

Summary
Background
An association between chronic hyperaldosteronism and medullary nephrocalcinosis has rarely been made, with only a handful of cases described in literature.
Case Report
We describe five cases of hyperaldosteronism with a long- standing history in whom associated medullary nephrocalcinosis was established.
Conclusions
We infer that a chronic hyperaldosteronic status, whether primary or secondary, is a causal factor in the etiopathogenesis of medullary nephrocalcinosis. This article illustrates and summarizes various postulated theories that support our proposed association between hyperaldosteronism and nephrocalcinosis. We conclude that chronic hyperaldosteronism should be included as one of the causes of nephrocalcinosis and that our case series emphasizes the need of a well-organized retrospective study to prove it further.
MeSH Keywords: Hyperaldosteronism, Hypercalciuria, Nephrocalcinosis
Background
Nephrocalcinosis refers to diffuse calcification in the renal parenchyma (generally, in the renal pyramids), and this condition must be differentiated from nephrolithiasis in which calcium deposition occurs within the lumen of the collecting system, ureter, and bladder. Based on location, nephrocalcinosis can be divided into the cortical type – classically seen with acute tubular necrosis, and the medullary type – seen with several metabolic disorders. In nephrology, the term nephrocalcinosis is more or less synonymous with the medullary type, which is around 20 times more common than the cortical type [1] While the term nephrocalcinosis was first coined in association with hyperparathyroidism, a number of conditions have been linked to this condition since its original descriptions like medullary sponge kidney, renal tubular acidosis, papillary necrosis due to diabetes, sickle cell anaemia and tuberculosis [1–3]. The diagnosis is purely radiological and can be demonstrated on plain radiographs, ultrasound or computed tomography. Basic pathophysiology of this condition includes sustained hypercalcemia and hypercalciuria, tubular delivery of calcium to the interstitium and subsequent deposition based on local conditions including the pH and inhibitory ions [1]. Aldosterone is reported to facilitate urinary calcium excretion and induce negative calcium balance [3,4]. It can thus be conjectured that a chronic, long- standing hyperaldosteronic state can cause super- saturation and crystallization of calcium in the interstitium and induce nephrocalcinosis. Very few cases exist in literature linking hyperaldosteronism with nephrocalcinosis [3–6].
In the text to follow, we present a series of five unique cases in which an organic cause for chronic hyperaldosteronic state was found; and concomitant medullary nephrocalcinosis was appreciated on renal imaging. Postulated theories and a review of literature follow a detailed description of the clinical cases. We hope to provide substantiating clinical and radiologically relevant information so that our proposed theory of chronic hyperaldosteronism causing medullary nephrocalcinosis can be validated.
Case Report
Case 1
A 52-year-old lady presented with a long- standing history of intermittent headache, palpitations, vague episodes of muscle cramping, and progressive bilateral lower limb weakness. On examination, the patient had an elevated blood pressure (170/110 mm Hg) and the power of both lower limbs was grade 3. A screening abdominal ultrasound revealed a heterogeneous hypoechoic left suprarenal mass with vascularity within (Figure 1A, 1B). Increased diffuse hyperechogenicity without shadowing was also noted in the region of the renal pyramids (Figure 1A, 1B) suggestive of medullary nephrocalcinosis. Laboratory investigation revealed hypokalemia (K+ of 1.8 mmol/L) and the plasma aldosterone concentration (PAC) to plasma renin activity (PRA) ratio, PAC/PRA, was elevated (42.8) thus confirming the diagnosis of primary hyperaldosteronism. She underwent a left radical adrenalectomy and the histopathology was suggestive of an adrenal cortical carcinoma.
Figure 1.
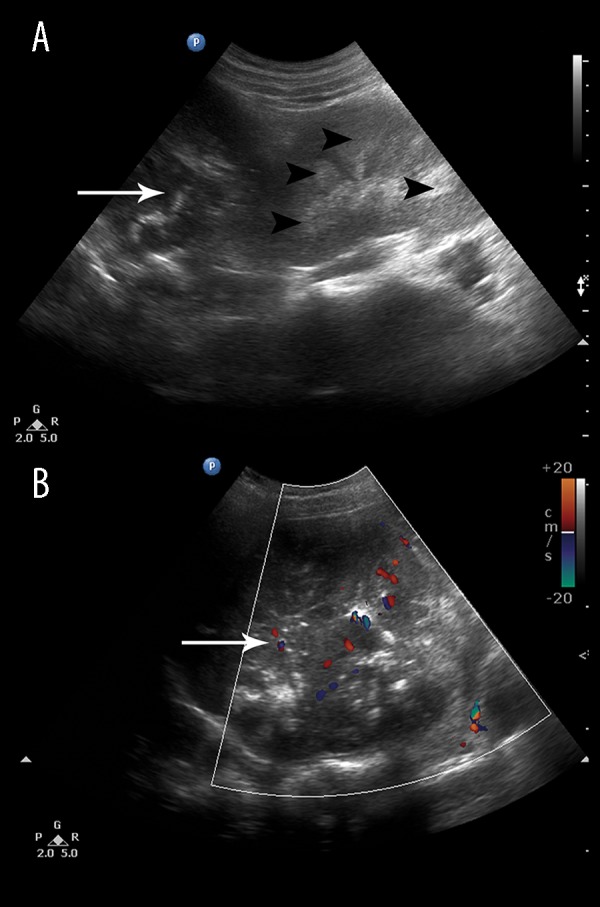
A 52-year-old lady with primary hyperaldosteronism due to adrenal cortical carcinoma. Abdominal ultrasound shows a large heterogeneous suprarenal mass (white arrow) (A) with vascularity within (B) on Doppler, which proved to be an adrenal cortical carcinoma on histopathology. Note the associated medullary nephrocalcinosis (black arrowheads).
Case 2
A 28-year-old hypertensive (blood pressure of 178/98 mm Hg) female complaining of recurrent episodes of cramping muscle pain and weakness for the past 2 years was referred to our department for an abdominal CT scan which demonstrated diffuse enlargement of bilateral adrenal glands. Incidentally, associated bilateral medullary nephrocalcinosis was also noted (Figure 2). On biochemical investigations, the patient was found to be hypokalemic (potassium 2.9 mEq/L). Hormonal evaluation of the patient revealed the following: – high serum aldosterone: 360 pg/mL (normal reference value: 37 240 pg/ml) and suppressed PRA: 0.1 ng/mL/h (normal reference value: 0.15–2.33 ng/mL/h), thus confirming a primary hyperaldosteronism state with associated medullary nephrocalcinosis.
Figure 2.
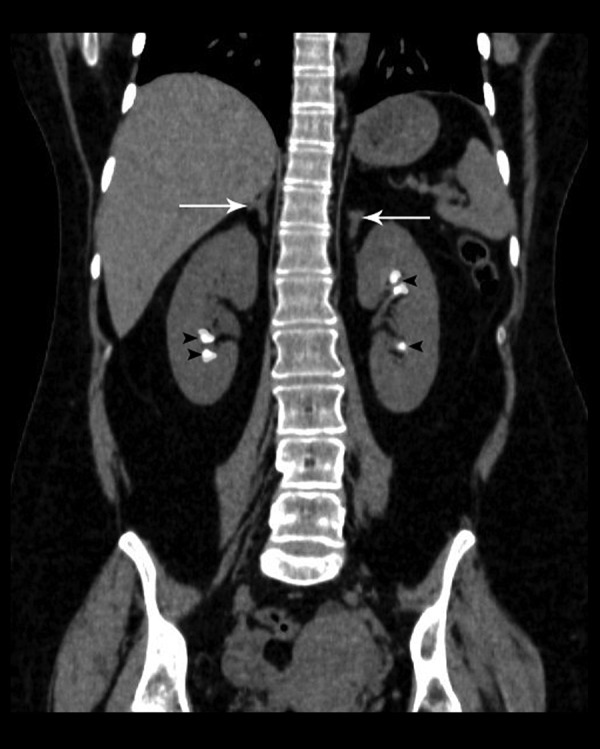
A 28-year-old female with primary hyperaldosteronism and associated medullary nephrocalcinosis. Unenhanced CT scan of the abdomen, coronal reformatted image, shows bilateral adrenal hyperplasia (white arrows) and associated bilateral medullary nephrocalcinosis (black arrowheads).
Case 3
A 61-year-old lady presented with a long- standing dry cough, easy fatigability and polyuria. She gave a history of significant weight loss over the past month (>10% of body weight). On examination, she was tachypnoeic (30/min) and febrile (38.5°C). A chest x-ray demonstrated an ill-defined opacity in the left lower zone and a CT scan of the chest confirmed the presence of a nodular mass lesion with calcification in the left main bronchus which showed homogenous intense enhancement on contrast administration (Figure 3A, 3B). Subsequently performed bronchoscopic biopsy confirmed a bronchial carcinoid tumour. CT of the abdomen showed the presence of medullary nephrocalcinosis (Figure 4). The patient was hypokalemic (2.9 mmol/L) with hyperreninemic hyperaldosteronism; Renin concentration was 34 ng/ml (reference value 0.3–3 ng/ml) and plasma aldosterone concentration was 97 ng/dl (reference value of 0.9–13 ng/dl). Thus, a paraneoplastic syndrome due to a renin- secreting pulmonary carcinoid with subsequent secondary hyperaldosteronism, hypokalemia and associated nephrocalcinosis was diagnosed.
Figure 3.
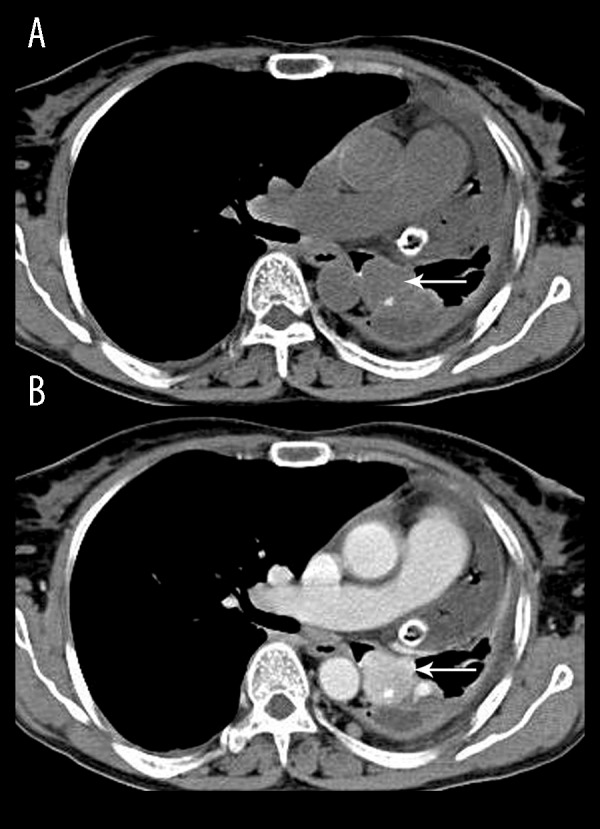
A 61-year-old lady with a paraneoplastic syndrome due to a renin- secreting pulmonary carcinoid with subsequent secondary hyperaldosteronism, hypokalemia and associated nephrocalcinosis. Axial CT of the chest, unenhanced (A) shows a nodular mass lesion (white arrow) with calcification in the left main bronchus which showed homogenous intense enhancement on contrast administration (B). Also noted, post- biopsy hydropneumothorax and intercostal drain in situ.
Figure 4.
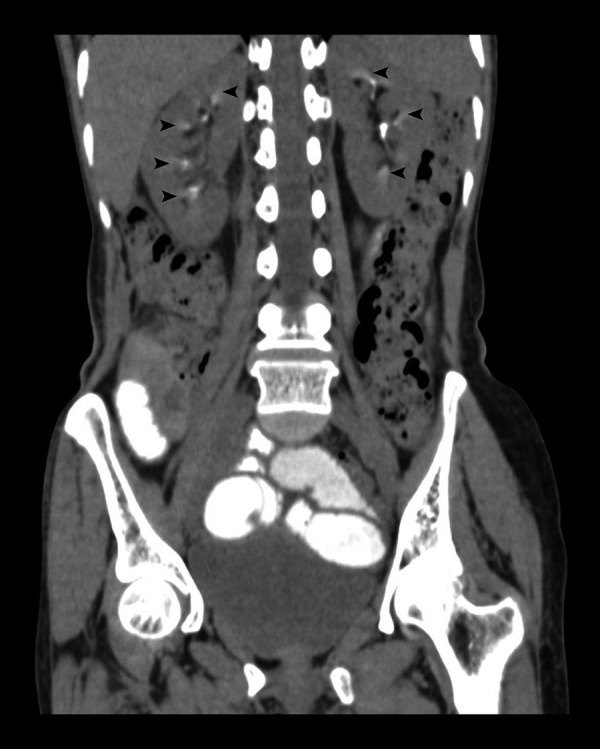
A 61-year-old lady with a paraneoplastic syndrome due to a renin- secreting pulmonary carcinoid with subsequent secondary hyperaldosteronism, hypokalemia and associated nephrocalcinosis. CT abdomen, unenhanced, coronal sections, of the same patient shows the presence of medullary nephrocalcinosis in bilateral kidneys (black arrowheads).
Case 4
A 22-year-old male presented with a 6-month history of a gradually progressing testicular pain and lumpish feeling in bilateral scrotal sacs. The patient also complained of muscle fatigue and progressive bilateral lower limb weakness. General examination showed bilaterally enlarged testicles with scrotal sac pigmentation and excess body hair. The patient was a known case of congenital adrenal hyperplasia (CAH) of the 11 β-hydroxylase deficiency type diagnosed at birth. At the time of presentation, the patient was afebrile and hypertensive (blood pressure of 170/100 mm Hg). S crotal ultrasound was performed which revealed multiple hypoechoic areas in both testes which showed marked vascularity on Doppler examination (Figure 5). An abdominal ultrasound was performed, which showed hyperechoic foci in bilateral renal medullary pyramids (Figure 6) suggestive of medullary nephrocalcinosis. Based on the clinical history and ultrasound features, a diagnosis of congenital adrenal cell rests was made in a known case of CAH. Laboratory investigations revealed a hypokalemic state (K+ level of 1.5 mmol/L). The laboratory investigations were as follows – ACTH level of 340 pg/ml (normal – <60 pg/ml), 11-deoxycortisol: 37 (N: 0 to 1 ng/ml); androstenedione: 25 (N: 0.5 to 2 ng/ml); total testosterone: 700 (N: 240 to 800 ng/dl); renin concentration was 1.2 ng/ml (reference value 0.3–3 ng/ml), and plasma aldosterone concentration was 76 ng/dl (reference value of 0.9–13 ng/dl). Those findings were consistent with our findings of CAH due to 11β-hydroxylase deficiency. Our patient thus displayed an unusual association of nephrocalcinosis with hyperaldosteronism in a case of CAH due to 11β-hydroxylase deficiency.
Figure 5.

A 22-year-old male with unusual association of nephrocalcinosis with hyperaldosteronism in a case of CAH due to 11β-hydroxylase deficiency. Testicular adrenal cell rests seen on a scrotal ultrasound as multiple hypoechoic areas in bilateral testes (white arrowheads) with marked vascularity on Doppler.
Figure 6.
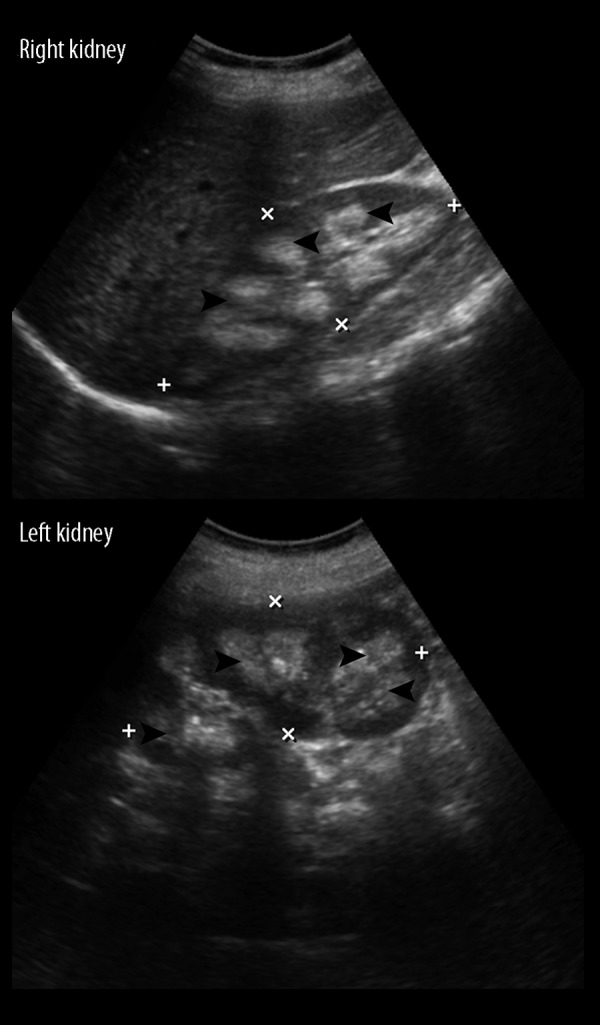
A 22-year-old male with unusual association of nephrocalcinosis with hyperaldosteronism in a case of CAH due to 11β-hydroxylase deficiency. An abdominal ultrasound of the same patient demonstrated bilateral hyperechoic medullary renal pyramids (black arrowheads) suggestive of nephrocalcinosis.
Case 5
A 17-year-old male presented with a 6-hour history of sudden onset weakness of the left half of the body. On examination his blood pressure measured 164/92 mm Hg and the power of the left upper and lower limb was grade 2. A thorough general examination revealed excess facial, body, and pubic hair with bilaterally enlarged testes. An emergency CT scan of the brain was asked for in view of sudden-onset weakness which revealed acute bleeding in the right gangliocapsular region most likely of hypertensive etiology. A screening abdominal ultrasound showed bilateral medullary nephrocalcinosis with enlarged adrenals (Figure 7). A scrotal ultrasound was performed in view of macroorchidism and showed presence of multiple hypoechoeic lesions with increased vascularity throughout both testicular parenchymas indicative of testicular adrenal cell rests (Figure 8). The CT scan of the abdomen showed bilaterally enlarged adrenals (Figure 9A) and confirmed the presence of testicular masses (Figure 9B). A diagnosis of CAH was suspected based on the history of hypertension, virilising features and imaging findings, and was confirmed on laboratory investigations: K+ of 1.8 mmol/L; Na+ – 140 mmol/L; ACTH level of 410 pg/ml (normal – <60 pg/ml); total testosterone: 563 (N: 210 to 790 ng/dl); renin concentration was 1 ng/ml (reference value 0.3–3 ng/ml), and plasma aldosterone concentration was 98 ng/dl (reference value of 1–14 ng/dl). We thus highlight incidentally diagnosed medullary nephrocalcinosis in a case of CAH due to 11β-hydroxylase deficiency with testicular adrenal cell rests.
Figure 7.
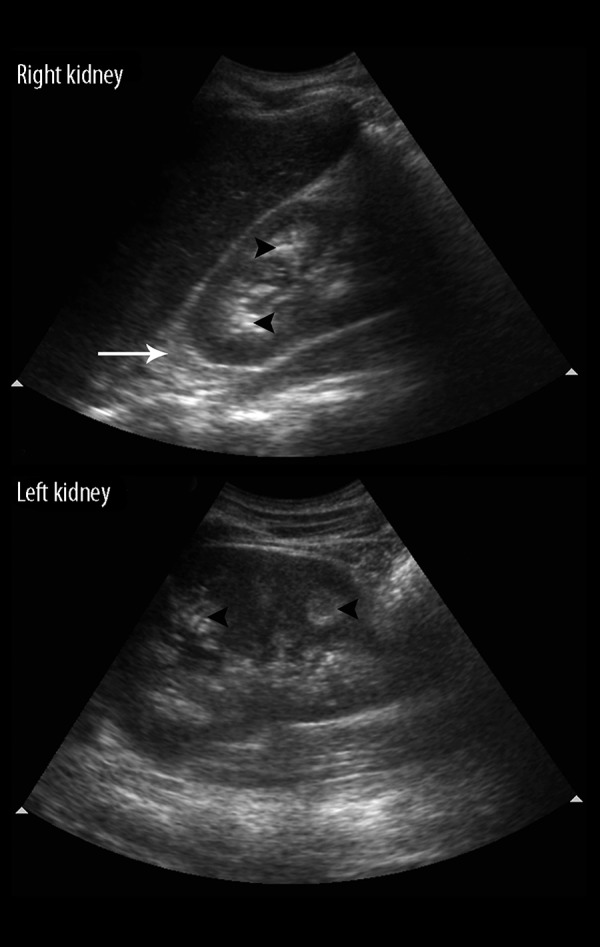
A 17-year-old male with incidentally diagnosed medullary nephrocalcinosis in a case of CAH due to 11β-hydroxylase deficiency with testicular adrenal cell rests. An abdominal ultrasound, showing hyperechoic foci in bilateral renal medullary pyramids suggestive of medullary nephrocalcinosis (black arrowheads). Note the associated enlarged adrenal (white arrow) as expected in congenital adrenal hyperplasia.
Figure 8.

A 17-year-old male with incidentally diagnosed medullary nephrocalcinosis in a case of CAH due to 11β-hydroxylase deficiency with testicular adrenal cell rests. Scrotal ultrasound shows multiple multiple hypoechoic areas in both testes with marked vascularity on Doppler examination suggestive of congenital adrenal cell rests.
Figure 9.
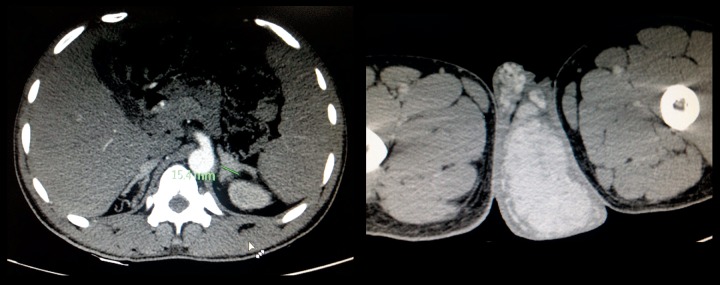
A 17-year-old male with incidentally diagnosed medullary nephrocalcinosis in a case of CAH due to 11β-hydroxylase deficiency with testicular adrenal cell rests. Axial CT scan of the same patient shows bilaterally enlarged adrenals (A) and enlarged testes showing intense contrast enhancement suggestive of testicular adrenal rest cells (B) consistent with congenital adrenal hyperplasia.
Discussion
Nephrocalcinosis is the term used to describe pathological renal parenchymal calcification and may be either dystrophic or metastatic. In the dystrophic type, calcium deposition occurs haphazardly anywhere in the cortex or medulla, in necrotic or ischemic renal tissue, in absence of derangements in calcium metabolism; seen in relation to renal abscesses, tumours, hematomas or tuberculosis. On the other hand, the term metastatic calcification is used when the deposition of calcium salts occurs in normal renal parenchyma secondary to derangements in calcium metabolism, caused by hyperparathyroidism, renal tubular acidosis and renal failure [1,2]. Based on the distribution, metastatic nephrocalcinosis may either be cortical (5%) or medullary (95%). Causes of cortical and medullary nephrocalcinosis are enlisted below (Table 1). Due to the concentrating function of the loops of Henle in the medulla, medullary calcinosis is 20 times more common than cortical nephrocalcinosis. An explanation for the higher incidence of medullary deposition of calcium is based on the Anderson Carr Randall theory which postulates that there is a high concentration of calcium in the fluid surrounding the medullary tubules which drains in the lymphatic vessels. When this amount exceeds the lymphatic drainage due to any cause of hypercalciuria, calcium deposition takes place in the renal papillae and the medullary margins in the interstitium, causing the classical calcium depository pattern of nephrocalcinosis. With disease progression, there is symmetrical medullary calcification with sparing of the cortex [1]. Thus, overt hypercalcemia or hypercalciuria secondary to any cause plays a fundamental role in the pathophysiology of nephrocalcinosis.
Table 1.
Causes of cortical and medullary nephrocalcinosis.
| Causes of cortical nephrocalcinosis [1] | Causes of medullary nephrocalcinosis [1–3] | |
|---|---|---|
| Common causes | Less common causes | |
| Chronic glomerulonephritis | Hyperparathyroidism | Bartter’s disease |
| Chronic pyelonephritis | Renal tubular acidosis type I | Sarcoidosis |
| Toxaemia of pregnancy | Medullary sponge kidney | Cushing’s syndrome |
| Rejected renal transplants | Dent’s disease | |
| Drugs like arsenic | Vitamin D intoxication | |
| Ethylene glycol poisoning | Oculocerebral syndrome | |
| Milk alkali syndrome | ||
| Idiopathic hypercalciuria | ||
| Liddle’s syndrome | ||
| Lowe’s syndrome | ||
| Primary hyperoxaluria types 1 and 2 | ||
| X-linked hypophosphatemia | ||
| Williams’ syndrome | ||
| Wilson’s disease | ||
| Prolonged use of diuretics | ||
Primary hyperaldosteronism (hyporeninemic hyperaldosteronism) is attributed to adrenal causes while secondary hyperaldosteronism (also called hyperreninism or hyperreninemic hyperaldosteronism) is due to over activity of the renin- angiotensin system. Renin- producing tumors, juxtaglomerular cell tumors and renal artery stenosis all lead to increased renin level and this produces secondary hyperaldosteronism.
Nephrocalcinosis associated with hyperaldosteronism is rare, with only a handful of cases described in literature [3–6]. Hyperaldosteronism is known to facilitate Ca+ excretion in urine [3,4]. Calcium excretion is closely related to Na+ excretion in urine; for every 100 mEq/dl increment in Na+ excretion, there is a 40-mg/dl rise in Ca+ excretion [3]. In hyperaldosteronism, there is a reduced reabsorption of Na+ from the aldosterone – insensitive tubular sites, causing subsequent hypercalciuria [3]. C.W Yang et al. proposed an alternative theory for nephrocalcinosis due to hyperaldosteronism [6]. According to them, chronic hypokalemia seen in cases of hyperaldosteronism can cause a tubulointerstitial injury. This is associated with elevated ammoniagenesis and subsequent renal damage through ammonia- activated compliment pathways. The medullary cyst formation, interstitial inflammation and medullary calcification may all be related to the ammonia- mediated nephropathy [6]. Schwedler et al. proposed an alternative theory for nephrocalcinosis in chronic hypokalemic states [7]. They suggested that metabolic alkalosis associated with hypokalemic states decreases calcium phosphate or oxalate solubility in alkaline urine, predisposing to calcinosis. An additional theory postulated by E. Tantisattamo et al. states that aldosterone and deoxycorticosterone- mediated hypertension was associated with hypocalcaemia, secondary hyperparathyroidism, and secondary nephrocalcinosis. Furthermore, hyperaldosterone – induced phosphaturia and hypocitraturia are additional risk factors for nephrolithiasis [8]. These various theories that have been postulated, linking chronic hyperaldosterone status and nephrocalcinosis are enumerated below (Table 2).
Table 2.
Various postulated theories linking chronic hyperaldosterone status and nephrocalcinosis.
| Various postulated theories linking chronic hyperaldosterone status and nephrocalcinosis |
| • Hyperaldosteronism-induced reduced reabsorption of Na+ from the aldosterone-insensitive tubular sites and subsequent hypercalciuria [3] |
| • Ammonia-mediated nephropathy secondary to hypokalemia [6] |
| • Metabolic alkalosis associated with hypokalemic states causing decreased calcium phosphate or oxalate solubility in the alkaline urine and thus predisposing to calcinosis [7] |
| • Aldosterone-mediated hypertension causing hypocalcaemia, secondary hyperparathyroidism and secondary nephrocalcinosis [8] |
| • Hyperaldosterone-induced phosphaturia [8] |
| • Hyperaldosterone-induced hypocitraturia [8] |
All cases reviewed in literature, describe the association of primary hyperaldosteronism with nephrocalcinosis. Whether secondary hyperaldosteronism is a risk factor of nephrocalcinosis and nephrolithiasis remains unclear [8], with not a single case documented in literature to the best of our knowledge. We described an unusual spectrum of nephrocalcinosis associated with varied causes of hyperaldosteronism – not just primary adrenal causes.
Case 1 highlights an aldosterone -secreting adrenocortical carcinoma with associated medullary nephrocalcinosis. Adrenocortical carcinoma (ACC) is a rare tumour with an estimated incidence of 0.7 to 2/million population/year [9]. Functional tumours are seen in around 60% of the cases [9–11] and while cortisol accounts for the main steroid that is secreted (50%), aldosterone is secreted in less than 2% of the cases [10,11]; as seen in our first case.
Case 1 and 2 highlight the association of nephrocalcinosis with primary aldosteronism (PA). A total of 60% of cases of PA are attributed to bilateral adrenal hyperplasia as seen in our second case [12]. Extremely rare causes of PA include unilateral adrenal hyperplasia (1%), ectopic aldosterone – producing adenoma (1%) and adrenocortical carcinoma (1%); highlighted in our first case [12,13]. It is noteworthy that most patients with primary aldosteronism (PA) are normokalemic [12]. Associated hypokalemia is seen in only 9 37% of cases [12,14]. Interestingly, both our cases of PA (case 1 and 2) had hypokalemia, which substantiates the existing theory by C.W. Yang et al. [6] that probably hypokalemia could play a major role in the pathogenesis of hyperaldosterone – induced nephrocalcinosis.
Carcinoid syndrome is a paraneoplastic syndrome seen very rarely in cases of pulmonary carcinoid (less than 2% cases) [15]. Renin- secreting paraneoplastic syndromes in association with neuroendocrine tumours are rare and are seen in association with small cell lung carcinomas, paragangliomas, and carcinoids [16]. While most bronchial carcinoids secrete 5-HT [15], our third case highlights a renin – secreting bronchial carcinoid causing hyperreninemic hyperaldosteronism, aldosterone – mediated hypokalemia, and nephrocalcinosis. Such a case of secondary hyperaldosteronism- induced nephrocalcinosis has not been described in literature to the best of our knowledge.
In our final two cases (case 4 and 5) we bring forward an unusual association of medullary nephrocalcinosis with congenital adrenal hyperplasia (CAH), not yet documented in literature, to the best of our knowledge. CAH is a rare autosomal recessive disorder characterised by decreased production of glucocorticoids. An increase in the ACTH levels leads to hyperplasia of ACTH – sensitive tissue at sites like the testes, leading to testicular masses called testicular adrenal rest tumours (TARTS) [17,18]. The 11β-hydroxylase deficiency accounts for 5–8% of the causes of congenital adrenal hyperplasia [19]. Decreased 11β-hydroxylase activity in our patient led to lowered conversion of 11-deoxycortisol to cortisol and 11-deoxycorticosterone to corticosterone, with subsequent ACTH stimulation. Increased secretion of ACTH leads to overstimulation of the adrenal cortex and primary adrenal rest tissue and produces an excess of proximal cortisol precursors, increased mineralocorticoids and sex steroids [19]. While in our fourth patient presented to us predominantly for the testicular symptoms, our final case had an unusual presentation in the form of hypertensive bleeding and who was retrospectively diagnosed to have CAH. TARTS and hyperaldosterone status were established in both cases, and medullary nephrocalcinosis was only incidentally diagnosed. The excess ACTH stimulation of TARTS in our final two cases was responsible for increased aldosterone activity.
Conclusions
Our study highlights a rare gamut of cases and provides evidence that chronically elevated aldosterone levels could possibly be responsible for increasing the incidence of medullary nephrocalcinosis. Screening abdominal ultrasound is an important investigation. We postulate that aldosterone- mediated hypokalemia, seen in all our cases, may be a prime causal factor in this etiopathogenesis. We also infer that it is probably a prolonged hyperaldosteronic state that induces changes of nephrocalcinosis. We thus add to the already existing literature and provide evidence that chronic hyperaldosteronism secondary to any cause, not just primary aldosteronism, may be a potential risk factor of nephrocalcinosis. We conclude that chronic hyperaldosteronism should be included as one of the causes of nephrocalcinosis and that our case series emphasizes the need of a well-organized retrospective study to prove it further.
References
- 1.Rumack CM, Wilson SR, Charboneau JW, Levine D. Diagnostic Ultraound. 4th edition. I. Mosby; pp. 346–48. Part II; Chapter 9. [Google Scholar]
- 2.Miller NL, Humphreys MR, Coe FL, et al. Nephrocalcinosis: re-defined in the era of endourology. Urol Res. 2010;38(6):421–27. doi: 10.1007/s00240-010-0328-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yasuda K, Sasaki K, Yamato M, et al. A case of nephrocalcinosis associated with primary aldosteronism. Intern Med. 2012;51(6):625–27. doi: 10.2169/internalmedicine.51.6543. [DOI] [PubMed] [Google Scholar]
- 4.Kabadi UM. Renal calculi in primary hyperaldosteronism. Postgrad Med J. 1995;71(839):561–62. doi: 10.1136/pgmj.71.839.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shey J, Cameron MA, Sakhaee K, Moe OW. Recurrent calcium nephrolithiasis associated with primary aldosteronism. Am J Kidney Dis. 2004;44(1):e7–e12. doi: 10.1053/j.ajkd.2004.03.037. [DOI] [PubMed] [Google Scholar]
- 6.Yang CW, Kim SY, Kim YS, et al. Nephrocalcinosis associated with primary aldosteronism. Nephron. 1994;68:507–8. doi: 10.1159/000188318. [DOI] [PubMed] [Google Scholar]
- 7.Schwedler SB, Grone EF, Luft FC. Chronic hypokalaemia and nephrocalcinosis. NDT Plus. 2009;2(4):314–17. doi: 10.1093/ndtplus/sfp047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tantisattamo E, Francis TB. Primary hyperaldosteronism as a risk factor for recurrent nephrolithiasis. Kidney Res Clin Pract. 2012;31(2):A78. [Google Scholar]
- 9.Huang C-J, Wang T-H, Lo Y-H, et al. Adrenocortical carcinoma initially presenting with hypokalemia and hypertension mimicking hyperaldosteronism: a case report. BMC Res Notes. 2013;6:405. doi: 10.1186/1756-0500-6-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chowdhury PS, Nayak P, Gurumurthy S, David D. Aldosterone and cortisol co-secreting bifunctional adrenal cortical carcinoma: A rare event. Indian J Urol. 2014;30(3):339–41. doi: 10.4103/0970-1591.134248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peppa M, Pikounis V, Papaxoinis G, et al. Adrenocortical carcinoma secreting cortisol, androgens and aldosterone: a case report. Cases J. 2009;2:8951. doi: 10.4076/1757-1626-2-8951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aronova A, Iii TJF, Zarnegar R. Management of hypertension in primary aldosteronism. World J Cardiol. 2014;6(5):227–33. doi: 10.4330/wjc.v6.i5.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Panchani R, Goyal A, Varma T, et al. Adrenal incidentalomas: A collection of six interesting cases and brief review of literature. Indian J Endocrinol Metab. 2012;16(Suppl 2):S378–81. doi: 10.4103/2230-8210.104100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cooray MSA, Bulugahapitiya US, Peiris DN. Rhabdomyolysis: A rare presentation of aldosterone-producing adenoma. Indian J Endocrinol Metab. 2013;17(Suppl 1):S237–39. doi: 10.4103/2230-8210.119583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tryfon S, Parisis V, Ioannis K, et al. Excessive muscle paralysis due to pulmonary carcinoid -a case report. Clin Med Insights Case Rep. 2012;5:43–48. doi: 10.4137/CCRep.S9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaltsas G, Androulakis II, de Herder WW, Grossman AB. Paraneoplastic syndromes secondary to neuroendocrine tumours. Endocr Relat Cancer. 2010;17(3):R173–93. doi: 10.1677/ERC-10-0024. [DOI] [PubMed] [Google Scholar]
- 17.Kaynar M, Sönmez MG, Unlü Y, et al. Testicular adrenal rest tumor in 11-Beta-hydroxylase deficiency driven congenital adrenal hyperplasia. Korean J Urol. 2014;55(4):292–94. doi: 10.4111/kju.2014.55.4.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karakuş E, Azılı MN, Tiryaki T. Testicular adrenal rest tumor mimicking leydig cell tumor in a patient with congenital adrenal hyperplasia. APSP J Case Rep. 2014;5(1):10. [PMC free article] [PubMed] [Google Scholar]
- 19.Tosatti R, Júnior, de Souza HS, Tosatti A. Hiperplasia supra-renal congênita por deficiência de 11-β-hidroxilase. Arq Bras Cardiol. 2005;85(6):421–24. doi: 10.1590/s0066-782x2005001900008. [in Portuguese] [DOI] [PubMed] [Google Scholar]