (Hz – heterozygous; db – diabetic). *P < 0.05; **P < 0.01. Significantly different compared with db + Ang-(1–7), †P < 0.05; ††P < 0.01, calculated using one-way anova.
BM-derived EPC count was significantly lower in db/db mice than in non-diabetic controls. Following administration of Ang-(1–7) in db/db mice, EPC counts were increased. Co-administration of Ang-(1–7) with PD123319, A-779, L-NAME or icatibant, but not losartan, blocked the effects of Ang-(1–7) (Figure 6A; whereas treatment with the inhibitors alone had no effect, Supporting Information Fig. S4).
In order to contribute to tissue vasculogenesis, EPCs need to migrate from the BM into the circulation. Similar to results seen in the BM, levels of circulating EPCs were decreased in db/db mice (Figure 6B), and administration of Ang-(1–7) resulted in a significant increase in circulating EPCs. Again, co-administration of Ang-(1–7) with PD123319, A-779, L-NAME or icatibant blocked the effects of Ang-(1–7) treatment.
Discussion and conclusions
Ang-(1–7) has been recently shown to improve heart function in different animal models of heart failure including myocardial infarction, diabetic rats with cardiomyopathy and metabolic syndrome rat model (Giani et al., 2010; Marques et al., 2011; Singh et al., 2011). These studies demonstrated reduced cardiac hypertrophy and fibrosis, which are also hallmarks of T2DM-related cardiomyopathies. Recent studies by the Oudit group also showed improvement of heart and kidney function in db/db mice with advanced disease (Mori et al., 2014a,b,). In these studies, 5-month-old mice were implanted with an osmotic pump and treated for 28 days. In agreement with these studies, we have shown that short-term treatment (2 weeks) with Ang-(1–7) prevents progression of heart and kidney dysfunction even in the early stage of diabetes.
Our results show that heart and kidney function was impaired in db/db mice, and administration of Ang-(1–7) resulted in significant improvements in multiple measures of cardiac and renal function. The increase in cardiac output observed in diabetic mice treated with Ang-(1–7) appears to be in part due to an increase in contractility expressed as fractional shortening, whereas improvement of glomerular health in kidneys seems to be attributed to a decrease in OS.
In patients with T2DM, left ventricle hypertrophy is often a major cause of the long-term decrease in cardiac function. In this study, young diabetic mice had significant increase in cardiomyocyte area and width, without increase in heart weight or apoptosis, which suggests an early stage of cardiomyopathy. Two of the main mechanisms leading to cardiomyocyte hypertrophy are Ang-II activation of the AT1 receptor and increase in inflammation (van Empel and De Windt, 2004). This may in part help to explain the mechanism by which Ang-(1–7) reduced cardiomyocyte hypertrophy in db/db mice. First, Ang-(1–7) signalling through the MAS1 receptor opposes many of the actions of Ang-II, which could result in decreased signalling via the AT1 receptor. Evaluation of the inflammatory cell infiltration in the heart confirmed an increase in number of neutrophils in the diabetic mice with a reversal upon treatment with Ang-(1–7).
Cardiac function is also regulated by the vascular system. The changes in cardiomyocyte size and blood vessel density after Ang-(1–7) treatment may, in part, be due to its effects on the BM. Previously, we showed that treatment with Ang-(1–7) decreased OS, increased NO production and decreased nitration in the BM of db/db mice (Mordwinkin et al., 2012). Increasing EPC and MSC counts, which are known to be decreased in diabetic patients (Tepper et al., 2002; Fadini et al., 2005), could reduce the risk of vascular disease by enhanced angiogenesis and repair of the damaged cardiac tissue. Our data show that similar to circulating EPC, BM-derived EPC and MSC are also significantly reduced in db/db mice. The effect of Ang-(1–7) on progenitor cells was blocked by multiple inhibitors suggesting a complex signalling that involves MAS1 and AT2 receptors as well as NO and bradykinin. While the exact mechanism is not clear, NO is known to act as a molecular signal resulting in the proliferation and migration of EPC and MSC from the BM niche into the circulation (Aicher et al., 2003). The decreased NO levels observed in diabetes could therefore prevent the mobilization of EPC. Our data show that inhibition of NO production blocks Ang-(1–7)-induced increase in progenitor cell number. This confirms an important role of NO in proliferation and migration of EPC and MSC.
We have also shown that OS significantly contributes to diabetic nephropathy. Damage to proteins, DNA and lipids due to increased OS leads to glomerular fibrosis and mesangial expansion, as well as an increase in endothelial permeability and overall impairment of filtration function (Kashihara et al., 2010). Increased levels of eNOS expression have been described in multiple disease models including high glucose-exposed human aortic cells (Cosentino et al., 1997) and a rat model of myocardial infarction (Bauersachs et al., 1999). Even though NADPH oxidase is usually the primary source of superoxide, several studies suggest that eNOS can be involved in production of superoxide under certain pathological conditions, including diabetes (Cosentino et al., 1997; Guzik et al., 2002). Overexpression of eNOS may result in uncoupling of this enzyme and formation of superoxide rather than NO, contributing to increase in OS. As no changes in the expression of p22-phox (NADPH subunit) were seen, we hypothesize that increased levels of superoxide come from eNOS rather than NADPH oxidase. Peroxynitrite, which can be formed from superoxide and NO, reacts with tyrosine residues causing irreversible damage to proteins (Yang et al., 2009). In fact, Bouloumié et al. (1997) showed that impaired endothelial function is associated with increased eNOS expression, superoxide production and nitrotyrosine appearance. What is more, both active (phosphorylation at Ser1177) and inactive (phosphorylation at Thr495) forms of eNOS were increased in the kidneys of diabetic animals. Phosphorylation of uncoupled eNOS at either Ser1177 or Thr495 has no effect on its activity (Chen et al., 2008). We hypothesize that increased levels of both phospho-eNOS forms in diabetic animals represent, at least in part, an uncoupled fraction of whole eNOS, which is reflected in increased nitrotyrosine formation. In contrast, increased levels of eNOS phosphorylation at Ser1177 in db/db mice treated with Ang-(1–7) did not result in elevated nitrotyrosine staining, suggesting that this increase represents the coupled fraction of eNOS. While losartan, PD123319, L-NAME and icatibant did not alter the effects of Ang-(1–7) treatment, co-administration with A-779 resulted in an increase of both phosphorylated forms of eNOS, which suggests that this signalling occurs primarily through activation of the MAS1 receptor. We believe that the increase of phosphorylation on Thr495 in diabetic animals treated with saline or combination of Ang-(1–7) and A-779 might be a compensatory response to OS. Consistent with decrease in eNOS expression, Ang-(1–7) also reduced the extent of tyrosine nitration and thus contributed to the decrease in OS.
In this study, we showed beneficial effects of Ang-(1–7) treatment on cardiovascular and renal function in a murine model of type 2 diabetes (db/db). Even though the mice were relatively young and the treatment lasted only 2 weeks, the effects on physiological heart and kidney function were profound. Kidney tissue showed OS injury perhaps through eNOS uncoupling, which was attenuated with Ang-(1–7). We identified multiple possible mechanisms of Ang-(1–7) action. Treatment decreased cardiomyocyte hypertrophy, fibrosis and inflammatory cell infiltration in the heart. Vascularization of the heart was also improved, which correlated with increased numbers of BM residing and circulating endothelial and MSCs. We also identified receptors mediating Ang-(1–7) action. The main receptor involved in the effects of Ang-(1–7) is the MAS1 receptor but others, such as AT2 receptors, might also play a role.
Acknowledgments
This study was funded by the National Institutes of Health (Grant No. 5R01HL082722-02).
Glossary
- Ang-(1–7)
angiotensin-(1–7)
- Ang-II
angiotensin II
- BM
bone marrow
- eNOS
endothelial NOS
- EPC
endothelial progenitor cell
- MSC
mesenchymal stem cells
- OS
oxidative stress
- RAS
renin–angiotensin system
- T2DM
type 2 diabetes mellitus
Author contributions
A. M. P. and N. M. M. performed the research and wrote the manuscript. C. J. M. and S. S. J. helped with performing the experiments and design of the study. K. E. R. contributed funding and work space, helped with study design and data analysis.
Conflict of interest
The authors have nothing to disclose.
Supporting Information
Figure S1 Effects of Ang-(1–7) on cardiomyocyte hypertrophy.
Figure S2 Effects of Ang-(1–7) on glomerular hypertrophy and mesangial expansion.
Figure S3 Phosphorylation of eNOS and formation of nitrotyrosine in the kidneys.
Figure S4 Effects of inhibitors on bone marrow EPC in diabetic animals.
Figure S5 EPC flow cytometry analysis.
References
- Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, et al. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med. 2003;9:1370–1376. doi: 10.1038/nm948. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, McGrath JC, et al. The concise guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol. 2013a;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, McGrath JC, et al. The concise guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013b;170:1449–1458. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauersachs J, Bouloumie A, Fraccarollo D, Hu K, Busse R, Ertl G. Endothelial dysfunction in chronic myocardial infarction despite increased vascular endothelial nitric oxide synthase and soluble guanylate cyclase expression: role of enhanced vascular superoxide production. Circulation. 1999;100:292–298. doi: 10.1161/01.cir.100.3.292. [DOI] [PubMed] [Google Scholar]
- Benter IF, Yousif MHM, Anim JT, Cojocel C, Diz DI. Angiotensin-(1–7) prevents development of severe hypertension and end-organ damage in spontaneously hypertensive rats treated with L-NAME. Am J Physiol Heart Circ Physiol. 2006;290:H684–H691. doi: 10.1152/ajpheart.00632.2005. [DOI] [PubMed] [Google Scholar]
- Benter IF, Yousif MHM, Cojocel C, Al-Maghrebi M, Diz DI. Angiotensin-(1–7) prevents diabetes-induced cardiovascular dysfunction. Am J Physiol Heart Circ Physiol. 2007;292:H666–H672. doi: 10.1152/ajpheart.00372.2006. [DOI] [PubMed] [Google Scholar]
- Bosnyak S, Jones ES, Christopoulos A, Aguilar M-I, Thomas WG, Widdop RE. Relative affinity of angiotensin peptides and novel ligands at AT1 and AT2 receptors. Clin Sci. 2011;121:297–303. doi: 10.1042/CS20110036. [DOI] [PubMed] [Google Scholar]
- Bouloumié A, Bauersachs J, Linz W, Schölkens BA, Wiemer G, Fleming I, et al. Endothelial dysfunction coincides with an enhanced nitric oxide synthase expression and superoxide anion production. Hypertension. 1997;30:934–941. doi: 10.1161/01.hyp.30.4.934. [DOI] [PubMed] [Google Scholar]
- Burnier M, Zanchi A. Blockade of the renin-angiotensin-aldosterone system: a key therapeutic strategy to reduce renal and cardiovascular events in patients with diabetes. J Hypertens. 2006;24:11–25. doi: 10.1097/01.hjh.0000191244.91314.9d. [DOI] [PubMed] [Google Scholar]
- Capobianco S. Endothelial progenitor cells as factors in neovascularization and endothelial repair. World J Cardiol. 2010;2:411–420. doi: 10.4330/wjc.v2.i12.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro CH, de Santos RASD, Ferreira AJ, Bader M, Alenina N, de Almeida AP. Evidence for a functional interaction of the angiotensin-(1–7) receptor Mas with AT1 and AT2 receptors in the mouse heart. Hypertension. 2005;46:937–942. doi: 10.1161/01.HYP.0000175813.04375.8a. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control. 2014. National Diabetes Statistics Report [Online]. Available at: http://www.cdc.gov/diabetes/ (accessed 7/9/2014)
- Chen C-A, Druhan LJ, Varadharaj S, Chen Y-R, Zweier JL. Phosphorylation of endothelial nitric-oxide synthase regulates superoxide generation from the enzyme. J Biol Chem. 2008;283:27038–27047. doi: 10.1074/jbc.M802269200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-H, Lin S-J, Lin F-Y, Wu T-C, Tsao C-R, Huang P-H, et al. High glucose impairs early and late endothelial progenitor cells by modifying nitric oxide-related but not oxidative stress-mediated mechanisms. Diabetes. 2007;56:1559–1568. doi: 10.2337/db06-1103. [DOI] [PubMed] [Google Scholar]
- Cosentino F, Hishikawa K, Katusic ZS, Lüscher TF. High glucose increases nitric oxide synthase expression and superoxide anion generation in human aortic endothelial cells. Circulation. 1997;96:25–28. doi: 10.1161/01.cir.96.1.25. [DOI] [PubMed] [Google Scholar]
- Ebermann L, Spillmann F, Sidiropoulos M, Escher F, Heringer-Walther S, Schultheiss H-P, et al. The angiotensin-(1–7) receptor agonist AVE0991 is cardioprotective in diabetic rats. Eur J Pharmacol. 2008;590:276–280. doi: 10.1016/j.ejphar.2008.05.024. [DOI] [PubMed] [Google Scholar]
- van Empel VPM, De Windt LJ. Myocyte hypertrophy and apoptosis: a balancing act. Cardiovasc Res. 2004;63:487–499. doi: 10.1016/j.cardiores.2004.02.013. [DOI] [PubMed] [Google Scholar]
- Fadini GP, Agostini C, Avogaro A. Endothelial progenitor cells and vascular biology in diabetes mellitus: current knowledge and future perspectives. Curr Diabetes Rev. 2005;1:41–58. doi: 10.2174/1573399052952640. [DOI] [PubMed] [Google Scholar]
- Giani JF, Muñoz MC, Mayer MA, Veiras LC, Arranz C, Taira CA, et al. Angiotensin-(1–7) improves cardiac remodeling and inhibits growth-promoting pathways in the heart of fructose-fed rats. Am J Physiol Heart Circ Physiol. 2010;298:H1003–H1013. doi: 10.1152/ajpheart.00803.2009. [DOI] [PubMed] [Google Scholar]
- Giani JF, Burghi V, Veiras LC, Tomat A, Muñoz MC, Cao G, et al. Angiotensin-(1–7) attenuates diabetic nephropathy in Zucker diabetic fatty rats. Am J Physiol Renal Physiol. 2012;302:F1606–F1615. doi: 10.1152/ajprenal.00063.2012. [DOI] [PubMed] [Google Scholar]
- Guzik TJ, Mussa S, Gastaldi D, Sadowski J, Ratnatunga C, Pillai R, et al. Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation. 2002;105:1656–1662. doi: 10.1161/01.cir.0000012748.58444.08. [DOI] [PubMed] [Google Scholar]
- Hadi AM, Mouchaers KTB, Schalij I, Grunberg K, Meijer GA, Vonk-Noordegraaf A, et al. Rapid quantification of myocardial fibrosis: a new macro-based automated analysis. Cell Oncol (Dordr) 2011;34:343–354. doi: 10.1007/s13402-011-0035-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer SN, Ferrario CM, Chappell MC. Angiotensin-(1–7) contributes to the antihypertensive effects of blockade of the renin-angiotensin system. Hypertension. 1998;31:356–361. doi: 10.1161/01.hyp.31.1.356. [DOI] [PubMed] [Google Scholar]
- Iyer SN, Averill DB, Chappell MC, Yamada K, Allred AJ, Ferrario CM. Contribution of angiotensin-(1–7) to blood pressure regulation in salt-depleted hypertensive rats. Hypertension. 2000;36:417–422. doi: 10.1161/01.hyp.36.3.417. [DOI] [PubMed] [Google Scholar]
- Kashihara N, Haruna Y, Kondeti VK, Kanwar YS. Oxidative stress in diabetic nephropathy. Curr Med Chem. 2010;17:4256–4269. doi: 10.2174/092986710793348581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto A, Asahara T, Losordo DW. Transplantation of endothelial progenitor cells for therapeutic neovascularization. Cardiovasc Radiat Med. 2002;3:221–225. doi: 10.1016/s1522-1865(03)00082-9. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, et al. Recommendations for chamber quantification. Eur J Echocardiogr. 2006;7:79–108. doi: 10.1016/j.euje.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Marques FD, Ferreira AJ, Sinisterra RDM, Jacoby BA, Sousa FB, Caliari MV, et al. An oral formulation of angiotensin-(1–7) produces cardioprotective effects in infarcted and isoproterenol-treated rats. Hypertension. 2011;57:477–483. doi: 10.1161/HYPERTENSIONAHA.110.167346. [DOI] [PubMed] [Google Scholar]
- Marwick TH. Diabetic heart disease. Heart. 2006;92:296–300. doi: 10.1136/hrt.2005.067231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGavock JM, Victor RG, Unger RH, Szczepaniak LS American College of Physicians and the American Physiological Society. Adiposity of the heart, revisited. Ann Intern Med. 2006;144:517–524. doi: 10.7326/0003-4819-144-7-200604040-00011. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2006;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- Mordwinkin NM, Meeks CJ, Jadhav SS, Espinoza T, Roda N, diZerega GS, et al. Angiotensin-(1–7) Administration Reduces Oxidative Stress in Diabetic Bone Marrow. Endocrinology. 2012;153:2189–2197. doi: 10.1210/en.2011-2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori J, Patel VB, Abo Alrob O, Basu R, Altamimi T, DesAulniers J, et al. Angiotensin 1–7 ameliorates diabetic cardiomyopathy and diastolic dysfunction in db/db mice by reducing lipotoxicity and inflammation. Circ Heart Fail. 2014a;7:327–339. doi: 10.1161/CIRCHEARTFAILURE.113.000672. [DOI] [PubMed] [Google Scholar]
- Mori J, Patel VB, Ramprasath T, Alrob OA, DesAulniers J, Scholey JW, et al. Angiotensin 1–7 mediates renoprotection against diabetic nephropathy by reducing oxidative stress, inflammation, and lipotoxicity. Am J Physiol Renal Physiol. 2014b;306:F812–F821. doi: 10.1152/ajprenal.00655.2013. [DOI] [PubMed] [Google Scholar]
- Navarro JF, Mora C. Diabetes, inflammation, proinflammatory cytokines, and diabetic nephropathy. Scientificworldjournal. 2006;6:908–917. doi: 10.1100/tsw.2006.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabelo LA, Alenina N, Bader M. ACE2-angiotensin-(1–7)-Mas axis and oxidative stress in cardiovascular disease. Hypertens Res. 2011;34:154–160. doi: 10.1038/hr.2010.235. [DOI] [PubMed] [Google Scholar]
- Sarnak MJ, Levey AS, Schoolwerth AC, Coresh J, Culleton B, Hamm LL, et al. Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American heart association councils on kidney in cardiovascular disease, high blood pressure research, clinical cardiology, and epidemiology and prevention. Circulation. 2003;108:2154–2169. doi: 10.1161/01.CIR.0000095676.90936.80. [DOI] [PubMed] [Google Scholar]
- Schena FP. Pathogenetic mechanisms of diabetic nephropathy. J Am Soc Nephrol. 2005;16:S30–S33. doi: 10.1681/asn.2004110970. [DOI] [PubMed] [Google Scholar]
- Singh K, Singh T, Sharma PL. Beneficial effects of angiotensin (1–7) in diabetic rats with cardiomyopathy. Ther Adv Cardiovasc Dis. 2011;5:159–167. doi: 10.1177/1753944711409281. [DOI] [PubMed] [Google Scholar]
- Tepper OM, Galiano RD, Capla JM, Kalka C, Gagne PJ, Jacobowitz GR, et al. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. 2002;106:2781–2786. doi: 10.1161/01.cir.0000039526.42991.93. [DOI] [PubMed] [Google Scholar]
- Thum T, Fraccarollo D, Schultheiss M, Froese S, Galuppo P, Widder JD, et al. Endothelial nitric oxide synthase uncoupling impairs endothelial progenitor cell mobilization and function in diabetes. Diabetes. 2007;56:666–674. doi: 10.2337/db06-0699. [DOI] [PubMed] [Google Scholar]
- Tracy RE, Sander GE. Histologically measured cardiomyocyte hypertrophy correlates with body height as strongly as with body mass index. Cardiol Res Pract. 2011;2011:658958. doi: 10.4061/2011/658958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai T-H, Chai H-T, Sun C-K, Yen C-H, Leu S, Chen Y-L, et al. Obesity suppresses circulating level and function of endothelial progenitor cells and heart function. J Transl Med. 2012;10:1–12. doi: 10.1186/1479-5876-10-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters PE, Gaspari TA, Widdop RE. Angiotensin-(1–7) acts as a vasodepressor agent via angiotensin II type 2 receptors in conscious rats. Hypertension. 2005;45:960–966. doi: 10.1161/01.HYP.0000160325.59323.b8. [DOI] [PubMed] [Google Scholar]
- Weekers L, Bouhanick B, Hadjadj S, Gallois Y, Roussel R, Pean F, et al. Modulation of the renal response to ACE inhibition by ACE insertion/deletion polymorphism during hyperglycemia in normotensive, normoalbuminuric type 1 diabetic patients. Diabetes. 2005;54:2961–2967. doi: 10.2337/diabetes.54.10.2961. [DOI] [PubMed] [Google Scholar]
- Westerweel PE, Teraa M, Rafii S, Jaspers JE, White IA, Hooper AT, et al. Impaired endothelial progenitor cell mobilization and dysfunctional bone marrow stroma in diabetes mellitus. PLoS ONE. 2013;8:e60357. doi: 10.1371/journal.pone.0060357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf G, Ritz E. Combination therapy with ACE inhibitors and angiotensin II receptor blockers to halt progression of chronic renal disease: pathophysiology and indications. Kidney Int. 2005;67:799–812. doi: 10.1111/j.1523-1755.2005.00145.x. [DOI] [PubMed] [Google Scholar]
- Yang YM, Huang A, Kaley G, Sun D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am J Physiol Heart Circ Physiol. 2009;297:H1829–H1836. doi: 10.1152/ajpheart.00230.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousif MHM, Dhaunsi GS, Makki BM, Qabazard BA, Akhtar S, Benter IF. Characterization of Angiotensin-(1–7) effects on the cardiovascular system in an experimental model of type-1 diabetes. Pharmacol Res. 2012;66:269–275. doi: 10.1016/j.phrs.2012.05.001. [DOI] [PubMed] [Google Scholar]
- Zablocki D, Sadoshima J. Angiotensin II and oxidative stress in the failing heart. Antioxid Redox Signal. 2013;19:1095–1109. doi: 10.1089/ars.2012.4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effects of Ang-(1–7) on cardiomyocyte hypertrophy.
Figure S2 Effects of Ang-(1–7) on glomerular hypertrophy and mesangial expansion.
Figure S3 Phosphorylation of eNOS and formation of nitrotyrosine in the kidneys.
Figure S4 Effects of inhibitors on bone marrow EPC in diabetic animals.
Figure S5 EPC flow cytometry analysis.