

Abstract
Osteosa rcoma is an aggressive malignant neoplasm that exhibits osteoblastic differentiation and produces malignant osteoid. The aim of this study was to find feature genes associated with osteosarcoma and correlative gene functions which can distinguish cancer tissues from non-tumor tissues. Gene expression profile GSE14359 was downloaded from Gene Expression Omnibus (GEO) database, including 10 osteosarcoma samples and 2 normal samples. The differentially expressed genes (DEGs) between osteosarcoma and normal specimens were identified using limma package of R. DAVID was applied to mine osteosarcoma associated genes and analyze the GO enrichment on gene functions and KEGG pathways. Then, corresponding protein-protein interaction (PPI) network of DEGs was constructed based on the data collected from STRING datasets. Principal component of top10 DEGs and PPI network of top 20 DEGs were further analyzed. Finally, transcription factors were predicted by uploading the two groups of DEGs to TfactS database. A total of 437 genes, including 114 up-regulated genes and 323 down-regulated genes, were filtered as DEGs, of which 46 were associated with osteosarcoma by Disease Module. GO and KEGG pathway enrichment analysis showed that genes mainly affected the process of immune response and the development of skeletal and vascular system. The PPI network analysis elucidated that hemoglobin and histocompatibility proteins and enzymes, which were associated with immune response, were closely associated with osteosarcoma. Transcription factors MYC and SP1 were predicted to be significantly related to osteosarcoma. The discovery of gene functions and transcription factors has the potential to use in clinic for diagnosis of osteosarcoma in future. In addition, it will pave the way to studying mechanism and effective therapies for osteosarcoma.
Keywords: Osteosarcoma, DEGs, PPI network, GO enrichment, transcription factors
Introduction
Osteosarcoma (OS), the most common and primary malignant bone tumor, is a painful health burden and a deadly disorder [1]. It arises around the metaphysis of tubular long bones that exhibits osteoblastic differentiation, and generates immature bone [2,3]. Femur, tibia and humerus are the other most common sites of OS [3]. Pain is the most common early symptom of OS and can even lead to fracture of the affected bone. The frequency of OS is higher in males than in females and slightly higher frequent in Blacks and Hispanics than Caucasians [4]. Osteosarcoma is the most common primary malignant bone tumor in children and young adults [5]. More than 80% of children with osteogenic sarcoma (OS) relapse and 35% to 40% of them die within the first 2 years after diagnosis due to relapse [6].
Although in the past osteosarcoma was a lethal disease, the development of chemotherapy in last 30 years has raised the 5-year survival to 75% [7]. At present, the available standard treatment is complete radical surgery combined with multiagent chemotherapy regimens [8]. Using chemotherapy before surgery gives us the opportunity to save the limb in these patients. So chemotherapy is now accepted as the standard preoperative option. But the type of chemotherapy is yet in controversy with the majority of regimens. Treatment of osteosarcoma remains a challenging issue. In addition, early diagnosis of cancer is an urgent need. The lack of understanding of the molecular mechanism, the research in screening biomarkers at the early stage has been hinderedt. Additionally, the exact etiology of OS is unclear because of the complex molecular mechanism of tumor development as well. It is reported to be associated with a variety of risk factors including age, sex, genetic and familial factors which contribute to the progression of OS [9]. Thus, it is necessary to understand the detailed mechanisms of tumorigenicity and metastasis for early diagnose and novel therapeutic approaches of osteosarcoma [10].
Genetic aberrations have been reported as an important factor that may contribute to osteosarcoma pathogenesis. It was reported that many transcription factors such as Twist, Snail1, Slug and Zeb family induce epithelial to mesenchymal transition by downregulating E-cadherin [11]. It was also found that miR-195 levels in sera from osteosarcoma patients were significantly lower than those in healthy controls [12], and IL-11Rα was highly expressed in osteosarcoma [13]. Therefore all the detected biomarkers associated with osteosarcoma may be useful for screening osteosarcoma and can predict poor prognosis.
Bioinformatics analysis, an effective way to identify interactions between DNAs and proteins in vivo, has become very popular in recent years [14]. In this paper, it was utilized to identify potential target genes and transcription factors, and the gene functions in osteosarcoma were analyzed to understand the potential biological process of osteosarcoma progress, which has the potential to use in clinic for treatment of osteosarcoma in future. However, more investigations are necessary for better understanding of the roles of MYC and SP1 in osteosarcoma. It may provide insight into tumor formation and malignant progression, as well as provide a basis for innovative therapeutic approaches and diagnostic markers for osteosarcoma.
Materials and methods
Microarray data
The gene expression profile GSE14359 was extracted from GEO (Gene Expression Omnibus) database including 10 Osteosarcoma tissue samples and 2 normal tissue samples. Platform information was GPL96.
Data preprocessing
The probe-level data in CEL files were converted into expression value matrix by eReadAffy function [15] in R Affy package and performed background correction and quartile data normalization by the robust multiarray average (RMA) [16] algorithm with defaulted parameters. Data distribution was presented as box graph. The R/Bioconductor package and chip annotation platform were used to generate gene accession number, and the probes without annotation were filtered.
DEGs screening and hierarchical cluster analysis
DEGs between osteosarcoma and normal tissues were identified by t-test based on samr package [17] in R language. The genes changed for more than 2 times in gene expression were selected and q-value < 0.1 was used as the cut-off criterion. In order to ensure that the screened DEGs can be good characterizations of osteosarcoma and normal tissues, hierarchy cluster analysis was performed and cluster dendrogram was constructed. In hierarchical cluster analysis, Pearson coefficient was used in sample cluster analysis and Spearman coefficient in gene expression analysis. Cluster dendrogram was constructed to verify the grouping condition of the original data and filter out those unreasonable clusters.
Re-screening of DEGs and function enrichment analysis
Gene expression profile was rebuilt after hierarchical cluster analysis and data filtering. The limma package [12] in R was used to identify DEGs between osteosarcoma samples and normal osteoblasts after data filtering. The adjusted P-value < 0.05 and |logFC| > 2 were used as the cut-off criterion. DAVID [18] was applied to mine osteosarcoma associated genes and analyze the GO enrichment on gene functions and KEGG pathways. In this study, osteosarcoma associated genes from DEGs were mined by Disease Module. The Annotation Module was used to analyze the enrichment of the interested genes in each GO function module or KEGG pathway, and the FDR (false discovery rate) less than 0.05 was used as the cut-off criterion. All DEGs were mapped onto STRING [19] database to construct the protein-protein interaction pairs.
Principal component analysis (PCA) of top10 DEGs
In order to distinguish osteosarcoma tissues from normal osteoblasts, ten significantly up-regulated DEGs were screened for principal component analysis. PCA is a mathematical algorithm [15] which can not only reduce the data dimension but also concentrate the majority of variables. Based on principal component identification, a direction was firstly found, along which the data distributed was named as the maximum to reduce data dimension. Through PCA, we can choose several variables instead of thousands of variables to classify the samples.
PPI network analysis of top20 DEGs
In order to study the interactions among the Top20 DEGs in osteosarcoma group, The Top20 DEGs were mapped onto STRING database to build the protein-protein interaction pairs. PPI pairs with reliability score higher than 0.4 were screened to construct PPI network of top20 DEGs.
Transcription factor prediction
TfactS database [8] collects target genes of transcription factors after tests. After uploading the two groups of DEGs to TfactS database, four indexes: p-value, q-value, E-value and FDR were utilized to indicate transcription factors enrichment. Only when the values of all the four indicators were less than 0.05, target genes of transcription factors could be considered as significant enrichment.
Results
Data preprocessing
The expression profile data were firstly preprocessed then analyzed by Affy package in R language. Total of 13104 genes were screened. Cassette figures before and after data standardization was shown in Figure 1. That all the black lines in the figure are almost on the same straight line reveals a good standardization degree.
Figure 1.
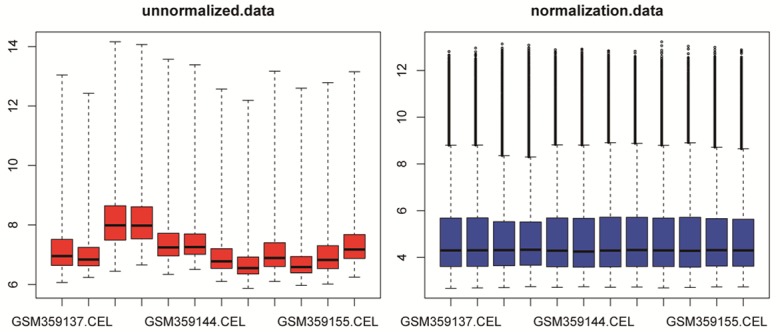
Cassette figures of data distribution. The horizontal axis stands for sample names while the vertical axis represents the expression value. The black line in the cassette is the median of each data group, and the data standardization degree can be judged by its distribution. That all the black lines in the figure are almost on the same straight line reveals a good standardization degree.
Differentially expressed genes (DEGs) screening and hierarchical cluster analysis
Total 1608 genes were selected as DEGs, and 545 up-regulated genes and 1063 down-regulated genes were included. Hierarchy cluster analysis indicated that the 10 osteosarcoma samples distributed in osteosarcoma sample cluster and the 2 normal samples in normal sample cluster (Figure 2). The result revealed that grouping was reasonable and the data can be directly applied to further analysis.
Figure 2.
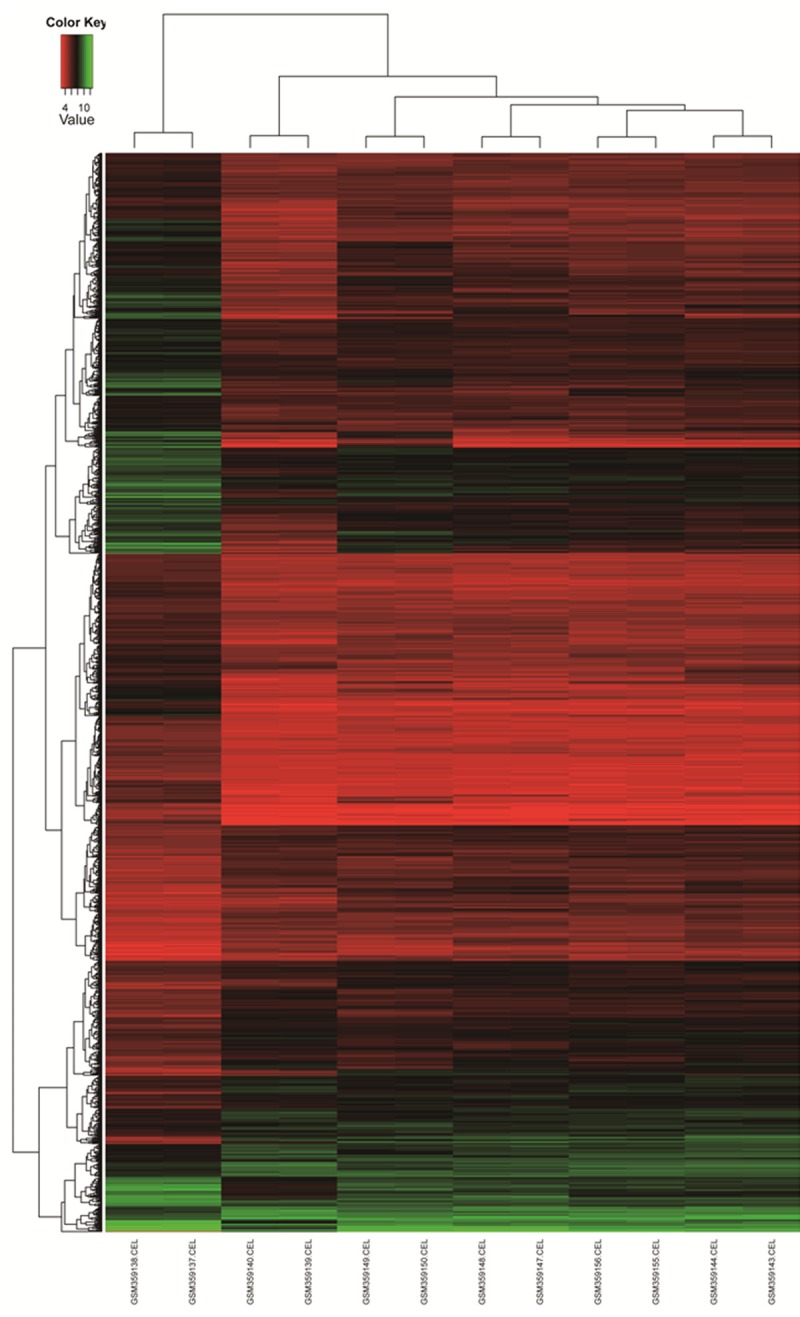
Hierarchical cluster dendrogram of DEGs. The horizontal axis represents sample names. GSM359137 and GSM359138 are normal tissue samples. GSM359139, GSM359140, GSM359143, GSM359144, GSM359147~GSM359150, GSM359155 and GSM359156 are osteosarcoma samples. The left vertical axis shows clusters of DEGs, and the above horizontal axis shows clusters of samples. Red represents up-regulated genes and green represents down-regulated genes.
Re-screening of DEGs and function enrichment analysis
A total of 437 genes were re-screened as DEGs, including 114 up-regulated genes and 323 down-regulated genes. DAVID was used to analyze all the re-screened DEGs. Total 46 osteosarcoma associated genes were mined by Disease Module (Table 1). GO and KEGG pathway enrichment analysis showed that the up-regulated genes mainly enriched in the process of immune response, and the down-regulated genes mainly enriched in the development of skeletal and vascular system (Figure 3) (Tables S1 and S2).
Table 1.
Osteosarcoma associated genes from DEGs
| Cancer | Genes |
|---|---|
| Osteosarcoma | CD36, Fas, MAD2, ngo1, TIMP3, Akr1c1, akr1c3, BIRC5, CTSB, cav1, cav2, Ccl2, CXCL12, C1qa, CDKN1A, CYP1B1, eqfr, ephx1, fn1, GAS1, Gdf15, iqfbp3, iqfbp7, IL6, LGALS3, LEPR, hla-dpa1, HLA-DPB1, HLA-DQB1, HLA-DPB3, hla-drb1, MMP13, Mmp2, Mmp9, PECAM1, RGS2, RECK, RNASE1, rnh1, SPP1, SERPINE1, TDG,hla-dga1, PRKDC, sod2, Thbs1 |
Figure 3.
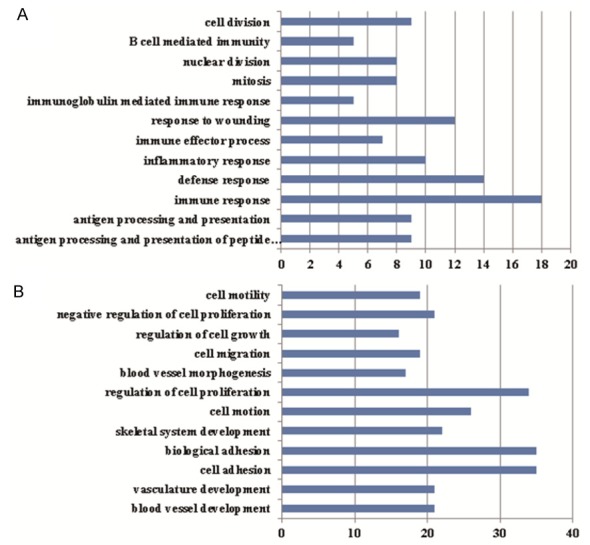
Gene Ontology enrichment analyses. A. Gene Ontology enrichment of up-regulated genes of osteosarcoma; B. Gene Ontology enrichment of down-regulated genes of osteosarcoma. The horizontal axis represents the number of enriched genes. The vertical axis represents the Gene Ontology, and P values increase from bottom to top.
Construction of protein-protein interaction (PPI) network of DEGs
All DEGs were mapped onto STRING database to construct the PPI network (Figure 4), and 323 genes were identified to be able to integrate to the network.
Figure 4.
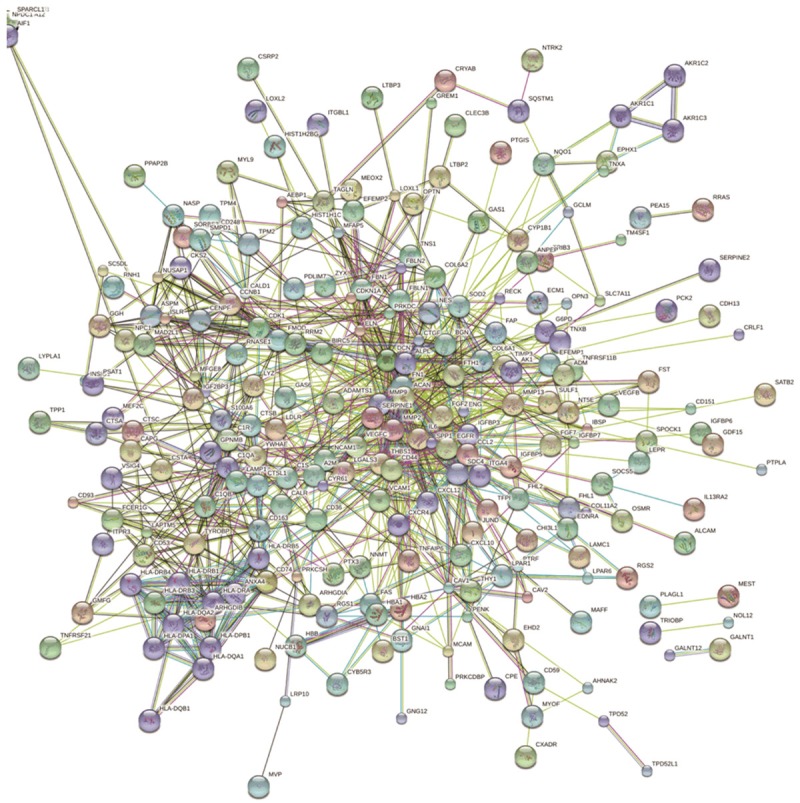
PPI network of DEGs. The nodes represent the genes and the edges represent the corresponding PPI pairs. Total 323 genes were integrated to the network.
Principal component analysis (PCA) of top10 DEGs
Ten significantly up-regulated DEGs were screened for principal component analysis. It was shown in Figure 5 that the top 10 DEGs can directly distinguish osteosarcoma tissues from normal tissues. The first principal component explained 77.95% of the variance in 10 variables, and the second principal component explained 15.38% of the variance. The interpretation degree of the cumulative variance was 93.33%.
Figure 5.
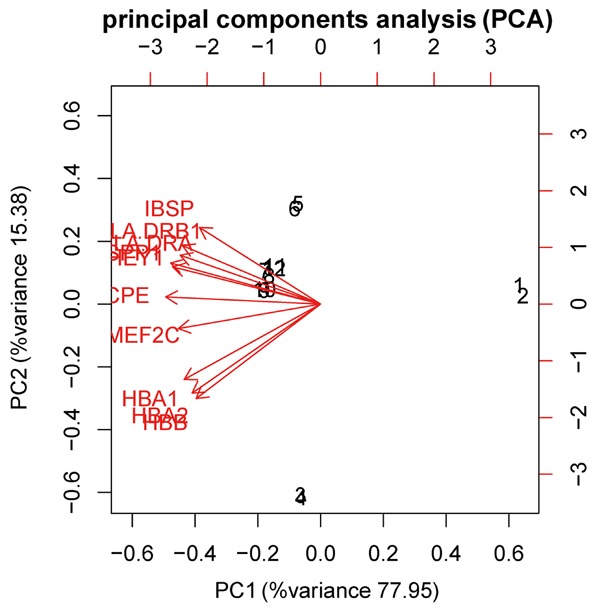
Principal component analysis (PCA) of top10 DEGs. The horizontal axis represents the first principal component scores of each sample, and the vertical axis represents the second principal component scores. The first principal component explained 77.95% of the variance in 10 variables, and the second principal component explained 15.38% of the variance. The interpretation degree of the cumulative variance was 93.33%. Number 1 and 2 represent the normal samples (2 in total), and number 3-12 represent osteosarcoma tissue samples (10 in total). The red line represents the impact on osteosarcoma tissues of the genetic variables among which the 10 genes were most significantly expressed.
PPI network analysis of top20 DEGs
The Top20 DEGs (Table 2) were mapped onto STRING database to construct the protein-protein interaction network (Figure 6). Six DEGs (CPE, WIF1, FABP5, MEF2C, HEY1, and A2M) failed to form the PPI pairs. The PPI network mainly included the histocompatibility complex forming network associated with immune response process and hemoglobin interacting network. The former mainly involved the histocompatibility proteins (such as CD74, HLA, etc.) and enzymes (such as tyrosine kinase, metallopeptidase, etc.), and the latter mainly involved the interactions of the hemoglobin subunits.
Table 2.
Significantly top20 DEGs in osteosarcoma tissues
| Gene | Gene description |
|---|---|
| SPP1 | secreted phosphoprotein 1 |
| HBA1 | Hemoglobin, alpha 1 |
| HBB | hemoglobin, beta |
| HBA2 | hemoglobin, alpha 2 |
| HLA-DRB1 | major histocompatibility complex, class II, DR beta 1 |
| IBSP | integrin-binding sialoprotein |
| HLA-DRA | major histocompatibility complex, class II, DR alpha |
| CPE | carboxypeptidase E |
| HEY1 | hes-related family bHLH transcription factor with YRPW motif 1 |
| MEF2C | Myocyte enhancer factor 2C |
| MMP9 | matrix metallopeptidase 9 |
| HLA-DPA1 | major histocompatibility complex, class II, DP alpha 1 |
| WIF1 | WNT inhibitory factor 1 |
| HEY1 | hes-related family bHLH transcription factor with YRPW motif 1 |
| FABP5 | fatty acid binding protein 5 |
| A2M | alpha-2-macroglobulin |
| SATB2 | SATB homeobox 2 |
| LAPTM5 | lysosomal protein transmembrane 5 |
| C1QA | complement component 1, q subcomponent, A chain |
| TYROBP | TYRO protein tyrosine kinase binding protein |
| CD74 | CD74 molecule, major histocompatibility complex, class II invariant chain |
Figure 6.
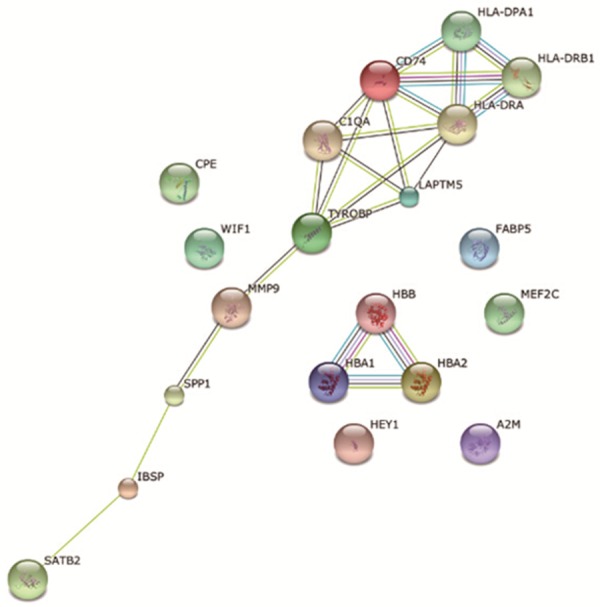
PPI network of top20 DEGs. The nodes represent the genes and the edges represent the corresponding PPI pairs. Six DEGs (CPE, WIF1, FABP5, MEF2C, HEY1, and A2M) failed to form the PPI pairs.
Transcription factor prediction
After uploading the two groups of DEGs to TfactS database, total 123 transcription factors were identified, including 78 up-regulated transcription factors and 100 down-regulated ones, and 55 transcription factors were mutual in the two groups (Figure 7). The MYC and SP1 transcription factors had the largest number of target genes (Table 3).
Figure 7.
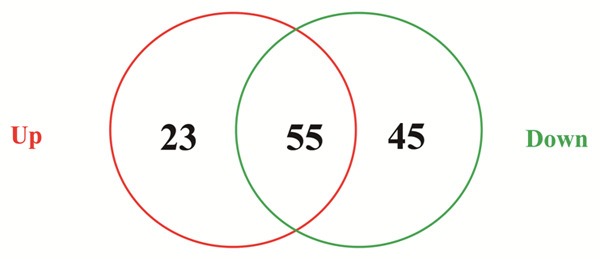
Data sets of transcription factors in osteosarcoma tissues. The red circle represents the number of up-regulated transcription factors, and the green circle represents the number of down-regulated ones. The intersection of the two circles means the number of mutual transcription factors of the two groups.
Table 3.
Corresponding genes of Transcription factor MYC and SP1
| Transcription factor | Genes |
|---|---|
| MYC | RGS2, NCAM1, CXCR4, CKS2, CTSC, CCNB1, TPD52, HLA-DPB1 |
| SP1 | CD163, GGH, CSRP2, NES, MMP9, SPP1, NCAM1 |
Discussion
Osteosarcoma remains a devastating disease, and it is reported to be the eighth leading form of childhood cancer with an incidence of 4.4 per million [4]. Its treatment is still a major challenge in oncology, so the clear understanding of its mechanism is necessary for the development of novel therapeutic strategies. In this paper, we identified the potential target genes and their functions to understand the potential biological process of osteosarcoma progression. It can be concluded that osteosarcoma progression was closely associated with the immune response and the development of skeletal and vascular system. Also, histocompatibility proteins (such as CD74, HLA, etc.) and enzymes (such as tyrosine kinase, metallopeptidase, etc.), which were related to immune response, and hemoglobin played important roles in osteosarcoma development. Besides, transcription factors MYC and SP1 were predicted to be significantly related to osteosarcoma.
Tyrosine kinase is critical for transducing intracellular signaling cascades for various immune recognition receptors, such as the B-cell receptor and the Fc receptor [20]. The activated receptor tyrosine kinases devote to in vitro phenotypes which are involved in metastatic potential: motility, colony formation, and cell growth [21]. The activation of receptor tyrosine kinase gives rise to enhanced proliferation, survival, and even metastasis, therefore, it has developed as target for cancer diagnoses and therapies [12]. Four receptor tyrosine kinases (Axl, EphB2, FGFR2, and Ret) have been identified in osteosarcoma and may serve as targets for novel therapeutics [8]. Hemoglobin is the iron-containing oxygen-transport metalloprotein in the red blood cells of all vertebrates and its concentration measurement is among the most commonly performed blood tests, usually as part of a complete blood count (http://en.wikipedia.org/wiki/Hemoglobin-cite_note-1). Increased fetal hemoglobin levels were related to neoplastic diseases [22]. Previous study has convinced that patients treated for osteosarcoma had the potential to develop hematological abnormalities mimicking early myelodysplastic syndrome [23]. Therefore, it is essential to monitor hematological changes of patients recovering from osteosarcoma.
Transcription factor MYC (myelocytomatosis viral oncogene) is a multifunctional, nuclear phosphoprotein that plays a role in cell cycle progression, apoptosis and cellular transformation. Malfunctions in MYC have also been found in carcinoma of the cervix, colon, breast, lung and stomach. It was found that the expressions of MYC was negatively correlated with apoptotic index of osteosarcoma tissue, was not correlated with pathological types of osteosarcoma, and was closely related to prognosis of the patients (Wu et al., 2012). The present study demonstrates that MYC overexpression promotes osteosarcoma cell invasion, probably via activation of MEK-ERK pathway (Han et al., 2012). According to our result, cyclin-dependent kinase-associated protein (CKS2) gene was regulated by the transcription factor MYC. In patients who developed muscle-invasive cancer, CKS2 gene showed significantly increased expression after, compared with before, invasion (Chen et al., 2011). Therefore, the CKS2 gene may also be a potential biomarker for predicting osteosarcoma in early stage.
Transcription factor SP1 (specificity protein 1) is involved in many cellular processes, including cell differentiation, cell growth, apoptosis, immune responses, response to DNA damage, and chromatin remodeling [4]. SP1 performs a significant role in regulating important biological processes such as DNA repair, cell growth, differentiation, and apoptosis [24]. The level of the transcription factor SP1 may exert an effect in osteosarcoma. Based on our result, SPP1 gene was regulated by the transcription factor SP1. It has been found that SPP1 was not necessary for osteosarcoma progression and may be related to inflammatory response and bone remodeling, which will function as a good biomarker [25].
Conclusions
In summary, our data provides a comprehensive bioinformatics analysis of genes and pathways which may be involved in the progression of osteosarcoma. Total 437 DEGs was obtained, and protein-protein interaction networks of these DEGs were constructed. And 46 genes were associated with osteosarcoma. GO and KEGG pathway enrichment analysis showed that genes mainly affected the process of immune response and the development of skeletal and vascular system. Histocompatibility proteins, enzymes and hemoglobin were closely associated with osteosarcoma. Furthermore, we predicted the association of MYC and SP1 with osteosarcoma. The top ten up-regulated genes in osteosarcoma tissue can be used to distinguish cancer samples from normal specimen. Our discovery may be useful in investigating the complex interacting mechanisms underlying the disease, and provides a new strategy in the medical therapy of osteosarcoma. However, further experiments are still needed to confirm our result.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Moore DD, Luu HH. Osteosarcoma. Cancer Treat Res. 2014;162:65–92. doi: 10.1007/978-3-319-07323-1_4. [DOI] [PubMed] [Google Scholar]
- 2.Miao J, Wu S, Peng Z, Tania M, Zhang C. MicroRNAs in osteosarcoma: diagnostic and therapeutic aspects. Tumour Biol. 2013;34:2093–2098. doi: 10.1007/s13277-013-0940-7. [DOI] [PubMed] [Google Scholar]
- 3.Kundu ZS. Classification, imaging, biopsy and staging of osteosarcoma. Indian J Orthop. 2014;48:238–246. doi: 10.4103/0019-5413.132491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ottaviani G, Jaffe N. The epidemiology of osteosarcoma. Cancer Treat Res. 2009;152:3–13. doi: 10.1007/978-1-4419-0284-9_1. [DOI] [PubMed] [Google Scholar]
- 5.Samimi MA, Mirkheshti N, Pazouki A. Assessing the percent of necrosis after neoadjuvant chemotherapy with 24 hr infusional cisplatin/ 3 days doxorubicin intermittent with ifosfamide-doxorubicin for osteosarcoma. Int J Hematol Oncol Stem Cell Res. 2014;8:5–8. [PMC free article] [PubMed] [Google Scholar]
- 6.Aung L, Tin AS, Quah TC, Pho RW. Osteogenic sarcoma in children and young adults. Ann Acad Med Singapore. 2014;43:305–313. [PubMed] [Google Scholar]
- 7.Anninga JK, Gelderblom H, Fiocco M, Kroep JR, Taminiau AH, Hogendoorn PC, Egeler RM. Chemotherapeutic adjuvant treatment for osteosarcoma: where do we stand? Eur J Cancer. 2011;47:2431–2445. doi: 10.1016/j.ejca.2011.05.030. [DOI] [PubMed] [Google Scholar]
- 8.Rettew AN, Getty PJ, Greenfield EM. Receptor tyrosine kinases in osteosarcoma: not just the usual suspects. Adv Exp Med Biol. 2014;804:47–66. doi: 10.1007/978-3-319-04843-7_3. [DOI] [PubMed] [Google Scholar]
- 9.Calvert GT, Randall RL, Jones KB, Cannon-Albright L, Lessnick S, Schiffman JD. At-risk populations for osteosarcoma: the syndromes and beyond. Sarcoma. 2012;2012:152382. doi: 10.1155/2012/152382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lucas MC, Tan SL. Small-molecule inhibitors of spleen tyrosine kinase as therapeutic agents for immune disorders: will promise meet expectations? Future Med Chem. 2014;6:1811–1827. doi: 10.4155/fmc.14.126. [DOI] [PubMed] [Google Scholar]
- 11.Yang G, Yuan J, Li K. EMT transcription factors: implication in osteosarcoma. Med Oncol. 2013;30:697. doi: 10.1007/s12032-013-0697-2. [DOI] [PubMed] [Google Scholar]
- 12.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 13.Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 14.Conley AB, Jordan IK. Computational Biology of Transcription Factor Binding. Springer; 2010. Identification of Transcription Factor Binding Sites Derived from Transposable Element Sequences Using ChIP-seq; pp. 225–240. [DOI] [PubMed] [Google Scholar]
- 15.Gautier L, Cope L, Bolstad BM, Irizarry RA. Affy-analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 16.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 17.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 19.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–D568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lohmann DJ, Hasle H. Hematological changes mimicking myelodysplastic syndrome following treatment for osteosarcoma. J Pediatr Hematol Oncol. 2014 doi: 10.1097/MPH.0000000000000229. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 21.Lee BH, Ryu PD, Lee SY. Serum starvationinduced voltage-gated potassium channel Kv7.5 expression and its regulation by Sp1 in canine osteosarcoma cells. Int J Mol Sci. 2014;15:977–993. doi: 10.3390/ijms15010977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pan W, Dong Z, Meng W, Zhang W, Li T, Li C, Zhang B, Chen L. Improvement of influenza vaccine strain A/Vietnam/1194/2004 (H5N1) growth with the neuraminidase packaging sequence from A/Puerto Rico/8/34. Hum Vaccin Immunother. 2012;8:252–259. doi: 10.4161/hv.18468. [DOI] [PubMed] [Google Scholar]
- 23.Olivares-Esquer JJ, Ortiz-Lazcano S, Aguirre-Gas H, Cervantes-Osorio LF, Gonzalez-Llaven J. [Increased fetal hemoglobin levels in neoplastic diseases. Premlinary report] . Arch Invest Med (Mex) 1975;6:413–418. [PubMed] [Google Scholar]
- 24.Dalla-Torre CA, Yoshimoto M, Lee CH, Joshua AM, de Toledo SR, Petrilli AS, Andrade JA, Chilton-MacNeill S, Zielenska M, Squire JA. Effects of THBS3, SPARC and SPP1 expression on biological behavior and survival in patients with osteosarcoma. BMC Cancer. 2006;6:237. doi: 10.1186/1471-2407-6-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raychaudhuri S, Stuart JM, Altman RB. Principal components analysis to summarize microarray experiments: application to sporulation time series. Pac Symp Biocomput. 2000:455–466. doi: 10.1142/9789814447331_0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.