

Abstract
The purpose of this review is to stimulate new ideas regarding low-dose environmental mixtures and carcinogens and their potential to promote invasion and metastasis. Whereas a number of chapters in this review are devoted to the role of low-dose environmental mixtures and carcinogens in the promotion of invasion and metastasis in specific tumors such as breast and prostate, the overarching theme is the role of low-dose carcinogens in the progression of cancer stem cells. It is becoming clearer that cancer stem cells in a tumor are the ones that assume invasive properties and colonize distant organs. Therefore, low-dose contaminants that trigger epithelial–mesenchymal transition, for example, in these cells are of particular interest in this review. This we hope will lead to the collaboration between scientists who have dedicated their professional life to the study of carcinogens and those whose interests are exclusively in the arena of tissue invasion and metastasis.
Introduction
Tissue invasion and metastasis is one of the six hallmarks of cancer originally detailed by Hanahan et al. (1). In their 2011 article, Hanahan et al. (2) noted the enormous advances that had been made since their original article. They noted that the molecular mechanisms that drive this hallmark are indeed complex and present several knowledge gaps in our understanding of cancer as a whole. Considering the carcinomas that constitute almost 90% of cancers, upon oncogenic transformation, the process begins with the downregulation of E-cadherin that holds the epithelial cells together as a society of cells that are well differentiated and otherwise quiescent (3) as depicted in Figure 1. Concomitant with this downregulation of E-cadherin is the conversion of the epithelial cells to mesenchymal cells in a process commonly known as EMT or epithelial–mesenchymal transition (4). Some studies reported in this review intimate that low-dose exposure to some environmental carcinogens may accelerate this transition (5). The transcription factors that control EMT such as snail, slug, Twist and Zeb1/2 are some of the best characterized signaling molecules in biology (6,7). It is known that this process is also accelerated by chronic inflammation mediated by nuclear factor kappa B (NF-κB) (8). During the process of EMT, a number of inflammatory cells are attracted to the growing tumor mass (8). Other contributing factors may also be low-dose environmental contaminants that drive the transcription of NF-κB and exacerbate the process (9,10).
Figure 1.
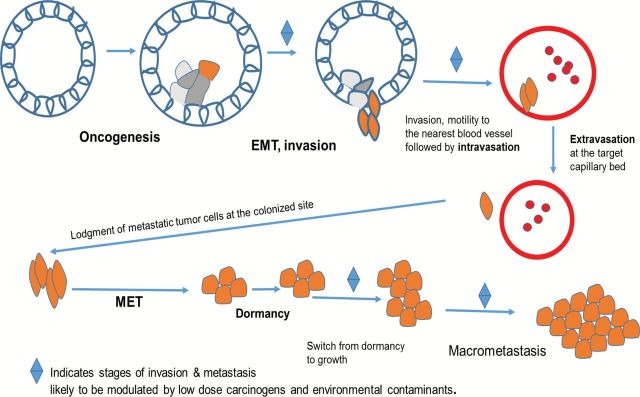
Key steps of invasion and metastasis.
Upon attaining the mesenchymal characteristics, the tumor cells are able to move out of the confines of their natural environment, aided by cross talk between them and stromal cells resulting in the secretion of matrix degrading enzymes such as matrix metalloproteinases (MMPs) (11). Naturally, environmental chemicals that mediate the activation of these enzymes or drive their synthesis will likewise contribute to the process of tissue invasion (12). Other invasion-mediating molecules include hepatocyte growth factor secreted mainly by tumor-associated fibroblasts and signals the metastatic cells to move upon their interactions with their cell surface receptors cMet (13). The metastatic cells are then attracted by chemokines and move to the nearest blood vessel or lymphatic vessel where they complete the process of intravasation and are then transported to the capillary bed in their colonized site or new home (14). Upon reaching this destination, they then undergo extravasation where they come out of the capillaries or lymphatic vessels, most likely again following the cues emanating from the chemokines in their new microenvironments.
To survive in their new home, they may revert back and assume the cuboidal morphology of epithelial cells undergoing the reversal of EMT otherwise known as mesenchymal–epithelial transition or MET (15). At this point, they may remain dormant for a very long time until such time that the conditions for their division and growth are favorable. These conditions are currently not well defined. Could it be that at this time low concentrations of environmental disruptors and or carcinogens may be required to switch these cells from their dormant to proliferative state? Do they at this time receive signals from the primary tumors to stop dividing until the primary tumor is removed by resection? Lastly, low-dose environmental contaminants that can trigger transient or sustained increases of intracellular calcium should be considered as significant drivers of tissue invasion and metastasis. Such increases would favor cellular motility and invasion of extracellular matrices by the metastatic cells (16). Increases in [Ca2+] are connected to rapid secretion of exosomes that have been shown to mediate cellular motility and invasion (17). Cellular exosomes may also be required in the preparation of metastatic niches (18). These are fertile areas for future cancer research.
This review is structured first with a brief review of chemical, biological and physical agents that are known to cause invasion and metastasis, secondly, with an overview of several mechanisms including the EMT, the MMPs, the galectins and the aryl hydrocarbon receptor (AHR) and the substances that contribute to these processes and thirdly, application of these principles to cancers in specific organs including the prostate, oral, head and neck and breast. Finally, we review the evidence of cross talk between the processes of invasion and metastasis and the other pathways of tumor progression.
Agents modulating the invasion and metastasis pathways
Many cancer researchers who began their careers as graduate students in early 1970s to mid-1980s were introduced to the field of cancer through the door of ‘Carcinogenesis’. The studies mainly revolved around carcinogenic insult to DNA, giving rise to DNA adducts followed by promotion of the transformed cells (19,20). The preferred cell culture models were rodent fibroblasts treated under the two-stage initiation promotion protocols (21). Upon the perfection of the techniques for culturing human epithelial cells and the discovery of tumor oncogenes beginning in 1980s (22–25), the emphasis in cancer research quickly shifted to molecular mechanisms of cancer progression. From this point onward, carcinogens as mediators of the initiation of mutations of ‘proto-oncogenes’ and ‘tumor suppressor genes’ became a lower research priority. In this review, we will look at recent data showing that low doses of group 1 carcinogens impinge on signaling pathways germane to tissue invasion and metastasis.
Numerous chemical carcinogens, in addition to their tumorigenic properties, may also possess proinvasive and prometastatic abilities. Taking into account that some of these chemicals are associated with the lifestyle and/or ubiquitously represented in the environment, such chemical carcinogens have received much attention in the last decade.
Polycyclic compounds
Cigarette smoke
One of the achievements of the era of ‘Carcinogenesis’ was the realization that cigarette smoke contains many carcinogens that initiate lung cancer and other chronic illnesses (26,27). Those studies unequivocally informed policy makers in the industrialized world to vigorously ban smoking in public spaces (28). Whereas it is generally assumed that cigarette-based carcinogens only initiate lung carcinogenesis, there are compelling data indicating that low-dose tobacco carcinogens such as in second-hand smoke can exacerbate or promote lung and breast cancers (29,30). For example, the tobacco-related carcinogen nitrosamine 4-(methylnitrosamino)-1-butanone (NNK) is a major component of cigarette smoke that is derived from nicotine. NNK is considered a group 1 carcinogen according to the International Agency for Research on Cancer (IARC). Studies by Shen et al. (31) demonstrated that NNK promotes migration and invasion of lung cancer cells through activation of cSrc/PKCɭ/focal adhesion kinase (FAK) loop. The carcinogen NNK binds and activates α7nAChRs, which in turn activates Src which then phosphorylates PKC and FAK, signaling molecules that are major regulators of tissue invasion and metastasis (32). The activation of PKC by NNK could explain why cigarette smoke may drive the metastasis of other malignancies such as breast cancer (30).
Tobacco smoke
Tobacco smoke is considered one of the most widespread known carcinogens and is a cause of ~5 million deaths annually worldwide (33,34). It is a complex chemical mixture that contains over 4800 compounds of which at least 70 are known carcinogens (35). Tobacco smoke is a primary risk factor for a number of human cancers, including lung and breast cancers, and is associated with both genetic and epigenetic alterations reviewed in refs (34,36). Recent reports also indicate that tobacco smoke components may not only be the cause of cancer but may also be involved in tumor invasiveness and metastasis. For instance, mice injected with mammary tumor cells develop a higher number of pulmonary metastatic tumors upon exposure to cigarette smoke (37,38). Nagaraj et al. (38) report that cigarette smoke condensate increases the invasiveness of UM-SCC-101A and MDA-686Tu oral carcinoma cells by activation of B-, D-, and L-cathepsins. Interestingly, suppression of these cathepsins by specific chemical inhibitors resulted in significant decrease of the invasion process in vitro (38). In epidemiological studies, female smokers were reported to be at a higher risk of developing pulmonary metastatic disease (39).
Benzo(a)pyrene
Benzo(a)pyrene (BaP) is a polycyclic aromatic hydrocarbon (PAH) and a known environmental carcinogen found in automobile exhaust, food and importantly is one of the major components of the tobacco smoke. BaP is capable of causing tumors in experimental models and has been considered a class 1 human carcinogen (IARC). Studies indicate that BaP may also stimulate the invasive and metastatic potential of tumors. Using the human lung adenocarcinoma CL5 cells, Ueng et al. (40) reported that treatment with BaP increases invasiveness by targeting fibroblast growth factor-9 (FGF-9). Interestingly, BaP was detected in the breast milk, suggesting that it reaches the breast and, thus, may exert its effects in this organ (41). Indeed, subsequent studies indicated that BaP holds a significant metastatic potential in breast cancer. For instance, exposure of MDA-MB-231 human breast cancer cells to BaP increased the invasive abilities of these cells and was primarily associated with increased COX-II expression and prostaglandin E2 output (42).
Food preparation
It is generally accepted that carcinogenic heterocyclic amines can be produced during food preparation. Of particular concern is 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), one of the most abundant of these amines that has been shown to induce breast, colon and prostate tumors in experimental animals (43). Epidemiological studies suggest that consumption of well-done meat is associated with esophagus, stomach, colon, lung and breast cancers (43,44). Recent studies also implicate PhIP as a chemical with invasive potential. It has been reported that exposure of MCF-7 and T47D human breast cancer cells to PhIP can promote the invasive behavior of breast cancer cells (45). This phenomenon is thought to be associated with its potent estrogenic abilities (45).
Organochlorine compounds
Organochlorines (OCs) are ubiquitous environmental chemicals with a long (10–20 years or more) half-life possessing significant health threats to the human population. Their persistence in the environment and the estrogenic properties that the majority of OC possess are of concern regarding their carcinogenic effects, especially in breast tissue. Indeed, several animal studies confirmed that such OC as 1,1-dichloro-2,2-bis(4-chlorophenyl)ethylene can alter cellular proliferation and differentiation in the mammary gland and promote the growth of mammary tumors (46,47). Epidemiological studies have also confirmed these findings, showing a significant trend between the exposure to OC and breast cancer risks (48). Additionally, some studies indicate that persistent exposure to OC may also result in more aggressive forms of breast cancer. Demers et al. (49) report that the probability of lymph-node invasion among breast cancer cases significantly increased with exposure to 1,1-dichloro-2,2-bis(4-chlorophenyl)ethylene. The authors also indicate that the 1,1-dichloro-2,2-bis(4-chlorophenyl)ethylene exposure was associated with a dose-related increased relative risk of exhibiting both lymph-node metastasis and large tumors. The mechanisms of this invasive and metastatic potential are not clearly understood but may be chemical dependent. For example, the enhanced metastatic potential of polychlorinated biphenyls (PCBs) in breast cancer cells was linked to activation of Rho-associated kinase (50). Another study, performed with widespread OC pesticide hexachlorobenzene, suggested involvement of c-Src/HER1/STAT5B and HER1/ERK1/2 signaling pathways (51). Further studies are clearly needed to delineate the enigmatic mechanisms that underline the invasive and metastatic potential of these chemical carcinogens.
Other organic compounds
Alcohol
Alcohol consumption is considered a significant risk factor for the development of hepatocellular carcinoma. Acetaldehyde, the by-product of ethanol metabolism, has been shown to activate NF-κB and activator protein-1 (AP-1), JNK/beta-TrCP and p38 signaling pathways, resulting in elevation of MMP9 gene expression and increased invasive potential in hepatocarcinoma cells (9). Other studies by Meng et al. (52) demonstrated that ethanol stimulated the invasion and migration of a breast carcinoma cell line MDA-MB-231 through the downregulation of E-cadherin.
Alkylating agents
Alkylating agents are routinely used as chemotherapeutic agents particularly for leukemia (53). It was reported in a case study that the treatment for chronic myelogenous leukemia with the alkylating agent chlorambucil may have initiated and/or promoted the development of hepatocellular carcinoma in a 55-year-old man (54). Another case involved the development of skin (cutaneous) metastasis of primary gall bladder tumors following treatment with chlorambucil for leukemia (55). These studies suggest that chlorambucil has the potential to drive tumorigenesis of secondary malignancies and so its use in the treatment of leukemias should be revisited.
Heavy metals
Hexavalent chromium
Hexavalent chromium [Cr(VI)] is a potent human mutagen and carcinogen, widely used in industrial activities and present in fossil fuel and cigarette smoke (56–58). Studies by Ding et al. (5) have demonstrated that chromium is capable of inducing EMT via reactive oxygen species (ROS), hallmarks of tissue invasion and metastasis, subsequent to oncogenic transformation in human lung epithelial cells. Other studies by Wang et al. (59) demonstrated that arsenic [As(III)] and hexavalent chromium promote colorectal tumorigenesis partly through ROS-mediated Wnt/β-catenin signaling pathway.
Cadmium
Cadmium has been classified as a group 1 carcinogen and has been shown to be directly associated with tumors of the testes, prostate, adrenal, lung and hematopoietic system in rodents (60). Although the mechanism by which cadmium transforms cells are not well established and epidemiologic human studies are inconclusive (60), an early event associated with cadmium ion nephrotoxicity is the alteration of the properties of adherent junctions and tight junctions attributed to Ca2+ displacement leading to disruption of E-cadherin (61). Subsequent studies demonstrated that Cd2+ induces Wnt signaling in kidney proximal tubule cells (62). In support of this invasion-related mechanism, in vitro evidence indicates that Cd2+ ions enhance tumor progression (63–65).
Nickel
Nickel is another group 1 carcinogen. In a study conducted using Fisher and Hooded rats, intramuscular injection with Ni3S2 induced tumors ranging from well-differentiated rhabdomyosarcomas to mesenchymal tumors. Interestingly the tumors that developed in Hooded rats were more prone to metastasis compared with Fisher rats (66). More recent studies demonstrated that nickel chloride induced the generation of ROS which in turn induced EMT in human bronchial epithelial cells and inhibited E-cadherin expression by upregulating the hypermethylation of the promoter (67). The influence of known carcinogens on pathways relevant to tissue invasion and metastasis is depicted in Figure 2.
Figure 2.

Tissue invasion and metastatic pathways influenced by group 1 carcinogens.
Biological agents modulating the invasion and metastasis pathways
Helicobacter pylori
It is well established that Helicobacter pylori is one of the key initiating carcinogens in gastric cancers (68). However, continuous exposure to the bacteria is also key in driving the progression of gastric cancer (69). Specifically, it has been demonstrated that H.pylori activates Ras, NF-κB and AP-1 (10,70). These are signaling mechanisms that are involved in tissue invasion and metastasis (71,72). Therefore, one of the approaches that may be helpful in controlling gastric cancers is to eliminate H.pylori (69). Other studies have shown the elevation of metastasis relevant enzymes, MMP3 and MMP7, in H.pylori-infected gastric cancer patients (73).
Aflatoxins
Aflatoxins (AFs) produced by fungi Aspergillus fluvus and Aspergillus parasiticus are well-established liver carcinogens that mainly form DNA adducts (74). Contamination due to aflatoxins is restricted primarily to developing countries, with children known to be more sensitive than adults. A study by Ubagai et al. (74) demonstrated the effects of AFB1 on the proliferation, invasion and metastatic capacity during hepatocarcinogenesis. They showed that AFB1 exposure led to the elevated gene expression of insulin-like growth factor 2 and insulin-like growth factor receptor.
Mechanisms of invasion and metastasis
In the following sections, we describe mechanisms that are important in the processes of invasion and metastasis. These include the EMT, the matrix metalloproteases, the galectins, self-renewal pathways of stem cells and the AHR. For each of these mechanisms, a series of examples of the ways that chemical agents disrupt the pathway are described.
The epithelial–mesenchymal transition
The EMT as first described by developmental biologists is the morphological change that epithelial cells undergo at specific sites during embryonic development, resulting in more migratory cells (75). EMT has also been characterized in epithelial cancers, where tumor cells at the invasive front undergo this transition to promote invasion, migration and subsequent metastasis (76,77). Studies have shown that during EMT, epithelial cells expressing keratin intermediate filaments, desmosomes and adherens junction proteins repress genes encoding these cell adhesion proteins (such as E-cadherin) and modify the type of intermediate filaments expressed (76,77). This is accompanied by acquisition of a mesenchymal genotype that includes vimentin, N-cadherin and osteopontin expression, synthesis of extracellular matrix molecules such as fibronectin and certain types of collagen and a flattened phenotype. These cells subsequently become more migratory, express gelatinase to become more invasive and traverse underlying basement membrane. Following EMT, the cells may differentiate into other cell types or revert back to epithelial cells (75,76,78). EMT can be induced by extracellular matrix molecules such as collagen, soluble factors such as epidermal growth factor (EGF), scatter factor/hepatocyte growth factor, members of the transforming growth factor-beta (TGF-β) and FGF family (79). Certain master genes can regulate the entire EMT process. These include Snail and Twist transcription factors that can downregulate E-cadherin by binding several E-boxes located in the promoter region (76,80).
Cigarette smoking is a risk factor for lung cancer and can also promote lung cancer aggressiveness (81–84). BaP is a PAH released as a by-product of incomplete combustion or burning of organic (carbon-containing) items, such as cigarettes, gasoline and wood. Studies have shown that BaP induces EMT in lung cancer cells and accelerates disease progression in patients with lung cancer (85). Specifically, BaP increased messenger RNA expression of mesenchymal markers such as Twist and decreased expression of E-cadherin in A-549 bronchiol alveolar carcinoma cell line, and subsequent removal of BaP was not able to reverse these effects (85). Strikingly, E-cadherin expression is reduced in 42% of lung cancer patients (86). Another study has associated Twist expression as a useful prognostic marker for patients with bladder cancer and its expression seems to be correlated to the tobacco status of the patients (87).
Nicotine, a constituent of cigarettes that is absorbed into the body during smoking, has also been found to induce invasion and EMT and may contribute to the progression of breast and lung cancers (88). Additionally, nicotine was shown to increase colon cancer cell migration through upregulation of α7-nAChR and fibronectin, a mesenchymal marker for EMT (89). Furthermore, COX-2 signal was involved in the induction of fibronectin (89). Interplay between cancer stem-like cells (CSCs) and EMT has been proposed to promote tumorigenesis and metastasis in oral squamous cell carcinoma (OSCC). Yu et al. (90) demonstrated that nicotine enhanced the stemness and EMT property in oral epithelial cells, which could be reduced by targeting Snail, thus suggesting that therapeutic targeting of Snail might offer a new strategy for the treatment of OSCC patients with smoking habit (91). Therefore, smoking may play a role in the progression of several cancers through induction of the EMT process.
Heavy metals have been associated with cancer due to their abilities to bind DNA, causing mutations and DNA damage. Smokers have higher levels of heavy metals such as cadmium (Cd) than non-smokers. Cd may also be consumed through contaminated food or water. Chronic Cd exposure may induce renal fibrosis and/or cancer and it has been reported that Cd induced EMT markers Twist, fibronectin and collagen I via upregulation of the Wnt pathway in mouse kidney cells (92).
Although the association between heavy metals and EMT is scant, there are more studies showing an association between radiation and EMT in cancer. Ionizing radiation has been shown to induce EMT in cervical, colorectal, lung and breast cancer cells (93–96). For example, irradiation of cervical cancer cells with accumulative dose of 75 Gy induced EMT mediated by p65 (93). Furthermore, doses as low as 2 Gy induced EMT in six different cancer cell lines from different human organs, via upregulation of TGF-β (94). Additionally, irradiation of non-malignant human mammary epithelial cells with 2 Gy and cotreatment with TGF-β led to EMT mediated in part by mitogen-activated protein kinase (MAPK) activation (97). Therefore, it may be important to look into suppressing EMT markers in patients that undergo chemoradiotherapy or who have been exposed to low doses of ionizing radiation.
Pesticides have also been associated with cancer progression. One study showed that human hepatocytes cultured with the pesticides, OC or 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), underwent EMT, which may account in part for the carcinogenic and/or fibrogenic nature of pesticides (98).
It is well recognized that human cancer development is associated with chronic inflammation, and ROS released by inflammatory cells may result in DNA damage (99,100). It has also been reported that spontaneous generation of the ROS, hydrogen peroxide, in tumor tissue was associated with clinical stage in small cell lung cancer and squamous cell carcinoma patients (101). ROS, particularly superoxide and hydrogen peroxide, are not only harmful metabolic by-products of oxidative stress but are also intracellular signaling molecules. For example, ROS can lead to the activation of NF-κB and AP-1 transcription factors, which play a role in cell proliferation, differentiation and morphogenesis (102). Additionally, ROS has been associated with EMT; TGF-β was shown to induce EMT via upregulation of hydrogen peroxide and MAPK signaling in proximal tubular epithelial cells (103). It has been published that Snail can induce ROS (hydrogen peroxide and superoxide) in prostate cancer (PCa) cells (104). Certain compounds and natural products that antagonize ROS have been reported to revert EMT. Prostaglandin D2 was shown to exhibit antioxidant property and inhibit TGF-β1-mediated ROS production and EMT in Madin–Darby canine kidney epithelial cells (105). Antioxidants such as epigallocatechin-3-gallate, the bioactive polyphenol component of green tea, was also able to revert EMT in breast and oral cancer cells (106,107), whereas resveratrol, a polyphenolic compound of grapes and red wine, could also revert EMT in breast, melanoma and lung cancer cells (108–110).
MMPs in tumor progression and metastasis
MMPs are a class of primarily extracellularly targeted zinc-dependent endopeptidases that were initially characterized by their ability to proteolyze various components of the extracellular matrix (111). MMPs are associated with a variety of normal and pathological conditions including tumorigenesis (112,113). MMP activity first identified was that of collagenolytic activity associated with absorption of a tadpole tail during metamorphosis (114). However, MMPs have long been associated with tumor progression (115). Although MMP levels are typically low in non-diseased, quiescent tissue, MMPs levels increase and correlate with disease progression. A role for MMPs in tissue invasion was initially proposed following studies demonstrating cleavage of basement membrane by those MMPs with gelatinolytic activity (MMP2 and MMP9). Due to the association of MMP upregulation correlating with increasingly progressed tumors, a function for MMPs as removal of extracellular matrix barrier to facilitate cellular invasion and metastasis was proposed (112,113). Over time, the discovery of non-extracellular matrix MMP substrates grew, documenting MMP-dependent regulation of growth factor availability, adhesion factors and chemokine gradients (112). This led to an appreciation of additional MMP roles throughout tumorigenesis, from regulation of tumor cell growth and survival to influence over tumor stromal events such as angiogenesis and inflammation (116). On the other hand, current studies that importantly include the use of mice with genetic ablation of single MMP family members has revealed both pro- and antitumorigenic roles for MMPs during tumorigenesis (117). In retrospect, this is unsurprising as MMPs are important regulators of normal processes such as wound healing and tissue repair and remodeling (113). Indeed, many of the same processes upregulated during wound healing are exploited during tumorigenesis, which is why tumors are at times referred to as non-healing wound pathologies (118). This section focuses on the influence of environmental chemical pollutants on regulating MMP expression and/or activity. As chronic exposure to low-level chemicals is more apt to work as a promoter of tumorigenesis, the regulation of MMPs by chemical pollutants in a variety of inflammation and tissue damage models is included.
MMPs are a subset of the family of metal-dependent proteases, the metzincins (111,119). MMP activity is highly regulated. All MMP family members share a conserved catalytic domain (HEXX-HXX-GXXH) that coordinates a zinc ion at the active site to achieve hydrolysis (111,119). An N-terminal leader sequence directs the MMPs for either extracellular secretion or as a membrane bound molecule. MMPs are formed as inactive zymogens. A conserved sequence in the prodomain (PRCG(V/N)PD) contains a cysteine residue that binds to the zinc ion in the catalytic domain and maintains enzymatic latency (111,119,120). Proteolytic processing of the prodomains opens the catalytic site for substrate binding. Activity is then downregulated through interactions with endogenous inhibitors. The majority of MMPs also contain a hemopexin domain that provides additional protein–protein contacts and facilitates substrate specificity and interactions with naturally occurring inhibitors, the tissue inhibitors of MMPs (TIMPs) (121). A cysteine within TIMP forms a non-covalent interaction with the zinc ion in the catalytic site of the MMP. This strategy is the basis of many synthetic inhibitors of MMPs. In addition, plasma α-2 macroglobulin serves as a non-specific inhibitor of MMPs (111). Likewise, MMP activity can be reduced through interactions with serine protease inhibitors, such as α1-antitrypsin. Some MMPs can be inhibited by reverse-inducing cysteine-rich protein (RECK) (122). Furthermore, MMPs can be modified by ROS (120).
MMP expression is another important regulatory control mechanism (123,124). Most MMPs are expressed at low levels, if at all, in quiescent tissues. During times of active tissue remodeling, MMP expression can be rapidly induced. Most MMPs are extracellularly secreted enzymes. However, some can be stored in secretory vesicles, for example, MMP9 in macrophages and MMP8 (neutrophil collagenase) in mast cells. MMP expression can be induced by a variety of growth factors or cytokines, such as EGF, platelet-derived growth factor, keratinocyte growth factor (KGF), vascular endothelial growth factor, tumor necrosis factor alpha, bFGF, hepatocyte growth factor and TGF-β (123,124). The promoters of the various MMP family members contain a number of transcriptional binding sites including AP-1 and AP-2, PEA-3, NF-κB, STAT and TCF sites (124). The various combinations of response elements in individual MMP family members confer some specificity of gene regulation, with coregulated MMPs tending to share similar promoter elements. Overall, any perturbation of regulatory processes, either signaling processes governing expression of MMP or modifying native inhibitors of MMP, will impact the overall balance of active MMP to act on its downstream substrates and influence cellular processes.
MMPs and environmental pollutants
Air particulates and gas pollutants have wide spread effects on lung and the cardiovascular systems. Some of the resultant inflammation and subsequent tissue damage may be linked to deregulated MMP expression either through initiating MMP expression and/or recruitment of inflammatory cells with granular release of MMPs at the tissue site. Ozone exposure has been reported to initiate an inflammatory response in a rat model by increasing alveolar macrophages and MMP2 and MMP9 levels and in a mouse model by increasing neutrophil infiltration and MMP9 levels in bronchoalveolar lavage fluid (125,126). In another mouse model study, MMP2 expression was increased only when lungs were coexposed to urban air pollutant (EHC93) and ozone, but not to single exposure of either alone (127). Furthermore, iNos-deficient animals show an even greater sensitivity to ozone inhalation resulting in even higher levels of neutrophil infiltrate and MMP9 levels over wild-type counterparts (125). However, MMPs are a part of normal wound repair processes. A protective role for MMP9 in response to ozone-induced lung injury comes from work with MMP9-deficient animals. MMP9 null animals increased lung tissue damage in response to inhalation of ozone and with enhanced proliferation and alveolar epithelial thickening and alveolor vacuolization (128). Furthermore, absence of MMP9 led to an increase in ozone-induced inflammation as measured by infiltrating neutrophils in lavage fluid. However, absence of MMP7 resulted in no change in ozone-dependent inflammation or lung injury (128). On the other hand, deregulated MMP12, also known as macrophage elastase, may play a more lung tissue damaging role in response to air pollutants. Extensive repeated exposure to ozone of 6 weeks, but not 3 weeks, led to a chronic inflammation accompanied by epithelial damage and alveolar enlargement. This tissue damage was associated with an increase in caspase-3 and MMP12 expression (129). This relationship is not surprising, as MMP12 has been implicated in other chronic obstructive pulmonary disease models, and importantly absence of MMP12 abrogates murine emphysema induced by cigarette smoke (130).
Other types of combustion materials impact lung and cardiovascular health. In a wood-smoke-induced emphysema guinea pig model, increased levels of MMP1, MMP2 and MMP9 were noted in lung tissue (131). Inhalation of gasoline engine exhaust by ApoE null mice induced vascular remodeling and increases in MMP2, MMP9 and TIMP-2 and of endothelin-1, a mediator of atherosclerosis (132). These effects translated to humans since human volunteers exposed to whole diesel emissions had elevated MMP9 and ET-1 in plasma (132). Using this same mouse, the vascular effects in a side-by-side comparative study using a large panel of air pollutants (whole combustion emissions, secondary organic aerosols, single gas contaminants in spent fuels) were examined (133). Contrary to the wood-smoke-induced emphysema guinea pig model, no effect on MMP9 expression was noted following wood smoke inhalation of ApoE null animals, although there was an increase in aortic gelatinolytic activity. It should be noted that the Campen et al. study challenged mice for 1 week, whereas Ramos et al. challenged animals up to 7 months with wood smoke (131,133). Likewise, particulate matter had no effect in this study (133). Gasoline exhaust and monoxide gas products CO and NO elicited increases in MMP9, TIMP-2, endothelin-1 and also to an increase in aortic gelatinolytic activities (133).
Diesel exhaust particles (DEPs) are a major contributor to air particle pollutants. DEP has been shown to induce MMP1 in human lung epithelial cells in vitro and in vivo using a mouse model (134,135). DEP-dependent MMP1 induction initiates a ras-raf-ERK1/2 signaling cascade that is dependent on scaffolding by B-arrestin (135) and has also been shown to upregulate NOX4, a homolog of reduced nicotinamide adenine dinucleotide phosphate oxidase (134). Reduction of NOX4 or β-arrestin using RNAi knockdown blunted DEP-induced MMP1 expression (134,135). In contrast, Doornaert et al. (136) showed a decrease in MMP1 expression in human bronchial epithelial cells challenged with DEPs; however, this study used levels of DEP that were cytotoxic to cells. In addition to DEP effects on airway epithelium, the methanol extract of DEP can activate neutrophils leading to an upregulation of MMP9 (137).
Sulfur dioxide (SO2) is a common contaminant in automobile fumes. SO2 dampened mucocillary clearance in a frog palate injury model challenged with sodium metabisulphite, which forms SO2 upon contact with water. Furthermore, this SO2 injury model was accompanied by an elevation in MMP9 (138). Following the ban of lead from gasoline, other oxygenates have been added to gasoline to increase the octane rating and to reduce carbon monoxide. Methyl tert-butyl ether, a commonly used stabilizer, has been banned from some states due to contaminating drinking water. Using a zebrafish model, animals were challenged with low levels of the related ethers, ethyl tert-butyl ether and tertiary amyl methyl ether. Both chemicals led the developmental abnormalities including effects on vascular and craniofacial development (139). The alterations were associated with reduced transcriptional levels of WNT ligands and MMP9, with ethyl tert-butyl ether and tertiary amyl methyl ether exposure and additionally reduction in MMP2 transcripts with tertiary amyl methyl ether exposure (139).
In addition to reactive chemical pollutants, there are also particulates that damage tissue. Amorphous silica (i.e. non-crystalline) has been regarded as a less toxic form of crystalline silica; however, growing evidence suggests that it can induce lung inflammation and initiate lung tumors in rats (140). Male A/J were exposed by intratracheal instillation to ultrafine amorphous silica. Amorphous silica induced a dose-dependent short-term fibrosis that was reversible (141). This fibrosis was accompanied by an increased expression of the inflammatory cytokines, interleukin (IL)-4, IL-10, IL-13 and interferon-γ and also MMP2, MMP9, MMP10 and TIMP-1. The induced cytokine and MMPs levels were reversible as well except for continued upregulation of MMP2 and interferon-γ (141).
Other chemical pollutants find their way into waterways. Tetrabromobisphenol A (TBBPA) is a brominated flame retardant identified as an endocrine disruptor that is commonly found accumulating in the atmosphere and in water environments. Toxicity of TBBPA and its metabolites were assessed in an embryonic zebrafish model. TBBPA exposure was embryonic lethal resulting in developmental alterations such as tail malformations, edema and reduced heart rates (142). This TBBPA-dependent developmental defects were accompanied by MMP2, MMP9 and MMP13 expression with a concomitant increase in the ability to degrade gelatin and collagen type I (142).
From these studies, it is clear that chemical pollutants in air and water enhance MMP expression, a result that is not unexpected as these chemicals induce inflammation and tissue damage. What is less certain is whether MMP expression is a cause or consequence of subsequent tissue damage and more importantly for the focus of this review, the supporting role that these MMPs play in invasion and metastasis. More focused attention in this area is needed to elucidate the role of MMPs in this important health arena.
Galectins
Galectins are a family of animal β-galactoside lectins characterized by conserved sequence elements in their carbohydrate-binding domain (CRD). They have emerged as key players in inflammation and tumor progression by displaying intracellular and extracellular activities. Galectin family consists of three types of proteins: the ‘prototypical’ galectins (galectins 1, 2, 5, 7, 10, 11, 13, 14 and 15) have a single CRD and are capable of homodimerization. The second subclass of galectins (4, 6, 8, 9 and 12) contains tandem repeats in which two homologous CRDs are separated by a ‘linker’ within a single protein. The third class, represented by galectin-3, is characterized by a proline-rich collagen-like domain near the N-terminal domain in addition to the CRD (143). Galectin-3 is currently the most reliable marker for thyroid cancer progression (144). Galectins also play significant roles in signal transduction, inhibition of cell receptor internalization, induction of T-cell apoptosis and induction of angiogenesis (145–150) due to their ability to bind to glycosylated proteins such as EGFR, integrins, Neural Cell Adhesion Molecule (NCAM), fibronectin and laminin extracellularly (145,151–153). Galectins also play significant roles when expressed intracellularly. For example, galectin-1 and galectin-3 bind to members of the serine (S)-rich and arginine (R)-rich splicing factor family (SR proteins) and form spliceosome complexes within the nucleus (154,155). Deregulation of galectins has been found in multiple cancers such as breast, thyroid, colorectal and melanoma, where they inhibit tumor cell apoptosis, promote angiogenesis, tumor growth and enhance metastasis (150,156–158).
NNK, a potent tobacco carcinogen, most likely contributes to the induction of human lung cancer in combination with PAHs and is strongly related to human pulmonary adenocarcinoma. NNK is known to be well activated in the lung, with production of methylating and pyridyloxobutylating agents that attack DNA and cause mutations (159). The incidence of NNK-induced lung cancer was significantly reduced in gal3−/− compared with gal3+/+ mice. The differences between the NNK-induced lung tumors in both wild-type and knockout mice showed alteration in genes related to cellular growth and differentiation, immune response, Wnt/β-catenin and platelet-derived growth factor/EGF signaling pathways (160). These results suggest that disrupted galectin-3 may attenuate lung carcinogenesis due to its regulatory role in B-cell receptor, ERK/MAPK and peroxisome proliferator-activated receptor (PPAR) signaling pathways.
Mendonça et al. (161) used 4-nitroquinoline-1-oxide-treated galectin-3+/+ and galectin-3−/− mice as models of oral carcinogenesis. Their findings suggested that malignant transformation of tongue epithelium is associated with increased p-GSK3β-Ser9 expression in galectin-3+/+ animals, but not in galectin-3−/− mice.
PAHs are a large group of ubiquitous environmental contaminants formed by the incomplete combustion of carbon compounds such as automobile exhaust, tobacco smoke and coal tar. PAH adducts have been associated with carcinogenesis due to multiple alterations in gene expression. Hooven et al. (162) have shown altered expression of heat shock proteins, cytoskeletal proteins, DNA-associated proteins and glycolytic and mitochondrial proteins in breast cancer cells MCF-7 treated with BaP, dibenzo[a]pyrene and coal tar extract. Interestingly, expression of galectin-3 was also increased in these treatments.
Synthetic pyrethroids are the most prominent insecticides for treatment and control of indoor pests such as cockroaches, houseflies and mosquitoes. In humans and animals, pyrethroid uptake occurs mainly through the skin, eyes, by inhalation or by ingestion. Cypermethrin is classified as a possible human carcinogen with limited evidence of carcinogenicity in animals, but no evidence of carcinogenicity in humans. George et al. (163) reported an increase in the expression levels of cancer-related proteins carbonic anhydrase 3, heat shock protein-27, galectin-7, S100A9 and A100A11 and downregulation of superoxide dismutase in the skin of mice treated with cypermethrin.
In a study conducted by Kayser et al. (164), asbestos fiber concentration in the lung parenchyma as a result of exposure to asbestos was studied in a lung cancer cohort. An increased asbestos concentration resulted in alteration of glycol-histochemical features of alveolar lining and reduced survival. Presence of increased asbestos was correlated to galectin-1-binding and the presence of epitopes for natural immunoglobulin G subfractions with selectivity to alpha-galactisides and alpha-mannosides.
Chromium VI is a known human carcinogen. Its exposure occurs via inhalation or ingestion in drinking water. The sources of its release in the environment include tanning, corrosion inhibition, plating, glassware cleaning solutions, wood preservatives, safety match manufacture, metal finishing and pigments (165). In a study by Tsao et al. (166), rats were fed with Na2Cr2O7 at concentrations ranging from 250 to 1250 p.p.m. Expression of c-myc and galetin-1 increased, whereas the expression of p53 and Rho-GDIα decreased in the stomach and colon of treated rats.
Self-renewal pathways of stem/progenitor cells
The biological effect of low-dose chemicals on human physiology and its association with the emergence of disease in utero and/or postnatal life is now accepted as a known phenomenon (167,168). Also widely accepted is the concept of adult stem cells being involved in maintaining tissue hemostasis throughout life (169). Recent advances in stem cell research suggest that normal stem and early progenitor cells may be a direct target of carcinogens, therefore becoming the ‘cells of origin’ for cancer initiation and progression during all phases of one’s life from the in utero stage to neonatal, adolescent, adult and geriatric phases (170,171). It is hypothesized that short or chronic exposure of low-dose carcinogens may alter the self-renewal pathways of stem cells preventing them to enter the quiescent state that follows the regeneration process, thereby enhancing the probability of multistage oncogenic events that result in transformed stem cells. This may involve genetic mutation and/or epigenetic alterations of adult tissue-specific stem cells leading to the transformation of a small number of progenitor cells, which by repeated asymmetric division would yield progenies of daughter cells destined either toward differentiation or self-renewal (169,172,173). This process could result in the formation of a heterogenous tumor consisting of a bulk of differentiated cells, and a small population of cancer cells possessing properties of differentiation and self-renewal, commonly known as CSCs (174). The CSCs have been shown to have the ability to initiate and sustain cancer growth (175). Several developmental pathways, including Notch, Wnt, Hedgehog, Janus kinase and activation of transcription factor (JAK/STAT) and TGF-β, have been shown to be crucial for the regulation of the self-renewal and maintenance of CSCs (176). Recent data suggest that CSCs are plastic as their expression changes with tumor progression and depends on the signals received from the tumor microenvironment (175). Although conventional therapies used for cancer treatment eradicate the majority of cancer cells within the tumor, most patients eventually develop drug-resistant recurrent cancers that remain incurable by the current treatment strategies. It is now becoming increasingly evident that most recurrent cancers are the by-product of residual CSCs that survive therapy and continuously seed and maintain the growth of new tumors (175).
The presence of low-dose carcinogens in the form of inorganic arsenic in drinking water (170), pesticides (177–179), benzene (from cigarette smoke, gasoline vapors, automobile exhausts and contaminated water and soil) (180), endocrine-disrupting compounds such as bisphenol A (BPA) and thalate, a synthetic monomer present in plastic containers, linings of food and beverage cans, dental materials, cosmetics and numerous other household products (172,181,182) have been linked to urogenital, prostatic, endometrial, breast and male and female genital neoplasms (183). Although the role of the above low-dose carcinogens in the emergence of CSCs has not been shown in humans, their involvement has been documented in experimental animals (172,181–183). One such example is arsenic-induced cancer. A series of studies on different strains of mice exposed to inorganic arsenic via maternal drinking water have demonstrated dose-related increases in lung adenocarcinomas, ovarian tumors, uterine and oviduct preneoplasia in female offsprings, whereas male offsprings demonstrated similar dose-related increases in hepatocellular carcinoma and adrenal adenomas (184). In this context, arsenic has been shown to affect the stem cell dynamics and differentiation in various cell lines and animal models (170,185–187). For example, in vitro exposure of epidermal cells to low doses of arsenic maintained the proliferative potential of epidermal keratinocytes, thereby decreasing their exit from the germinative compartment under conditions that promote differentiation of untreated cells (188). Chronic in vitro exposure of prostate epithelial stem cells to low doses of arsenic resulted in the transformation into a pluripotent CSC phenotype due to early depletion resulting from aberrant differentiation and subsequent reactivation of self-renewal-associated genes such as Oct4, BMI-1, ABCG2, Notch-1, p63 and sonic hedgehog (186). This alteration in self-renewal pathways occurred concurrently with the loss of tumor suppressor phosphatase and tensin homolog (PTEN), which was important for the acquisition of a malignant phenotype, leading to tumors enriched in CSCs in nude mice (189). This suggests that arsenic exposure has the ability to trap stem cells in an activated state of self-renewal forcing an abundance of CSCs with an acquired malignant phenotype (170). This is particularly relevant during the course of organogenesis in an in utero state when stem cells are generally greater in number and are more active, increasing the probability of oncogenic events occurring in stem cells (170). These data support the concept that low-dose chemical exposure of carcinogens reprogram and/or transform tissue-specific stem cells, providing them with abilities to initiate, develop and sustain tumor growth. Hence, under the in utero scenario, an increased number of transformed stem/progenitor cells in response to low-dose carcinogens would increase cancer risk by the shear presence of more cells available for transformation. These transformed, slowly proliferating stem cells may remain dormant in utero but get activated by an unknown secondary stimulus during the childhood, adulthood or geriatric phases of human life resulting in cancer.
In addition to arsenic-induced cancer, benzene toxicity has recently been proposed to develop through interactions of benzene metabolites with hematopoietic stem cells (180). Moreover, both animal experiments and epidemiological data suggest that fetal exposure to endocrine-disrupting compounds (a class of environmental toxicants that interfere with the endocrine signaling pathway that include BPA, phthalate, pesticides, PCBs, dioxins, etc.) increase the incidence of adult breast and PCas in the general population (181,182). It has been hypothesized that epigenetic reprogramming of endocrine sensitive, tissue-specific adult stem cells may be the direct targets of endocrine-disrupting compounds, therefore resulting in the predisposition to cancer in adult life and/or upon aging (182). In addition to exposure to carcinogens, CSC-like phenotypes have also been observed in tumors exposed to radiotherapy (190) and chemotherapy (191). This could indicate the acquisition of stemness genes in tumors in response to environmental stressors/carcinogens (192).
CSCs and epithelial plasticity
The mere definition that CSCs are the only self-renewing population in the tumor that are capable of seeding a new tumor suggests that CSCs could also be responsible for initiating metastasis (175). This notion has been further strengthened by the link between CSCs and EMT and identification of similar pathways that regulate CSCs and EMT (193,194). Tumor cells undergo EMT to disseminate from the primary tumor mass (195) and MET to colonize at remote sites (196). During EMT and MET, putative CSCs embedded in primary and\or metastatic tumors, become ‘migratory cancer stem cells’ (M-CSCs) when stimulated by environmental signals (175,197). These M-CSCs are thought to be responsible for tumor invasion, metastasis and drug resistance (197). Acquisition of EMT in cancer cells may occur to induce motility in response to adverse environmental changes, and this phenomenon only occurs in a population of cancer cells adaptive to the changed microenvironment after the facilitation of stem cell-like characteristics. In the M-CSC concept, a decisive step for tumor cells to migrate to the tumor–host interface may trigger aberrant signals that may also lead to the induction of EMT (192). According to this concept, most of the M-CSC-like cells are located at the invading edge of the tumors or in the circulation. In this context, few studies have demonstrated a distinct population of migratory stem cells coexpressing stem cell and motility markers at the invasive front of tumors, and patients with tumors with the CSC and motility phenotypes were shown to have an increased incidence of the metastatic disease (198,199). In addition, frequent expression of CSCs and EMT markers in the circulating tumor cells of cancer patients have also been reported (200). In this context, it should be mentioned that OC and endosulfan that are present in pesticides have been shown to induce EMT in human primary cultured hepatocytes and HepG2 liver cells (98,201), suggesting that these environmental carcinogens have the ability to dedifferentiate hepatic cells.
Conclusions
From the practical perspective, it is evident that human cells are predisposed to malignant transformation in response to short or chronic exposure to environmental carcinogens and that epigenetic changes that occur either in in utero or in tissue-specific adult stem cells may have future roles in the developmental process of CSCs and their associated tissue-specific cancers. However, additional efforts are required in defining and establishing the role of CSCs in response to low-dose carcinogen-associated cancers in utero and/or postnatal life. The potential pathways leading to the prospective events that occur during low-dose carcinogen-induced malignant transformation and the involvement of CSC-like cells are depicted in Figure 3.
Figure 3.

Modulation of cancer stem cells and epithelial plasticity by low dose environmental carcinogens.
The aryl hydrocarbon receptor
In order for dormant benign tumor to progress to invasive cancer with more malignant characteristics such as invasion and distant metastasis, the cells of origin undergo major transformation that includes alteration of their epithelial cells into a more motile fibroblastic shape, EMT and increase their expression and secretion of proteases that degrade their surrounding extracellular matrix. Both of these processes are multifactorial and involve regulation of expression of multiple genes and their protein products. Since the triggers for these processes are largely unknown, it is possible that exposure to environmental chemicals either singly or in mixtures is capable of inducing invasive properties. There is a substantial body of literature that links these processes with a group of environmental chemicals that include PAH and halogenated aromatic hydrocarbon.
PAH are ubiquitous by-products of combustion and they include benzopyrene, 3-methylcholanthrene, dimethyl benzo anthracine, etc. Halogenated aromatic hydrocarbons are by-products of incineration, manufacturing processes and vehicle exhaust that have contaminated the food supply (including crops, meat, sea food and dairy products). They include polychlorinated dibenzodioxins, polychlorinated dibenzofurans, planar PCBs and polybrominated biphenyls. The majority of these chemicals are linked to causing cancers in addition to disruption of many other biological processes (e.g. endocrine disruption), and some of them are regulated by IARC and US Environmental Protection Agency (USEPA) as known human carcinogens.
Many of these chemicals invariably function through an intracellular receptor protein, the AHR (202). Binding of these chemicals to AHR leads to its activation to a transcription factor that enhances the expression of multiple genes, including those encoding for cytochrome P450 enzymes (CYP1A1, CYP1A2 and CYP1B1). These enzymes metabolize PAH and some endogenous substrates (e.g. estrogens) into mutagenic intermediates, thus leading to cancer initiation (203–205). The AHR is historically known for mediating cancer initiation by DNA damage through its role in inducing these enzymes. Current evidence suggest major roles in promoting normal and neoplastic cell growth (206–209) and in malignant progression (210–216); however the exact mechanisms for these effects are poorly understood.
TCDD is most often utilized as it is the most potent non-genotoxic AHR agonist. TCDD has been extensively studied as a tumor promoter, which functions by expanding preneoplastic lesions (217), but it can also influence the stage of initiation by facilitating survival of genotoxically injured cells (218).
Role of chemical-activated AHR on the stage of cancer progression
The predominant findings across multiple cancer sites reveal that exposure to many of these persistent AHR ligands leads to increase in cancer progression by enhancing tissue invasion and metastasis (219,220). However, there are some reports that indicate contradictory findings. Here, we will examine the reports on the influence of the AHR on factors regulating cancer invasion and metastasis, both in the presence and absence of exogenous AHR ligands.
Activation of AHR by TCDD induces expression and activities of MMP1, MMP2 and MMP9 in transformed A2058 melanoma cells and increase invasiveness in cell culture, but not in the normal human melanocytes (213). TCDD has also been shown to increase MMP1 expression in keratinocytes (221).
Studies using immortalized mouse mammary fibroblasts from AHR-null mice have shown that they had decreased migration in culture, which was associated with an increase in stress fiber formation and low efficiency to induce lamellipodia (212). Lack of AHR in these cells inhibited signaling pathways that regulate cell migration, including lower activation of FAK, protein kinase B/AKT (PKB/AKT), mitogen-activated kinase, ERK1 and Rac-1.
The involvement of the ligand-activated AHR in cell plasticity and mobility is supported by studies using TCDD. Treatment of mammary carcinoma MCF-7 cells with TCDD resulted in cell scattering, increased cell surface, appearance of lamellipodia-like protrusions and subsequent higher cell motility. These changes were associated with cytoskeleton reorganization, mainly through vinculin and actin redistribution and were accompanied by changes in the expression of several genes, especially, a decrease in E-cadherin. Concomitantly, TCDD activates c-Jun N-terminal kinases (JNKs), which is found to be required for dioxin-mediated effects on cell morphology and mobility (222). The metastasis marker HEF1/NEDD9/CAS-L was implicated as an essential player in AHR-regulated cell plasticity (223). HEF1/NEDD9/CAS-L is a multifunctional docking protein involved in integrin-based signaling that notably affects cell motility and oncogenic transformation (224,225). HEF1 interacts with FAK and the Src family of tyrosine kinases, two critical regulators of focal adhesion (226). As a result, HEF1/NEDD9/CAS-L regulates migratory processes as demonstrated in a melanoma cell line (227); moreover, in several human cancers such as melanoma, glioblastoma (228) and lung tumors (229), increased HEF1/NEDD9/CAS-L expression was found to correlate with the metastasis potential of those tumors.
Closely relevant studies showed that PCB treatment of MCF7 and MDA-MB23 human breast cancer cell lines increased the Rho-associated kinase activity leading to increased cell motility in both the non-metastatic and metastatic cells line. In xenograft mouse model, PCBs significantly advanced disease progression, leading to enhanced capability of metastatic breast cancer cells to metastasize to bone, lung and liver (50).
Testing whether exposure to chemical could disrupt endothelial integrity and increase the transendothelial migration of tumor cells, investigators exposed human microvascular endothelial cell 1 to PCB 104, a representative of highly ortho-substituted non-coplanar PCB congeners, and tested their effect on the endothelial permeability and transendothelial migration of MDA-MB-231 breast cancer cells. They reported that PCB 104 induced endothelial hyperpermeability and markedly increased transendothelial migration of MDA-MB-231 cells. These effects were associated with overexpression of vascular endothelial growth factor through PI3K, but independent of NF-κB pathways (230). In a follow-up study, this team also reported that PCB exposure of endothelial cells stimulated transendothelial migration of tumor cells through upregulation of MMP3. The study provided evidence that PCB can activate EGFR and JAK3 in a closely coordinated and cross-dependent fashion. Activated EGFR and JAK3 stimulate in concert kinases c-JNK and ERK1/2 as well as increase DNA-binding activity of AP-1 and polyoma virus enhancer activator protein 3, leading to transcriptional upregulation of MMP3 expression (231). Dimethyl benzo anthracine, a genotoxic PAH mammary carcinogen, was tested for its ability to promote a more invasive mesenchymal phenotype. The dimethyl benzo anthracine-treated mammary tumor rel-3983 cell line exhibited an increased rate of proliferation, displayed growth to a higher cell density and acquired the ability to grow in soft agar and in Matrigel compared with control cells. The cells lost E-cadherin expression, reflecting their mesenchymal phenotype, and they exhibited increased NF-κB binding and higher levels of the NF-κB-transactivating subunits c-Rel, RelA and RelB, which seemed functional as judged by induction of c-Myc and vimentin, products of two NF-κB target genes (210).
As discussed above, TCDD was shown by many laboratories to promote invasion and metastasis; however, in this interesting report, it was shown that TCDD does that through a completely different mechanism from what was established previously. In this report, it is shown that TCDD induces mitochondrial dysfunction, stress signaling and tumor invasion by a mechanism similar to that described for mtDNA-depleted cells. Treatment of C2C12 cells with TCDD disrupted mitochondrial transmembrane potential in a time-dependent fashion and inhibited mitochondrial transcription and translation. TCDD also increased cytosolic [Ca2+]c and RyR1-specific Ca2+ release. These changes were associated with increased calcineurin levels and activation of calcineurin-sensitive NF-κB/Rel (IκBα-dependent) factors. These findings reveal that TCDD may promote tumor progression in vivo by directly targeting mitochondrial transcription and induction of mitochondrial stress signaling (232).
Exogenous ligand-independent regulation of cancer progression by AHR
The AHR appears to play a role in modulating cancer progression, independent of ligand activation.
In the absence of environmental chemical ligands, antagonism of high constitutively active levels of AHR in cells isolated from patients with head and neck squamous cell carcinoma (HNSCC) blocked the constitutive activation of the AHR. Treatment of these cell lines with the AHR antagonists 6,2′,4′-trimethoxyflavone, or the more potent GNF351, decreased migration and invasion of HNSCC cells (233). This antagonism approach has the potential for inclusion in the strategies for combating the effect of complex chemical mixtures.
Independent of exogenous ligands, the loss of AHR in normal keratinocytes increased migration, reduced levels of epithelial markers E-cadherin and β-catenin and increased expression of mesenchymal markers Snail, Slug/Snai2, vimentin, fibronectin and α-smooth muscle actin (234).
Contradictory effects of TCDD and related chemicals on cancer progression
The majority of these reports are on breast cancer and they stem from the original finding examining long-term feeding of TCDD, where female rats developed tumors in multiple organs except in mammary glands, which appeared protected by TCDD treatment. Later investigations using breast cancer cell lines uncovered that this was due to antiestrogenic effect of TCDD and is AHR dependent. One group observed that in the presence of dioxin, the AHR inhibits proliferation in mammary tumor cells and suppresses the ability of these cells to invade normal tissue (235). In a more recent report, it was observed that activation of AHR by various environmental and natural ligands inhibits invasive and metastatic features of human breast cancer cells and promotes their differentiation (236). Activation of the AHR by TCDD inhibits mammary tumor metastasis in a syngeneic mouse model of breast cancer (237). In addition, the activation of AHR by the flavonoid, β-naphthoflavone, suppressed invasion of esophageal squamous cell carcinoma cell lines (238).
In summary, the AHR appears to be capable of promoting and inhibiting carcinogenesis depending on the tissue and the level of tumor progression examined. Clearly, additional in vivo studies are needed to examine whether the AHR can be utilized as a target for cancer treatment. In particular, different classes of ligands will need to be examined for their effects on tumor growth and survival.
Insights and perspectives
Most of the doses of chemicals used in the studies discussed above are relatively high doses and usually single exposure. Therefore, one has to consider the environmental relevance, where ideally we are exposed to low doses and cumulative exposure. However, this should be easily managed since in the case of this group of chemicals we are dealing with a single receptor protein that mediates the toxic (carcinogenic) effects for all these chemicals.
Effects of chemicals on mechanisms of invasion and metastasis in specific organs
Next, we review the evidence for the role of exposures to environmental contaminants in various organs including the breast, prostate and oral, head and neck.
Breast cancer
Increasing incidence of early stage breast cancer in high-frequency regions, due in part to detection of early in situ lesions through widespread population mammographic screening (239), presents new challenges for the medical oncologist. Better treatment and detection and improvements in breast reconstructive surgery have paradoxically led to an undesirable situation where women get treated for a disease they may never actually develop. This is due to our current inability to distinguish those in situ lesions that will progress to invasive cancer from those that will remain confined or never metastasize. Some are of the opinion that the most effective strategy to reduce breast cancer incidence is simply to stop mass screening (240). Since Beatson’s observation in 1896 (241), estrogen has been known to be instrumental in progression of breast cancer and may also be an initiating factor. Its pharmacological antagonism has been at the forefront of therapeutic efforts. However, although many other risk factors have since been identified in addition to the influence of physiological circulating estrogen, relatively little importance has been placed on exposure to ‘environmental estrogens’ as a significant factor. This includes compounds such as BPA in hard plastics and lining of cans, nonylphenol in cleaning products, benzophenones found in sunblocks, perfumes, soaps and printer toner and pesticides such as dichlorodiphenyldichloroethylene. Although difficult to establish clear causal associations between breast cancer risk and exposure to these and many other substances, nevertheless, they have been detected in significant amounts in breast cancer patients (242). Ibarluzea et al. (243) found an increased risk in postmenopausal women to be associated with total effective xenoestrogen burden (TEXB-alpha) as well as with pesticides aldrin and lindane. Another study (244) demonstrated that BPA can induce neoplastic transformation in cells. In a recent editorial, Darbre et al. (245) made the interesting observation that an estrogenic stimulus could be reached by multiple combinations (from diverse lifestyle exposures) in the absence of a high level of any one particular compound. They highlight various issues of low-dose chronic effects, synergism and timing of exposure, as well as complementation of mechanistic effects of these agents, reflected in the complex nature of carcinogenesis. Phytoestrogens are another source derived from plant foods such as soybeans, tofu, whole grains, fruits and vegetables and certain spices and herbs. Paradoxically they may have anticancer properties. Genistein, for example, appears to have biphasic effects in vitro, stimulating growth at low concentrations and inhibiting at higher levels (246).
For the majority of women with high levels of tumor estrogen receptor (ER), endocrine therapy has been the mainstay of breast cancer treatment for several decades (247,248) but resistance to antiestrogens (249), briefly compensated by use of aromatase inhibitors (250,251) to reduce extraovarian peripheral estrogen production, poses significant problems.
Several mechanisms have been postulated to account for both de novo and acquired resistance, some of which involve activation of alternative growth factor pathways (252,253). In efforts to address this issue, we also established cellular models with functional loss of ER, which display insensitivity to estrogen/tamoxifen (254). In these cells, we observed a phenotypic change corresponding to an EMT, a phenomenon that has been increasingly reported to be associated with tumor progression, invasion and metastasis (255–259) but for the first time here linked to endocrine resistance (260). Meantime several groups have also established associations between EMT and acquisition of stem cell characteristics (261). The concept of stem cells present in adult tissues with capacity for self-renewal by asymmetric division as well as potential to differentiate into replacement cells has a number of attractions and has been extrapolated into a functional role in the progression of cancers. It is already well established that heterogeneity of tumors facilitates their drug-resistant properties (262). EMT and the reverse process of MET (operative in reestablishment of extravasating tumor cells at secondary sites) could be akin to the developmental programs in embryogenesis. Moreover, wound healing following tissue injury involves recruitment of mesenchymal stem cells that could be acquired through MET. Could cancer be viewed as an abnormality of tissue repair?
In 1994, Lapidot et al. (263) demonstrated that transplanting a rare population of immature CD34+CD38− leukemic cells could generate tumors in SCID mice. Since then several studies have shown the presence of a very minor component of solid malignancies that have a capacity for regeneration (264–268) of further neoplasms that subsequently assume the heterogeneity of the parent tumors from which they originated. This small population, possibly numbering only in hundreds, is characterized by a variety of cell surface markers as well as pluripotent embryonic cell markers including the transcriptional activators Klf-4, Oct 4, Nanog and Sox2 (269) and several cluster differentiation (CD) markers. The identification of CSCs by distinct marker profiles could be of clinical utility. Subpopulations of CD133+ cells (a hematopoietic stem cell marker) in brain tumors (265) as well as colon and pancreatic cancers (198,267) possess tumor-initiating capability. Other characteristic CSC markers include epithelial cell adhesion molecule and aldehyde dehydrogenase 1 (ALDH1) in hepatocellular, breast and ovarian cancer (270,271).
The first cancer-initiating cells with CSC-like properties that were identified were isolated from breast tumors; these exhibited a CD44+,CD24−/low phenotype (272). Xenotransplantation of just 100 such cells into NOD/SCID mice could generate tumors, whereas tens of thousands of cells with other phenotypes failed to do so. Breast cancer cell lines with high CD44+/CD24− and expressing basal/mesenchymal or myoepithelial but not luminal markers were selectively more invasive in nude mice (273); however, this was restricted to only a subset of such cells, as similarly reported for a minor fraction of CD44+/CD24−-enriched cells isolated from mammospheres of breast cancer cells (274). Clearly the phenotype needs further definition; just 20 cells with the CD44+/CD24−/ALDH+ phenotype were more tumorigenic than CD44+/CD24− or ALDH+ only cells (256). Coexpression of genes involved in cell motility, chemotaxis, angiogenesis, migration and invasion has been observed in CD44+/CD24− or ALDH+ cells (which overexpress IL-8 receptor CXCR1) and also in CD44+/CD24− cells that express the cell surface protein C receptor, CD201 (275–279). A variety of other signals that promote a metastatic CSC phenotype are extensively discussed in a recent review (280). It would seem that having a phenotype similar to stem cells confers metastatic ability.
A characteristic feature of metastatic tumors is the loss of critical cellular adhesion proteins. The complex signaling network that leads to EMT, involving Snail, Slug and Zeb1, Zeb2/SIP1, Twist, E47 and others, that interact with histone deacetylase and other corepressors on the E-box elements of its promoter, ultimately lead to repression of E-cadherin transcription, and thence to dissolution of cellular junctions, thereby promoting metastasis. Many lines of such evidence point toward a remarkable similarity between processes of metastasis, EMT and those characteristics of stem cells during embryonic development and particularly of CSCs. Several reports have observed that cells undergoing EMT have a CSC-like behavior (261,281,282). Upstream initiators of EMT such as TGF-β, Wnt and Notch are also instrumental in the transformation to the CD44+/CD24− state resembling CSCs. The question arises as to whether differentiated epithelial cells revert to CSCs or whether it is CSCs that acquire EMT traits. Alternatively, Lim et al. (283) have proposed that mesenchymal-like tumors originate from a mesenchymal progenitor cell. This is based on their observation that (the more aggressive) basal subtype breast cancers (which exhibit a mesenchymal gene expression profile) (284) with a BRCA1 mutation actually have a profile similar to that of normal luminal progenitor cells. In this scenario, it is not necessary to hypothesize the de- or transdifferentiation of epithelial cancer cells into the migratory type via EMT. However, preponderance of evidence favors the emergence of CSCs through EMT as prerequisites for metastasis.
Interestingly, CSCs derived from triple-negative (i.e. ER−PR−erbB2−) human breast tumor samples implanted into mice mammary fat pads gave rise, in all animals, to lung metastases (with similar expression profile to parental cells) (279). ER-silenced cells that are endocrine resistant, with increased propensity for invasion and an EMT signature with the mesenchymal characteristic phenotype of CD44+/CD24− (254), also suggest a link between EMT, CSCs, metastasis and loss of ER function and chemoresistance, although a capability to form increased metastases in vivo has not yet been demonstrated. Other studies also suggest a link between acquisition of chemoresistance and stem cell features (285). Thus, even the less aggressive luminal breast cancers with HER2/neu amplification and CSC characteristic of high CD44, low CD24 show drug resistance (286), with chemotherapy actually increasing the high CD44, low CD24 population in resistant tumors.
Certainly there appears to be some common underlying features between these various processes (287) and in all cases they refer to a very small but obviously highly relevant population of cells that logically would seem to be the most optimum therapeutic target. Exploiting molecular differences from the bulk tumor cells, CD133, CD44 or epithelial cell adhesion molecule could be targets of antibody-directed cell killing (288). Another interesting approach is the strategy of reversing EMT either to restore endocrine sensitivity/susceptibility or to inhibit cancer metastasis (289) with reexpression of E-cadherin being a principal objective. Similarly, it should also be possible to push tumors with CSCs into a more differentiated epithelial type less prone to metastasis and vulnerable to cell killing with agents such as salinomycin (290) or histone deacetylase inhibitors such as trichostatin A or vorinostat that are already undergoing clinical trials (291).
Another interesting observation concerns the antidiabetic drug metformin, which is reported to repress CD24 in a triple-negative breast cancer cell line (292) and, in combination with doxorubicin, to eliminate CSCs and repress breast tumors in mice (293). Moreover, CD44+CD24− CSCs in trastuzumab-resistant HER2+ breast cancer cell lines display selective sensitivity to low doses of metformin, which also acts synergistically with trastuzumab to repress proliferation and survival of CSCs (294). Furthermore, metformin effectively delayed acquisition of EMT-induced stem cell phenotype as well as blocking formation of self-renewing mammospheres through inhibition of Zeb1, Twist, Slug and TGF-β, the prototypic mediators of EMT. This is another use of a drug that is proving to be very ubiquitous. It may lead the way to a new strategy of specifically targeting the cells within the bulk of tumors that are destined to be the progenitors of new cancers (295) surviving through the therapeutic net whether it be by developing endocrine resistance, undergoing EMT or arising out of the cancer stem cell niche—or indeed all three.
Breast cancers are divided into five biological subtypes based on gene expression profiles: luminal A, luminal B, ErbB2, normal-like and basal-like (296). Basal-like breast cancer (BLBC) has similar gene expression patterns with myoepithelial/basal cells, which is preferentially negative for the expression of ER, progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2) and positive for basal markers (EGFR, CK5/6 and CK14) (297,298). BLBC contains many EMT markers and possesses abundant stem cell-like traits (193,299,300) and associates with an aggressive clinical history, development of recurrence, distant metastasis and shorter survival (298,301). However, BLBC does not respond to the targeted therapy, such as anti-ER and anti-HER2 (302,303), and has poor response to chemotherapy. Therefore, identification of the relevant molecular targets in BLBC is urgently needed before meaningful therapies could be achieved.
The molecular drivers that are responsible for the aggressive phenotype of BLBC remain unclear. With analysis of the gene expression pattern of BLBC, many molecules that may serve as specific therapeutic targets have been investigated. Here, we will highlight recent discoveries that have led to new insights into the role and mechanism of BRCA1, p53 and Snail in aggressive basal-like subtype of breast cancer.
BRCA1 and BLBC.
BRCA1 as tumor suppressor plays a central role in maintaining genome integrity (304). Women with inactivating mutations in BRCA1 have high rate of developing breast cancer in their lifetime. BLBC is particularly related to BRCA1 mutations, and about three-quarters of BRCA1-mutated cancers display basal-like phenotype (305). In sporadic basal-like cancers, somatic mutation of the BRCA1 gene has rarely been found (306). However, decreased levels of BRCA1 in messenger RNA and protein have been widely observed in BLBC. Recent studies have demonstrated that suppression of BRCA1 is associated with transcription regulation in sporadic BLBC. For example, inhibitor of DNA binding 4, which functions as a dominant-negative regulator of basic helix-loop-helix transcription factors, leads to the downregulation of BRCA1 expression (307). Significantly, a higher expression of inhibitor of DNA binding 4 is observed in BLBC with mutant BRCA1 (308). In addition, epigenetic regulations also play important roles in BRCA1 silencing in sporadic BLBC. Aberrant methylation of BRCA1 promoter is found in 11–14% of sporadic breast cancer (309). Metaplastic breast cancer (a rare subtype of BLBC) has an extremely high BRCA1 promoter methylation rate of 63% (308). The expression profile of sporadic tumors with BRCA1 methylation has a very similar profile with BRCA1-mutated tumors (305), indicating the importance of BRCA1 methylation in BLBC.
BRCA1 plays a critical role in normal mammary gland development and is required for the conversion of ER-negative cells to ER-positive. BRCA1 regulates cell fate and is involved in stem cell regulation, indicating that BRCA1 silencing may be required for the differentiation of ER-negative stem/progenitor. This speculation is supported by findings in a mouse model that demonstrated somatic loss of BRCA1 and p53 cause highly proliferative, poorly differentiated, ER-negative mammary tumor and show increased expression of basal-like markers.
p53 and BLBC
Mutant p53 is present in 82% of BLBC (310). Recent studies have shown that mutant p53 contributes to EMT, which is a process by which epithelial cells lose intercellular adhesion and cellular polarity, gain plasticity in cell migration and invasion (311,312). Several possible mechanisms are elucidated for mutant p53-mediated EMT. First, mutant p53 induces EMT by enhancing the function of EMT inducers, Twist1 and Slug (313,314). Second, mutant p53 can bind to and sequester the function of p63, thus facilitating TGF-β-induced EMT and metastasis (315). Third, mutant p53 overrides wild-type p53-mediated inhibition of hyaluronic acid receptor (CD44) (316).
Tumor cells preferentially utilize anaerobic glycolysis to generate energy, a process called Warburg effect, which provides potential survival advantages conferred by increased glycolytic flux and decreased oxidative phosphorylation (317). Interestingly, BLBC are more glycolytic than other subtypes (318). Mutant p53 promotes differentiating cells to survive by inducing a metabolic shift toward glycolysis. p53 is involved in many points in both glycolysis and oxidative phosphorylation (OXPHOS). p53 influences glycolitic flux by regulating glycolysis-related molecules (PGM, GLUT1, GLUT4 and TIGAR) (319,320) and promotes mitochondrial activity by activating OXPHOS pathway (321). For example, on one hand, p53 protein induces the expression of TIGAR, which lowers the level of fructose-2,6-bisphosphate and thus decreases glycolysis (319). On the other hand, p53 facilitates the rate of oxygen consumption through its downstream mediator, SCO2, a critical cytochrome c oxidase for regulating oxygen utilization in cells (321). Loss of p53 switches OXPHOS to glycolysis through the disruption of TIGAR and COX functions and thus significantly enhances the Warburg effect. Mutant p53 contributes survival and proliferation by this metabolic alteration, which at least partially accounts for the vulnerability to therapeutic intervention in BLBC.
Snail and BLBC
As a key transcriptional repressor of E-cadherin, Snail—which controls EMT, stem cell phenotypes and therapeutic responsiveness (322,323)—is highly expressed in BLBC (318,324–326), making it a candidate master regulator of the BLBC phenotype. Recently, the epigenetic regulation mediated by Snail in EMT has been intensively studied. Snail recruits several chromatin-modifying enzymes, including LSD1, G9a, Suv39H1, DNMTs, HDAC1/2 and PRC2 to the E-cadherin promoter for transcriptional silencing. These studies strongly demonstrate the central role of Snail on epigenetic silencing of E-cadherin by recruiting multiple repressor complexes to the E-cadherin promoter. Importantly, these Snail-mediated epigenetic modifications, such as H3K9me2/3 and DNA methylation, are enriched at the E-cadherin promoter in BLBC (324,327).
E-cadherin is a known transcriptional target of Snail. However, other targets, particularly those in regulating metabolic process, remain unclear. Our recent study shows that FBP1, a rate-limiting enzyme in gluconeogenesis, is a direct target of Snail. Snail binds to the FBP1 promoter to shut down its expression (318). Clinically, FBP1 is specifically lost in BLBC but not in other subtypes of breast cancer (318). Loss of FBP1 expression enhances the Warburg effect by enhanced glycolysis, increased glycolytic intermediates for biosynthesis and decreased mitochondrial activity, which is consistent with the notion that the Warburg effect positively correlates with aggressive behaviors seen in BLBC (318). In addition, our study also demonstrates that loss of FBP1 synergizes with loss of E-cadherin to facilitate the conversion from luminal to basal-like phenotype (318).
Snail not only induces EMT but also contributes to the acquisition of CSCs properties. However, how Snail-induced EMT contributes to the CSC trait in BLBC remains unclear. Loss of FBP1 by Snail repression results in the increased CSC characteristics (318). Our study demonstrates that loss of FBP1, which promotes metabolic switch from OXPHOS to the Warburg effect, causes decreased mitochondrial ROS (318). The low level of ROS shifts the interaction of β-catenin toward TCF4 instead of FOXO3a, which is consistent with the contention that BLBC cells contain β-catenin activation signature (328). Together, these data provide new insight into the cross talk of the Snail-FBP1 axis-mediated metabolic reprogramming with CSC-like phenotypes that associated with the clinical aggressive BLBC.
Perspective
Molecular subtyping in breast cancer has generated important impetus to deepening our understanding of the molecular characteristic of BLBC. At present, significant advances have been made in our understanding of the molecular mechanism of BLBC, such as dysfunction of BRCA1, mutant p53 and overexpression of Snail. These molecular specificities provide new candidates for therapeutic targets of BLBC. However, a number of fundamental questions, such as screening of targeted molecule inhibitors and evaluation of therapeutic effect, remain to be explored before arriving at novel treatment strategies that can target these molecules to achieve more efficient and selective killing of BLBC cells.
Prostate cancer
PCa is the second leading cause of cancer-related deaths in American men over the age of 60 and also the most commonly diagnosed cancer in Western men (329). In 2013, there were ~238 590 new cases diagnosed with 29 720 deaths in the USA. The formation and development of PCa normally proceed from the enlargement of prostate gland, invasive adenocarcinoma around tissues to distant metastasis in other organs. Surgery and radiation therapy are the two main modalities for the treatment of PCa patients with local adenocarcinoma at the early stages, whereas chemotherapy, including androgen-deprivation therapy, is used for PCa patients with distant metastasis at advanced stages. Tremendous advancements have been made by clinicians and scientists to reduce the incidence and mortality of PCa; however, many PCa patients still die of castration-resistant PCa. Two main questions still remaining to be addressed: (i) the exact driving mechanisms of initiation and development of PCa, including castration-resistant PCa and (ii) the impact of environmental factors including chemical carcinogens in the mechanisms that drive PCa. Understanding the roles of environmental factors including chemical carcinogens in PCa will certainly help us know the malignant effects or the correlations.
Roles of oncogenic signaling pathways in PCa
Several reports have shown that aberrant regulation of multiple oncogenic signaling pathways play essential roles in the initiation, development and progression of PCa (330). Deletions and mutations of tumor suppressors and activations of oncogenes are frequently found in PCa specimens. In vitro and in vivo animal models have shown that PCa can be induced by deletions and mutations of tumor suppressors and/or activations of oncogenes. As such, it is well accepted that the genetic alterations of tumor suppressor and oncogene genes drive PCa. Synergistic collaborations of tumor suppressors with oncogenes significantly accelerated PCa progression. For example, loss of tumor suppressor p53 accelerates Pten-deficient tumorigenesis (331), activation of K-Ras accelerated Pten-deficient tumorigenesis (332) and NF-κB accelerates c-Myc tumorigenesis (333). Furthermore, emerging evidence reveals that cancers, including PCa, are driven by sustained dysregulation of multiple signaling pathways including tumor suppressors and oncogenes at genetic and epigenetic levels. Currently, most studies are focused on defining the functional roles and mechanisms of tumor suppressors and oncogenes in PCa using genetics and molecular biology approaches. The information from these studies indeed reveals the oncogenic impact of these essential genes in given biological systems when they are deleted and mutated in an aberrant manner. The knowledge is very important and valuable, which certainly provides us with the solid bases to develop potent anticancer drugs to control PCa. Many effective chemotherapies have been generated to target androgen receptor, Akt/mTOR and MET signaling pathways in PCa. Despite these great advancements, it is still unclear about the exact causes on alterations of tumor suppressors and oncogenes in human PCa at genetic and epigenetic levels. Emerging evidence revealed that aberrations of histone modifications play critical roles in advanced PCa including distant metastasis and castration-resistant PCa (334,335). It is unclear whether and how environmental factors contribute to epigenetic alterations including histone modifications. Needless to say, the information will be important and helpful in driving down the incidence PCa.
Effects of environmental factors on PCa
Reports from various sources including environmental studies, epidemiology and laboratory experiments support the fact that environmental factors play important roles in the incidence of various forms of cancers including PCa (336–338). For example, PCa incidence in World Trade Center rescue and recovery workers was significantly increased compared with non-registered populations, suggesting that exposure to hazardous substances and carcinogens induces PCa (339,340). Elevated incidences of PCa were also found in fire fighters, chimney sweepers and pesticide workers (341–343). Recent studies indicated the consumption of fat and red meat significantly increased the risk of PCa malignancy (344–346). Heterocyclic amines and PAHs generated from frying, grilling and barbecuing contribute to advanced PCa (347), likely through DNA mutagenesis (348). PhIP induced an increase in phosphorylation of both MAPK/ERK kinase 1/2 and ERKs to promote PCa progression. These results indicate that chemical carcinogens are also contributing to PCa through regulation of essential signaling pathways (349). In addition, environmental factors and chemical carcinogens can cause chronic inflammation in prostate glands, which also plays a promoting role in PCa development (350). Despite these important observations, the defined impact of specific chemical carcinogens in etiological relevance to PCa in humans is still unclear. A big experimental challenge is that the prostates of animal models are inaccessible to direct administration of chemical carcinogens. Chemical carcinogens administered via routine routes such as gavage and IP are rarely detected in prostate glands (351). So far, few groups have successfully demonstrated the effects of chemical carcinogens on prostate carcinogenesis in rat models (352). N-nitrosobis (2-oxopropyl)-amine and N-nitrosobis (2-hydroxypropyl) amine can induce PCa in rats (353). Certainly, additional investigations will provide more insights into understanding roles of chemical carcinogens in PCa.
PCa prevention
Since PCa is the combined consequence of genetic alterations and environmental factors, it is possible that the incidence of this malignancy can be reduced with appropriate approaches to neutralize the environmental effects, at least. Studies showed that many natural products in plants and vegetables display their anticancer and chemoprevention potentials for various cancers including PCa (354,355). Their anticancer and chemoprevention functions are normally executed through suppression of cell cycle, inhibition of oncogenic activation and anti-inflammatory activity caused by chemical carcinogens. For example, the consumption of soybeans is beneficial due to the isoflavones that reduce PCa risk by inhibiting the cell proliferation and transformation (356). Green tea has cancer prevention potentials as it contains polyphenols that may mediate androgen receptor activity and signaling pathways (357). Due to the variations of bioactivities of these anticancer compounds in plants, standardized applications may be developed to achieve the chemoprevention effects for PCa control (358). Furthermore, additional studies should be performed to further define the chemoprevention mechanisms.
Taken together, we have demonstrated that low levels of carcinogens and other environmental disruptors can modulate pathways that are relevant in tumor invasion and metastasis in tumors ranging from breast to prostate. Certainly more work is needed in this line of investigation since there are more questions than answers as depicted in Figure 4. Little is known regarding the impact of low-dose environmental contaminants/disruptors on tumor stem cells that are responsible for seeding metastatic tumors.
Fig. 4.

Potential impact of environmental disruptors on the key pathways of tissue invasion and metastasis. The disruptive pathways supported by experimental evidence are denoted by +, whereas those for which there are no strong experimental evidence are denoted by ? +.
Oral, head and neck cancer
HNSCC and metastasis
HNSCC continues to be a disfiguring and deadly disease. It is the most common malignancy of the upper digestive tract, affecting ~48 000 individuals in the USA annually with a 5-year disease-specific survival of a dismal 56%, which has not improved over the past three decades (Surveillance, Epidemiology, and End Results, SEER, National Cancer Institute). This high mortality associated with HNSCC is considerably worse than rates for breast, cervical, prostate and colorectal cancers (American Cancer Society, 2011). HNSCC are marked by their aggressiveness and invasiveness and despite improvements in the local control of HNSCC, many patients continue to die due to the metastatic disease (359). Metastatic behavior is critical to survival, since patients with distant disease have a 5-year survival rate that is three times less than those with nodal metastases (360,361). Therapeutic options for advanced metastatic HNSCC are limited and unsuccessful (362,363). Elucidation of the molecular mechanisms controlling HNSCC metastasis is critical to improving clinical outcomes.
Oral cancer and metastasis
Approximately two-thirds of the oral cancer occur in the oral cavity (lip, tongue, floor of mouth, palate, gingival and alveolar mucosa, buccal mucosa), whereas the remainder occurs in the oropharynx (360). OSCC is the most prevalent and aggressive epithelial tumor of the head and neck region, with the poorest clinical outcome: in the USA alone, ~100 new cases are daily diagnosed, while one person dies from oral cancer every hour of every day (359,364–366). Worldwide the problem is far greater with new cases annually exceeding 640 000 (oralcancerfoundation.com). Primarily a men’s illness, affecting six men for every woman, over the past 10 years that ratio has alarmingly become 2:1 (367). Traditional risk factors such as tobacco use and alcohol consumption have a greater multiplicative effect and account for the majority of HNSCC (368). However, these established risk factors do not account for ~40% of oral cavity cases. It has been suggested that yet unknown genetic, occupational, or nutritional factors could influence risk in this age group (369). Interestingly, large epidemiological studies have found that age (<45 years) correlates with the development of distant metastasis in patients with HNSCC (370). Even younger age (<40 years) has been found relevant in oral cancer (371). OSCC displays a wide range of metastatic behavior that cannot be predicted by tumor size, standard histology or even individual gene or protein expression/activity (360,372). Despite the clinically obvious heterogeneity of OSCC, there are currently no means of predicting individual tumor behavior (372). Even small primary tumors of the oral cavity have a propensity to metastasize to cervical nodes, mandating that the majority of patients, even those with no clinical or radiographic evidence of nodal metastases, undergo some form of neck treatment either for staging or therapeutic purposes. Accurate prediction of metastasis in OSCC would have an immediate clinical impact through avoidance of unnecessary treatment of patients at low risk with appropriate direction of resources toward aggressive treatment of patients at high risk of having metastatic disease.
Genetic, antigenic and phenotypic changes in HNSCC metastasis
Deregulation of several pathways has been implicated in metastatic dissemination in head and neck area (372–377), making HNSCC a complex malignancy not yet fully understood. There is strong evidence that pathways’ activation may occur in response to tobacco, alcohol, stress, dietary agents, obesity, infectious agents, irradiation and environmental stimuli and that some transcription factors are constitutively activated in HNSCC (72,377). The mechanism underlying the invasion of carcinoma cells leading to tumor dissemination involves the EMT of cells with high tumorigenic potential (378,379); it is also known that EMT endows epithelial cells with invasive and stem cell properties (193). Cancer stem cells seem to localize at the invasive fronts of the HNSCC in the proximity of the blood vessels (380). We have recently shown that CSCs are important to the development of metastasis from oral, head and neck tumors (381,382). It has been hypothesized that CSCs may be relatively resistant to conventional chemotherapy and radiotherapy, owing to high expression of drug transporters, relative cell cycle quiescence, high levels of DNA repair machinery and resistance to apoptosis (363,383). The squamous epithelium covering the oral mucosa relies on epithelial stem cells for tissue renewal (384). CSCs and normal stem cells share similarities. The deregulation of pathways that control the self-renewal of normal stem cells, including Wnt, Notch and Hedgehog, leads to tumorigenesis in rodent models and also plays an important role in human carcinogenesis including HNSCC (385–390). Being long-lived, both CSCs and stem cells of normal tissues can be the targets of environmental carcinogens leading to the accumulation of consecutive genetic changes although several protective mechanisms have evolved to ensure the genetic integrity of the stem cell compartment in any given tissue (383).
Chemical carcinogens and environmental factors in HNSCC/OSCC
The head and neck region is an environment challenged by a large variety of insults, including pathogens and viruses, foods and chemicals. The significant increase in global industrialization and urbanization has changed the characteristics of our environment particularly over the past few decades and it is estimated that as many as two-thirds of all cancer cases are now linked to environmental causes (National Cancer Institute and National Institute of Environmental Health Sciences, 2003). The IARC has identified intake of alcoholic beverages, betel quid chewing with tobacco and cigarette smoking as human carcinogens, with various target organs from the head and neck area, including the oral cavity, pharynx, larynx and esophagus (374,391–398). Several chemicals have been recognized to increase human cancer risk in various subtypes of HNSCC, e.g. esophageal cancer (PAHs contamination of the food); oral cancer and esophageal cancer (exposure to heavy metals including arsenic, chromium, nickel and cadmium); oral, naso- and hypopharyngeal cancer (exposure to formaldehyde) (395,399–411). It is also known that radium, via oral exposure, can cause bone, head and nasal passage tumors in humans reviewed by Brugge et al. (412). Emerging evidence indicates iron as potential causative agent of various types of cancers with a proven role in tumor metastasis; high dietary iron seems to increase cancer risk via a mechanistic link with signaling in cancer cells, particularly the Wnt pathway (413). High levels of various metals including iron have been detected in the serum, hair and nails of patients with head and neck cancer (414). Chemical carcinogenesis involves a complex series of events, the earliest of which typically includes DNA damage. Evolution of DNA methylation has allowed cells to respond to environmental cues in a flexible, yet stable manner, by properly regulating the response at the molecular level. However, dysregulation of DNA methylation can lead to hyper- or hypomethylation of the promoters of critical genes, contributing to various diseases including HNSCC. Several reports have identified hypermethylation or hypomethylation in the promoters of key genes involved in oral, head and neck cancer (415–418) and have also demonstrated unequivocally that the vast majority of human HNSCC tumors contain multiple mutations (419,420). It has been shown that variability in DNA methylation exists within subtypes of HNSCC and is influenced by environmental factors such as diet (421,422). The potential interactions of chemical carcinogens and other environmental factors with the metastatic pathways that are altered in OSCC/HNSCC are under investigation (423). Promising epidemiologic studies, basic research and clinical investigations indicate that a diet rich in cruciferous vegetables and micronutrients may increase carcinogen metabolism, induce apoptosis and reduce the risk of developing head and neck tumors (424,425).
Pathogens and viruses in HNSCC/OSCC
A growing body of evidence implicates human oral bacteria (over 700 bacterial species inhabit the oral cavity) in the etiology of oral cancers and epidemiological studies consistently report increased risks of these cancers in patients with periodontal disease or tooth loss; furthermore, oral bacteria may activate alcohol and smoking-related carcinogens locally or systematically through chronic inflammation (426). Human papillomavirus (HPV) has recently emerged as the primary etiologic factor particularly for tumors developed in the tongue and oropharynx that are also associated with younger age at diagnosis; 65–85% of the oropharyngeal cancers diagnosed this year in the USA are HPV related with 3-year failure rates of 30–36% (427–434). The incidence of HPV(+) oropharyngeal cancers has increased by 225% from 1988 to 2004 in the USA; their annual number is expected to surpass the annual number of cervical cancers by the end of this year, 2014 (World Health Organization—IARC report, 2007). Consequently, unique pathologic profiles have emerged that are consistent with the changing incidence of HNSCC (435–437). Patients with HPV(+) head and neck cancer have a distinct risk profile, associated with a less remarkable history of tobacco and alcohol use (438,439), a more beneficial micronutrient profile (440) and improved survival compared with those with HPV(−) tumors (441–444). Notably, a significant subset (20–30%) of HPV(+) tumors fails to respond to treatment, recur locally or spread distantly. Studies conducted at the University of Michigan have made significant contributions for the understanding of the impact of HPV infection on the pathobiology of HNSCC and response to therapy (442–444). However, the mechanisms involved in these processes are not understood, and given the evolving epidemiology, there is a growing controversy over the optimal strategy for oropharynx cancer treatment (443,445).
Challenges to the integrity of the oral mucosa
The mucosal lining of the oral cavity and esophagus functions to protect the underlying tissues and organs against mechanical and chemical insults, including microorganisms and toxins that may be present in the oropharynx, or ingested antigens and carcinogens (446). There is an effective barrier in their stratified squamous epithelia and materials with various chemical properties cross this barrier by different routes; the intercellular compartment of the surface layer of the epithelium is the predominant route that can be rapidly saturated; subsequently, the absorbed materials will diffuse into the deeper layers at a rate that is more dependent on the reservoir capacity of loading than on the duration of surface exposure, suggesting that significant amounts can be absorbed through oral mucosa even after short exposure. Remarkably, the epithelium is constantly replaced with a rapid clearance of surface cells, which therefore may act as a protective mechanism (446).
Of the total surface of the oral lining, ~25% is keratinized resembling that of the epidermis covering the skin in regions subject to mechanical forces (masticatory mucosa of the gingiva and hard palate), 60% is the non-keratinized lining mucosa in the regions requiring flexibility to accommodate chewing, speech or swallowing (floor of the mouth, buccal regions, esophagus, etc.), with the remaining 15% as the specialized mucosa (dorsum of the tongue) that can be represented as a mosaic of keratinized and non-keratinized epithelium (447). The turnover of the oral mucosa is faster in the lining than in the masticatory regions, thus challenges to the integrity of the oral mucosa will affect in particular the more rapidly proliferating areas and the lining regions will suffer first (448–450). DNA is most vulnerable during mitosis, and it has been shown that mitotic activity can be affected by a number of factors, such as time of day, stress and inflammation (446,449). It is also becoming evident that the association between non-keratinocytes and keratinocytes in skin and oral mucosa represents a finely balanced relationship in which cytokines represent the controlling factors (451). There is an increasing wealth of data in support of a molecular cross talk between inflammation and tumor progression with several inflammatory mediators that have also been shown to mediate metastatic development (452). Recent in vitro and clinical studies at the University of Michigan have identified various proinflammatory and tumor-promoting soluble cytokines as good indicators of metastatic progression and survival in OSCC/HNSCC (453,454). The microenvironment is increasingly recognized as relevant in the process of metastasis as it is the immunity (455–457). Several studies indicated that HNSCC is strongly associated with alterations in the immune system, leading many to postulate that progression of HNSCC tumors is linked to immune evasion or failure of the immune system to fight the cancer (458–462). Other immunological host factors have been shown to be relevant to the process of metastasis, e.g. deficiency in natural killer (NK) T cells that has been reported to result in poorer outcomes in HNSCC, along with other immune system dysfunctions that have been described in HNSCC, including reduced circulating dendritic cells and reduced HLA-DR expressing in circulating tumor cells, diminished NK and NK-T activity and numbers in peripheral blood, etc. (458,463,464). Furthermore, it is known that incidence of some cancers is increased in immunocompromised patients and in the elderly owing to immunosenescence (465,466); the age-accumulative effects of exposure to carcinogens and environmental factors could also count against it (404).
Circadian rhythms and HNSCC/OSCC
Poor sleep pattern, poor dietary habits, stress and a sedentary lifestyle have been early recognized as able to increase cancer risk and to modify carcinogenic potency in HNSCC patients (368,467–470). In industrialized societies, changes in lifestyles lead to frequent disruption of endogenous circadian rhythm (sleep duration, pattern and quality, meals timing and characteristics, less direct social interaction) in ~50% of the human population, which contributes to increased cancer development worldwide (471). Disruption of circadian rhythms in animal models confirmed increased tumor development (410). Recent literature suggests key roles of clock genes in cancer progression in humans. Expression of critical cancer-related genes such as c-myc and p53 has been reported to follow a circadian rhythm in vivo (472–474). Loss of function of circadian genes leads to deregulated DNA damage and neoplastic growth resulting in oncogenic activation, uncontrolled cell proliferation and increased tumor development in mice; notably, beta-catenin, the key player of the Wnt pathway, has been identified as direct target gene of the clock genes (474). It has been demonstrated that there is a clear circadian rhythm for buccal epithelium (449). Despite the scarcity of data in regard with the implications of clock genes in oral epithelium homeostasis (449), overall the existing findings suggest that detection of the asynchronous regulation of clock genes and their targets might allow for a more accurate assessment of cancer risk or monitoring of tumor progression. Additional studies have confirmed p53, cyclin D1 and NF-κB as direct targets of the circadian clock genes (472,474,475) as well as increased incidence of cancers in people with disrupted circadian rhythms (476); however, the direct links between aberrant circadian clock gene expression and human malignancies remain largely unknown. It has been postulated that these disturbed circadian rhythms in people doing shift work is rather associated with the downregulation of tumor suppressor genes during wake times, and thus, it is more likely that the tumorigenic factors exert their impact during vulnerable (of low protective genes) phases of diminished tumor suppressor gene regulation, which could persist even if the individual sleeps regularly during night times again (477). Limited data are available in regard with the clock genes disturbances in oral, head and neck carcinogenesis (379,478,479). Downregulation of several circadian clock genes has been recently reported in patients with HNSCC that correlated with more advanced cancer stages, larger tumor size, deeper tumor invasion and poor patient survival, providing the first evidence of a possible association of circadian clock genes with HNSCC pathogenesis (480). These emerging clinical evidences showed promise for more effective chronobiology-based individualized treatment strategies in HNSCC patients (481,482). It will be interesting to explore if diurnal versus nocturnal exposure to carcinogens and environmental factors may have different biological consequences. Our head and neck cancer group at the University of Michigan have begun characterization of the biology of the subset of patients who die of distant metastases using molecular biomarkers, cellular immune parameters, nutritional and health behavior and epigenetic-based studies, in parallel with the investigation of new chemopreventive regiments that particularly target the metastatic dissemination as foundation for novel mechanism-based therapies (379,440,483). Recent investigations have tried to determine the predictors of poor quality sleep in patients with head and neck cancer (484) and how healthy behaviors (a proper daily life routine including physical activity, diet, sleep, etc.) influence inflammatory mediators and affect survival in these patients (453,485,486). Future studies focused on better understanding of the consequences of exposure to chemical carcinogens and environmental factors as it relates to oral, head and neck carcinomas are needed. Such knowledge will positively impact the assessment of cancer risk, cancer prevention, early detection and clinical management of HNSCC in the future.
Cross talk between tissue invasion–metastasis and the other hallmarks of cancer
Given that the carcinogenicity of low-dose exposures to chemical mixtures in any given tissue will likely depend on simultaneous instigation of several important tumor promotion mechanisms and the disruption of several important defense mechanisms, it was felt that a better way of visualizing the potential synergies of combinations of chemicals will ultimately involve a thorough review of disruptive actions across the full range of mechanisms that are known to be relevant in cancer biology. Accordingly, we undertook a thorough cross-validation activity to illustrate the importance of the prioritized target sites for disruption that this team has identified (i.e. across multiple aspects of cancer’s biology) and to illustrate the extent to which the prototypical chemical disruptors that we identified (i.e. also disruptive to other mechanisms that are also relevant to carcinogenesis). The validation data are summarized in Tables 1 and 2.
Table 1.
Cross-validation of target pathways
| Tissue invasion and metastasis priority targets | Genetic instability | Angiogenesis | Sustained proliferative signaling | Evasion of antigrowth signalling | Resistance to cell death | Replicative immortality | Immune system evasion | Tumor-promoting inflammation | Tumor microenvironment | Deregulated metabolism |
|---|---|---|---|---|---|---|---|---|---|---|
| Cyclooxygenase expression and stimulation of calcium signaling in migration | + (487) | + (488) | + (at least the COX-2 part) (489) | + (490–492) | + (at least the COX-2 part) (493) | − (494–496) | + (497,498) | + (489,499, 500) | + (501) | 0 |
| cSrc/Her1/STAT5B/ ERK1/2 | 0/+ (502), + (503), − (504) | 0 | − (505) | 0 | + (506–508) | 0 | 0 | + (509,510) | + (509) | 0 |
| EMT, catenin-Wnt pathway | 0/+ (511,512) | 0 | + (513) | − (514) | −/+ (515,516) | + (517,518) | 0 | + (519,520) | + (521) | + (522) |
| MMP9 activation | 0 | − (523) | + (524) | + (525–527) | −/+ (528–531) | 0 | + (532) | + (533) | + (521) | 0 |
| EMT | 0 | 0 | 0 | + (534–542) | −/+ (515,516) | + (543,544) | 0 | + (545) | + (546) | + (547) |
| Clock genes- mediated metastasis | 0 | 0 | + (548,549) | + (550) | + (at least the clock genes part, not the metastasis) (551) | − (552,553) | 0 | + (554,555) | 0 | + (556) |
Target pathways for tumor invasion and metastasis were cross-validated for effects in other cancer-associated pathways. Targets that were found to have opposing actions in a particular pathway (i.e. anticarcinogenic) were denoted using ‘−’ effects, whereas targets that were found to have promoting actions in a particular pathway (i.e. carcinogenic) were denoted using ‘+’. In instances where reports on relevant actions in other cancer-associated pathways were mixed (i.e. reports showing both procarcinogenic potential and anticarcinogenic potential), the term ‘−/+’ was used. Finally, in instances where no literature support was found to document the relevance of a target in a particular aspect of cancer’s biology, we documented this as ‘0’.
Table 2.
Cross-validation of disruptors
| Tissue invasion and metastasis disruptors | Deregulated metabolism | Replicative immortality | Tumor-promoting inflammation | Resistance to cell death | Genetic instability | Immune system evasion | Angiogenesis | Tumor microenvironment | Evasion of antigrowth signaling | Sustained proliferative signaling |
|---|---|---|---|---|---|---|---|---|---|---|
| BPA | + (557–560) | + (561) | + (562) | −/+ (563–566) | + (567) | 0 | + (568) | 0 | + (563) | + (569) |
| Hexachlorobenzene | + (570–572) | 0 | + (510,573) | − (574,575) | + (576) | 0 | − (577) | 0 | + (578) | + (579) |
| Iron | 0 | −/+ (580–582) | + (583) | − (584) | + (585) | + (586) | +/− | + (587) | 0 | + (413) |
| SO2 | 0 | 0 | + (588,589) | − (590,591) | + (592) | 0 | + Patent application number: 20100080843 | 0 | 0 for carcinogenesis but + (593) | + (593) |
| Phthalates | + (594–596) | 0 | + (594,596) | −/+ (595,597–600) | + (601) | 0 | + (602) | 0 | + (603) | + (604,605) |
| Biorhythms | − (606) | 0 | + (607) | 0 | + (472) | 0 | − (608) | 0 | 0 | + (609) |
Disruptors of tumor invasion and metastasis were cross-validated for effects in other cancer-associated pathways. Disruptors that were found to have opposing actions in a particular pathway (i.e. anticarcinogenic) were denoted using ‘−’ effects, whereas disruptors that were found to have promoting actions in a particular cancer-associated pathway (i.e. carcinogenic) were denoted using ‘+’. In instances where reports on relevant actions in other cancer-associated pathways were mixed (i.e. reports showing both procarcinogenic potential and anticarcinogenic potential), the term ‘−/+’ was used. Finally, in instances where no literature support was found to document the relevance of a chemical in a particular aspect of cancer’s biology, we documented this as ‘0’.
Metastasis is a complex process that is defined as the ability of cancerous cells to migrate to and reside in tissues distant to the primary site. In almost all cancers, metastasis capacity is what defines their aggressiveness. The metastatic process is complicated and involves many key steps and the dysregulation of many pivotal cellular pathways. Although invasion is a hallmark of all cancers, metastatic capabilities vary between different cancers and within each type of cancer. For example, although basal cell carcinoma is known for its invasiveness, its ability to metastasize is limited, whereas other cancers like lung and pancreatic attain an aggressive metastatic phenotype more rigorously. Previous work has shown that metastasis is an inherent characteristic of most cancers, which paved the way for the detection of metastatic capabilities by analyzing the genetic and epigenetic profiles of the primary tumor. Therefore, metastasis is not all or non-phenomenon; it represents a continuum where some cells may be very proficient at it, to others, perhaps within the same tumor, which metastasize poorly. In its basic characteristic, a metastatic cell has to dissociate from the primary body of cancer, invade its surrounding normal cells, invade through lymphatic and vascular channels, survive the hostile circulation, extravagate, occupy and survive within foreign tissues. This multistep process constitutes the hallmark of cancer metastasis and it involves several conserved and pivotal cellular capabilities such as deregulated metabolism, replicative immortality, tumor-promoting inflammation, resistance to cell death, evasion of antigrowth signaling, angiogenesis, genetic instability, immune system evasion, sustained proliferative signaling and the tumor microenvironment (Tables 1 and 2).
To illustrate our cross-validation, we have selected cyclooxygenase expression and stimulation of calcium signaling in migration (Table 1), which have been proposed as a priority mechanism of interest in tumor invasion and metastasis, were found complementary in genetic instability (610), angiogenesis (611), evasion of antigrowth signaling (611–613), sustained proliferative signaling (614), resistance to cell death (615), immune system evasion (497,616), tumor-promoting inflammation (499,500,614) and tumor microenvironment (501). No known relationship is reported in deregulated metabolism. Interestingly, calcium is believed to bind to telomerase complex and inhibits its activity with a contrary role through replicative immortality pathways (617,618). The selected prototypical disruptor influencing tumor invasion and metastasis BPA is striking (Table 2). Treatment of cell lines with BPA has been shown to result in a number of cellular and molecular changes across tissue types, which are also concentration dependent. However, there is no one clear molecular target of BPA and the role of BPA in human disease is controversial. In our validation, BPA resulted complementary in most of the hallmarks like angiogenesis (568), evasion of antigrowth signaling (563), replicative immortality (561), tumor-promoting inflammation (562), genetic instability (567), deregulated metabolism, sustained proliferative signaling (569) and partial complementary in resistance to cell death (564–566). No known studies with BPA were found in immune system evasion and tumor microenvironment.
The cases of cyclooxygenase expression and stimulation of calcium signaling and BPA reveal a significant degree of synergy between tumor invasion and metastasis and most of the hallmarks in carcinogenesis. Nevertheless, the validation process showed frequently not known relationship for the other targets/pathways influencing tumor invasion and metastasis (Table 1). At this stage, the identification of solid correlations between these environmental chemicals, tumor invasion and metastasis and the network of cancer hallmarks is indeed a challenging process. Nevertheless, these data suggest that more studies are needed to understand the mechanism of tumor invasion and metastasis. Finding a way to block these pathways would be a great step toward new therapeutic approaches (single or in combination).
Funding
National Institute of Environmental Health Sciences (R13ES023276); National Institute on Minority Health and Health Disparities-NIMHD (G12 MD007586 to J.O.), Fondazione Cariplo (2011-0370 to C.M.), Kuwait Institute for the Advancement of Sciences (2011-1302-06 to F.al-M), Grant University Scheme (RUGs) Ministry of Education Malaysia (04-02-12-2099RU to R.A.H), Italian Ministry of University and Research (2009FZZ4XM_002 to A.A.), The University of Florence (ex60%2012 to A.A.), US Public Health Service Grants (R01 CA92306, R01 CA92306-S1, R01 CA113447 to R.R.), Department of Science and Technology, Government of India (SR/FT/LS-063/2008 to N.S.).
Acknowledgements
Due to limitation of space we could not cite the contributions of all the outstanding investigators in the fields of carcinogenesis, tissue invasion and metastasis. We apologize to those whose articles were not cited.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations:
- AHR
aryl hydrocarbon receptor
- ALDH
aldehyde dehydrogenase
- AP-1
activator protein-1
- BaP
benzo(a)pyrene
- BLBC
basal-like breast cancer
- BPA
bisphenol A
- CD
cluster differentiation
- CRD
carbohydrate-binding domain
- CSC
cancer stem-like cell
- DEP
diesel exhaust particle
- EGF
epidermal growth factor
- EMT
epithelial–mesenchymal transition
- ER
estrogen receptor
- FAK
focal adhesion kinase
- HNSCC
head and neck squamous cell carcinoma
- HPV
human papillomavirus
- IARC
International Agency for Research on Cancer
- IL
interleukin
- MAPK
mitogen-activated protein kinase
- M-CSC
migratory cancer stem cell
- MET
mesenchymal–epithelial transition
- MMP
matrix metalloproteinase
- NF-κB
nuclear factor kappa B
- NNK
nitrosamine 4-(methylnitrosamino)-1-butanone
- OC
organochlorine
- OSCC
oral squamous cell carcinoma
- OXPHOS
oxidative phosphorylation
- PAH
polycyclic aromatic hydrocarbon
- PCa
prostate cancer
- PCB
polychlorinated biphenyl
- PhIP
2-amino-1-methyl-6-phenylimidazo[4,5-b] pyridine
- ROS
reactive oxygen species
- SO2
sulfur dioxide
- TBBPA
tetrabromobisphenol A
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- TGF-β
transforming growth factor-beta
- TIMP
tissue inhibitors of MMP.
References
- 1. Hanahan D., et al. (2000) The hallmarks of cancer. Cell, 100, 57–70. [DOI] [PubMed] [Google Scholar]
- 2. Hannahan D., et al. (2011) Hallmarks of cancer: the next generation. Cell, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 3. Berx G., et al. (2009) Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb. Perspect. Biol., 1, a003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Klymkowsky M.W., et al. (2009) Epithelial-mesenchymal transition: a cancer researcher’s conceptual friend and foe. Am. J. Pathol., 174, 1588–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ding S.Z., et al. (2013) Epithelial-mesenchymal transition during oncogenic transformation induced by hexavalent chromium involves reactive oxygen species-dependent mechanism in lung epithelial cells. Toxicol. Appl. Pharmacol., 269, 61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Micalizzi D.S., et al. (2010) Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J. Mammary Gland Biol. Neoplasia, 15, 117–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang J., et al. (2008) Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev. Cell, 14, 818–829. [DOI] [PubMed] [Google Scholar]
- 8. Kawata M., et al. (2012) TGF-beta-induced epithelial-mesenchymal transition of A549 lung adenocarcinoma cells is enhanced by pro-inflammatory cytokines derived from RAW 264.7 macrophage cells. J. Biochem., 151, 205–216. [DOI] [PubMed] [Google Scholar]
- 9. Hsiang C.Y., et al. (2007) Acetaldehyde induces matrix metalloproteinase-9 gene expression via nuclear factor-kappaB and activator protein 1 signaling pathways in human hepatocellular carcinoma cells: association with the invasive potential. Toxicol. Lett., 171, 78–86. [DOI] [PubMed] [Google Scholar]
- 10. Seo J.H., et al. (2004) Helicobacter pylori in a Korean isolate activates mitogen-activated protein kinases, AP-1, and NF-kappaB and induces chemokine expression in gastric epithelial AGS cells. Lab. Invest., 84, 49–62. [DOI] [PubMed] [Google Scholar]
- 11. Nagase H., et al. (2006) Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res., 69, 562–573. [DOI] [PubMed] [Google Scholar]
- 12. McCormick J.M., et al. (2010) Embryonic exposure to tetrabromobisphenol A and its metabolites, bisphenol A and tetrabromobisphenol A dimethyl ether disrupts normal zebrafish (Danio rerio) development and matrix metalloproteinase expression. Aquat. Toxicol., 100, 255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen H.C., et al. (1998) Tyrosine phosphorylation of focal adhesion kinase stimulated by hepatocyte growth factor leads to mitogen-activated protein kinase activation. J. Biol. Chem., 273, 25777–25782. [DOI] [PubMed] [Google Scholar]
- 14. Al-Mehdi A.B., et al. (2000) Intravascular origin of metastasis from the proliferation of endothelium-attached tumor cells: a new model for metastasis. Nat. Med., 6, 100–102. [DOI] [PubMed] [Google Scholar]
- 15. Chang C.C., et al. (2013) Connective tissue growth factor activates pluripotency genes and mesenchymal-epithelial transition in head and neck cancer cells. Cancer Res., 73, 4147–4157. [DOI] [PubMed] [Google Scholar]
- 16. Joeckel E., et al. High calcium concentration in bones promotes bone metastasis in renal cell carcinomas expressing calcium-sensing receptor. Mol. Cancer, 13, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Watson K., et al. Fetuin-A triggers the secretion of a novel set of exosomes in detached tumor cells that mediate their adhesion and spreading. FEBS Lett., 586, 3458–3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jung T., et al. (2009) CD44v6 dependence of premetastatic niche preparation by exosomes. Neoplasia, 11, 1093–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Slaga T.J. (1984) Multistage skin carcinogenesis: a useful model for the study of the chemoprevention of cancer. Acta Pharmacol. Toxicol. (Copenh), 55(suppl 2), 107–124. [DOI] [PubMed] [Google Scholar]
- 20. Randerath K., et al. (1989) Use of the 32P-postlabelling assay to study transplacental carcinogens and transplacental carcinogenesis. IARC Sci. Publ., 189–205. [PubMed] [Google Scholar]
- 21. Chouroulinkov I., et al. (1978) Two-stage (initiation-promotion) carcinogenesis in vivo and in vitro . Bull. Cancer, 65, 255–264. [PubMed] [Google Scholar]
- 22. Yuspa S.H. (1986) Cellular and molecular mechanisms of carcinogenesis in lining epithelia. Symp. Fundam. Cancer Res., 39, 3–15. [PubMed] [Google Scholar]
- 23. Stampfer M.R., et al. (1993) Culture systems for study of human mammary epithelial cell proliferation, differentiation and transformation. Cancer Surv., 18, 7–34. [PubMed] [Google Scholar]
- 24. Soule H.D., et al. (1990) Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res., 50, 6075–6086. [PubMed] [Google Scholar]
- 25. Murray M.J., et al. (1981) Three different human tumor cell lines contain different oncogenes. Cell, 25, 355–361. [DOI] [PubMed] [Google Scholar]
- 26. Wald N.J. (1976) Mortality from lung cancer and coronary heart-disease in relation to changes in smoking habits. Lancet, 1, 136–138. [DOI] [PubMed] [Google Scholar]
- 27. Hoffmann D., et al. (1982) Tobacco specific N-nitrosamines: occurrence and bioassays. IARC Sci. Publ., 309–318. [PubMed] [Google Scholar]
- 28. Warner K.E. (1989) Implications of a nicotine-free society. J. Subst. Abuse, 1, 359–368. [PubMed] [Google Scholar]
- 29. Gopalakrishna R., et al. (1994) Tobacco smoke tumor promoters, catechol and hydroquinone, induce oxidative regulation of protein kinase C and influence invasion and metastasis of lung carcinoma cells. Proc. Natl Acad. Sci. USA, 91, 12233–12237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Murin S., et al. (2001) Cigarette smoking and the risk of pulmonary metastasis from breast cancer. Chest, 119, 1635–1640. [DOI] [PubMed] [Google Scholar]
- 31. Shen J., et al. (2012) NNK promotes migration and invasion of lung cancer cells through activation of c-Src/PKCiota/FAK loop. Cancer Lett., 318, 106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wu W.S. (2006) The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev., 25, 695–705. [DOI] [PubMed] [Google Scholar]
- 33. Proctor R.N. (2004) The global smoking epidemic: a history and status report. Clin. Lung Cancer, 5, 371–376. [DOI] [PubMed] [Google Scholar]
- 34. Taioli E. (2008) Gene-environment interaction in tobacco-related cancers. Carcinogenesis, 29, 1467–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hoffmann D., et al. (2001) The less harmful cigarette: a controversial issue. a tribute to Ernst L. Wynder. Chem. Res. Toxicol., 14, 767–790. [DOI] [PubMed] [Google Scholar]
- 36. Koturbash I., et al. (2011) Role of epigenetic events in chemical carcinogenesis-a justification for incorporating epigenetic evaluations in cancer risk assessment. Toxicol. Mech. Methods, 21, 289–297. [DOI] [PubMed] [Google Scholar]
- 37. Murin S., et al. (2004) The effect of cigarette smoke exposure on pulmonary metastatic disease in a murine model of metastatic breast cancer. Chest, 125, 1467–1471. [DOI] [PubMed] [Google Scholar]
- 38. Nagaraj N.S., et al. (2007) Cigarette smoke condensate increases cathepsin-mediated invasiveness of oral carcinoma cells. Toxicol. Lett., 170, 134–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Murin S., et al. (2001) Cigarette smoking and the risk of pulmonary metastasis from breast cancer. Chest, 119, 1635–1640. [DOI] [PubMed] [Google Scholar]
- 40. Ueng T.-H., et al. Potential roles of fibroblast growth factor-9 in the benzo (a) pyrene-induced invasion in vitro and the metastasis of human lung adenocarcinoma. Arch. Toxicol., 84, 651–660. [DOI] [PubMed] [Google Scholar]
- 41. Madhavan N.D., et al. (1995) Polycyclic aromatic hydrocarbons in placenta, maternal blood, umbilical cord blood and milk of Indian women. Hum. Exp. Toxicol., 14, 503–506. [DOI] [PubMed] [Google Scholar]
- 42. Miller M.E., et al. (2005) Benzo-[a]-pyrene increases invasion in MDA-MB-231 breast cancer cells via increased COX-II expression and prostaglandin E2 (PGE2) output. Clin. Exp. Metastasis, 22, 149–156. [DOI] [PubMed] [Google Scholar]
- 43. Felton J.S., et al. (2004) Impact of environmental exposures on the mutagenicity/carcinogenicity of heterocyclic amines. Toxicology, 198, 135–145. [DOI] [PubMed] [Google Scholar]
- 44. Felton J.S., et al. (2002) Human exposure to heterocyclic amine food mutagens/carcinogens: relevance to breast cancer. Environ. Mol. Mutagen., 39, 112–118. [DOI] [PubMed] [Google Scholar]
- 45. Lauber S.N., et al. (2004) The cooked food derived carcinogen 2-amino-1-methyl-6-phenylimidazo [4, 5-b] pyridine is a potent oestrogen: a mechanistic basis for its tissue-specific carcinogenicity. Carcinogenesis, 25, 2509–2517. [DOI] [PubMed] [Google Scholar]
- 46. Brown N.M., et al. (1995) Xenoestrogens alter mammary gland differentiation and cell proliferation in the rat. Environ. Health Persp., 103, 708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Robison A.K., et al. (1985) DDT supports the growth of an estrogen-responsive tumor: mammary tumor growth; pesticides. Toxicol. Lett., 27, 109–113. [DOI] [PubMed] [Google Scholar]
- 48. Wolff M.S., et al. (1993) Blood levels of organochlorine residues and risk of breast cancer. J. Natl Cancer Inst., 85, 648–652. [DOI] [PubMed] [Google Scholar]
- 49. Demers A., et al. (2000) Risk and aggressiveness of breast cancer in relation to plasma organochlorine concentrations. Cancer Epidemiol. Biomarkers Prev., 9, 161–166. [PubMed] [Google Scholar]
- 50. Liu S., et al. Polychlorinated biphenyls (PCBs) enhance metastatic properties of breast cancer cells by activating Rho-associated kinase (ROCK). PLoS One, 5, e11272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pontillo C.A., et al. (2013) Action of hexachlorobenzene on tumor growth and metastasis in different experimental models. Toxicol. Appl. Pharmacol, 268, 331–342. [DOI] [PubMed] [Google Scholar]
- 52. Meng Q., et al. (2000) Stimulation of cell invasion and migration by alcohol in breast cancer cells. Biochem. Biophys. Res. Commun., 273, 448–453. [DOI] [PubMed] [Google Scholar]
- 53. Foon K.A., et al. (2010) Changing paradigms in the treatment of chronic lymphocytic leukemia. Leukemia, 24, 500–511. [DOI] [PubMed] [Google Scholar]
- 54. Katoh M., et al. (1994) Hepatocellular carcinoma with splenic metastasis developing after 16 years of chemotherapy for chronic myelogenous leukemia: a case report. Jpn. J. Clin. Oncol., 24, 111–115. [PubMed] [Google Scholar]
- 55. Tanriverdi O., et al. (2013) Cutaneous metastasis of gallbladder adenocarcinoma in a patient with chronic lymphocytic leukemia: a case report and review of the literature. Ann. Dermatol., 25, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gibb H.J., et al. (2000) Lung cancer among workers in chromium chemical production. Am. J. Ind. Med., 38, 115–126. [DOI] [PubMed] [Google Scholar]
- 57. O’Brien T.J., et al. (2003) Complexities of chromium carcinogenesis: role of cellular response, repair and recovery mechanisms. Mutat. Res., 533, 3–36. [DOI] [PubMed] [Google Scholar]
- 58. Nickens K.P., et al. (2010) Chromium genotoxicity: a double-edged sword. Chem. Biol. Interact., 188, 276–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang X., et al. (2012) Arsenic and chromium in drinking water promote tumorigenesis in a mouse colitis-associated colorectal cancer model and the potential mechanism is ROS-mediated Wnt/beta-catenin signaling pathway. Toxicol. Appl. Pharmacol., 262, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Koyama H., et al. (2002) [Low dose exposure to cadmium and its health effects (1). Genotoxicity and carcinogenicity]. Nihon Eiseigaku Zasshi, 57, 547–555. [DOI] [PubMed] [Google Scholar]
- 61. Prozialeck W.C., et al. (1999) Interaction of cadmium (Cd(2+)) with a 13-residue polypeptide analog of a putative calcium-binding motif of E-cadherin. Biochim. Biophys. Acta, 1451, 93–100. [DOI] [PubMed] [Google Scholar]
- 62. Chakraborty P.K., et al. (2010) Cadmium induces Wnt signaling to upregulate proliferation and survival genes in sub-confluent kidney proximal tubule cells. Mol. Cancer, 9, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Abshire M.K., et al. (1996) In vitro exposure to cadmium in rat L6 myoblasts can result in both enhancement and suppression of malignant progression in vivo . Carcinogenesis, 17, 1349–1356. [DOI] [PubMed] [Google Scholar]
- 64. Haga A., et al. (1996) Enhanced invasiveness of tumour cells after host exposure to heavy metals. Eur. J. Cancer, 32A, 2342–2347. [DOI] [PubMed] [Google Scholar]
- 65. Haga A., et al. (1997) Invasive properties of cadmium-resistant human fibrosarcoma HT-1080 cells. Cancer Biochem. Biophys., 15, 275–284. [PubMed] [Google Scholar]
- 66. Yamashiro S., et al. (1980) Nickel sulphide-induced rhabdomyosarcomata in rats. Acta Pathol. Jpn., 30, 9–22. [DOI] [PubMed] [Google Scholar]
- 67. Wu C.H., et al. (2012) Nickel-induced epithelial-mesenchymal transition by reactive oxygen species generation and E-cadherin promoter hypermethylation. J. Biol. Chem., 287, 25292–25302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gaddy J.A., et al. (2013) High dietary salt intake exacerbates Helicobacter pylori-induced gastric carcinogenesis. Infect. Immun., 81, 2258–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bhandari A., et al. (2012) Helicobacter pylori in gastric malignancies. Curr. Gastroenterol. Rep., 14, 489–496. [DOI] [PubMed] [Google Scholar]
- 70. Lu H., et al. (2005) Regulation of interleukin-6 promoter activation in gastric epithelial cells infected with Helicobacter pylori . Mol. Biol. Cell, 16, 4954–4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Neuzillet C., et al. (2013) Targeting the Ras-ERK pathway in pancreatic adenocarcinoma. Cancer Metastasis Rev., 32, 147–162. [DOI] [PubMed] [Google Scholar]
- 72. Prasad S., et al. (2010) NF-kappaB and cancer: how intimate is this relationship. Mol. Cell. Biochem., 336, 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yeh Y.C., et al. (2010) Elevated serum matrix metalloproteinase-3 and -7 in H. pylori-related gastric cancer can be biomarkers correlating with a poor survival. Dig. Dis. Sci., 55, 1649–1657. [DOI] [PubMed] [Google Scholar]
- 74. Ubagai T., et al. (2010) Aflatoxin B1 modulates the insulin-like growth factor-2 dependent signaling axis. Toxicol. In Vitro, 24, 783–789. [DOI] [PubMed] [Google Scholar]
- 75. Hay E.D. (1995) An overview of epithelio-mesenchymal transformation. Acta Anat. (Basel), 154, 8–20. [DOI] [PubMed] [Google Scholar]
- 76. Boyer B., et al. (2000) Induction and regulation of epithelial-mesenchymal transitions. Biochem. Pharmacol., 60, 1091–1099. [DOI] [PubMed] [Google Scholar]
- 77. Thiery J.P. (2002) Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer, 2, 442–454. [DOI] [PubMed] [Google Scholar]
- 78. Savagner P., et al. (1994) Alternative splicing in fibroblast growth factor receptor 2 is associated with induced epithelial-mesenchymal transition in rat bladder carcinoma cells. Mol. Biol. Cell, 5, 851–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Savagner P. (2001) Leaving the neighborhood: molecular mechanisms involved during epithelial-mesenchymal transition. Bioessays, 23, 912–923. [DOI] [PubMed] [Google Scholar]
- 80. Kang Y., et al. (2004) Epithelial-mesenchymal transitions: twist in development and metastasis. Cell, 118, 277–279. [DOI] [PubMed] [Google Scholar]
- 81. Videtic G.M., et al. (2003) Continued cigarette smoking by patients receiving concurrent chemoradiotherapy for limited-stage small-cell lung cancer is associated with decreased survival. J. Clin. Oncol., 21, 1544–1549. [DOI] [PubMed] [Google Scholar]
- 82. Tammemagi C.M., et al. (2004) Smoking and lung cancer survival: the role of comorbidity and treatment. Chest, 125, 27–37. [DOI] [PubMed] [Google Scholar]
- 83. Yoshino I., et al. (2006) Smoking status as a prognostic factor in patients with stage I pulmonary adenocarcinoma. Ann. Thorac. Surg., 81, 1189–1193. [DOI] [PubMed] [Google Scholar]
- 84. Rice D., et al. (2003) The risk of second primary tumors after resection of stage I nonsmall cell lung cancer. Ann. Thorac. Surg., 76, 1001–1007; discussion 1007–8. [DOI] [PubMed] [Google Scholar]
- 85. Yoshino I., et al. (2007) Induction of epithelial-mesenchymal transition-related genes by benzo[a]pyrene in lung cancer cells. Cancer, 110, 369–374. [DOI] [PubMed] [Google Scholar]
- 86. Kase S., et al. (2000) Expression of E-cadherin and beta-catenin in human non-small cell lung cancer and the clinical significance. Clin. Cancer Res., 6, 4789–4796. [PubMed] [Google Scholar]
- 87. Fondrevelle M.E., et al. (2009) The expression of Twist has an impact on survival in human bladder cancer and is influenced by the smoking status. Urol. Oncol., 27, 268–276. [DOI] [PubMed] [Google Scholar]
- 88. Dasgupta P., et al. (2009) Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int. J. Cancer, 124, 36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wei P.L., et al. (2011) Nicotine enhances colon cancer cell migration by induction of fibronectin. Ann. Surg. Oncol., 18, 1782–1790. [DOI] [PubMed] [Google Scholar]
- 90. Yu M.A., et al. Nicotine promotes acquisition of stem cell and epithelial-to-mesenchymal properties in head and neck squamous cell carcinoma. PLoS One, 7, e51967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yu C.C., et al. (2013) Enhancement of cancer stem-like and epithelial-mesenchymal transdifferentiation property in oral epithelial cells with long-term nicotine exposure: reversal by targeting SNAIL. Toxicol. Appl. Pharmacol., 266, 459–469. [DOI] [PubMed] [Google Scholar]
- 92. Chakraborty P.K., et al. (2010) Chronic cadmium exposure induces transcriptional activation of the Wnt pathway and upregulation of epithelial-to-mesenchymal transition markers in mouse kidney. Toxicol. Lett., 198, 69–76. [DOI] [PubMed] [Google Scholar]
- 93. Yan S., et al. (2013) Low-dose radiation-induced epithelial-mesenchymal transition through NF-kappaB in cervical cancer cells. Int. J. Oncol., 42, 1801–1806. [DOI] [PubMed] [Google Scholar]
- 94. Zhou Y.C., et al. (2011) Ionizing radiation promotes migration and invasion of cancer cells through transforming growth factor-beta-mediated epithelial-mesenchymal transition. Int. J. Radiat. Oncol. Biol. Phys., 81, 1530–1537. [DOI] [PubMed] [Google Scholar]
- 95. Kawamoto A., et al. (2012) Radiation induces epithelial-mesenchymal transition in colorectal cancer cells. Oncol. Rep., 27, 51–57. [DOI] [PubMed] [Google Scholar]
- 96. Ho J.N., et al. (2010) Bcl-XL and STAT3 mediate malignant actions of gamma-irradiation in lung cancer cells. Cancer Sci., 101, 1417–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Andarawewa K.L., et al. (2007) Ionizing radiation predisposes nonmalignant human mammary epithelial cells to undergo transforming growth factor beta induced epithelial to mesenchymal transition. Cancer Res., 67, 8662–8670. [DOI] [PubMed] [Google Scholar]
- 98. Zucchini-Pascal N., et al. (2012) Organochlorine pesticides induce epithelial to mesenchymal transition of human primary cultured hepatocytes. Food Chem. Toxicol., 50, 3963–3970. [DOI] [PubMed] [Google Scholar]
- 99. Ames B.N. (1989) Mutagenesis and carcinogenesis: endogenous and exogenous factors. Environ. Mol. Mutagen., 14( suppl 16), 66–77. [DOI] [PubMed] [Google Scholar]
- 100. O’Byrne K.J., et al. (2001) Chronic immune activation and inflammation as the cause of malignancy. Br. J. Cancer, 85, 473–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zieba M., et al. (2000) Comparison of hydrogen peroxide generation and the content of lipid peroxidation products in lung cancer tissue and pulmonary parenchyma. Respir. Med., 94, 800–805. [DOI] [PubMed] [Google Scholar]
- 102. Gabbita S.P., et al. (2000) Redox regulatory mechanisms of cellular signal transduction. Arch. Biochem. Biophys., 376, 1–13. [DOI] [PubMed] [Google Scholar]
- 103. Rhyu D.Y., et al. (2005) Role of reactive oxygen species in TGF-beta1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J. Am. Soc. Nephrol., 16, 667–675. [DOI] [PubMed] [Google Scholar]
- 104. Barnett P., et al. (2011) Snail-mediated regulation of reactive oxygen species in ARCaP human prostate cancer cells. Biochem. Biophys. Res. Commun., 404, 34–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Zhang A., et al. (2006) Prostaglandin D2 inhibits TGF-beta1-induced epithelial-to-mesenchymal transition in MDCK cells. Am. J. Physiol. Renal Physiol., 291, F1332–F1342. [DOI] [PubMed] [Google Scholar]
- 106. Belguise K., et al. (2007) Activation of FOXO3a by the green tea polyphenol epigallocatechin-3-gallate induces estrogen receptor alpha expression reversing invasive phenotype of breast cancer cells. Cancer Res., 67, 5763–5770. [DOI] [PubMed] [Google Scholar]
- 107. Chen P.N., et al. (2011) Epigallocatechin-3 gallate inhibits invasion, epithelial-mesenchymal transition, and tumor growth in oral cancer cells. J. Agric. Food Chem., 59, 3836–3844. [DOI] [PubMed] [Google Scholar]
- 108. Vergara D., et al. (2011) Resveratrol inhibits the epidermal growth factor-induced epithelial mesenchymal transition in MCF-7 cells. Cancer Lett., 310, 1–8. [DOI] [PubMed] [Google Scholar]
- 109. Chen M.C., et al. (2012) Resveratrol inhibits LPS-induced epithelial-mesenchymal transition in mouse melanoma model. Innate Immun., 18, 685–693. [DOI] [PubMed] [Google Scholar]
- 110. Wang H., et al. (2013) Resveratrol inhibits TGF-beta1-induced epithelial-to-mesenchymal transition and suppresses lung cancer invasion and metastasis. Toxicology, 303, 139–46. [DOI] [PubMed] [Google Scholar]
- 111. Nagase H., et al. (2006) Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res., 69, 562–573. [DOI] [PubMed] [Google Scholar]
- 112. McCawley L.J., et al. (2001) Matrix metalloproteinases: they’re not just for matrix anymore! Curr. Opin. Cell Biol., 13, 534–540. [DOI] [PubMed] [Google Scholar]
- 113. Page-McCaw A., et al. (2007) Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol., 8, 221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Brinckerhoff C.E., et al. (2002) Matrix metalloproteinases: a tail of a frog that became a prince. Nat. Rev. Mol. Cell Biol., 3, 207–214. [DOI] [PubMed] [Google Scholar]
- 115. Egeblad M., et al. (2002) New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer, 2, 161–174. [DOI] [PubMed] [Google Scholar]
- 116. Coussens L.M., et al. (2002) Inflammation and cancer. Nature, 420, 860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Decock J., et al. (2011) Matrix metalloproteinases: protective roles in cancer. J. Cell. Mol. Med., 15, 1254–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Dvorak H.F. (1986) Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med., 315, 1650–1659. [DOI] [PubMed] [Google Scholar]
- 119. Woessner J.F., et al. (2000) Matrix Metalloproteinases and TIMPs. Oxford University Press, Inc, New York, NY. [Google Scholar]
- 120. Ra H.J., et al. (2007) Control of matrix metalloproteinase catalytic activity. Matrix Biol., 26, 587–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Bourboulia D., et al. (2010) Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): Positive and negative regulators in tumor cell adhesion. Semin. Cancer Biol., 20, 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Nagase H. (1998) Stromelysins 1 and 2. In Parks W.C., Mecham R.P. (eds), Matrix Metalloproteinases. Academic Press, San Diego, pp. 43–84. [Google Scholar]
- 123. Visse R., et al. (2003) Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ. Res., 92, 827–839. [DOI] [PubMed] [Google Scholar]
- 124. Yan C., et al. (2007) Regulation of matrix metalloproteinase gene expression. J. Cell. Physiol., 211, 19–26. [DOI] [PubMed] [Google Scholar]
- 125. Kenyon N.J., et al. (2002) Susceptibility to ozone-induced acute lung injury in iNOS-deficient mice. Am. J. Physiol. Lung Cell. Mol. Physiol., 282, L540–L545. [DOI] [PubMed] [Google Scholar]
- 126. Sunil V.R., et al. (2012) Classical and alternative macrophage activation in the lung following ozone-induced oxidative stress. Toxicol. Appl. Pharmacol., 263, 195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Thomson E., et al. (2005) Differential regulation of the lung endothelin system by urban particulate matter and ozone. Toxicol. Sci., 88, 103–113. [DOI] [PubMed] [Google Scholar]
- 128. Yoon H.K., et al. (2007) Protective role of matrix metalloproteinase-9 in ozone-induced airway inflammation. Environ. Health Perspect., 115, 1557–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Triantaphyllopoulos K., et al. (2011) A model of chronic inflammation and pulmonary emphysema after multiple ozone exposures in mice. Am. J. Physiol. Lung Cell. Mol. Physiol., 300, L691–L700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Hautamaki R.D., et al. (1997) Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science, 277, 2002–2004. [DOI] [PubMed] [Google Scholar]
- 131. Ramos C., et al. (2009) Increase of matrix metalloproteinases in woodsmoke-induced lung emphysema in guinea pigs. Inhal. Toxicol., 21, 119–132. [DOI] [PubMed] [Google Scholar]
- 132. Lund A.K., et al. (2009) Vehicular emissions induce vascular MMP-9 expression and activity associated with endothelin-1-mediated pathways. Arterioscler. Thromb. Vasc. Biol., 29, 511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Campen M.J., et al. (2010) A comparison of vascular effects from complex and individual air pollutants indicates a role for monoxide gases and volatile hydrocarbons. Environ. Health Perspect., 118, 921–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Amara N., et al. (2007) Diesel exhaust particles induce matrix metalloprotease-1 in human lung epithelial cells via a NADP(H) oxidase/NOX4 redox-dependent mechanism. Am. J. Physiol. Lung Cell. Mol. Physiol., 293, L170–L181. [DOI] [PubMed] [Google Scholar]
- 135. Li J., et al. (2009) Diesel exhaust particles activate the matrix-metalloproteinase-1 gene in human bronchial epithelia in a beta-arrestin-dependent manner via activation of RAS. Environ. Health Perspect., 117, 400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Doornaert B., et al. (2003) Negative impact of DEP exposure on human airway epithelial cell adhesion, stiffness, and repair. Am. J. Physiol. Lung Cell. Mol. Physiol., 284, L119–L132. [DOI] [PubMed] [Google Scholar]
- 137. Matsuzaki T., et al. (2006) Effects of diesel exhaust particles on human neutrophil activation. Exp. Lung Res., 32, 427–439. [DOI] [PubMed] [Google Scholar]
- 138. O’Brien D.W., et al. (2004) A mechanism of airway injury in an epithelial model of mucociliary clearance. Respir. Res., 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Bonventre J.A., et al. (2012) Craniofacial abnormalities and altered wnt and mmp mRNA expression in zebrafish embryos exposed to gasoline oxygenates ETBE and TAME. Aquat. Toxicol., 120–121, 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Mohr U., et al. (2006) Pulmonary tumor types induced in Wistar rats of the so-called “19-dust study”. Exp. Toxicol. Pathol., 58, 13–20. [DOI] [PubMed] [Google Scholar]
- 141. Choi M., et al. (2008) Transient pulmonary fibrogenic effect induced by intratracheal instillation of ultrafine amorphous silica in A/J mice. Toxicol. Lett., 182, 97–101. [DOI] [PubMed] [Google Scholar]
- 142. McCormick J.M., et al. (2010) Embryonic exposure to tetrabromobisphenol A and its metabolites, bisphenol A and tetrabromobisphenol A dimethyl ether disrupts normal zebrafish (Danio rerio) development and matrix metalloproteinase expression. Aquat. Toxicol., 100, 255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Barondes S.H., et al. (1994) Galectins. Structure and function of a large family of animal lectins. J. Biol. Chem., 269, 20807–20810. [PubMed] [Google Scholar]
- 144. Shankar J., et al. (2012) Coordinated expression of galectin-3 and caveolin-1 in thyroid cancer. J. Pathol., 228, 56–66. [DOI] [PubMed] [Google Scholar]
- 145. Boscher C., et al. (2011) Glycosylation, galectins and cellular signaling. Curr. Opin. Cell Biol., 23, 383–392. [DOI] [PubMed] [Google Scholar]
- 146. Cedeno-Laurent F., et al. (2012) Galectin-1 research in T cell immunity: past, present and future. Clin. Immunol., 142, 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Lagana A., et al. (2006) Galectin binding to Mgat5-modified N-glycans regulates fibronectin matrix remodeling in tumor cells. Mol. Cell Biol., 26, 3181–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Markowska A.I., et al. (2011) Galectin-3 protein modulates cell surface expression and activation of vascular endothelial growth factor receptor 2 in human endothelial cells. J. Biol. Chem., 286, 29913–29921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Merlin J., et al. (2011) Galectin-3 regulates MUC1 and EGFR cellular distribution and EGFR downstream pathways in pancreatic cancer cells. Oncogene, 30, 2514–2525. [DOI] [PubMed] [Google Scholar]
- 150. Nangia-Makker P., et al. (2000) Galectin-3 induces endothelial cell morphogenesis and angiogenesis. Am. J. Pathol., 156, 899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Di Lella S., et al. (2011) When galectins recognize glycans: from biochemistry to physiology and back again. Biochemistry, 50, 7842–7857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Lau K.S., et al. (2008) N-Glycans in cancer progression. Glycobiology, 18, 750–760. [DOI] [PubMed] [Google Scholar]
- 153. van Kooyk Y., et al. (2008) Protein-glycan interactions in the control of innate and adaptive immune responses. Nat. Immunol., 9, 593–601. [DOI] [PubMed] [Google Scholar]
- 154. Haudek K.C., et al. (2010) SR proteins and galectins: what’s in a name? Glycobiology, 20, 1199–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Park J.W., et al. (2001) Association of galectin-1 and galectin-3 with Gemin4 in complexes containing the SMN protein. Nucleic Acids Res., 29, 3595–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Barrow H., et al. (2011) Serum galectin-2,-4, and-8 are greatly increased in colon and breast cancer patients and promote cancer cell adhesion to blood vascular endothelium. Clin. Cancer Res., 17, 7035–7046. [DOI] [PubMed] [Google Scholar]
- 157. Glinsky V.V., et al. (2009) Synthetic galectin-3 inhibitor increases metastatic cancer cell sensitivity to taxol-induced apoptosis in vitro and in vivo . Neoplasia (New York, NY), 11, 901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Mourad-Zeidan A.A., et al. (2008) Expression profiling of Galectin-3-depleted melanoma cells reveals its major role in melanoma cell plasticity and vasculogenic mimicry. Am. J. Pathol., 173, 1839–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Nettesheim P. (1991) Cells of origin of primary pulmonary neoplasms in mice. Exp. Lung Res., 17, 215–217. [DOI] [PubMed] [Google Scholar]
- 160. Abdel-Aziz H.O., et al. (2008) Targeted disruption of the galectin-3 gene results in decreased susceptibility to NNK-induced lung tumorigenesis: an oligonucleotide microarray study. J. Cancer Res. Clin. Oncol., 134, 777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Mendonça D.F., et al. (2012) The inactive form of glycogen synthase kinase-3Î2 is associated with the development of carcinomas in galectin-3 wild-type mice, but not in galectin-3-deficient mice. Int. J. Clin. Exp. Pathol., 5, 547. [PMC free article] [PubMed] [Google Scholar]
- 162. Hooven L.A., et al. (2008) Proteomic analysis of MCF-7 cells treated with benzo[a]pyrene, dibenzo[a,l]pyrene, coal tar extract, and diesel exhaust extract. Toxicology, 249, 1–10. [DOI] [PubMed] [Google Scholar]
- 163. George J., et al. (2011) Cypermethrin exposure leads to regulation of proteins expression involved in neoplastic transformation in mouse skin. Proteomics, 11, 4411–4421. [DOI] [PubMed] [Google Scholar]
- 164. Kayser K., et al. (2000) Association of concentration of asbestos and asbestos-like fibers with the patient’s survival and the binding capacity of lung parenchyma to galectin-1 and natural alpha-galactoside- and alpha-mannoside-binding immunoglobulin G subfractions from human serum. Pathol. Res. Pract., 196, 81–87. [DOI] [PubMed] [Google Scholar]
- 165. Barceloux D.G. (1999) Chromium. J. Toxicol. Clin. Toxicol., 37, 173–194. [DOI] [PubMed] [Google Scholar]
- 166. Tsao D.A., et al. (2010) The expression of RKIP, RhoGDI, galectin, c-Myc and p53 in gastrointestinal system of Cr (VI)-exposed rats. J. Appl. Toxicol., 31, 730–740. [DOI] [PubMed] [Google Scholar]
- 167. Trosko J.E., et al. (2005) The emperor wears no clothes in the field of carcinogen risk assessment: ignored concepts in cancer risk assessment. Mutagenesis, 20, 81–92. [DOI] [PubMed] [Google Scholar]
- 168. Jones L.A., et al. (1995) Effects of estrogenic chemicals on development. Environ. Health Perspect., 103( suppl 7), 63–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Li L., et al. (2006) Normal stem cells and cancer stem cells: the niche matters. Cancer Res., 66, 4553–4557. [DOI] [PubMed] [Google Scholar]
- 170. Tokar E.J., et al. (2011) Arsenic, stem cells, and the developmental basis of adult cancer. Toxicol. Sci., 120( suppl 1), S192–S203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Trosko J.E. (2003) The role of stem cells and gap junctional intercellular communication in carcinogenesis. J. Biochem. Mol. Biol., 36, 43–48. [DOI] [PubMed] [Google Scholar]
- 172. Soto A.M., et al. (2008) Neoplasia as development gone awry: the role of endocrine disruptors. Int. J. Androl., 31, 288–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Kang K.S., et al. (2011) Stem cells in toxicology: fundamental biology and practical considerations. Toxicol. Sci., 120(suppl 1), S269–S289. [DOI] [PubMed] [Google Scholar]
- 174. Vermeulen L., et al. (2012) The developing cancer stem-cell model: clinical challenges and opportunities. Lancet Oncol., 13, e83–e89. [DOI] [PubMed] [Google Scholar]
- 175. Medema J.P. (2013) Cancer stem cells: the challenges ahead. Nat. Cell Biol., 15, 338–344. [DOI] [PubMed] [Google Scholar]
- 176. Takebe N., et al. (2011) Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat. Rev. Clin. Oncol., 8, 97–106. [DOI] [PubMed] [Google Scholar]
- 177. Meyer T.E., et al. (2007) A case-control study of farming and prostate cancer in African-American and Caucasian men. Occup. Environ. Med., 64, 155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Mahajan R., et al. (2006) Phorate exposure and incidence of cancer in the agricultural health study. Environ. Health Perspect., 114, 1205–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Bassil K.L., et al. (2007) Cancer health effects of pesticides: systematic review. Can. Fam. Physician, 53, 1704–1711. [PMC free article] [PubMed] [Google Scholar]
- 180. Wang L., et al. (2012) Stem cell and benzene-induced malignancy and hematotoxicity. Chem. Res. Toxicol., 25, 1303–1315. [DOI] [PubMed] [Google Scholar]
- 181. Soto A.M., et al. (2008) Does breast cancer start in the womb? Basic Clin. Pharmacol. Toxicol., 102, 125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Hu W.Y., et al. (2012) Actions of estrogens and endocrine disrupting chemicals on human prostate stem/progenitor cells and prostate cancer risk. Mol. Cell. Endocrinol., 354, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Singh S., et al. (2012) Epigenetic effects of environmental chemicals bisphenol a and phthalates. Int. J. Mol. Sci., 13, 10143–10153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Waalkes M.P., et al. (2003) Transplacental carcinogenicity of inorganic arsenic in the drinking water: induction of hepatic, ovarian, pulmonary, and adrenal tumors in mice. Toxicol. Appl. Pharmacol., 186, 7–17. [DOI] [PubMed] [Google Scholar]
- 185. Patterson T.J., et al. (2007) Arsenite and insulin exhibit opposing effects on epidermal growth factor receptor and keratinocyte proliferative potential. Toxicol. Appl. Pharmacol., 221, 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Tokar E.J., et al. (2010) Arsenic exposure transforms human epithelial stem/progenitor cells into a cancer stem-like phenotype. Environ. Health Perspect., 118, 108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Waalkes M.P., et al. (2008) Arsenic exposure in utero exacerbates skin cancer response in adulthood with contemporaneous distortion of tumor stem cell dynamics. Cancer Res., 68, 8278–8285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Patterson T.J., et al. (2005) Arsenite maintains germinative state in cultured human epidermal cells. Toxicol. Appl. Pharmacol., 207, 69–77. [DOI] [PubMed] [Google Scholar]
- 189. Dubrovska A., et al. (2009) The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc. Natl Acad. Sci. USA, 106, 268–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Baumann M., et al. (2008) Exploring the role of cancer stem cells in radioresistance. Nat. Rev. Cancer, 8, 545–554. [DOI] [PubMed] [Google Scholar]
- 191. Rich J.N., et al. (2007) Chemotherapy and cancer stem cells. Cell Stem Cell, 1, 353–355. [DOI] [PubMed] [Google Scholar]
- 192. Ahmed N., et al. (2010) Epithelial mesenchymal transition and cancer stem cell-like phenotypes facilitate chemoresistance in recurrent ovarian cancer. Curr. Cancer Drug Targets, 10, 268–278. [DOI] [PubMed] [Google Scholar]
- 193. Mani S.A., et al. (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell, 133, 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Hollier B.G., et al. (2009) The epithelial-to-mesenchymal transition and cancer stem cells: a coalition against cancer therapies. J. Mammary Gland Biol. Neoplasia, 14, 29–43. [DOI] [PubMed] [Google Scholar]
- 195. Guarino M., et al. (2007) The role of epithelial-mesenchymal transition in cancer pathology. Pathology, 39, 305–318. [DOI] [PubMed] [Google Scholar]
- 196. Ocaña O.H., et al. (2012) Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell, 22, 709–724. [DOI] [PubMed] [Google Scholar]
- 197. Brabletz T., et al. (2005) Opinion: migrating cancer stem cells—an integrated concept of malignant tumour progression. Nat. Rev. Cancer, 5, 744–749. [DOI] [PubMed] [Google Scholar]
- 198. Hermann P.C., et al. (2007) Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell, 1, 313–323. [DOI] [PubMed] [Google Scholar]
- 199. Fodde R., et al. (2007) Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr. Opin. Cell Biol., 19, 150–158. [DOI] [PubMed] [Google Scholar]
- 200. Aktas B., et al. (2009) Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res., 11, R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Peyre L., et al. (2012) Effects of endosulfan on hepatoma cell adhesion: epithelial-mesenchymal transition and anoikis resistance. Toxicology, 300, 19–30. [DOI] [PubMed] [Google Scholar]
- 202. Beischlag T.V., et al. (2008) The aryl hydrocarbon receptor complex and the control of gene expression. Crit. Rev. in Eukaryot. Gene Expr., 18, 207–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Nebert D.W., et al. (1972) Genetic regulation of aryl hydrocarbon hydroxylase induction in the mouse. Fed. Proc., 31, 1315–1325. [PubMed] [Google Scholar]
- 204. Poland A.P., et al. (1974) Genetic expression of aryl hydrocarbon hydroxylase activity. Induction of monooxygenase activities and cytochrome P1-450 formation by 2,3,7,8-tetrachlorodibenzo-p-dioxin in mice genetically “nonresponsive” to other aromatic hydrocarbons. J. Biol. Chem., 249, 5599–5606. [PubMed] [Google Scholar]
- 205. Nebert D.W., et al. (1991) Human AH locus polymorphism and cancer: inducibility of CYP1A1 and other genes by combustion products and dioxin. Pharmacogenetics, 1, 68–78. [DOI] [PubMed] [Google Scholar]
- 206. Ray S.S., et al. (2004) Dioxin-induced immortalization of normal human keratinocytes and silencing of p53 and p16INK4a. J. Biol. Chem., 279, 27187–27193. [DOI] [PubMed] [Google Scholar]
- 207. Puga A., et al. (2002) Role of the aryl hydrocarbon receptor in cell cycle regulation. Chem. Biol. Interact., 141, 117–130. [DOI] [PubMed] [Google Scholar]
- 208. Elizondo G., et al. (2000) Altered cell cycle control at the G(2)/M phases in aryl hydrocarbon receptor-null embryo fibroblast. Mol. Pharmacol., 57, 1056–1063. [PubMed] [Google Scholar]
- 209. Brooks J., et al. (2011) Malignant transformation of mammary epithelial cells by ectopic overexpression of the aryl hydrocarbon receptor. Curr. Cancer Drug Targets, 11, 654–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Shin S.R., et al. (2006) 7,12-dimethylbenz(a)anthracene treatment of a c-rel mouse mammary tumor cell line induces epithelial to mesenchymal transition via activation of nuclear factor-kappaB. Cancer Res., 66, 2570–2575. [DOI] [PubMed] [Google Scholar]
- 211. Miller M.E., et al. (2005) Benzo-[a]-pyrene increases invasion in MDA-MB-231 breast cancer cells via increased COX-II expression and prostaglandin E2 (PGE2) output. Clin. Exp. Metastasis, 22, 149–156. [DOI] [PubMed] [Google Scholar]
- 212. Mulero-Navarro S., et al. (2005) Immortalized mouse mammary fibroblasts lacking dioxin receptor have impaired tumorigenicity in a subcutaneous mouse xenograft model. J. Biol. Chem., 280, 28731–28741. [DOI] [PubMed] [Google Scholar]
- 213. Villano C.M., et al. (2006) 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) induces matrix metalloproteinase (MMP) expression and invasion in A2058 melanoma cells. Toxicol. Appl. Pharmacol., 210, 212–224. [DOI] [PubMed] [Google Scholar]
- 214. Côme C., et al. (2004) Roles of the transcription factors snail and slug during mammary morphogenesis and breast carcinoma progression. J. Mammary Gland Biol. Neoplasia, 9, 183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Belguise K., et al. (2007) Green tea polyphenols reverse cooperation between c-Rel and CK2 that induces the aryl hydrocarbon receptor, slug, and an invasive phenotype. Cancer Res., 67, 11742–11750. [DOI] [PubMed] [Google Scholar]
- 216. Goode G.D., et al. (2013) Knockdown of aberrantly upregulated aryl hydrocarbon receptor reduces tumor growth and metastasis of MDA-MB-231 human breast cancer cell line. Int. J. Cancer, 133, 2769–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Pitot H.C., et al. (1980) Quantitative evaluation of the promotion by 2,3,7,8-tetrachlorodibenzo-p-dioxin of hepatocarcinogenesis from diethylnitrosamine. Cancer Res., 40, 3616–3620. [PubMed] [Google Scholar]
- 218. Stinchcombe S., et al. (1995) Inhibition of apoptosis during 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated tumour promotion in rat liver. Carcinogenesis, 16, 1271–1275. [DOI] [PubMed] [Google Scholar]
- 219. Donnell E.F., et al. The aryl hydrocarbon receptor mediates leflunomide-induced growth inhibition of melanoma cells. PLoS One, 7, e40926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. O’Donnell E., et al. The aryl hydrocarbon receptor mediates raloxifene-induced apoptosis in estrogen receptor-negative hepatoma and breast cancer cells. Cell Death Dis., 5, e1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Murphy K.A., et al. (2004) Interaction between the aryl hydrocarbon receptor and retinoic acid pathways increases matrix metalloproteinase-1 expression in keratinocytes. J. Biol. Chem., 279, 25284–25293. [DOI] [PubMed] [Google Scholar]
- 222. Diry M., et al. (2006) Activation of the dioxin/aryl hydrocarbon receptor (AhR) modulates cell plasticity through a JNK-dependent mechanism. Oncogene, 25, 5570–5574. [DOI] [PubMed] [Google Scholar]
- 223. Bui L.C., et al. (2009) Nedd9/Hef1/Cas-L mediates the effects of environmental pollutants on cell migration and plasticity. Oncogene, 28, 3642–3651. [DOI] [PubMed] [Google Scholar]
- 224. Astier A., et al. (1997) Association of the Cas-like molecule HEF1 with CrkL following integrin and antigen receptor signaling in human B-cells: potential relevance to neoplastic lymphohematopoietic cells. Leuk. Lymphoma, 28, 65–72. [DOI] [PubMed] [Google Scholar]
- 225. Izumchenko E., et al. (2009) NEDD9 promotes oncogenic signaling in mammary tumor development. Cancer Res., 69, 7198–7206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. van Seventer G.A., et al. (2001) Focal adhesion kinase regulates beta1 integrin-dependent T cell migration through an HEF1 effector pathway. Eur. J. Immunol., 31, 1417–1427. [DOI] [PubMed] [Google Scholar]
- 227. Kim M., et al. (2006) Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell, 125, 1269–1281. [DOI] [PubMed] [Google Scholar]
- 228. Natarajan M., et al. (2006) HEF1 is a necessary and specific downstream effector of FAK that promotes the migration of glioblastoma cells. Oncogene, 25, 1721–1732. [DOI] [PubMed] [Google Scholar]
- 229. Ji H., et al. (2007) LKB1 modulates lung cancer differentiation and metastasis. Nature, 448, 807–810. [DOI] [PubMed] [Google Scholar]
- 230. Eum S.Y., et al. (2004) VEGF regulates PCB 104-mediated stimulation of permeability and transmigration of breast cancer cells in human microvascular endothelial cells. Exp. Cell Res., 296, 231–244. [DOI] [PubMed] [Google Scholar]
- 231. Eum S.Y., et al. (2006) Interplay between epidermal growth factor receptor and Janus kinase 3 regulates polychlorinated biphenyl-induced matrix metalloproteinase-3 expression and transendothelial migration of tumor cells. Mol. Cancer Res., 4, 361–370. [DOI] [PubMed] [Google Scholar]
- 232. Biswas G., et al. (2008) Dioxin-mediated tumor progression through activation of mitochondria-to-nucleus stress signaling. Proc. Natl Acad. Sci. USA, 105, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. DiNatale B.C., et al. (2012) Ah receptor antagonism represses head and neck tumor cell aggressive phenotype. Mol. Cancer Res., 10, 1369–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Rico-Leo E.M., et al. (2013) Dioxin receptor expression inhibits basal and transforming growth factor beta-induced epithelial-to-mesenchymal transition. J Biol Chem, 288, 7841–7856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Greenlee W.E., et al. (2001) Molecular basis of dioxin actions: evidence supporting chemoprotection. Toxicol. Pathol., 29, 6–7. [DOI] [PubMed] [Google Scholar]
- 236. Hall J.M., et al. (2010) Activation of the aryl-hydrocarbon receptor inhibits invasive and metastatic features of human breast cancer cells and promotes breast cancer cell differentiation. Mol. Endocrinol., 24, 359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Wang T., et al. (2011) Activation of the aryl hydrocarbon receptor by TCDD inhibits mammary tumor metastasis in a syngeneic mouse model of breast cancer. Toxicol. Sci., 124, 291–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Zhang J., et al. (2012) Activation of aryl hydrocarbon receptor suppresses invasion of esophageal squamous cell carcinoma cell lines. Tumori, 98, 152–157. [DOI] [PubMed] [Google Scholar]
- 239. Bleyer A., et al. Effect of three decades of screening mammography on breast-cancer incidence. N. Engl. J. Med., 367, 1998–2005. [DOI] [PubMed] [Google Scholar]
- 240. Gøtzsche P.C. Time to stop mammography screening? CMAJ: Can. Med. Assoc. J., 183, 1957. [Google Scholar]
- 241. Beatson G.T., et al. (1896) On the treatment of inoperable cases ofcarcinoma of the mamma: suggestions for a new method of treatment, with illustrative cases. Lancet, 148, 104–107. [PMC free article] [PubMed] [Google Scholar]
- 242. Barr L., et al. (2011) Measurement of paraben concentrations in human breast tissue at serial locations across the breast from axilla to sternum. J. Appl. Toxicol., 32, 219–232. [DOI] [PubMed] [Google Scholar]
- 243. Ibarluzea J.S., et al. (2004) Breast cancer risk and the combined effect of environmental estrogens. Cancer Causes Control, 15, 591–600. [DOI] [PubMed] [Google Scholar]
- 244. Fernandez S.V., et al. (2010) Estrogen and xenoestrogens in breast cancer. Toxicol. Pathol., 38, 110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Darbre P.D., et al. (2012) Environmental oestrogens and breast cancer: long-term low-dose effects of mixtures of various chemical combinations. J. Epidemiol. Community Health, 67, 203–205. [DOI] [PubMed] [Google Scholar]
- 246. Magee P.J., et al. (2004) Phyto-oestrogens, their mechanism of action: current evidence for a role in breast and prostate cancer. Br. J. Nutr., 91, 513–531. [DOI] [PubMed] [Google Scholar]
- 247. Jordan V.C., et al. (2007) Development and evolution of therapies targeted to the estrogen receptor for the treatment and prevention of breast cancer. Steroids, 72, 7–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Cheung K.L. (2007) Endocrine therapy for breast cancer: an overview. Breast, 16, 327–343. [DOI] [PubMed] [Google Scholar]
- 249. Schiff R., et al. (2003) Breast cancer endocrine resistance how growth factor signaling and estrogen receptor coregulators modulate response. Clinical Cancer Research, 9, 447s–454s. [PubMed] [Google Scholar]
- 250. Johnston S.R.D., et al. (2003) Aromatase inhibitors for breast cancer: lessons from the laboratory. Nat. Rev. Cancer, 3, 821–831. [DOI] [PubMed] [Google Scholar]
- 251. Brodie A., et al. (2011) Adaptive changes result in activation of alternate signaling pathways and acquisition of resistance to aromatase inhibitors. Clin. Cancer Res., 17, 4208–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Musgrove E.A., et al. (2009) Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer, 9, 631–643. [DOI] [PubMed] [Google Scholar]
- 253. Zilli M., et al. (2009) Molecular mechanisms of endocrine resistance and their implication in the therapy of breast cancer. Biochim. Biophys. Acta, 1795, 62–81. [DOI] [PubMed] [Google Scholar]
- 254. Al Saleh S., et al. (2011) Estrogen receptor silencing induces epithelial to mesenchymal transition in human breast cancer cells. PLoS One, 6, e20610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Thiery J.P., et al. (2009) Epithelial-mesenchymal transitions in development and disease. Cell, 139, 871–890. [DOI] [PubMed] [Google Scholar]
- 256. Trimboli A.J., et al. (2008) Direct evidence for epithelial-mesenchymal transitions in breast cancer. Cancer Res., 68, 937–945. [DOI] [PubMed] [Google Scholar]
- 257. Brabletz T., et al. (2005) Migrating cancer stem cells-an integrated concept of malignant tumour progression. Nat. Rev. Cancer, 5, 744–749. [DOI] [PubMed] [Google Scholar]
- 258. Sphyris N., et al. (2009) The importance of the epithelial-mesenchymal transition in breast cancer. Curr. Breast Cancer Rep., 1, 229–237. [Google Scholar]
- 259. Tse J.C., et al. (2007) Mechanisms of metastasis: epithelial-to-mesenchymal transition and contribution of tumor microenvironment. J. Cell. Biochem., 101, 816–829. [DOI] [PubMed] [Google Scholar]
- 260. Al Saleh S., et al. (2011) Signalling pathways involved in endocrine resistance in breast cancer and associations with epithelial to mesenchymal transition (Review). Int. J. Oncol., 38, 1197. [DOI] [PubMed] [Google Scholar]
- 261. Mani S.A., et al. (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell, 133, 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Zhou B.-B.S., et al. (2009) Tumour-initiating cells: challenges and opportunities for anticancer drug discovery. Nat. Rev. Drug Discov., 8, 806–823. [DOI] [PubMed] [Google Scholar]
- 263. Lapidot T., et al. (1994) A cell initiating human acute myeloid leukaemia after transplantation into SCID mice Nature, 367, 645–648. [DOI] [PubMed]
- 264. Prince M.E., et al. (2007) Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl Acad. Sci. USA, 104, 973–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Singh S.K., et al. (2004) Identification of human brain tumour initiating cells. Nature, 432, 396–401. [DOI] [PubMed] [Google Scholar]
- 266. Beier D., et al. (2007) CD133(+) and CD133(-) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res., 67, 4010–4015. [DOI] [PubMed] [Google Scholar]
- 267. O’Brien C.A., et al. (2006) A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature, 445, 106–110. [DOI] [PubMed] [Google Scholar]
- 268. Ricci-Vitiani L., et al. (2006) Identification and expansion of human colon-cancer-initiating cells. Nature, 445, 111–115. [DOI] [PubMed] [Google Scholar]
- 269. Zhao W., et al. Embryonic stem cell markers. Molecules, 17, 6196–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Ginestier C., et al. (2007) ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell, 1, 555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Yamashita T., et al. (2009) EpCAM-positive hepatocellular carcinoma cells are tumor initiating cells with stem/progenitor cell features. Gastroenterology, 136, 1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Al-Hajj M., et al. (2003) Prospective identification of tumorigenic breast cancer cells. Proc. Natl Acad. Sci. USA, 100, 3983–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Sheridan C., et al. (2006) CD44+/CD24-breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res., 8, R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Fillmore C.M., et al. (2008) Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res., 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Croker A.K., et al. (2009) High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J. Cell. Mol. Med., 13(8B), 2236–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Shipitsin M., et al. (2007) Molecular definition of breast tumor heterogeneity. Cancer Cell, 11, 259–273. [DOI] [PubMed] [Google Scholar]
- 277. Bonuccelli G., et al. (2009) Caveolin-1 (P132L), a common breast cancer mutation, confers mammary cell invasiveness and defines a novel stem cell/metastasis-associated gene signature. Am. J. Pathol., 174, 1650–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Charafe-Jauffret E., et al. (2009) Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res., 69, 1302–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Liu H., et al. (2010) Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc. Natl Acad. Sci. USA, 107, 18115–18120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Velasco-Velázquez M.A., et al. The role of breast cancer stem cells in metastasis and therapeutic implications. Am J. Pathol., 179, 2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Morel A.P., et al. (2008) Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One, 3, e2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Santisteban M., et al. (2009) Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res., 69, 2887–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Lim E., et al. (2009) Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med., 15, 907–913. [DOI] [PubMed] [Google Scholar]
- 284. Sørlie T., et al. (2001) Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl Acad. Sci. USA, 98, 10869–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Voulgari A., et al. (2009) Epithelial mesenchymal transition in cancer metastasis: mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer, 1796, 75–90. [DOI] [PubMed] [Google Scholar]
- 286. Li X., et al. (2008) Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl Cancer Inst., 100, 672–679. [DOI] [PubMed] [Google Scholar]
- 287. Hennessy B.T., et al. (2009) Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res., 69, 4116–4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Deonarain M.P., et al. (2009) Antibodies targeting cancer stem cells: a new paradigm in immunotherapy? MAbs, 1, 12–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Yan W., et al. (2009) GATA3 inhibits breast cancer metastasis through the reversal of epithelial-mesenchymal transition. J. Biol. Chem., 285, 14042–14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Gupta P.B., et al. (2009) Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell, 138, 645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Lane A.A., et al. (2009) Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol., 27, 5459–5468. [DOI] [PubMed] [Google Scholar]
- 292. Vazquez-Martin A., et al. (2011) The anti-diabetic drug metformin suppresses the metastasis-associated protein CD24 in MDA-MB-468 triple-negative breast cancer cells. Oncol. Rep., 25, 135–140. [PubMed] [Google Scholar]
- 293. Hirsch H.A., et al. (2009) Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res., 69, 7507–7511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Vazquez-Martin A., et al. The anti-diabetic drug metformin suppresses self-renewal and proliferation of trastuzumab-resistant tumor-initiating breast cancer stem cells. Breast Cancer Res. Treat., 126, 355–364. [DOI] [PubMed] [Google Scholar]
- 295. Frank N.Y., et al. The therapeutic promise of the cancer stem cell concept. J. Clin. Invest., 120, 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Vargo-Gogola T., et al. (2007) Modelling breast cancer: one size does not fit all. Nat. Rev. Cancer, 7, 659–672. [DOI] [PubMed] [Google Scholar]
- 297. Fadare O., et al. (2008) Clinical and pathologic aspects of basal-like breast cancers. Nat. Clin. Pract. Oncol., 5, 149–159. [DOI] [PubMed] [Google Scholar]
- 298. Rakha E.A., et al. (2008) Basal-like breast cancer: a critical review. J. Clin. Oncol., 26, 2568–2581. [DOI] [PubMed] [Google Scholar]
- 299. Hennessy B.T., et al. (2009) Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res., 69, 4116–4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Sarrió D., et al. (2008) Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res., 68, 989–997. [DOI] [PubMed] [Google Scholar]
- 301. Kreike B., et al. (2007) Gene expression profiling and histopathological characterization of triple-negative/basal-like breast carcinomas. Breast Cancer Res., 9, R65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Schneider B.P., et al. (2008) Triple-negative breast cancer: risk factors to potential targets. Clin. Cancer Res., 14, 8010–8018. [DOI] [PubMed] [Google Scholar]
- 303. Yehiely F., et al. (2006) Deconstructing the molecular portrait of basal-like breast cancer. Trends Mol. Med., 12, 537–544. [DOI] [PubMed] [Google Scholar]
- 304. Venkitaraman A.R. (2002) Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell, 108, 171–182. [DOI] [PubMed] [Google Scholar]
- 305. Sorlie T., et al. (2003) Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl Acad. Sci. USA, 100, 8418–8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Kwei K.A., et al. (2010) Genomic instability in breast cancer: pathogenesis and clinical implications. Mol. Oncol., 4, 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Roldán G., et al. (2006) Tumoral expression of BRCA1, estrogen receptor alpha and ID4 protein in patients with sporadic breast cancer. Cancer Biol. Ther., 5, 505–510. [DOI] [PubMed] [Google Scholar]
- 308. Turner N.C., et al. (2007) BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene, 26, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 309. Rice J.C., et al. (2000) Methylation of the BRCA1 promoter is associated with decreased BRCA1 mRNA levels in clinical breast cancer specimens. Carcinogenesis, 21, 1761–1765. [DOI] [PubMed] [Google Scholar]
- 310. Sørlie T., et al. (2001) Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl Acad. Sci. USA, 98, 10869–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Kalluri R., et al. (2009) The basics of epithelial-mesenchymal transition. J. Clin. Invest., 119, 1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Thiery J.P., et al. (2009) Epithelial-mesenchymal transitions in development and disease. Cell, 139, 871–890. [DOI] [PubMed] [Google Scholar]
- 313. Wang S.P., et al. (2009) p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat. Cell Biol., 11, 694–704. [DOI] [PubMed] [Google Scholar]
- 314. Kogan-Sakin I., et al. (2011) Mutant p53(R175H) upregulates Twist1 expression and promotes epithelial-mesenchymal transition in immortalized prostate cells. Cell Death Differ., 18, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Adorno M., et al. (2009) A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell, 137, 87–98. [DOI] [PubMed] [Google Scholar]
- 316. Bossi G., et al. (2008) Conditional RNA interference in vivo to study mutant p53 oncogenic gain of function on tumor malignancy. Cell Cycle, 7, 1870–1879. [DOI] [PubMed] [Google Scholar]
- 317. Koppenol W.H., et al. (2011) Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer, 11, 325–337. [DOI] [PubMed] [Google Scholar]
- 318. Dong C., et al. (2013) Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell, 23, 316–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Bensaad K., et al. (2006) TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell, 126, 107–120. [DOI] [PubMed] [Google Scholar]
- 320. Vousden K.H., et al. (2009) p53 and metabolism. Nat. Rev. Cancer, 9, 691–700. [DOI] [PubMed] [Google Scholar]
- 321. Matoba S., et al. (2006) p53 regulates mitochondrial respiration. Science, 312, 1650–1653. [DOI] [PubMed] [Google Scholar]
- 322. Shirley S.H., et al. (2010) The skinny on Slug. Mol. Carcinog., 49, 851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Nieto M.A. (2002) The snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell Biol., 3, 155–166. [DOI] [PubMed] [Google Scholar]
- 324. Dong C., et al. (2012) G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J. Clin. Invest., 122, 1469–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Dong C., et al. (2013) Interaction with Suv39H1 is critical for Snail-mediated E-cadherin repression in breast cancer. Oncogene, 32, 1351–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Geradts J., et al. (2011) Nuclear Snail1 and nuclear ZEB1 protein expression in invasive and intraductal human breast carcinomas. Hum. Pathol., 42, 1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Dong C., et al. (2013) Interaction with Suv39H1 is critical for Snail-mediated E-cadherin repression in breast cancer. Oncogene, 32,1351–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. DiMeo T.A., et al. (2009) A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res., 69, 5364–5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Siegel R., et al. (2013) Cancer statistics, 2013. CA. Cancer J. Clin., 63, 11–30. [DOI] [PubMed] [Google Scholar]
- 330. Shen M.M., et al. (2010) Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev., 24, 1967–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Chen Z., et al. (2005) Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature, 436, 725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Iwanaga K., et al. (2008) Pten inactivation accelerates oncogenic K-ras-initiated tumorigenesis in a mouse model of lung cancer. Cancer Res., 68, 1119–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333. Jin R.J., et al. (2008) The nuclear factor-kappaB pathway controls the progression of prostate cancer to androgen-independent growth. Cancer Res., 68, 6762–6769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. Seligson D.B., et al. (2005) Global histone modification patterns predict risk of prostate cancer recurrence. Nature, 435, 1262–1266. [DOI] [PubMed] [Google Scholar]
- 335. Ellinger J., et al. (2012) Global histone H3K27 methylation levels are different in localized and metastatic prostate cancer. Cancer Invest., 30, 92–97. [DOI] [PubMed] [Google Scholar]
- 336. Dix D. (2003) On the role of genes relative to the environment in carcinogenesis. Mech. Ageing Dev., 124, 323–332. [DOI] [PubMed] [Google Scholar]
- 337. Soto A.M., et al. (2010) Environmental causes of cancer: endocrine disruptors as carcinogens. Nat. Rev. Endocrinol., 6, 363–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Sathiakumar N., et al. (2011) A review of epidemiologic studies of triazine herbicides and cancer. Crit. Rev. Toxicol., 41(suppl 1), 1–34. [DOI] [PubMed] [Google Scholar]
- 339. Li J., et al. (2012) Association between World Trade Center exposure and excess cancer risk. JAMA, 308, 2479–2488. [DOI] [PubMed] [Google Scholar]
- 340. Solan S., et al. (2013) Cancer incidence in world trade center rescue and recovery workers, 2001–2008. Environ. Health Perspect., 121, 699–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Evanoff B.A., et al. (1993) Mortality and incidence of cancer in a cohort of Swedish chimney sweeps: an extended follow up study. Br. J. Ind. Med., 50, 450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Clapp R.W., et al. (2008) Environmental and occupational causes of cancer: new evidence 2005–2007. Rev. Environ. Health, 23, 1–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343. Demers P.A., et al. (1994) Cancer incidence among firefighters in Seattle and Tacoma, Washington (United States). Cancer Causes Control, 5, 129–135. [DOI] [PubMed] [Google Scholar]
- 344. Punnen S., et al. (2011) Impact of meat consumption, preparation, and mutagens on aggressive prostate cancer. PLoS One, 6, e27711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Giovannucci E., et al. (1993) A prospective study of dietary fat and risk of prostate cancer. J. Natl Cancer Inst., 85, 1571–1579. [DOI] [PubMed] [Google Scholar]
- 346. Joshi A.D., et al. (2012) Red meat and poultry, cooking practices, genetic susceptibility and risk of prostate cancer: results from a multiethnic case-control study. Carcinogenesis, 33, 2108–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347. John E.M., et al. (2011) Meat consumption, cooking practices, meat mutagens, and risk of prostate cancer. Nutr. Cancer, 63, 525–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Knize M.G., et al. (2005) Formation and human risk of carcinogenic heterocyclic amines formed from natural precursors in meat. Nutr. Rev., 63, 158–165. [DOI] [PubMed] [Google Scholar]
- 349. Creton S.K., et al. (2007) The cooked meat carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine activates the extracellular signal regulated kinase mitogen-activated protein kinase pathway. Cancer Res., 67, 11455–11462. [DOI] [PubMed] [Google Scholar]
- 350. De Marzo A.M., et al. (2007) Inflammation in prostate carcinogenesis. Nat. Rev. Cancer, 7, 256–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Smith E.R., et al. (1981) Uptake and secretion of carcinogenic chemicals by the dog and rat prostate. Prog. Clin. Biol. Res., 75B, 131–163. [PubMed] [Google Scholar]
- 352. Bosland M.C. (1992) Animal models for the study of prostate carcinogenesis. J. Cell. Biochem. Suppl., 16H, 89–98. [DOI] [PubMed] [Google Scholar]
- 353. Pour P.M. (1983) Prostatic cancer induced in MRC rats by N-nitrosobis(2-oxopropyl)-amine and N-nitrosobis(2-hydroxypropyl)amine. Carcinogenesis, 4, 49–55. [DOI] [PubMed] [Google Scholar]
- 354. Juge N., et al. (2007) Molecular basis for chemoprevention by sulforaphane: a comprehensive review. Cell. Mol. Life Sci., 64, 1105–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Deep G., et al. (2007) Chemopreventive efficacy of silymarin in skin and prostate cancer. Integr. Cancer Ther., 6, 130–145. [DOI] [PubMed] [Google Scholar]
- 356. Xu R., et al. (2013) Inhibition effects and induction of apoptosis of flavonoids on the prostate cancer cell line PC-3 in vitro . Food Chem., 138, 48–53. [DOI] [PubMed] [Google Scholar]
- 357. Bailey H.H., et al. (2013) Green tea polyphenols and cancer chemoprevention of genitourinary cancer. Am. Soc. Clin. Oncol. Educ. Book, 2013, 92–6. [DOI] [PubMed] [Google Scholar]
- 358. Nambiar D., et al. (2013) Advances in prostate cancer chemoprevention: a translational perspective. Nutr. Cancer, 65(suppl 1), 12–25. [DOI] [PubMed] [Google Scholar]
- 359. Jemal A., et al. (2011) Global cancer statistics. CA. Cancer J. Clin., 61, 69–90. [DOI] [PubMed] [Google Scholar]
- 360. Shah J., et al. (2003) Oral Cancer. Martin Dunitz Press, London. [Google Scholar]
- 361. de Bree R., et al. (2012) Distant metastases mfrom head and neck squamous cell carcinoma. Part II. Diagnosis. Oral Oncol., 48, 780–786. [DOI] [PubMed] [Google Scholar]
- 362. Haigentz M., Jr, et al. (2012) Distant metastases from head and neck squamous cell carcinoma. Part III. Treatment. Oral Oncol., 48, 787–793. [DOI] [PubMed] [Google Scholar]
- 363. Chinn S.B., et al. (2012) The role of head and neck squamous cell carcinoma cancer stem cells in tumorigenesis, metastasis, and treatment failure. Front. Endocrinol. (Lausanne), 3, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364. Arbes S.J., Jr, et al. (1999) Factors contributing to the poorer survival of black Americans diagnosed with oral cancer (United States). Cancer Causes Control, 10, 513–523. [DOI] [PubMed] [Google Scholar]
- 365. Canto M.T., et al. (2003) Oral cancer examinations among U.S. Hispanics in 1998. J. Cancer Educ., 18, 48–52. [DOI] [PubMed] [Google Scholar]
- 366. Horowitz A.M., et al. (1998) Maryland adults’ knowledge of oral cancer and having oral cancer examinations. J. Public Health Dent., 58, 281–287. [DOI] [PubMed] [Google Scholar]
- 367. Eliassen A.M., et al. (2013) Head and neck squamous cell carcinoma in pregnant women. Head Neck, 35, 335–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368. Cruz G.D., et al. (2002) Oral cancer knowledge, risk factors and characteristics of subjects in a large oral cancer screening program. J. Am. Dent. Assoc., 133, 1064–1071; quiz 1094. [DOI] [PubMed] [Google Scholar]
- 369. Hashibe M., et al. (2009) Interaction between tobacco and alcohol use and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Cancer Epidemiol. Biomarkers Prev., 18, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Garavello W., et al. (2006) Risk factors for distant metastases in head and neck squamous cell carcinoma. Arch. Otolaryngol. Head. Neck Surg., 132, 762–766. [DOI] [PubMed] [Google Scholar]
- 371. Liao C.T., et al. (2006) Higher distant failure in young age tongue cancer patients. Oral Oncol., 42, 718–725. [DOI] [PubMed] [Google Scholar]
- 372. Myers J. (2010) Oral Cancer Metastasis. Soringer Sciences, LLC, New York, NY. [Google Scholar]
- 373. Chambers A.F., et al. (2002) Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer, 2, 563–572. [DOI] [PubMed] [Google Scholar]
- 374. Hillbertz N.S., et al. (2012) Viral and molecular aspects of oral cancer. Anticancer Res., 32, 4201–4212. [PubMed] [Google Scholar]
- 375. Izzo J.G., et al. (2006) Pretherapy nuclear factor-kappaB status, chemoradiation resistance, and metastatic progression in esophageal carcinoma. Mol. Cancer Ther., 5, 2844–2850. [DOI] [PubMed] [Google Scholar]
- 376. Pannone G., et al. (2010) WNT pathway in oral cancer: epigenetic inactivation of WNT-inhibitors. Oncol. Rep., 24, 1035–1041. [DOI] [PubMed] [Google Scholar]
- 377. Jackson-Bernitsas D.G., et al. (2007) Evidence that TNF-TNFR1-TRADD-TRAF2-RIP-TAK1-IKK pathway mediates constitutive NF-kappaB activation and proliferation in human head and neck squamous cell carcinoma. Oncogene, 26, 1385–1397. [DOI] [PubMed] [Google Scholar]
- 378. Iwatsuki M., et al. (2010) Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Sci., 101, 293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Papagerakis S., et al. (2012) Epithelial-mesenchymal interactions in oral cancer metastasis. In Ogbureke K.U. (ed), Oral Cancer. InTech, Rijeka, Croatia, pp. 373–388. [Google Scholar]
- 380. Krishnamurthy S., et al. (2012) Head and neck cancer stem cells. J. Dent. Res., 91, 334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381. Davis S.J., et al. (2010) Metastatic potential of cancer stem cells in head and neck squamous cell carcinoma. Arch. Otolaryngol. Head. Neck Surg., 136, 1260–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382. Czerwinski M.J., et al. (2013) In vitro evaluation of sialyl Lewis X relationship with head and neck cancer stem cells. Otolaryngol. Head. Neck Surg., 149, 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383. Prise K.M., et al. (2011) Concise review: stem cell effects in radiation risk. Stem Cells, 29, 1315–1321. [DOI] [PubMed] [Google Scholar]
- 384. Iglesias-Bartolome R., et al. (2013) Control of the epithelial stem cell epigenome: the shaping of epithelial stem cell identity. Curr. Opin. Cell Biol., 25, 162–169. [DOI] [PubMed] [Google Scholar]
- 385. Nickoloff B.J., et al. (2003) Notch signaling as a therapeutic target in cancer: a new approach to the development of cell fate modifying agents. Oncogene, 22, 6598–6608. [DOI] [PubMed] [Google Scholar]
- 386. Dolled-Filhart M., et al. (2006) Quantitative in situ analysis of beta-catenin expression in breast cancer shows decreased expression is associated with poor outcome. Cancer Res., 66, 5487–5494. [DOI] [PubMed] [Google Scholar]
- 387. Du J., et al. (2003) The nuclear localization of NFkappaB and p53 is positively correlated with HPV16 E7 level in laryngeal squamous cell carcinoma. J. Histochem. Cytochem., 51, 533–539. [DOI] [PubMed] [Google Scholar]
- 388. Al-Hajj M., et al. (2004) Self-renewal and solid tumor stem cells. Oncogene, 23, 7274–7282. [DOI] [PubMed] [Google Scholar]
- 389. Karhadkar S.S., et al. (2004) Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature, 431, 707–712. [DOI] [PubMed] [Google Scholar]
- 390. Klaus A., et al. (2008) Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer, 8, 387–398. [DOI] [PubMed] [Google Scholar]
- 391. IARC (1987) Overall Evaluations of Carcinogenicity: An Updating on IARC Monographs. IARC Monographs on the Evaluations of Carcinogenic Risks to Humans. Vol. 1–42 World Health Organization International Agency for Research on Cancer, Lyon, France. [Google Scholar]
- 392. IARC (1988) Alcohol Drinking. IARC Monographs on the Evaluations of Carcinogenic Risks to Humans. Vol. 44 World Health Organization International Agency for Research on Cancer, Lyon, France. [Google Scholar]
- 393. IARC (1990) Chromium, Nickel, and Welding. IARC Monographs on the Evaluations of Carcinogenic Risks to Humans. Vol. 49 World Health Organization International Agency for Research on Cancer, Lyon, France. [Google Scholar]
- 394. IARC (2003) Betel-quid and Areca-nut chewing and some areca-nut derived nitrosamines. IARC Monographs on the evaluations of carcinogenic risks to humans. Vol. 85 World Health Organization International Agency for Research on Cancer, Lyon. [PMC free article] [PubMed] [Google Scholar]
- 395. Chiang C., et al. (2012) Environmental factors identifies in the etiology of oral cancers in Taiwan. In Kue O. (ed), Oral Cancer. InTech, Rijeka, Croatia, pp. 107–128. [Google Scholar]
- 396. Blot W.J., et al. (1988) Smoking and drinking in relation to oral and pharyngeal cancer. Cancer Res., 48, 3282–3287. [PubMed] [Google Scholar]
- 397. Cancela M.D.E.C., et al. (2009) Alcohol intake and oral cavity cancer risk among men in a prospective study in Kerala, India. Community Dent. Oral Epidemiol., 37, 342–349. [DOI] [PubMed] [Google Scholar]
- 398. Choi S.Y., et al. (1991) Effect of cigarette smoking and alcohol consumption in the aetiology of cancer of the oral cavity, pharynx and larynx. Int. J. Epidemiol., 20, 878–885. [DOI] [PubMed] [Google Scholar]
- 399. Carpenter R.L., et al. (2013) Roles of EGFR, PI3K, AKT, and mTOR in heavy metal-induced cancer. Curr. Cancer Drug Targets, 13, 252–266. [DOI] [PubMed] [Google Scholar]
- 400. Li J.Y. (1982) Epidemiology of esophageal cancer in China. Natl Cancer Inst. Monogr., 62, 113–120. [PubMed] [Google Scholar]
- 401. Li M.H., et al. (1980) Recent progress in research on esophageal cancer in China. Adv. Cancer Res., 33, 173–249. [PubMed] [Google Scholar]
- 402. Miller E.C., et al. (1981) Searches for ultimate chemical carcinogens and their reactions with cellular macromolecules. Cancer, 47, 2327–2345. [DOI] [PubMed] [Google Scholar]
- 403. Miller J.A. (1970) Carcinogenesis by chemicals: an overview–G. H. A. Clowes memorial lecture. Cancer Res., 30, 559–576. [PubMed] [Google Scholar]
- 404. Poirier M.C. (2004) Chemical-induced DNA damage and human cancer risk. Nat. Rev. Cancer, 4, 630–637. [DOI] [PubMed] [Google Scholar]
- 405. Puñal-Riobóo J., et al. (2010) [Occupation as a risk factor for oral and pharyngeal cancer]. Acta Otorrinolaringol. Esp., 61, 375–383. [DOI] [PubMed] [Google Scholar]
- 406. Rheeder J.P., et al. (1994) Soil fertility factors in relation to oesophageal cancer risk areas in Transkei, southern Africa. Eur. J. Cancer Prev., 3, 49–56. [DOI] [PubMed] [Google Scholar]
- 407. Roth M.J., et al. (1998) Histopathologic changes seen in esophagectomy specimens from the high-risk region of Linxian, China: potential clues to an etiologic exposure? Hum. Pathol., 29, 1294–1298. [DOI] [PubMed] [Google Scholar]
- 408. Roth M.J., et al. (1998) High levels of carcinogenic polycyclic aromatic hydrocarbons present within food from Linxian, China may contribute to that region’s high incidence of oesophageal cancer. Eur. J. Cancer, 34, 757–758. [DOI] [PubMed] [Google Scholar]
- 409. Türkdoğan M.K., et al. (2003) Heavy metals in soil, vegetables and fruits in the endemic upper gastrointestinal cancer region of Turkey. Environ. Toxicol. Pharmacol., 13, 175–179. [DOI] [PubMed] [Google Scholar]
- 410. Yang C.S. (1980) Research on esophageal cancer in China: a review. Cancer Res., 40, 2633–2644. [PubMed] [Google Scholar]
- 411. Yuspa S.H., et al. (1988) Chemical carcinogenesis: from animal models to molecular models in one decade. Adv. Cancer Res., 50, 25–70. [DOI] [PubMed] [Google Scholar]
- 412. Brugge D., et al. Radium in the environment: exposure pathways and health effects. Rev. Environ. Health, 27, 1–17. [DOI] [PubMed] [Google Scholar]
- 413. Torti S.V., et al. (2013) Iron and cancer: more ore to be mined. Nat. Rev. Cancer, 13, 342–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414. Woźniak A., et al. (2013) [Head and neck cancer–history]. Przegl. Lek., 69, 1079–1083. [PubMed] [Google Scholar]
- 415. Sun W., et al. (2012) Detection of TIMP3 promoter hypermethylation in salivary rinse as an independent predictor of local recurrence-free survival in head and neck cancer. Clin. Cancer Res., 18, 1082–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416. Colacino J.A., et al. (2013) Comprehensive analysis of DNA methylation in head and neck squamous cell carcinoma indicates differences by survival and clinicopathologic characteristics. PLoS One, 8, e54742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417. Carvalho A.L., et al. (2006) Deleted in colorectal cancer is a putative conditional tumor-suppressor gene inactivated by promoter hypermethylation in head and neck squamous cell carcinoma. Cancer Res., 66, 9401–9407. [DOI] [PubMed] [Google Scholar]
- 418. Tan H.K., et al. (2008) Quantitative methylation analyses of resection margins predict local recurrences and disease-specific deaths in patients with head and neck squamous cell carcinomas. Br. J. Cancer, 99, 357–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419. Agrawal N., et al. (2011) Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science, 333, 1154–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420. Stransky N., et al. (2011) The mutational landscape of head and neck squamous cell carcinoma. Science, 333, 1157–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421. Sartor M.A., et al. (2011) Genome-wide methylation and expression differences in HPV(+) and HPV(-) squamous cell carcinoma cell lines are consistent with divergent mechanisms of carcinogenesis. Epigenetics, 6, 777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422. Colacino J.A., et al. (2012) Pretreatment dietary intake is associated with tumor suppressor DNA methylation in head and neck squamous cell carcinomas. Epigenetics, 7, 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423. Kanojia D., et al. (2006) 4-nitroquinoline-1-oxide induced experimental oral carcinogenesis. Oral Oncol., 42, 655–667. [DOI] [PubMed] [Google Scholar]
- 424. Fowke J.H. (2007) Head and neck cancer: a case for inhibition by isothiocyanates and indoles from cruciferous vegetables. Eur. J. Cancer Prev., 16, 348–356. [DOI] [PubMed] [Google Scholar]
- 425. Shklar G., et al. (1993) Oral cancer inhibition by micronutrients. The experimental basis for clinical trials. Eur. J. Cancer. B. Oral Oncol., 29B, 9–16. [DOI] [PubMed] [Google Scholar]
- 426. Ahn J., et al. (2012) Oral microbiome and oral and gastrointestinal cancer risk. Cancer Causes Control, 23, 399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427. Agrawal Y., et al. (2008) Oral human papillomavirus infection before and after treatment for human papillomavirus 16-positive and human papillomavirus 16-negative head and neck squamous cell carcinoma. Clin. Cancer Res., 14, 7143–7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428. Ang K.K., et al. (2010) Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med., 363, 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429. Chaturvedi A.K., et al. (2008) Incidence trends for human papillomavirus-related and -unrelated oral squamous cell carcinomas in the United States. J. Clin. Oncol., 26, 612–619. [DOI] [PubMed] [Google Scholar]
- 430. D’Souza G., et al. (2009) Oral sexual behaviors associated with prevalent oral human papillomavirus infection. J. Infect. Dis., 199, 1263–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431. Gillison M.L., et al. (2000) Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J. Natl Cancer Inst., 92, 709–720. [DOI] [PubMed] [Google Scholar]
- 432. Marur S., et al. (2010) HPV-associated head and neck cancer: a virus-related cancer epidemic. Lancet Oncol., 11, 781–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433. Pannone G., et al. (2011) The role of human papillomavirus in the pathogenesis of head & neck squamous cell carcinoma: an overview. Infect. Agent. Cancer, 6, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434. Hausen H. (2006) Infections Causing Human Cancer. Wiley-VCH Verlag, Weinheim, Germany. [Google Scholar]
- 435. Chenevert J., et al. (2012) Squamous cell carcinoma metastatic to neck from an unknown primary: the potential impact of modern pathologic evaluation on perceived incidence of human papillomavirus-positive oropharyngeal carcinoma prior to 1970. Laryngoscope, 122, 793–796. [DOI] [PubMed] [Google Scholar]
- 436. Vidal L., et al. (2008) Human papillomavirus in HNSCC: recognition of a distinct disease type. Hematol. Oncol. Clin. North Am., 22, 1125–1142, vii. [DOI] [PubMed] [Google Scholar]
- 437. Tang A., et al. (2013) Head and neck cancer stem cells: the effect of HPV, an in vitro and mouse study. Otolaryngol. Head Neck Surg., 142, 252–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438. Applebaum K.M., et al. (2007) Lack of association of alcohol and tobacco with HPV16-associated head and neck cancer. J. Natl Cancer Inst., 99, 1801–1810. [DOI] [PubMed] [Google Scholar]
- 439. Gillison M.L., et al. (2008) Distinct risk factor profiles for human papillomavirus type 16-positive and human papillomavirus type 16-negative head and neck cancers. J. Natl Cancer Inst., 100, 407–420. [DOI] [PubMed] [Google Scholar]
- 440. Arthur A.E., et al. Higher micronutrient intake is associated with human papillomavirus-positive head and neck cancer: a case-only analysis. Nutr. Cancer, 63, 734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441. Fakhry C., et al. (2008) Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J. Natl Cancer Inst., 100, 261–269. [DOI] [PubMed] [Google Scholar]
- 442. Kumar B., et al. (2008) EGFR, p16, HPV Titer, Bcl-xL and p53, sex, and smoking as indicators of response to therapy and survival in oropharyngeal cancer. J. Clin. Oncol., 26, 3128–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443. Maxwell J.H., et al. (2010) Tobacco use in human papillomavirus-positive advanced oropharynx cancer patients related to increased risk of distant metastases and tumor recurrence. Clin. Cancer Res., 16, 1226–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444. Worden F.P., et al. (2008) Chemoselection as a strategy for organ preservation in advanced oropharynx cancer: response and survival positively associated with HPV16 copy number. J. Clin. Oncol., 26, 3138–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445. Maxwell J.H., et al. (2010) HPV-positive/p16-positive/EBV-negative nasopharyngeal carcinoma in white North Americans. Head Neck, 32, 562–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446. Squier C.A., et al. (2001) Biology of oral mucosa and esophagus. J. Natl Cancer Inst. Monogr., 29, 7–15. [DOI] [PubMed] [Google Scholar]
- 447. Collins L.M., et al. (1987) The surface area of the adult human mouth and thickness of the salivary film covering the teeth and oral mucosa. J. Dent. Res., 66, 1300–1302. [DOI] [PubMed] [Google Scholar]
- 448. Squier C.A., et al. (1976) Human Oral Mucosa: Development, Structure and Function. Blackwell Scientific, Oxford. [Google Scholar]
- 449. Bjarnason G.A., et al. (1999) Circadian variation in the expression of cell-cycle proteins in human oral epithelium. Am. J. Pathol., 154, 613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450. Thomson P.J., et al. (1999) Mapping dynamic epithelial cell proliferative activity within the oral cavity of man: a new insight into carcinogenesis? Br. J. Oral Maxillofac. Surg., 37, 377–383. [DOI] [PubMed] [Google Scholar]
- 451. Feliciani C., et al. (1996) Keratinocytes and cytokine/growth factors. Crit. Rev. Oral Biol. Med., 7, 300–318. [DOI] [PubMed] [Google Scholar]
- 452. Pries R., et al. (2006) Cytokines in head and neck cancer. Cytokine Growth Factor Rev., 17, 141–146. [DOI] [PubMed] [Google Scholar]
- 453. Duffy S.A., et al. (2008) Health behaviors of head and neck cancer patients the first year after diagnosis. Head Neck, 30, 93–102. [DOI] [PubMed] [Google Scholar]
- 454. Shkeir O., et al. (2013) In vitro cytokine release profile: predictive value for metastatic potential in head and neck squamous cell carcinomas. Head Neck, 35, 1542–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455. Ostrand-Rosenberg S. (2008) Immune surveillance: a balance between protumor and antitumor immunity. Curr. Opin. Genet. Dev., 18, 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456. Rapidis A.D., et al. (2009) Immunotherapy of head and neck cancer: current and future considerations. J. Oncol., 2009, 346345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457. Whiteside T.L. (2008) The tumor microenvironment and its role in promoting tumor growth. Oncogene, 27, 5904–5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458. Accomando W.P., et al. (2012) Decreased NK cells in patients with head and neck cancer determined in archival DNA. Clin. Cancer Res., 18, 6147–6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459. Duray A., et al. (2010) Immune suppression in head and neck cancers: a review. Clin. Dev. Immunol., 2010, 701657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460. Kuss I., et al. (2004) Decreased absolute counts of T lymphocyte subsets and their relation to disease in squamous cell carcinoma of the head and neck. Clin. Cancer Res., 10, 3755–3762. [DOI] [PubMed] [Google Scholar]
- 461. Kuss I., et al. (2005) Imbalance in absolute counts of T lymphocyte subsets in patients with head and neck cancer and its relation to disease. Adv. Otorhinolaryngol., 62, 161–172. [DOI] [PubMed] [Google Scholar]
- 462. Wulff S., et al. (2009) Decreased levels of circulating regulatory NK cells in patients with head and neck cancer throughout all tumor stages. Anticancer Res., 29, 3053–3057. [PubMed] [Google Scholar]
- 463. Levy E.M., et al. (2011) Natural killer cells in human cancer: from biological functions to clinical applications. J. Biomed. Biotechnol., 2011, 676198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464. Molling J.W., et al. (2007) Low levels of circulating invariant natural killer T cells predict poor clinical outcome in patients with head and neck squamous cell carcinoma. J. Clin. Oncol., 25, 862–868. [DOI] [PubMed] [Google Scholar]
- 465. Euvrard S., et al. (2003) Skin cancers after organ transplantation. N. Engl. J. Med., 348, 1681–1691. [DOI] [PubMed] [Google Scholar]
- 466. Hakim F.T., et al. (2004) Aging, immunity and cancer. Curr. Opin. Immunol., 16, 151–156. [DOI] [PubMed] [Google Scholar]
- 467. Winn D.M. (1995) Diet and nutrition in the etiology of oral cancer. Am. J. Clin. Nutr., 61, 437S–445S. [DOI] [PubMed] [Google Scholar]
- 468. Lutz W.K. (1999) Carcinogens in the diet vs. overnutrition. Individual dietary habits, malnutrition, and genetic susceptibility modify carcinogenic potency and cancer risk. Mutat. Res., 443, 251–258. [DOI] [PubMed] [Google Scholar]
- 469. Zain R.B. (2001) Cultural and dietary risk factors of oral cancer and precancer–a brief overview. Oral Oncol., 37, 205–210. [DOI] [PubMed] [Google Scholar]
- 470. Karvonen-Gutierrez C.A., et al. (2008) Quality of life scores predict survival among patients with head and neck cancer. J. Clin. Oncol., 26, 2754–2760. [DOI] [PubMed] [Google Scholar]
- 471. Gery S., et al. (2010) Circadian rhythms and cancer. Cell Cycle, 9, 1097–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472. Fu L., et al. (2003) The circadian clock: pacemaker and tumour suppressor. Nat. Rev. Cancer, 3, 350–361. [DOI] [PubMed] [Google Scholar]
- 473. Fu L., et al. (2002) The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo . Cell, 111, 41–50. [DOI] [PubMed] [Google Scholar]
- 474. Hunt T., et al. (2007) Riding tandem: circadian clocks and the cell cycle. Cell, 129, 461–464. [DOI] [PubMed] [Google Scholar]
- 475. Spengler M.L., et al. (2012) Core circadian protein CLOCK is a positive regulator of NF-kappaB-mediated transcription. Proc. Natl Acad. Sci. USA, 109, E2457–E2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476. Schernhammer E.S., et al. (2003) Night-shift work and risk of colorectal cancer in the nurses’ health study. J. Natl Cancer Inst., 95, 825–828. [DOI] [PubMed] [Google Scholar]
- 477. Zieker D., et al. (2010) Circadian expression of clock- and tumor suppressor genes in human oral mucosa. Cell. Physiol. Biochem., 26, 155–166. [DOI] [PubMed] [Google Scholar]
- 478. Zheng L., et al. (2011) Expression of clock proteins in developing tooth. Gene Expr. Patterns, 11, 202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479. Zheng L., et al. (2012) Clock genes show circadian rhythms in salivary glands. J. Dent. Res., 91, 783–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480. Hsu C.M., et al. (2012) Altered expression of circadian clock genes in head and neck squamous cell carcinoma. Tumour Biol., 33, 149–155. [DOI] [PubMed] [Google Scholar]
- 481. Focan C., et al. (2003) [Chronobiological considerations for the management of human head and neck cancer]. Pathol. Biol. (Paris), 51, 201–203. [DOI] [PubMed] [Google Scholar]
- 482. Ohdo S. (2010) Chronotherapeutic strategy: rhythm monitoring, manipulation and disruption. Adv. Drug Deliv. Rev., 62, 859–875. [DOI] [PubMed] [Google Scholar]
- 483. Wansom D., et al. (2010) Correlation of cellular immunity with human papillomavirus 16 status and outcome in patients with advanced oropharyngeal cancer. Arch. Otolaryngol. Head Neck Surg., 136, 1267–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484. Shuman A.G., et al. (2010) Demographics and efficacy of head and neck cancer screening. Otolaryngol. Head Neck Surg., 143, 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485. Duffy L., et al. (2007) The lived experience of women with cancer: phenomenological findings expressed through poetry. Can. Oncol. Nurs. J., 17, 193–205. [DOI] [PubMed] [Google Scholar]
- 486. Duffy S.A., et al. (2013) Health behaviors predict higher interleukin-6 levels among patients newly diagnosed with head and neck squamous cell carcinoma. Cancer Epidemiol. Biomarkers Prev., 22, 374–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487. Lucci A., et al. (2009) Cyclooxygenase-2 expression in primary breast cancers predicts dissemination of cancer cells to the bone marrow. Breast Cancer Res. Treat., 117, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488. Xin X., et al. Targeting COX-2 and EP4 to control tumor growth, angiogenesis, lymphangiogenesis and metastasis to the lungs and lymph nodes in a breast cancer model. Lab. Invest., 92, 1115–1128. [DOI] [PubMed] [Google Scholar]
- 489. Li W., et al. Cyclooxygenase 2 promoted the tumorigenecity of pancreatic cancer cells. Tumour Biol., 35, 2271–2278. [DOI] [PubMed] [Google Scholar]
- 490. Tang H.Y., et al. (2006) Resveratrol-induced cyclooxygenase-2 facilitates p53-dependent apoptosis in human breast cancer cells. Mol. Cancer Ther., 5, 2034–2042. [DOI] [PubMed] [Google Scholar]
- 491. Subbaramaiah K., et al. (1999) Inhibition of cyclooxygenase-2 gene expression by p53. J. Biol. Chem., 274, 10911–10915. [DOI] [PubMed] [Google Scholar]
- 492. Corcoran C.A., et al. (2005) Cyclooxygenase-2 interacts with p53 and interferes with p53-dependent transcription and apoptosis. Oncogene, 24, 1634–1640. [DOI] [PubMed] [Google Scholar]
- 493. Sun Y., et al. (2002) Cyclooxygenase-2 overexpression reduces apoptotic susceptibility by inhibiting the cytochrome c-dependent apoptotic pathway in human colon cancer cells. Cancer Res., 62, 6323–6328. [PubMed] [Google Scholar]
- 494. Monteith G.R., et al. (2007) Calcium and cancer: targeting Ca2+ transport. Nat. Rev. Cancer, 7, 519–530. [DOI] [PubMed] [Google Scholar]
- 495. Rosenberger S., et al. (2007) A novel regulator of telomerase S100A8 mediates differentiation-dependent and calcium-induced inhibition of telomerase activity in the human epidermal keratinocyte line HaCaT. J. Biol. Chem., 282, 6126–6135. [DOI] [PubMed] [Google Scholar]
- 496. Liao C.H., et al. (2007) Nuclear translocation of telomerase reverse transcriptase and calcium signaling in repression of telomerase activity in human lung cancer cells by fungal immunomodulatory protein from Ganoderma tsugae. Biochem. Pharmacol., 74, 1541–1554. [DOI] [PubMed] [Google Scholar]
- 497. Garufi A., et al. (2012) Targeting COX-2/PGE(2) pathway in HIPK2 knockdown cancer cells: impact on dendritic cell maturation. PLoS One, 7, e48342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498. Rizzo M.T. Cyclooxygenase-2 in oncogenesis. Clin. Chim. Acta, 412, 671–687. [DOI] [PubMed] [Google Scholar]
- 499. Hibino T., et al. (2013) S100A9 is a novel ligand of EMMPRIN that promotes melanoma metastasis. Cancer Res., 73, 172–183. [DOI] [PubMed] [Google Scholar]
- 500. Chen Y.F., et al. (2013) Remodeling of calcium signaling in tumor progression. J. Biomed. Sci., 20, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501. Bierie B., et al. (2008) Transforming growth factor-beta regulates mammary carcinoma cell survival and interaction with the adjacent microenvironment. Cancer Res., 68, 1809–1819. [DOI] [PubMed] [Google Scholar]
- 502. Arriola E., et al. (2008) Genomic analysis of the HER2/TOP2A amplicon in breast cancer and breast cancer cell lines. Lab. Invest., 88, 491–503. [DOI] [PubMed] [Google Scholar]
- 503. Mallette F.A., et al. (2007) The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev., 21, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504. Lin Y.W., et al. (2003) Persistent activation of ERK1/2 by lead acetate increases nucleotide excision repair synthesis and confers anti-cytotoxicity and anti-mutagenicity. Carcinogenesis, 24, 53–61. [DOI] [PubMed] [Google Scholar]
- 505. Hampson P., et al. Kinetics of ERK1/2 activation determine sensitivity of acute myeloid leukaemia cells to the induction of apoptosis by the novel small molecule ingenol 3-angelate (PEP005). Apoptosis, 15, 946–55. [DOI] [PubMed] [Google Scholar]
- 506. Wada T., et al. (2004) Mitogen-activated protein kinases in apoptosis regulation. Oncogene, 23, 2838–2849. [DOI] [PubMed] [Google Scholar]
- 507. Pucci B., et al. (2009) ERK-1 MAP kinase prevents TNF-induced apoptosis through bad phosphorylation and inhibition of Bax translocation in HeLa cells. J. Cell. Biochem., 108, 1166–1174. [DOI] [PubMed] [Google Scholar]
- 508. Jiang Q., et al. (2013) ATF4 activation by the p38MAPK-eIF4E axis mediates apoptosis and autophagy induced by selenite in Jurkat cells. FEBS Lett., 587, 2420–2429. [DOI] [PubMed] [Google Scholar]
- 509. Pontillo C.A., et al. (2011) Activation of c-Src/HER1/STAT5b and HER1/ERK1/2 signaling pathways and cell migration by hexachlorobenzene in MDA-MB-231 human breast cancer cell line. Toxicol. Sci., 120, 284–96. [DOI] [PubMed] [Google Scholar]
- 510. Pontillo C.A., et al. (2013) Action of hexachlorobenzene on tumor growth and metastasis in different experimental models. Toxicol. Appl. Pharmacol., 268, 331–342. [DOI] [PubMed] [Google Scholar]
- 511. Segditsas S., et al. (2006) Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene, 25, 7531–7537. [DOI] [PubMed] [Google Scholar]
- 512. Thorstensen L., et al. (2005) Genetic and epigenetic changes of components affecting the WNT pathway in colorectal carcinomas stratified by microsatellite instability. Neoplasia, 7, 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513. Pez F., et al. (2013) Wnt signaling and hepatocarcinogenesis: molecular targets for the development of innovative anticancer drugs. J. Hepatol., 59, 1107–1117. [DOI] [PubMed] [Google Scholar]
- 514. Tell S., et al. (2006) The Wnt signaling pathway has tumor suppressor properties in retinoblastoma. Biochem. Biophys. Res. Commun., 349, 261–269. [DOI] [PubMed] [Google Scholar]
- 515. Kajiyama H., et al. (2007) Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int. J. Oncol., 31, 277–283. [PubMed] [Google Scholar]
- 516. Massagué J. (2008) TGFbeta in Cancer. Cell, 134, 215–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517. Hoffmeyer K., et al. (2012) Wnt/beta-catenin signaling regulates telomerase in stem cells and cancer cells. Science, 336, 1549–1554. [DOI] [PubMed] [Google Scholar]
- 518. Diala I., et al. (2013) Telomere protection and TRF2 expression are enhanced by the canonical Wnt signalling pathway. EMBO Rep., 14, 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519. Kavitha K., et al. (2013) Astaxanthin inhibits NF-kappaB and Wnt/beta-catenin signaling pathways via inactivation of Erk/MAPK and PI3K/Akt to induce intrinsic apoptosis in a hamster model of oral cancer. Biochim. Biophys. Acta, 1830, 4433–4444. [DOI] [PubMed] [Google Scholar]
- 520. Akaboshi S., et al. (2009) HMGA1 is induced by Wnt/beta-catenin pathway and maintains cell proliferation in gastric cancer. Am. J. Pathol., 175, 1675–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521. Kim J., et al. (2013) CK2 inhibitor CX-4945 blocks TGF-beta1-induced epithelial-to-mesenchymal transition in A549 human lung adenocarcinoma cells. PLoS One, 8, e74342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522. Wang Y., et al. (2013) The role of snail in EMT and tumorigenesis. Curr. Cancer Drug Targets, 13, 963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523. Rundhaug J.E. (2003) Matrix metalloproteinases, angiogenesis, and cancer: commentary re: A. C. Lockhart et al., Reduction of wound angiogenesis in patients treated with BMS-275291, a broad spectrum matrix metalloproteinase inhibitor. Clin. Cancer Res., 9: 00-00, 2003. Clin. Cancer Res., 9, 551–554. [PubMed] [Google Scholar]
- 524. Li L., et al. Role of microRNA-mediated MMP regulation in the treatment and diagnosis of malignant tumors. Cancer Biol. Ther., 14, 796–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525. Liu J., et al. (2006) Wild-type p53 inhibits nuclear factor-kappaB-induced matrix metalloproteinase-9 promoter activation: implications for soft tissue sarcoma growth and metastasis. Mol. Cancer Res., 4, 803–810. [DOI] [PubMed] [Google Scholar]
- 526. Choe G., et al. (2002) Active matrix metalloproteinase 9 expression is associated with primary glioblastoma subtype. Clin. Cancer Res., 8, 2894–2901. [PubMed] [Google Scholar]
- 527. Kim J.H., et al. (2010) Differential roles of matrix metalloproteinase-9 and -2, depending on proliferation or differentiation of retinoblastoma cells. Invest. Ophthalmol. Vis. Sci., 51, 1783–1788. [DOI] [PubMed] [Google Scholar]
- 528. Jin M.L., et al. (2014) Halofuginone induces the apoptosis of breast cancer cells and inhibits migration via downregulation of matrix metalloproteinase-9. Int. J. Oncol., 44, 309–318. [DOI] [PubMed] [Google Scholar]
- 529. Chintala S.K., et al. (2002) Deficiency in matrix metalloproteinase gelatinase B (MMP-9) protects against retinal ganglion cell death after optic nerve ligation. J. Biol. Chem., 277, 47461–47468. [DOI] [PubMed] [Google Scholar]
- 530. Ovechkin A.V., et al. (2005) Role of matrix metalloproteinase-9 in endothelial apoptosis in chronic heart failure in mice. J. Appl. Physiol. (1985), 99, 2398–2405. [DOI] [PubMed] [Google Scholar]
- 531. Park S.Y., et al. (2013) Pro-apoptotic and migration-suppressing potential of EGCG, and the involvement of AMPK in the p53-mediated modulation of VEGF and MMP-9 expression. Oncol. Lett., 6, 1346–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532. Fiore E., et al. (2002) Matrix metalloproteinase 9 (MMP-9/gelatinase B) proteolytically cleaves ICAM-1 and participates in tumor cell resistance to natural killer cell-mediated cytotoxicity. Oncogene, 21, 5213–5223. [DOI] [PubMed] [Google Scholar]
- 533. Vandooren J., et al. (2013) Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): the next decade. Crit. Rev. Biochem. Mol. Biol., 48, 222–272. [DOI] [PubMed] [Google Scholar]
- 534. Katsuno Y., et al. (2013) TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol., 25, 76–84. [DOI] [PubMed] [Google Scholar]
- 535. Miyazono K. (2009) Transforming growth factor-beta signaling in epithelial-mesenchymal transition and progression of cancer. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci., 85, 314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536. Wendt M.K., et al. (2009) Mechanisms of the epithelial-mesenchymal transition by TGF-beta. Future Oncol., 5, 1145–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 537. Wendt M.K., et al. TGF-beta stimulates Pyk2 expression as part of an epithelial-mesenchymal transition program required for metastatic outgrowth of breast cancer. Oncogene, 32, 2005–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538. Jacob J., et al. (2012) Retinoblastoma protein determines aggressiveness in triple-negative breast cancer. Expert Rev. Anticancer Ther., 12, 581–584. [DOI] [PubMed] [Google Scholar]
- 539. Arima Y., et al. (2008) Rb depletion results in deregulation of E-cadherin and induction of cellular phenotypic changes that are characteristic of the epithelial-to-mesenchymal transition. Cancer Res., 68, 5104–5112. [DOI] [PubMed] [Google Scholar]
- 540. Chang C.J., et al. (2011) p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat. Cell Biol., 13, 317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541. Termen S., et al. (2013) p53 regulates epithelial-mesenchymal transition induced by transforming growth factor beta. J. Cell. Physiol., 228, 801–13. [DOI] [PubMed] [Google Scholar]
- 542. Ren D., et al. (2013) Wild-type p53 suppresses the epithelial-mesenchymal transition and stemness in PC-3 prostate cancer cells by modulating miR145. Int. J. Oncol., 42, 1473–1481. [DOI] [PubMed] [Google Scholar]
- 543. Ansieau S., et al. (2008) Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell, 14, 79–89. [DOI] [PubMed] [Google Scholar]
- 544. Morel A.P., et al. (2012) EMT inducers catalyze malignant transformation of mammary epithelial cells and drive tumorigenesis towards claudin-low tumors in transgenic mice. PLoS Genet., 8, e1002723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545. Rhim A.D., et al. (2012) EMT and dissemination precede pancreatic tumor formation. Cell, 148, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546. Wu Y., et al. (2014) Molecular mechanisms underlying chronic inflammation-associated cancers. Cancer Lett., 345, 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547. Mimeault M., et al. Altered gene products involved in the malignant reprogramming of cancer stem/progenitor cells and multitargeted therapies. Mol. Aspects Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548. Fu L., et al. (2013) The circadian clock in cancer development and therapy. Prog. Mol. Biol. Transl Sci., 119, 221–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549. Li A., et al. (2013) Circadian gene Clock contributes to cell proliferation and migration of glioma and is directly regulated by tumor-suppressive miR-124. FEBS Lett., 587, 2455–2460. [DOI] [PubMed] [Google Scholar]
- 550. Miki T., et al. (2013) p53 regulates Period2 expression and the circadian clock. Nat. Commun., 4, 2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551. Lee J.H., et al. (2011) Regulation of apoptosis by the circadian clock through NF-kappaB signaling. Proc. Natl Acad. Sci. USA, 108, 12036–12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552. Kagawa Y. (2012) From clock genes to telomeres in the regulation of the healthspan. Nutr. Rev., 70, 459–471. [DOI] [PubMed] [Google Scholar]
- 553. Khapre R.V., et al. (2011) Circadian clock protein BMAL1 regulates cellular senescence in vivo . Cell Cycle, 10, 4162–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554. Summa K.C., et al. (2013) Disruption of the circadian clock in mice increases intestinal permeability and promotes alcohol-induced hepatic pathology and inflammation. PLoS One, 8, e67102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555. Yoshida K., et al. (2013) TNF-alpha modulates expression of the circadian clock gene Per2 in rheumatoid synovial cells. Scand. J. Rheumatol., 42, 276–280. [DOI] [PubMed] [Google Scholar]
- 556. Husse J., et al. (2012) Circadian clock genes Per1 and Per2 regulate the response of metabolism-associated transcripts to sleep disruption. PLoS One, 7, e52983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557. Jiang Y., et al. Prenatal exposure to bisphenol A at the reference dose impairs mitochondria in the heart of neonatal rats. J. Appl. Toxicol, 34, 1012–1022. [DOI] [PubMed] [Google Scholar]
- 558. Lee H.S., et al. (2012) Set, a putative oncogene, as a biomarker for prenatal exposure to bisphenol A. Asian Pac. J. Cancer Prev., 13, 2711–2715. [DOI] [PubMed] [Google Scholar]
- 559. Betancourt A.M., et al. (2012) Altered carcinogenesis and proteome in mammary glands of rats after prepubertal exposures to the hormonally active chemicals bisphenol a and genistein. J. Nutr., 142, 1382S–1388S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560. Fillon M. (2012) Getting it right: BPA and the difficulty proving environmental cancer risks. J. Natl Cancer Inst., 104, 652–655. [DOI] [PubMed] [Google Scholar]
- 561. Takahashi A., et al. (2004) Bisphenol A from dental polycarbonate crown upregulates the expression of hTERT. J. Biomed. Mater. Res. B. Appl. Biomater., 71, 214–221. [DOI] [PubMed] [Google Scholar]
- 562. Hassan Z.K., et al. (2012) Bisphenol A induces hepatotoxicity through oxidative stress in rat model. Oxid. Med. Cell. Longev., 2012, 194829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563. Dairkee S.H., et al. (2013) Bisphenol-A-induced inactivation of the p53 axis underlying deregulation of proliferation kinetics, and cell death in non-malignant human breast epithelial cells. Carcinogenesis, 34, 703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564. Terasaka H., et al. (2005) Cytotoxicity and apoptosis-inducing activity of bisphenol A and hydroquinone in HL-60 cells. Anticancer Res., 25, 2241–2247. [PubMed] [Google Scholar]
- 565. Xu J., et al. (2002) Bisphenol A induces apoptosis and G2-to-M arrest of ovarian granulosa cells. Biochem. Biophys. Res. Commun., 292, 456–462. [DOI] [PubMed] [Google Scholar]
- 566. Goodson W.H., III, et al. (2011) Activation of the mTOR pathway by low levels of xenoestrogens in breast epithelial cells from high-risk women. Carcinogenesis, 32, 1724–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567. Allard P., et al. (2010) Bisphenol A impairs the double-strand break repair machinery in the germline and causes chromosome abnormalities. Proc. Natl Acad. Sci. USA, 107, 20405–20410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568. Durando M., et al. (2011) Prenatal exposure to bisphenol A promotes angiogenesis and alters steroid-mediated responses in the mammary glands of cycling rats. J. Steroid Biochem. Mol. Biol., 127, 35–43. [DOI] [PubMed] [Google Scholar]
- 569. Pupo M., et al. (2012) Bisphenol A induces gene expression changes and proliferative effects through GPER in breast cancer cells and cancer-associated fibroblasts. Environ. Health Perspect., 120, 1177–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570. Billi de Catabbi S.C., et al. (2005) Hepatic arachidonic acid metabolism is disrupted after hexachlorobenzene treatment. Toxicol. Appl. Pharmacol., 204, 187–195. [DOI] [PubMed] [Google Scholar]
- 571. Chaufan G., et al. (2001) How does hexachlorobenzene treatment affect liver uroporphyrinogen decarboxylase? Int. J. Biochem. Cell Biol., 33, 621–630. [DOI] [PubMed] [Google Scholar]
- 572. Lelli S.M., et al. (2007) Hexachlorobenzene as hormonal disruptor–studies about glucocorticoids: their hepatic receptors, adrenal synthesis and plasma levels in relation to impaired gluconeogenesis. Biochem. Pharmacol., 73, 873–879. [DOI] [PubMed] [Google Scholar]
- 573. Michielsen C., et al. (2002) The environmental pollutant hexachlorobenzene causes eosinophilic and granulomatous inflammation and in vitro airways hyperreactivity in the Brown Norway rat. Arch. Toxicol., 76, 236–247. [DOI] [PubMed] [Google Scholar]
- 574. Chiappini F., et al. (2009) Hexachlorobenzene triggers apoptosis in rat thyroid follicular cells. Toxicol. Sci., 108, 301–310. [DOI] [PubMed] [Google Scholar]
- 575. Giribaldi L., et al. (2011) Hexachlorobenzene induces deregulation of cellular growth in rat liver. Toxicology, 289, 19–27. [DOI] [PubMed] [Google Scholar]
- 576. Canonero R., et al. (1997) Testing of p-dichlorobenzene and hexachlorobenzene for their ability to induce DNA damage and micronucleus formation in primary cultures of rat and human hepatocytes. Mutagenesis, 12, 35–39. [DOI] [PubMed] [Google Scholar]
- 577. Majkova Z., et al. (2010) The role of caveolae in endothelial cell dysfunction with a focus on nutrition and environmental toxicants. J. Cell. Mol. Med., 14, 2359–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578. De Coster S., et al. (2008) Pollutant effects on genotoxic parameters and tumor-associated protein levels in adults: a cross sectional study. Environ. Health, 7, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579. García M.A., et al. (2010) Hexachlorobenzene induces cell proliferation and IGF-I signaling pathway in an estrogen receptor alpha-dependent manner in MCF-7 breast cancer cell line. Toxicol. Lett., 192, 195–205. [DOI] [PubMed] [Google Scholar]
- 580. Mainous A.G., III, et al. (2013) Telomere length and elevated iron: the influence of phenotype and HFE genotype. Am. J. Hematol., 88, 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581. Uringa E.J., et al. (2007) RTEL1: an essential helicase for telomere maintenance and the regulation of homologous recombination. Nucleic Acids Res., 39, 1647–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582. Brown K.E., et al. (2007) Increased hepatic telomerase activity in a rat model of iron overload: a role for altered thiol redox state? Free Radic. Biol. Med., 42, 228–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583. Xue X., et al. (2013) Intestinal iron homeostasis and colon tumorigenesis. Nutrients, 5, 2333–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 584. Velez-Pardo C., et al. (1997) Dopamine and iron induce apoptosis in PC12 cells. Pharmacol. Toxicol., 80, 76–84. [DOI] [PubMed] [Google Scholar]
- 585. Prá D., et al. (2008) Genotoxicity and mutagenicity of iron and copper in mice. Biometals, 21, 289–297. [DOI] [PubMed] [Google Scholar]
- 586. Simon T., et al. (2011) Heme oxygenase and carbon monoxide as an immunotherapeutic approach in transplantation and cancer. Immunotherapy, 3, 15–18. [DOI] [PubMed] [Google Scholar]
- 587. Eckard J., et al. (2010) Effects of cellular iron deficiency on the formation of vascular endothelial growth factor and angiogenesis. Iron deficiency and angiogenesis. Cancer Cell Int., 10, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588. Yun Y., et al. (2011) SO(2) inhalation modulates the expression of pro-inflammatory and pro-apoptotic genes in rat heart and lung. J. Hazard. Mater., 185, 482–488. [DOI] [PubMed] [Google Scholar]
- 589. Xie J., et al. (2009) Effects of sulfur dioxide on expressions of p53, bax and bcl-2 in lungs of asthmatic rats. Inhal. Toxicol., 21, 952–957. [DOI] [PubMed] [Google Scholar]
- 590. Bai J., et al. (2010) Expression of caspase and apoptotic signal pathway induced by sulfur dioxide. Environ. Mol. Mutagen., 51, 112–122. [DOI] [PubMed] [Google Scholar]
- 591. Bai J., et al. (2005) Effects of sulfur dioxide on apoptosis-related gene expressions in lungs from rats. Regul. Toxicol. Pharmacol., 43, 272–279. [DOI] [PubMed] [Google Scholar]
- 592. Yadav J.S., et al. (1996) Effect of sulphur dioxide exposure on human chromosomes. Mutat. Res., 359, 25–29. [DOI] [PubMed] [Google Scholar]
- 593. Qin G., et al. (2010) Expression of oncogenes and tumor suppressor genes in lungs of rats exposed to sulfur dioxide and benzo(a)pyrene. Inhal. Toxicol., 22, 322–329. [DOI] [PubMed] [Google Scholar]
- 594. Valles E.G., et al. (2003) Role of the peroxisome proliferator-activated receptor alpha in responses to diisononyl phthalate. Toxicology, 191, 211–225. [DOI] [PubMed] [Google Scholar]
- 595. Rao K.N., et al. (1997) Hepatic hyperplasia and cancer in rats: metabolic alterations associated with cell growth. Gastroenterology, 113, 238–248. [DOI] [PubMed] [Google Scholar]
- 596. Wang Y.C., et al. (1912) Possible mechanism of phthalates-induced tumorigenesis. Kaohsiung J. Med. Sci., 28, S22–S27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597. Kim I.Y., et al. (2004) Phthalates inhibit tamoxifen-induced apoptosis in MCF-7 human breast cancer cells. J. Toxicol. Environ. Health A, 67, 2025–2035. [DOI] [PubMed] [Google Scholar]
- 598. Wang X., et al. (2010) [Effects of di-(2-ethylexyl) phthalate on apoptosis of cytotrophoblasts in early pregnancy]. Zhonghua Fu Chan Ke Za Zhi, 45, 411–414. [PubMed] [Google Scholar]
- 599. Lin C.H., et al. (2011) Activation of Trim17 by PPARgamma is involved in di(2-ethylhexyl) phthalate (DEHP)-induced apoptosis on Neuro-2a cells. Toxicol. Lett., 206, 245–251. [DOI] [PubMed] [Google Scholar]
- 600. Isenberg J.S., et al. (2001) Reversibility and persistence of di-2-ethylhexyl phthalate (DEHP)- and phenobarbital-induced hepatocellular changes in rodents. Toxicol. Sci., 64, 192–199. [DOI] [PubMed] [Google Scholar]
- 601. Kleinsasser N.H., et al. (2000) Phthalates demonstrate genotoxicity on human mucosa of the upper aerodigestive tract. Environ. Mol. Mutagen., 35, 9–12. [DOI] [PubMed] [Google Scholar]
- 602. Hsieh T.H., et al. (2012) n-Butyl benzyl phthalate promotes breast cancer progression by inducing expression of lymphoid enhancer factor 1. PLoS One, 7, e42750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 603. Lee H.-R., et al. (2014) The estrogen receptor signaling pathway activated by phthalates is linked with transforming growth factor-Î2 in the progression of LNCaP prostate cancer models. Int. J.Oncol., 45, 595–602. [DOI] [PubMed] [Google Scholar]
- 604. Hsieh T.H., et al. (2012) Phthalates induce proliferation and invasiveness of estrogen receptor-negative breast cancer through the AhR/HDAC6/c-Myc signaling pathway. FASEB J., 26, 778–787. [DOI] [PubMed] [Google Scholar]
- 605. Park M.A., et al. (2012) Cell growth of BG-1 ovarian cancer cells is promoted by di-n-butyl phthalate and hexabromocyclododecane via upregulation of the cyclin D and cyclin-dependent kinase-4 genes. Mol. Med. Rep., 5, 761–766. [DOI] [PubMed] [Google Scholar]
- 606. Kelleher F.C., et al. (2014) Circadian molecular clocks and cancer. Cancer Lett., 342, 9–18. [DOI] [PubMed] [Google Scholar]
- 607. Kadomatsu T., et al. (2013) A molecular clock regulates angiopoietin-like protein 2 expression. PLoS One, 8, e57921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 608. Montague P.R. (1993) Transforming sensory experience into structural change. Proc. Natl Acad. Sci. USA, 90, 6379–6380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 609. Savvidis C., et al. (2012) Circadian rhythm disruption in cancer biology. Mol. Med., 18, 1249–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 610. Singh B., et al. (2007) Cyclooxygenase-2 expression induces genomic instability in MCF10A breast epithelial cells. J. Surg. Res., 140, 220–226. [DOI] [PubMed] [Google Scholar]
- 611. Xin X., et al. (2012) Targeting COX-2 and EP4 to control tumor growth, angiogenesis, lymphangiogenesis and metastasis to the lungs and lymph nodes in a breast cancer model. Lab. Invest., 92, 1115–1128. [DOI] [PubMed] [Google Scholar]
- 612. Subbaramaiah K., et al. (1999) Inhibition of cyclooxygenase-2 gene expression by p53. J. Biol. Chem., 274, 10911–10915. [DOI] [PubMed] [Google Scholar]
- 613. Corcoran C.A., et al. (2005) Cyclooxygenase-2 interacts with p53 and interferes with p53-dependent transcription and apoptosis. Oncogene, 24, 1634–1640. [DOI] [PubMed] [Google Scholar]
- 614. Li W., et al. (2013) Cyclooxygenase 2 promoted the tumorigenecity of pancreatic cancer cells. Tumour Biol. [DOI] [PubMed] [Google Scholar]
- 615. Sun Y., et al. (2002) Cyclooxygenase-2 overexpression reduces apoptotic susceptibility by inhibiting the cytochrome c-dependent apoptotic pathway in human colon cancer cells. Cancer Res., 62, 6323–6328. [PubMed] [Google Scholar]
- 616. Rizzo M.T. (2011) Cyclooxygenase-2 in oncogenesis. Clin. Chim. Acta., 412, 671–687. [DOI] [PubMed] [Google Scholar]
- 617. Liao C.H., et al. (2007) Nuclear translocation of telomerase reverse transcriptase and calcium signaling in repression of telomerase activity in human lung cancer cells by fungal immunomodulatory protein from Ganoderma tsugae. Biochem. Pharmacol., 74, 1541–1554. [DOI] [PubMed] [Google Scholar]
- 618. Monteith G.R., et al. (2007) Calcium and cancer: targeting Ca2+ transport. Nat. Rev. Cancer, 7, 519–530. [DOI] [PubMed] [Google Scholar]