

Abstract
Background
Sargramostim, granulocyte–macrophage colony-stimulating factor (GM-CSF), a hematopoietic growth factor, stimulates cells of the intestinal innate immune system. Clinical trials show that Sargramostim induces clinical response and remission in patients with active Crohn's disease. To study the mechanism, we examined the effects of GM-CSF in the dextran sulphate sodium (DSS) induced acute colitis model. We hypothesized that GM-CSF may work through effects on dendritic cells (DCs).
Methods
Acute colitis was induced in Balb/c mice by administration of DSS in drinking water. Mice were treated with daily GM-CSF or PBS. To probe the role of plasmacytoid DCs (pDCs) in the response to GM-CSF, we further examine the effects of monoclonal antibody 440c, which is specific for a sialic acid-binding immunoglobulin (Ig)-like lectin expressed on pDCs.
Results
GM-CSF ameliorates acute DSS-induced colitis; resulting in significantly improved clinical parameters and histology. Microarray analysis showed reduced expression of pro-inflammatory genes including TNFα and IL1β; results further confirmed by real-time RT-PCR and serum Bio-plex analysis. GM-CSF treatment significantly expands pDCs and type 1 IFN production. Administration of mAb 440c completely blocked the therapeutic effect of GM-CSF. GM-CSF is also effective in RAG1−/− mice, demonstrating activity independent effects on T and B cells. IFN-β administration mimics the therapeutic effect of GM-CSF in DSS-treated mice. GM-CSF increases systemic and mucosal type 1 IFN expression and exhibits synergy with pDC activators, such as microbial CpG DNA.
Conclusions
GM-CSF is effective in the treatment of DSS colitis in a mechanism involving the 440c+ plasmacytoid DC population.
Introduction
Crohn's disease (CD) is a chronic inflammatory disorder of the gastrointestinal tract. Although the etiology is surely multifactorial and remains incompletely understood, the common endpoint in the disease is loss of tolerance to commensal gut flora and an aberrant or overactive adaptive immune response. Evidence suggests that activated macrophages, CD4+ Th1 cells and their products have a pivotal role in disease pathogenesis.1 Accordingly, most current therapies and investigational agents for CD are immunosuppressive in nature and target the T-cell response mediated by the adaptive immune system. The role of proinflammatory cytokines have also been implicated in exacerbation of CD.1 Therapeutic approaches targeting proinflammatory cytokines like TNF-α and IL-1, have been extensively investigated.2,3 More recent evidence suggests that defects in mucosal innate immune function may also have a critical role in CD.1 Support for the role of innate immune dysfunction in the development of CD includes: i) patients with genetic disorders of innate immunity, specifically quantitative or qualitative disorders of phagocytic cell populations, frequently develop GI inflammation that is indistinguishable from CD;4 ii) CSF therapy in these patients often leads to improvement of GI disease; iii) experimental defects of innate immune function can culminate in a chronic T-cell mediated enterocolitis; 5 iv) genetic and environmental risk factors for CD such as smoking negatively affect innate immune function6 and v) the discovery of an association between CD and mutations in genes important for innate immune elements (e.g. CARD15/NOD2, TLRs).7-9
We hypothesized that innate immune deficiency may be central to the pathogenesis of CD and, furthermore, that CSFs may have clinical utility in the treatment of CD through regulation or activation of the innate immunity. A recent Phase II, randomized, double-blind, placebo controlled trial of Sargramostim (yeast derived recombinant human GM-CSF) found that it was effective in the treatment of patients with moderately-to- severely active CD.10
However, the mechanism of action was not clear. GM-CSF is a hematopoietic growth factor that plays a pivotal role in the development and function of innate immune cells, including dendritic cells (DCs), macrophages and granulocytes. GM-CSF increases proliferation and maturation of neutrophils, monocytes and DCs, and is clinically used in the treatment of neutropenia.11 However, knockout mice lacking GM-CSF or GM- CSF receptors have normal steady state hematopoiesis. This suggests that the major physiologic function for GM-CSF may be to stimulate the function of effector cell populations. GM-CSF expands DCs when administered exogenously.12,13 DCs are antigen-presenting cells that provide a direct connection between innate and acquired immunity. Plasmacytoid DCs (pDCs) are the main producers of type 1 IFN in response to certain viral infections via TLR-9 stimulation and have recently been also found to exert an important regulatory role in the immune response. TLR-9 mediated type 1 IFN production also occurs with stimulation by bacterial or synthetically derived cytosine-phosphate-guanosine (CpG) motifs.14 Type 1 IFN has a number of functional effects that may be important in the regulation of intestinal immune responses.15-17 To better understand the mechanism of GM-CSF in human CD, we examined it's effects in a mouse model of inflammatory bowel disease (IBD); acute dextran sulfate sodium (DSS) colitis.18 Acute DSS colitis is a T cell–independent model as intestinal inflammation also develops in SCID mice and RAG1−/−.19-21 DSS treatment produces barrier breakdown and leads to well characterized histologic changes in the colon, including ulceration, infiltration of inflammatory cells into the lamina propria and focal crypt damage.18, 22 The DSS model provides a unique opportunity to examine therapeutic approaches targeting innate immune clearance. We hypothesized that GM-CSF regulates or activates the innate immune system and is responsible, in part, for the tolerogenic state characteristic of healthy mucosa. To probe the role of pDCs in the therapeutic response to GM-CSF, we utilized mAb 440c. Monoclonal Ab 440c recognize a previously uncharacterized member (Siglec-H) of the sialic acid-binding immunoglobulin (Ig)-like lectin (Siglect) family/ Siglec-H is selectively expressed on pDCs. Siglec-H associates with the adaptor DAP12 for signaling and blocks type I IFN expression following TLR stimulation.23, 24 Furthermore, to demonstrate that the therapeutic effects of GM-CSF are independent of adaptive immune elements, we examined its effects on DSS colitis in mice lacking mature T and B cells (RAG1−/− mice). The results of our studies show that GM-CSF is effective in the treatment of DSS-induced colitis, and this effect is dependant on 440c positive (440c+) pDCs and is independent of adaptive immune elements (T and B cells).
Materials and methods
Reagents
Pegylated-recombinant murine-GM-CSF was produced in Pichia pastoris and modified as previously reported.25 Recombinant murine IFN-β and mAb 440c were kindly donated by Drs. Ed Croze and Marco Colonna, respectively. Phosphorothiate-stabilized CpG 1018 (5’ TGACTGTGAACGTTCGAGATGA-3’) was purchased from IDT.
Animals
Specific pathogen free, female, 6-8 week old Balb/c mice, and 6-8 week old RAG1−/− mice on a Balb/c background were purchased from Jackson Laboratory. Animals were housed in the Washington University School of Medicine barrier facility, maintained on light/dark cycles of 12h and fed with a standard rodent chow diet. The Washington University Animal Studies Committee approved all experimental procedures.
Induction of DSS colitis
Previous studies have shown that a concentration of 5% (w/v) DSS (USB Corp.) in drinking water consistently induces colitis in Balb/c mice.18, 26 For RAG1−/− mice, 4 % (w/v-1) DSS was administered to induce a similar severity of colitis.15 Control mice received sterile water during the study period. Mice were monitored daily and weights were recorded. Some groups were treated with daily, 5 μg i.p. injections of pegylated recombinant murine GM-CSF. Groups treated with mAb 440c received 200 μg i.p. on alternate days. Another group was administered with murine recombinant type 1 IFN (mIFN-β) 100,000U/d, (i.p.) for 7 days. At the time of sacrifice, stool samples were subjected to occult blood testing using the Seracult kit (Propper Manufacturing), and bleeding score was determined as previously reported.22 After 7 days of exposure to DSS, mice were sacrificed following halothane (Halocarbon laboratories) inhalation. Their colons were excised from the cecum to the pelvic brim and their length was measured in centimeters. They were equally divided into proximal and distal colon and used for RNA isolation and histology, respectively.
Histological scoring
After colon excision, a portion of distal colon was fixed in 10% neutral formalin buffer (Sigma). Five-micrometer paraffin sections were cut transversely and stained with H&E for histological scoring by a pathologist blinded to the treatment groups using an established method.27
Microarray analysis
Microarray analysis was performed to identify genes significantly increased following acute DSS colitis. The total RNA isolated from the colon of control and DSS-treated mice were used for Agilent whole genome mouse arrays (Agilent technologies, CA). A primary statistical analysis of the array data was performed using the Significance Analysis of Microarrays (SAM) procedure.28 These analyses were performed on log-transformed mean signal intensities from five duplicate experiments. SAM assigns a gene-specific ‘t’ test based on changes in gene expression relative to the standard deviation of repeated measurements for that gene.
Numerical quantification of 440c+ cells in sections
Sections of spleens isolated from control, GM-CSF, GM-CSF+440c and 440c treated groups were subjected to immunohistochemical staining using mAb 440c+. In each group, a total of six high power fields (hpf) were evaluated from different corners of the slide. In each hpf, total cells and 440c+ (brown) cells were counted and the percentage of 440c+ cells was determined.
Real Time RT-PCR
Total RNA was isolated from the proximal colon using Trizol (Invitrogen) and subjected to mRNA purification using Oligotex mRNA mini kit (Qiagen). mRNA was then reverse transcribed with SuperScript II reverse transcriptase (Invitrogen Corp) in the presence of random hexamer primers (Invitrogen Corp). cDNA were used for real time RT-PCR analyses using Jumpstart Taq DNA polymerase (Sigma) and SYBR green nucleic acid stain. Mouse primers used in analysis were as follows: β-actin ATCATTGCTCTCCTG-AGCG-3’ and 5’-GCTGATCCACATCTGGAA-3’; TNF-α 5’-GACCCTCACACTCAGATCATCCTTCT-3’ and 5’-ACGCTGGCTCAGCCACTC-3’; IL1-β, 5’-TCGCTCAGGGTCACAAGAAA-3’ and 5’-CATCAGAGGCAAGGAGGAAAAC-3’;IL1-α, 5’- TCCAGGGCAGAGAGGGAGT and 5’-GGAACTTTGGCCATCTTGATTT-3’; IL-10, 5’- TGCAGCAGCTCAGAGGGTT-3’ and 5’-TGGCCACAGTTTTCAGGGAT-3’; IFN-α, 5’-TCTGATGCAGCAGGTGGG-3’ and 5’-AGGGCTCTCCAGAC-TTCTGCTCTG-3’; IFN-β, 5’-GCACTGGGTGGAATGAGACT-3’ and 5’- AGTGGAGAGCAGTTGAGGACA-3’; IDO, 5’-TCTGTGAGAAAGTTCCACCTCGCA-3’ and 5’-TTCCACATTTGAGGGCTCTTCCGA-3’.
Crossing threshold values for individual genes were normalized to β-actin expression.
Quantitation of Serum Cytokines
Following halothane-induced anesthesia of mice, blood samples were collected by retro-orbital puncture and allowed to clot for 1–2 hr at 37°C. They were centrifuged at 1,000 × g, 4°C to obtain serum and stored at −80°C. Bio-PlexTM multiple cytokine assays (BioRad Laboratories) were performed for quantitation of serum cytokine levels as per the manufacturer's instructions. Data was analyzed using Bio-Plex Manager Software (v 3.0) with 5 parameter curve fitting and assay precision.
Statistical analysis
All data were expressed as mean ± SEM. Statistical analysis of significance was determined by Student's ‘t’ test for unpaired data with Welch correction using Graph Pad Instat (Graph Pad Software Inc.). A P ≤ 0.05 was considered significant.
Results
GM-CSF reduced the severity of DSS colitis
Acute colitis was induced in Balb/c mice by treatment with 5% DSS in their drinking water for 7 days. DSS-treated animals exhibited weight loss and rectal bleeding similar to previous reports.18 Weight loss began around day 4 (Fig 1A) and became significant compared to untreated control animals by day 7 (−6.37% of baseline weight, P < 0.01). Treatment with GM-CSF significantly attenuated this weight loss (+0.5%, P < 0.01). Recently, others have observed a protective role for type 1 IFNs (α and β) in murine models of experimental colitis.15,29 The natural interferon-producing pDC is the major source of type 1 IFN following in vivo stimulation of TLR-9.23 To directly investigate whether the pDC is involved in the therapeutic response to GM-CSF, we utilized a novel mAb, 440c. Monoclonal Ab 440c binds to Siglec-H, a cell surface receptor exclusively present on the pDC.24 In vivo and in vitro, mAb 440c blocks the pDC functional response to TLR stimulation.23,24 When DSS treated animals received GM-CSF and mAb 440c (DSS+GM-CSF+mAb 440c), the therapeutic effects of GM-CSF was blocked (wt loss; −6.85%, P < 0.01). Treatment with DSS and mAb 440c alone (DSS+mAb 440c) resulted in disease activity and histopathologic scores not significantly different from treatment with DSS alone (data not shown). Two additional markers of colitis severity, rectal bleeding and colon shortening were also determined at necroscopy on day 7. Treatment with GM-CSF (DSS+GM-CSF) reduced both the rectal bleeding score and degree of colonic shortening compared with DSS alone (Fig 1B & C). The therapeutic effects of GM-CSF on these disease activity measures were also reversed by co-administration of mAb 440c (DSS+GM-CSF vs. DSS+GM-CSF+mAb 440c).
Figure 1.

GM-CSF treatment reduces severity of DSS colitis. Acute colitis was induced in Balb/c mice by administration of 5% DSS in drinking water for 7d. Experimental groups contained 5 mice and results are representative of 3 individual experiments. (A) Acute DSS colitis led to significant weight loss compared to controls by day 7. However, GM-CSF treatment (DSS+GM-CSF) significantly attenuated this weight loss. Administration of mAb 440c (DSS+GM-CSF+mAb 440c) reversed the therapeutic effect of GM-CSF. (B) At necroscopy, bleeding scores were determined using a Seracult kit. DSS colitis led to gastrointestinal bleeding, which was significantly reduced by GM-CSF treatment (DSS+GM-CSF). However, administration of mAb 440c (DSS+GM-CSF+mAb 440c) reversed the effect of GM-CSF. (C) DSS-induced inflammation leads to shortening of the colon. GM-CSF treatment (DSS+GM-CSF) significantly attenuated colon shortening. However, mAb 440c treatment (DSS+GM-CSF+mAb 440c) reversed the therapeutic effect of GM-CSF and led to a significant decrease in colon length. (D) Distal colon sections stained with H&E show normal crypt morphology in control mice. (E) DSS colitis led to inflammatory infiltrates and crypt damage. (F) GM-CSF treatment (DSS+GM-CSF) reduced infiltration of inflammatory cells and preserved crypt architecture. (G) Administration of mAb 440c (DSS+GM-CSF+mAb 440c) led to severe colitis with inflammatory infiltrate and crypt damage. All histological sections were presented in 20× magnification. Data are expressed as the mean ± SEM. Control vs. DSS: ** P < 0.01. DSS vs. DSS+GM-CSF: ## P < 0.01. DSS+GM-CSF vs. DSS+GM-CSF+mAb 440c: † P < 0.05; †† P < 0.01.
Segments of distal colon were stained with H&E for histopathological evaluation. Control mice had normal crypt morphology (Fig 1D), whereas comparable segments from DSS-treated mice had overt colitis with ulceration and inflammatory infiltrates (Fig 1E). GM-CSF-treated mice (DSS+GM-CSF) had significantly reduced inflammatory infiltrates, ulceration, and crypt damage (Fig 1F). Addition of mAb 440c (DSS+GM-CSF+mAb 440c) reversed the therapeutic effects observed with GM-CSF (Fig1G). Formal histopathologic scoring was performed utilizing a validated scoring system.27 DSS treatment led to a significant increase in histopathologic score (control, 0 ± 0 vs. DSS, 6.6 ± 2.2, P < 0.01). GM-CSF treatment significantly reduced histologic disease activity (DSS, 6.6 ± 2.2 vs. DSS+GM-CSF, 1.4 ± 0.6, P < 0.01). Finally, addition of mAb 440c blocked the therapeutic effect of GM-CSF (DSS+GM-CSF, 1.4 ± 0.6 vs. DSS+GM-CSF+440c, 5.0 ± 2.2, P < 0.01). These results demonstrate that GM-CSF significantly reduces the severity of DSS colitis in a manner functionally involving the pDC.
GM-CSF reduces the pro-inflammatory gene expression in DSS colitis
In an attempt to identify gene biomarkers for disease activity, replicate microarray analyses were performed to identify genes who's expression is significantly changed following the induction of acute DSS colitis. RNA was isolated from the colons of DSS-treated mice on day 7. Statistical analysis of microarrays identified 368 genes significantly upregulated in DSS colitis. Table 1 contains a subset of these genes known to be potentially important in the setting of IBD. Treatment with GM-CSF (DSS+GM-CSF) also was associated with decreases in most of these genes (data not shown). We validated a number of these genes known to be important in IBD by real time RT-PCR.26 GM-CSF treatment (DSS+GM-CSF vs. DSS) significantly blunted DSS-induced increases in pro-inflammatory cytokines: TNF-α (6.5 vs. 9.8 fold P < 0.05); IL1-β (3.5 vs. 13.9 fold, P < 0.05); IL1-α (8.4 vs. 29.9 fold, P < 0.05). Administration of mAb 440c (DSS+GM-CSF+mAb 440c) nullified the therapeutic effect of GM-CSF and induced a proinflammatory cytokine profile similar to that observed in animals treated with DSS alone (Fig 2A).
Table 1. Identification of biomarkers in DSS-treated mice by microarray.
Gene expression induced by acute DSS colitis
| GENE ACCESION | GENE DESCRIPTION | DSS vs. Con |
|---|---|---|
| NM_013693* | Tumor necrosis factor alpha (TNFα) | 5.0 |
| NM_008361* | Interleukin 1 beta (Il1β) | 8.7 |
| NM_008324 | Indole 2,3-dioxygenase (IDO) | 33.5 |
| M74294 | IL-1rn antagonist protein | 3.0 |
| NM_019440 | Interferon-g induced GTPase | 15.2 |
| NM_018851 | IFN-gamma induced (Mg11) | 7.1 |
| NM_008326 | Interferon inducible protein 1 (Ifi1) | 6.7 |
| NM_011610 | Tumor necrosis factor receptor superfamily, (Tnfrsf1b) | 4.1 |
| NM_011905 | Toll-like receptor 2 (Tlr2) | 2.2 |
| NM_015783 | Interferon-stimulated protein (15 kDa) | 12.4 |
| NM_011333 | Small inducible cytokine A2 (Scya2) | 8.6 |
| M31585 | Intercellular adhesion molecule 1 | 4.1 |
| NM_008176 | GRO1 oncogene (Gro1), | 18.3 |
Verified by Real Time RT-PCR
The significance analysis of microarray (SAM) procedure was used to identify genes significantly upregulated by DSS. SAM selected a total of 368 genes as differentially expressed between control and DSS group with a false discovery rate 2.39%. The average fold differences (five array experiments) of these genes are listed in the table. Only a few important genes are listed in this table. The complete set of genes is available as online supplemental material.
Figure 2.

GM-CSF treatment prevents the pro-inflammatory response induced by DSS colitis. (A) Following real time RT-PCR analysis of total RNA from proximal colon, GM-CSF treatment (DSS+GM-CSF) led to a significant inhibition of DSS-induced increases in expression of the pro-inflammatory cytokines TNF-α, IL1-β and IL1-α. However, administration of mAb 440c (DSS+GM-CSF+mAb 440c) nullified the effect of GM-CSF and exhibited a condition similar to DSS alone. (B) Serum cytokine levels were measured using the Bio-Plex protein assay. GM-CSF treatment inhibited DSS-induced increases in the levels TNF-α, IL-β and IL1-α. However, mAb 440c administration (DSS+GM-CSF+mAb 440c) blocked the anti-inflammatory effects of GM-CSF. Data are expressed as the mean ± SEM. Control vs. DSS: * P < 0.05; ** P < 0.01. DSS vs. DSS+GM-CSF: # P < 0.05; ## P < 0.01. DSS+GM-CSF vs. DSS+GM-CSF+mAb 440c: † P < 0.05.
To confirm the GM-CSF effects on proinflammatory cytokines at the protein level, serum samples were quantified using Bio-Plex assays (Fig 2B). As expected, compared to the cytokine levels observed in animals treated with DSS alone, GM-CSF treatment (DSS+GM-CSF) significantly attenuated the increases in several cytokines: TNF-α (168.7 vs. 258.1 pg/ml); IL1-β (9.7 vs. 35.9 pg/ml, P < 0.05) and IL1-α (28.8 vs. 113.4 pg/ml, P <0.01). In contrast, administration of mAb 440c (DSS+GM-CSF+mAb 440c) reversed the GM-CSF-mediated reductions in these proinflammatory cytokines. To exclude the non-specific function of Ig via Fc receptors we previously examined control groups containing isotype control antibodies in this model. These results were similar to those reported here (data not shown).
GM-CSF expands the interferon producing pDC (440c+)
The pDC is the major the source of systemic type 1 interferon secretion following TLR-9 stimulation.15,23 Since pDC production of type 1 IFN may contribute to the therapeutic effects of GM-CSF on DSS colitis, we postulated that GM-CSF may act by expanding the pDC cell population. Balb/c mice were treated with GM-CSF (5 μg/d, i.p.) for 5 days and tissue sections were subjected to immunohistochemical staining using mAb 440c (Fig 3D). GM-CSF treatment, significantly increased the percentage of 440c+ pDC in the spleen compared to untreated animals (4.3 vs. 0.5 %) (Fig 3B). Addition of mAb 440c (GM-CSF+mAb 440c) partially attenuated the effect of GM-CSF on the number of 440c+ splenic cells (Fig 3C). Animals treated with mAb 440c alone did not exhibit significant changes in 440c+ cells compared with controls (Fig 3D). These data show that GM-CSF treatment significantly expands splenic 440c+ cells (Fig 3E). Since assay of type 1 IFN in the serum is not particularly sensitive, we measured expression of type 1 IFN at the mRNA level in the spleen by real time RT-PCR.15 Significant increases in IFNα mRNA expression were observed following treatment with GM-CSF (Fig 3F). This effect was blocked following mAb 440c administration. Increases in IFN-β and indoleamine dioxygenase (IDO) were also observed after treatment with GM-CSF, however the differences did not reach statistical significance. Type 1 IFN and IDO cytokine levels were also increased in the intestine following GM-CSF treatment (data not shown).
Figure 3.

GM-CSF induces expansion of 440c+ pDCs in the spleen. Immunohistochemical staining with mAb 440c was used to identify 440c+ pDCs in frozen splenic sections (representative stained cell shown by arrow). (A) Few 440c+ pDCs in a control spleen. (B) GM-CSF treatment led to an increased number of 440c+ pDCs. (C) Administration of mAb 440c (GM-CSF+mAb 440c) reduced the GM-CSF-mediated increase in number of 440c+ pDCs. (D) No changes were observed with mAb 440c treatment alone. (E) Results were further confirmed by counting absolute numbers of splenocytes, including 440c stained cells, over multiple hpfs. Changes in the percentage of 440c+ pDCs were highly significant (F) Real time RT-PCR analysis of total RNA from spleen demonstrated a significant increase in expression of IFN-α mRNA following GM-CSF treatment. However, the GM-CSF-mediated increase was significantly blocked with mAb 440c administration (GM-CSF+mAb 440c). Although similar effects of GM-CSF treatment were also observed for the expression of IFN-β and IDO these changes were not statistically significant. All histological sections were presented in 20× magnification. Data are expressed as the mean ± SEM. Control vs. GM-CSF: * P < 0.05; ** P < 0.01. GM-CSF vs. GM-CSF+mAb 440c: # P < 0.05; ## P < 0.01. GM-CSF+mAb 440c vs. mAb 440c: † P < 0.05.
GM-CSF ameliorates DSS colitis in T and B-cell deficient mice
To explore whether GM-CSF effects are independent of the adaptive arm of the immune system, we examined the DSS model in T and B-cell deficient mice. Mice homozygous for a deletion in the recombination activating gene (RAG1−/−) lack mature T and B cells. H&E staining of colonic sections (Fig 4A) shows crypt damage and mucosal ulceration with DSS colitis, similar to that observed in wild-type mice. Treatment with GM-CSF (DSS+GM-CSF) resulted in decreased inflammation and maintenance of normal crypt architecture. Formal histopathologic scoring was performed by a pathologist blinded to the treatment groups. GM-CSF treatment resulted in a significant reduction in histopathologic disease activity: (control, 0 ± 0 vs. DSS, 16.1 ± 2.2; P < 0.01) and (DSS, 16.1 ± 2.2 vs. DSS+GM-CSF, 10.4 ± 2.7; P < 0.01). Clinical parameters (weight loss and rectal bleeding) also improved following GM-CSF treatment (data not shown).
Figure 4.

GM-CSF ameliorates DSS colitis in RAG 1 −/− mice. Acute DSS colitis was induced in T and B cell deficient RAG1−/− mice following administration of 4% DSS in drinking water for 7d. (A) H&E staining of distal colon sections showed crypt damage, inflammatory infiltrate and mucosal injury in DSS colitis. However, GM-CSF treatment (DSS+GM-CSF) restored crypt architecture similar to controls. (B) After DSS treatment, real time RT-PCR analysis of total RNA from proximal colon showed increases in expression of the pro-inflammatory cytokines TNF-α, IL1-β and IL1-α. However, GM-CSF treatment (DSS+GM-CSF) reduced expression of these cytokines. All histological sections were presented in 20× magnification. Data are expressed as the mean ± SEM. Control vs. DSS: * P < 0.05. DSS vs. DSS+GM-CSF: # P < 0.05; ## P < 0.01.
Real time RT-PCR analyses demonstrated significant DSS-induced increases in the expression of proinflammatory cytokines (Fig 4B). Compared to DSS treated animals, GM-CSF treatment (DSS+GM-CSF) attenuated the increase in these cytokines: TNF-α (2.4 vs. 6.07 fold); IL1-β (3.8 vs. 12.04 fold, P < 0.05) IL1-α (5.2 vs. 19.7 fold, P < 0.01). These results show that the therapeutic mechanism of action of GM-CSF in experimental colitis does not require the presence of mature T and B cells.
IFN-β treatment mimics the effects of GM-CSF in DSS colitis
Our data supports the hypothesis that GM-CSF works by effects on the 440c+ pDC. Since this cell is the major source of systemic type 1 IFN secretion following TLR9 stimulation, we next conducted an experiment to determine if administration of type 1 IFN would mimic the disease ameliorative effects of GM-CSF. Administration of IFN-β to animals treated with DSS resulted in a significant reduction in histopathological colitis (Fig 5A). Real time RT-PCR analyses from colonic RNA demonstrated that mice treated with IFN-β (DSS+IFN-β) had decreased expression of pro-inflammatory cytokines compared to DSS alone: TNF-α (4.4 vs. 9.8 fold, P < 0.05), IL1β- (9.3 vs. 13.9 fold) and IL1-α (1.6 vs. 29.9 fold, P < 0.05) (Fig 5B). Clinical improvement was observed with IFN-β treatment (data not shown). These results suggest that a critical role of the 440c+ pDC in the therapeutic response to GM-CSF may be local production of type 1 IFN.
Figure 5.
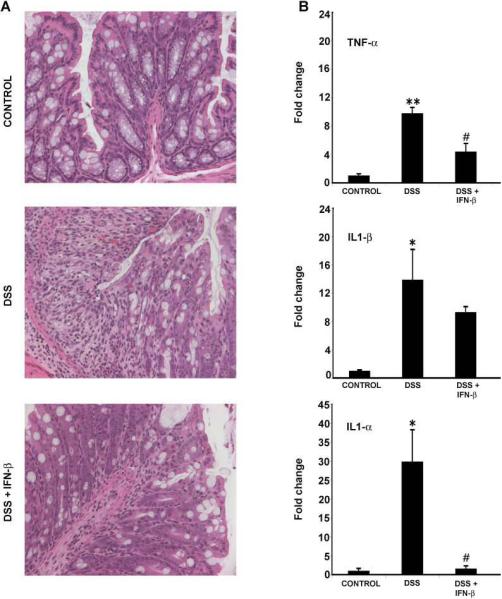
IFN-β treatment mimics the effect of GM-CSF in DSS colitis. Representative H&E staining of colonic sections demonstrate that treatment with IFN-β (100,000 U/d, i.p.) mimics the GM-CSF-mediated amelioration of DSS colitis. (A) Normal crypt architecture in control mice. DSS colitis resulted in crypt damage, infiltration of inflammatory cells and mucosal injury. However, IFN-β treatment (DSS+IFN-β) led to restoration of crypt architecture, an effect similar to that observed with GM-CSF treatment. (B) Following real time RT-PCR analysis, IFN-β treatment significantly inhibited DSS-induced increases in expression of the proinflammatory cytokines TNF-α, IL1-β and IL1-α. Again, these effects of IFN-β treatment mimicked those of GM-CSF. All histological sections were presented in 20× magnification. Data are expressed as the mean ± SEM. Control vs. DSS: * P < 0.05; ** P < 0.01. DSS vs. DSS+IFN-β: # P < 0.05.
GM-CSF treatment augments TLR-9 mediated type 1 IFN production
Studies have demonstrated that immunostimulatory DNA, (CpG DNA motifs) induce type 1 IFN production through TLR-9 stimulation in pDCs.15,17& 30 To explore whether GM-CSF has significant effects on the functional response to CpG DNA, we examined type 1 IFN production in the spleen and intestine after systemic stimulation with CpG. Balb/c mice were treated with GM-CSF for five days. 18h before sacrifice, 200 μg of CpG ODN 1018 was injected subcutaneously. Real time RT-PCR was performed to measure mRNA expression of type 1 IFNs (α and β), IDO and IL-10 in the spleen and small intestine of CpG and GM-CSF (CpG+GM-CSF) treated mice.
We observed that CpG treatment alone led to an increase of IFN-α (91.7 fold), IFN-β (33.9 fold), IDO (38.6 fold) and IL10 (18.2 fold) expression in the spleen (fig 6A). GM-CSF treatment significantly augmented expression of these cytokines following CpG treatment: IFN-α (91.7 vs. 633.3 fold); IFN-β (33.9 vs. 158.8 fold); IDO (38.6 vs. 171.6 fold); IL-10 (18.2 vs. 98.5 fold) (Fig 6A). Similar results were also observed in the small intestine (Fig 6B). These results suggest that GM-CSF augments the production of type 1 IFNs, and the anti-inflammatory molecules IDO and IL-10 following systemic administration of a TLR-9 agonist.
Figure 6.

GM-CSF treatment augments CpG-mediated Type 1 IFN production. Balb/c mice were treated with the TLR-9 agonist, CpG DNA (200 μg s.c.). After 18 h, mice were sacrificed and total RNA from spleen and intestine were subjected to real-time RT-PCR analysis. Panels (A) & (B) demonstrate the expression of proinflammatory cytokines in spleen and small intestine, respectively. The expression of IFN-α, IFN-β, IDO and IL-10 increased following CpG DNA treatment. However, mice treated with daily GM-CSF (5 μg, i.p.) for 5 d prior to sacrifice had further elevation in expression of these cytokines. Data are expressed as the mean fold change relative to control ± SEM.
Discussion
Although Crohn's disease (CD) was first described over 70 years ago, its pathogenesis remains incompletely understood. Observations and reports in human disease led us to hypothesize that defects of innate immunity may be central to the development of CD.4,31 We also hypothesized that CSFs would modulate disease activity through their effects on the number and function of innate immune cells. This led to phase I and II trials of GM-CSF in patients with moderately-to-severely active CD. Both trials showed clear evidence that treatment with GM-CSF was associated with a reduction of disease activity.10 Further data in human disease as well as animal models of colitis have supported the role of innate immune deficiency in the pathogenesis of CD.15,19& 32-34 Accordingly, we examined the effectiveness of GM-CSF administration in the mouse model of acute DSS colitis. GM-CSF has broad effects on cells involved with innate immunity, including macrophages, dendritic cells, neutrophils, and intestinal epithelium. GM-CSF regulates proliferation and differentiation of hematopoietic cells. GM-CSF is released by various cell types including T lymphocytes, macrophages, fibroblasts and endothelial cells. GM-CSF activates and enhances the production and survival of neutrophils, eosinophils, and macrophages, which play a key role in the innate immune response.25 A study by Fukuzawa et al,35 using single cell RT-PCR has demonstrated the local production of GM-CSF by the Paneth cell of the intestine. These Paneth cell are located at the bottom of the intestinal crypts and as well as in the epithelial cell lines. GM-CSF receptor β chain is present in both Paneth and non-Paneth cells of the small intestinal epithelium. Thus, GM-CSF secreted from Paneth cells may act locally by autocrine and paracrine mechanisms. GM-CSF in Paneth cells might also have a role in mucosal immunity of the small intestine by enhancing expression of costimulatory molecules, not only in typical antigen presenting cells, but also in epithelial cells of intestine crypts under certain pathologic conditions. GM-CSF regulates innate immune clearance of other mucosal surfaces including the lung.
To study the pathophysiology of Crohn's disease, unfortunately there is no animal model that completely recapitulates its cause and manifestations. The best animal model for investigating the mechanism of action for a therapeutic agent, is the model that recapitulates the therapeutic response. We chose DSS colitis because it is a model of disrupted epithelial barrier function and results in increased exposure of lamina propria innate immune elements to luminal microbes. One of the key mechanisms of DSS induced colitis is thought be a direct toxic effect on mucosal epithelia cells, but the exact mechanisms are still unknown. As DSS is considered to produce toxic effect on mucosal epithelia cells, the dysfunction of the mucosal barrier may be an initial event leading to mucosal inflammation in this model.36 This may be an excellent model for CD in which innate immune dysfunction may lead to impaired barrier function and microbial clearance. This mouse model has potential applicability to human disease and has been recommended for preclinical testing of new therapeutic methods for IBD.37, 38 To identify disease markers, microarray analysis was performed on colon samples from DSS –treated mice. Microarrays are capable of simultaneously measuring expression of thousands of genes in specimens from affected and normal and have the potential to provide information about disease pathogenesis not previously possible. In the present study, we have identified important proinflammatory cytokines and markers, such as TNF-α, which has been targeted by therapeutic agents in Crohn's disease.3 The present study demonstrates that GM-CSF leads to reduced severity of acute DSS colitis by multiple disease parameters. GM-CSF treatment was also associated with a reduction in the expression of proinflammatory cytokines. Previous work demonstrated that animals pretreated with bacteria-derived CpG motifs, are protected from development of DSS colitis.15-17& 39 This effect was shown to be mediated by type 1 IFN production through TLR-9 signaling, most likely by pDCs.15 Therefore, we hypothesized that the effect of GM-CSF in DSS colitis might also be mediated by effects on the pDC population. To test our hypothesis, we used a recently described mAb, 440c. This mAb 440c selectively recognizes murine pDCs and functionally blocks type 1 IFN production in response to TLR-9 ligation by CpG. Treatment with mAb 440c abolished the therapeutic effects of GM-CSF in DSS colitis, on clinical, histological parameters, and at the level of pro-inflammatory cytokine expression. This suggested that local type 1 IFN production by 440c+ pDCs may be central to the anti-inflammatory effect of GM-CSF in acute DSS colitis.
Previous reports had indicated that GM-CSF expands DC populations.40 Therefore, we examined the effect of GM-CSF on the numbers of splenic 440c+ pDCs by immunohistology. GM-CSF treatment significantly increased the numbers of 440c+ pDCs in the spleen compared with controls. GM-CSF treatment also increased IFN-α mRNA expression in the spleen. This effect was reversed by the addition of mAb 440c. To determine if the effects of GM-CSF are dependent on adaptive immune mechanisms, we examined RAG1−/− mice, which lack mature T and B cells. RAG1−/− mice developed DSS colitis similar to wild type mice. This data highlights the fact that the effects of GM-CSF in DSS colitis do not require mature T and B cells. Further studies are planned to examine the effect of GM-CSF in the adoptive transfer model (CD45RB high) transfer model to delineate the role of T cells. Our observation in RAG1−/− mice suggests that the mechanism of action of GM-CSF may be modulation of innate immune function as opposed to an alternate mechanism, such as increasing regulatory T cell populations. These findings are reminiscent of the data from Katakura et al, 15 in which the inhibition of DSS colitis by pretreatment with CpG was also observed in RAG1−/− mice. We also found that the therapeutic effects of GM-CSF (DSS+GM-CSF) in DSS colitis were mimicked by administration of IFN-β (DSS+IFN-β). We, therefore, investigated the role of GM-CSF in the induction of type 1 IFN production. Following systemic TLR-9 stimulation by CpG, there was increased expression of type 1 IFNs, IDO, and IL-10 mRNA in the spleen and ileum. This induction was augmented by pretreatment with GM-CSF. Mice treated with GM-CSF alone exhibited a slight increase in type 1 IFNs in ileum but not in spleen. This CpG-mediated induction is blocked with administration of mAb 440c. These results are consistent with previous studies demonstrating that mAb 440c blocks CpG stimulated type 1 IFN production.23, 24 These data support the assertion that it may be type 1 IFN production by pDCs that mediates the protective effects of GM-CSF in DSS colitis.
In summary, GM-CSF is effective in treating intestinal inflammation in human CD. In mouse models, it is also effective in preventing the development of colitis. Studies have reported that type 1 IFNs have multiple effects on the immune system including up-regulation of MHC class I and II, enhanced co-stimulatory expression, B cell expansion and differentiation, modulation of immunoglobulin production and synergy with IL-12, to enhance IFN γ production and augmentation of NK and CTL responses. Thus, type 1 IFN contributes to both innate and adaptive immunity.41,42 Clinical trials have been conducted with type 1 IFN for ulcerative colitis and Crohn's disease. An open study with IFN-α obtained a remission rate of 82% in patients with refractory ulcerative colitis after six months of treatment.43 Furthermore, a study in patients with Crohn's disease and concomitant herpes virus infection has shown that IFN-α can induce an antiviral reaction that was associated with reduced intestinal inflammation.44 A clinical study by Nikolaus et al,45 demonstrated that treatment with IFN-β in ulcerative colitis patients had achieved clinical response and remission in 50% and 30% of patients respectively. These clinical studies support the role of type 1 IFNs in the regulation of mucosal immunity.
Acknowledgements
We thank the Morphology core, Division of Gastroenterology for immunostaining protocols. We thank Elyse Dubno Grusky, Adelphi Inc for manuscript reviewing.
Supported by NIH grants (DK 60106, AI 069390, T 32 HD007507-09) and Washington University Digestive Disease Research Core (NIH P30 DK52574).
Abbreviations
- CD
Crohn's Disease
- pDC
plasmacytoid dendritic cell
- CpG
cytosine-phosphate-guanosine
- IBD
inflammatory bowel disease
- DSS
dextran sulphate sodium
- RAG
recombination activating gene
- IDO
indoleamine dioxygenase
- SAM
Significance Analysis of Microarrays
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hanauer SB. Inflammatory bowel disease: epidemiology, pathogenesis, and therapeutic opportunities. Inflamm Bowel Dis. 2006;12(Suppl 1):S3–9. doi: 10.1097/01.mib.0000195385.19268.68. [DOI] [PubMed] [Google Scholar]
- 2.Ferretti M, Casini-Raggi V, Pizarro TT, et al. Neutralization of endogenous IL-1 receptor antagonist exacerbates and prolongs inflammation in rabbit immune colitis. J Clin Invest. 1994;94:449–53. doi: 10.1172/JCI117345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stack WA, Mann SD, Roy AJ, et al. Randomised controlled trial of CDP571 antibody to tumour necrosis factor-alpha in Crohn's disease. Lancet. 1997;349:521–4. doi: 10.1016/s0140-6736(97)80083-9. [DOI] [PubMed] [Google Scholar]
- 4.Dieckgraefe BK, Korzenik JR, Husain A, et al. Association of glycogen storage disease 1b and Crohn disease: results of a North American survey. Eur J Pediatr. 2002;161(Suppl 1):S88–92. doi: 10.1007/s00431-002-1011-z. [DOI] [PubMed] [Google Scholar]
- 5.Takeda K, Clausen BE, Kaisho T, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- 6.Jantchou P, Monnet E, Carbonnel F. [Environmental risk factors in Crohn's disease and ulcerative colitis (excluding tobacco and appendicectomy)]. Gastroenterol Clin Biol. 2006;30:859–67. doi: 10.1016/s0399-8320(06)73333-4. [DOI] [PubMed] [Google Scholar]
- 7.Figueroa C, Peralta A, Herrera L, et al. NOD2/CARD15 and Toll-like 4 receptor gene polymorphism in Chilean patients with inflammatory bowel disease. Eur Cytokine Netw. 2006;17:125–30. [PubMed] [Google Scholar]
- 8.Franchimont D, Vermeire S, El Housni H. Deficient host-bacteria interactions in inflammatory bowel disease? The toll-like receptor (TLR)-4 Asp299gly polymorphism is associated with Crohn's disease and ulcerative colitis. Gut. 2004;53:987–92. doi: 10.1136/gut.2003.030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Heel DA, Ghosh S, Hunt KA, et al. Synergy between TLR9 and NOD2 innate immune responses is lost in genetic Crohn's disease. Gut. 2005;54:1553–7. doi: 10.1136/gut.2005.065888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korzenik JR, Dieckgraefe BK, Valentine JF, et al. Sargramostim for active Crohn's disease. N Engl J Med. 2005;352:2193–201. doi: 10.1056/NEJMoa041109. [DOI] [PubMed] [Google Scholar]
- 11.Ruef C, Coleman DL. Granulocyte-macrophage colony-stimulating factor: pleiotropic cytokine with potential clinical usefulness. Rev Infect Dis. 1990;12:41–62. doi: 10.1093/clinids/12.1.41. [DOI] [PubMed] [Google Scholar]
- 12.Daro E, Pulendran B, Brasel K, et al. Polyethylene glycol-modified GM-CSF expands CD11b(high)CD11c(high) but notCD11b(low)CD11c(high) murine dendritic cells in vivo: a comparative analysis with Flt3 ligand. J Immunol. 2000;165:49–58. doi: 10.4049/jimmunol.165.1.49. [DOI] [PubMed] [Google Scholar]
- 13.Daro E, Butz E, Smith J, et al. Comparison of the functional properties of murine dendritic cells generated in vivo with Flt3 ligand, GM-CSF and Flt3 ligand plus GM-SCF. Cytokine. 2002;17:119–30. doi: 10.1006/cyto.2001.0995. [DOI] [PubMed] [Google Scholar]
- 14.Facchetti F, Vermi W, Mason D, et al. The plasmacytoid monocyte/interferon producing cells. Virchows Arch. 2003;443:703–17. doi: 10.1007/s00428-003-0918-8. [DOI] [PubMed] [Google Scholar]
- 15.Katakura K, Lee J, Rachmilewitz D, et al. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J Clin Invest. 2005;115:695–702. doi: 10.1172/JCI22996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rachmilewitz D, Katakura K, Karmeli F, et al. Toll-like receptor 9 signaling mediates the anti-inflammatory effects of probiotics in murine experimental colitis. Gastroenterology. 2004;126:520–8. doi: 10.1053/j.gastro.2003.11.019. [DOI] [PubMed] [Google Scholar]
- 17.Rachmilewitz D, Karmeli F, Takabayashi K, et al. Immunostimulatory DNA ameliorates experimental and spontaneous murine colitis. Gastroenterology. 2002;122:1428–41. doi: 10.1053/gast.2002.32994. [DOI] [PubMed] [Google Scholar]
- 18.Okayasu I, Hatakeyama S, Yamada M, et al. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- 19.Dieleman LA, Ridwan BU, Tennyson GS, et al. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology. 1994;107:1643–52. doi: 10.1016/0016-5085(94)90803-6. [DOI] [PubMed] [Google Scholar]
- 20.Dieleman LA, Palmen MJ, Akol H, et al. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin Exp Immunol. 1998;114:385–91. doi: 10.1046/j.1365-2249.1998.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Araki A, Kanai T, Ishikura T, et al. MyD88-deficient mice develop severe intestinal inflammation in dextran sodium sulfate colitis. J Gastroenterol. 2005;40:16–23. doi: 10.1007/s00535-004-1492-9. [DOI] [PubMed] [Google Scholar]
- 22.Cooper HS, Murthy SN, Shah RS, et al. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–49. [PubMed] [Google Scholar]
- 23.Blasius A, Vermi W, Krug A, et al. A cell-surface molecule selectively expressed on murine natural interferon-producing cells that blocks secretion of interferon-alpha. Blood. 2004;103:4201–6. doi: 10.1182/blood-2003-09-3108. [DOI] [PubMed] [Google Scholar]
- 24.Blasius AL, Cella M, Maldonado J, et al. Siglec-H is an IPC-specific receptor that modulates type I IFN secretion through DAP12. Blood. 2006;107:2474–6. doi: 10.1182/blood-2005-09-3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sainathan SK, Tu L, Bishnupuri KS, et al. PEGylated murine Granulocyte-macrophage colony-stimulating factor: production, purification, and characterization. Protein Expr Purif. 2005;44:94–103. doi: 10.1016/j.pep.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 26.Melgar S, Karlsson A, Michaelsson E, et al. Acute colitis induced by dextran sulfate sodium progresses to chronicity in C57BL/6 but not in BALB/c mice: correlation between symptoms and inflammation. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1328–38. doi: 10.1152/ajpgi.00467.2004. [DOI] [PubMed] [Google Scholar]
- 27.Egger B, Procaccino F, Lakshmanan J, et al. Mice lacking transforming growth factor alpha have an increased susceptibility to dextran sulfate-induced colitis. Gastroenterology. 1997;113:825–32. doi: 10.1016/s0016-5085(97)70177-x. [DOI] [PubMed] [Google Scholar]
- 28.Tusher VG, Tibshirani R, Chu G, et al. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee J, Rachmilewitz D, Raz E. Homeostatic effects of TLR9 signaling in experimental colitis. Ann N Y Acad Sci. 2006;1072:351–5. doi: 10.1196/annals.1326.022. [DOI] [PubMed] [Google Scholar]
- 30.Obermeier F, Dunger N, Strauch UG, et al. Contrasting activity of cytosinguanosin dinucleotide oligonucleotides in mice with experimental colitis. Clin Exp Immunol. 2003;134:217–24. doi: 10.1046/j.1365-2249.2003.02288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Korzenik JR, Dieckgraefe BK. Is Crohn's disease an immunodeficiency? A hypothesis suggesting possible early events in the pathogenesis of Crohn's disease. Dig Dis Sci. 2000;45:1121–9. doi: 10.1023/a:1005541700805. [DOI] [PubMed] [Google Scholar]
- 32.Folwaczny C, Glas J, Torok HP. Crohn's disease: an immunodeficiency? Eur J Gastroenterol Hepatol. 2003;15:621–6. doi: 10.1097/00042737-200306000-00007. [DOI] [PubMed] [Google Scholar]
- 33.Hisamatsu T, Suzuki M, Reinecker HC, et al. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology. 2003;124:993–1000. doi: 10.1053/gast.2003.50153. [DOI] [PubMed] [Google Scholar]
- 34.Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603–6. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 35.Fukuzawa H, Sawada M, Kayahara T, et al. Identification of GM-CSF in Paneth cells using single-cell RT-PCR. Biochem Biophys Res Commun. 2003;312:897–902. doi: 10.1016/j.bbrc.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 36.Kitajima S, Takuma S, Morimoto M. Changes in colonic mucosal permeability in mouse colitis induced with dextran sulfate sodium. Exp. Anim. 1999;48:137–143. doi: 10.1538/expanim.48.137. [DOI] [PubMed] [Google Scholar]
- 37.Ohkawara T, Nishihira J, Takeda H, et al. Amelioration of dextran sulfate sodium-induced colitis by anti-macrophage migration inhibitory factor antibody in mice. Gastroenterology. 2002;123:256–70. doi: 10.1053/gast.2002.34236. [DOI] [PubMed] [Google Scholar]
- 38.Tessner TG, Cohn SM, Schloemann S, et al. Prostaglandins prevent decreased epithelial cell proliferation associated with dextran sodium sulfate injury in mice. Gastroenterology. 1998;115:874–82. doi: 10.1016/s0016-5085(98)70259-8. [DOI] [PubMed] [Google Scholar]
- 39.Obermeier F, Dunger N, Strauch UG, et al. CpG motifs of bacterial DNA essentially contribute to the perpetuation of chronic intestinal inflammation. Gastroenterology. 2005;129:913–27. doi: 10.1053/j.gastro.2005.06.061. [DOI] [PubMed] [Google Scholar]
- 40.Maraskovsky E, Brasel K, Teepe M, et al. Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand-treated mice: multiple dendritic cell subpopulations identified. J Exp Med. 1996;184:1953–62. doi: 10.1084/jem.184.5.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gibson SJ, Lindh JM, Riter TR, et al. Plasmacytoid dendritic cells produce cytokines and mature in response to the TLR7 agonists, imiquimod and resiquimod. Cell Immunol. 2002;218:74–86. doi: 10.1016/s0008-8749(02)00517-8. [DOI] [PubMed] [Google Scholar]
- 42.Bekeredjian-Ding IB, Wagner M, Hornung V, et al. Plasmacytoid dendritic cells control TLR7 sensitivity of naive B cells via type I IFN. J Immunol. 2005;174:4043–50. doi: 10.4049/jimmunol.174.7.4043. [DOI] [PubMed] [Google Scholar]
- 43.Sumer N, Palabiyikoglu M. Induction of remission by interferon-alpha in patients with chronic active ulcerative colitis. Eur J Gastroenterol Hepatol. 1995;7:597–602. [PubMed] [Google Scholar]
- 44.Ruther U, Nunnensiek C, Muller HA, et al. Interferon alpha (IFN alpha 2a) therapy for herpes virus-associated inflammatory bowel disease (ulcerative colitis and Crohn's disease). Hepatogastroenterology. 1998;45:691–9. [PubMed] [Google Scholar]
- 45.Nikolaus S, Rutgeerts P, Fedorak R, et al. Interferon beta-1a in ulcerative colitis: a placebo controlled, randomised, dose escalating study. Gut. 2003;52:1286–90. doi: 10.1136/gut.52.9.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]