

Background: Chemokine receptors CXCR4 and ACKR3 are deregulated in many diseases.
Results: Potent modulators of CXCR4 and ACKR3 were engineered by phage display of the chemokine CXCL12.
Conclusion: Requirements for high affinity receptor binding and activation are suggested from experimental data and molecular modeling.
Significance: These chemokine variants represent useful reagents, and the phage display strategy will facilitate further optimization of CXCR4 and ACKR3 modulators.
Keywords: C-X-C chemokine receptor type 4 (CXCR-4), chemokine, G protein-coupled receptor (GPCR), phage display, signaling, ACKR3, CXCL12, SDF-1a, chemokine receptors, experimental autoimmune encephalomyelitis (EAE)
Abstract
The chemokine CXCL12 and its G protein-coupled receptors CXCR4 and ACKR3 are implicated in cancer and inflammatory and autoimmune disorders and are targets of numerous antagonist discovery efforts. Here, we describe a series of novel, high affinity CXCL12-based modulators of CXCR4 and ACKR3 generated by selection of N-terminal CXCL12 phage libraries on live cells expressing the receptors. Twelve of 13 characterized CXCL12 variants are full CXCR4 antagonists, and four have Kd values <5 nm. The new variants also showed high affinity for ACKR3. The variant with the highest affinity for CXCR4, LGGG-CXCL12, showed efficacy in a murine model for multiple sclerosis, demonstrating translational potential. Molecular modeling was used to elucidate the structural basis of binding and antagonism of selected variants and to guide future designs. Together, this work represents an important step toward the development of therapeutics targeting CXCR4 and ACKR3.
Introduction
CXCR45 is a member of the chemokine subfamily of G protein-coupled receptors that functions through interaction with a small 68-amino acid ligand, CXCL12 (SDF-1) (1). CXCR4 is expressed on a broad range of cell types (e.g. T cells, monocytes, bone marrow stromal cells, and endothelial cells) (2, 3) where its interaction with CXCL12 results in classical G protein-coupled receptor signaling activities, including G protein and MAPK activation and recruitment of β-arrestin (4). In turn, these signaling events lead to physiological processes such as cell migration in the context of immune surveillance and inflammatory responses (5) as well as embryonic development where both CXCR4 and CXCL12 are critical for hematopoiesis, lymphogenesis, and cerebral development (6, 7).
In addition to CXCR4, CXCL12 also binds to the atypical chemokine receptor ACKR3 (previously called CXCR7 and RDC1) (8). Although the biological role of ACKR3 is not fully understood, it clearly functions as a scavenger of CXCL12 to establish CXCL12 gradients, and also modulates CXCR4 signaling (9–11).
CXCR4 and ACKR3 have attracted attention as therapeutic targets because of their involvement in inflammatory diseases (12), cancer progression and metastasis (13), and in the case of CXCR4, AIDS (14). Several studies have demonstrated that small molecule antagonists of CXCR4 (e.g. the bicyclam Plerixafor (AMD3100)) provide beneficial effects in multiple disease models (15–17). In 2008, Plerixafor gained Food and Drug Administration approval for mobilization of hematopoietic stem cell transplants in non-Hodgkin lymphoma and multiple myeloma (18), making CXCR4 the second chemokine receptor (in addition to CCR5) to be the target of a marketed drug. Small molecule inhibitors of ACKR3 are extensively studied because of their ability to block tumor reappearance in experimental models of glioblastoma multiforme (19). Finally, chemokine-based inhibitors also show therapeutic promise (20, 21); for example, P2G-CXCL12, an antagonist variant of CXCL12, was demonstrated to slow the progression of experimental autoimmune encephalomyelitis (EAE), a murine model of multiple sclerosis (21).
Chemokine N termini play a critical role in receptor binding and activation, and thus for many chemokines, N-terminal modifications result in altered affinity and/or activity (22). For example, CXCL12 N-terminal mutants K1R and P2G retain near WT binding affinity but have no ability to promote receptor signaling and thus serve as potent antagonists (23). The therapeutic utility of these variants in disease models (21, 24) provides proof of principle and calls for the development of additional chemokine variants with improved affinity and receptor selectivity, as well as better stability and resistance to inactivation by proteolysis. Additionally, for studying the phenomenon of biased receptor signaling and its implications in biology and disease, a panel of reagents with defined and diverse pharmacological properties is needed.
To rapidly engineer proteins with desired properties such as altered pharmacology (e.g. antagonism) or high affinity binding to a target, in vitro selection platforms such as phage display have proven extremely powerful (25–27). As chemokine affinity and pharmacology can be modified by minimal sequence changes in their N termini, they would seem an obvious scaffold for phage display. To this end, Hartley and co-workers identified N-terminally modified variants of the chemokine CCL5 (RANTES) that are effective against R5-tropic HIV by selecting sequence libraries against live cells expressing CCR5, a primary HIV co-receptor (20, 28, 29). We hypothesized that due to the roles of CXCR4 and ACKR3 in disease, modifications of CXCL12 might be expected to produce similarly important therapeutic leads. However, to our knowledge, phage display studies have not been reported for this chemokine. In retrospect, this is not surprising, as we encountered significant challenges in our initial endeavors with CXCL12.
Here, we present a series of high affinity CXCR4 antagonists obtained as a result of phage display with mutations focused on the CXCL12 N terminus. ACKR3 was also included in some selections with the aim of identifying dual or receptor-specific inhibitors. Although WT CXCL12 is 100-fold more potent in binding ACKR3 than CXCR4, the variants presented here range from those with affinities comparable with WT CXCL12 to those with higher affinity for CXCR4 (e.g. LGGG-CXCL12 is 10-fold more potent in binding CXCR4 than ACKR3). LGGG-CXCL12 was tested in EAE and showed efficacy despite being unoptimized as an antagonist or for pharmacokinetic and pharmacodynamic properties. Molecular modeling was used to elucidate the structural basis of the high affinity binding and antagonism of selected CXCL12 variants toward CXCR4.
Experimental Procedures
Cell Lines and Maintenance
HEK293 cells (ATCC) were cultured in Dulbecco's modified Eagle's medium (DMEM) + GlutaMAX (Life Technologies) supplemented with 10% fetal bovine serum (Life Technologies) and appropriate drugs. MDA-MD-231 cells (ATCC), a human mammary carcinoma cell line, and Jurkat (ATCC), a human T lymphocytic cell line, were cultured in RPMI 1640 medium (Life Technologies) supplemented with 10% fetal bovine serum (Life Technologies). For binding assays, HEK293 cells expressing high levels of CXCR4 or ACKR3 were generated using a tetracycline-inducible system as follows. HEK293 cells containing pcDNA6/TR and the pACMV-TetO vector were generous gifts from H. G. Khorana (Massachusetts Institute of Technology, Cambridge, MA). Human CXCR4 and ACKR3 were cloned into pACMV-TetO and transfected into HEK293 cells containing pcDNA6/TR with TransIT-LT1 reagent (Mirus Bio LLC) according to the manufacturer's instructions. Transfectants were selected and maintained in the presence of 700 μg/ml G418 (Life Technologies) and 5 μg/ml blasticidin. All cells were grown at 37 °C with 5% CO2.
Library Creation and Phage Methods
For phage experiments, two CXCL12 libraries were designed: in the “N-addition” library, the N terminus of CXCL12 was extended by one residue and randomized simultaneously with WT CXCL12 residues 1–3, whereas in the “N-truncation” library, the first four residues were deleted, and residues 5–8 were randomized. Each library was amplified in two steps, beginning with the constant region of CXCL12 (residues 4–68 for N-addition or residues 9–68 for N-truncation library) called the “stub fragment.” The purified stub fragment was used as a template for a forward degenerate library primer and a reverse primer. The degenerate primers utilized were 5′-ccgtggcccaggcggccnnknnknnknnkagcctgagctatcgctgcccg-3′ for the N-addition library and 5′-ccgtggcccaggcggccnnknnknnknnktgcccgtgccgcttctttg-3′ for the N-truncation library. The resulting product was cloned into the SfiI/SfiI site of the phagemid vector pComb3SS (a generous gift from C. F. Barbas, The Scripps Research Institute, La Jolla, CA) in-frame with and between the OmpA signal sequence and the phage coat protein g3p and transformed by electroporation into Escherichia coli K12 ER2738 cells. The library size was experimentally found to be 108 and 107 for the N-addition and N-truncation CXCL12 libraries, respectively, by calculating the number of clones after library transformation. Both libraries exceed the theoretical library diversity of ∼105 wherein each member is represented once. The resulting libraries were propagated by growing the bacterial cells in MOPS-buffered Superbroth with carbenicillin (200 μg/ml) and tetracycline (10 μg/ml) overnight at 30 °C. Library cryostocks were prepared from the overnight bacterial culture and stored at −80 °C in 20% glycerol.
Library cryostocks were utilized for phage generation. In the optimized method, production of CXCL12 N-terminally fused to the phage g3p protein (CXCL12-g3p) was initiated with 1 mm isopropyl β-d-thiogalactopyranoside for 2 h at room temperature (23 °C) before adding VCSM13 helper phage (Stratagene) for 30 min. This was done to initiate production of CXCL12-g3p fusion protein before the E. coli host export machinery became taxed with the production of phage coat proteins. The cells were collected by centrifugation and resuspended in fresh medium containing carbenicillin (200 μg/ml) and kanamycin (70 μg/ml). After overnight growth at 37 °C, cells were removed by centrifugation, and 4% (w/v) PEG 8000 and 3% (w/v) NaCl were added to the supernatant. Phage particles were collected by centrifugation (15,000 × g for 20 min), resuspended in phosphate-buffered saline (PBS), and filtered before use in panning experiments.
Sandwich ELISAs were carried out in a 96-well format using untreated Corning plates. The wells were coated with 20 ng/well anti-CXCL12 antibody (R&D Systems, catalog number AF-310-NA) in Tris-buffered saline (TBS) with an overnight incubation at 4 °C. The wells were incubated with phage for 1 h and rinsed five times with a mixture of TBS and 0.5% Tween 20 to remove unbound phage. HRP-conjugated anti-M13 phage antibody (GE Healthcare, catalog number 27-9421-01) was added at 1:500 dilution, incubated for 1 h, and washed. Absorbance at 405 nm was measured after addition of 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) with a SpectraMax M5 plate reader (Molecular Devices).
Each panning experiment comprised up to seven rounds of selection and amplification using HEK293 and MDA-MB-231 cells expressing various levels of CXCR4 and ACKR3. Selection experiments were performed in 60-mm cell culture dishes with cells at a confluence of 70–90%. Purified phage particles were allowed to bind their target receptors in PBS for 2 h at 37 °C. Cells were lifted from the plates with 1 mm EDTA in PBS and washed 3–10 times (3 times for initial low stringency panning rounds and 10 times for later high stringency rounds) with PBS followed by acid elution (0.2 mm glycine, pH 3.0). The eluate was neutralized and used to infect fresh E. coli K12 ER2738 cells for amplification, titering, and sequencing.
Cloning, Expression, and Purification of CXCL12 Proteins
CXCL12 was cloned into the NdeI/XhoI site of a pET21-based vector with an N-terminal His8 tag followed by an enterokinase recognition site. The desired mutations were introduced into the WT sequence using QuikChange site-directed mutagenesis (Stratagene). For protein production, the CXCL12 plasmid was transformed into E. coli BL21(DE3)pLysS cells. Cells were grown at 37 °C in Luria-Bertani medium, and protein expression was induced with isopropyl β-d-thiogalactopyranoside at midlog phase (A600 = 0.4) for 3–4 h. Cells were harvested by centrifugation, resuspended (10 mm Tris, pH 8.0 with 1 mm MgCl2), and frozen at −80 °C. Cell pellets were lysed by sonication, and inclusion bodies containing CXCL12 were collected by centrifugation and washed once (with 10 mm Tris, pH 8.0, 0.25% (w/v) deoxycholic acid) before being dissolved in denaturant (6 m guanidinium chloride, 100 mm sodium phosphate, 10 mm Tris, pH 8.0) and added in batch format to nickel-nitrilotriacetic acid resin (Qiagen). Refolding of CXCL12 variants was done by exchanging the resin into non-denaturing conditions (50 mm Tris, pH 8.0, 250 mm NaCl, 10 mm KCl, 400 mm l-arginine, 1 mm reduced glutathione, 0.1 mm oxidized glutathione) and allowing the refolding to proceed overnight at 4 °C. CXCL12 variants were eluted with imidazole and purified using reverse phase HPLC on a C18 column, frozen, and lyophilized. To remove the His8 tag, proteins were resuspended in buffer (10 mm Tris, pH 7.4 with 50 mm NaCl and 2 mm CaCl2) and treated with enterokinase (purified recombinantly) overnight at 37 °C. A final purification step was done by reverse phase HPLC; the CXCL12 proteins were then frozen, lyophilized, and stored at −80 °C.
Competitive Radioligand Binding Assays
Competition cell binding assays for CXCL12 variants were performed using scintillation proximity assay technology (GE Healthcare) with 125I-CXCL12 (PerkinElmer Life Sciences) as a tracer. Cells stably transfected with CXCR4 or ACKR3 were induced with 10 mm sodium butyrate and 2 μg/ml doxycycline for 24 h for high level receptor expression. Each assay point contained 20,000 cells, 800 μg of polyvinyl toluene wheat germ agglutinin scintillation proximity assay beads (PerkinElmer Life Sciences), 40 pm 125I-CXCL12, and increasing concentrations of cold competitor (CXCL12 variants). Assays were performed in quadruplicate in a 96-well plate format, and signal was recorded on a MicroBeta TriLux (PerkinElmer Life Sciences) after 2 h of equilibration at room temperature. Curves were fit to a one-site competitive binding model using OriginPro (OriginLab) to determine IC50 values. For CXCR4 binding studies, the concentration of tracer CXCL12 is significantly lower (50-fold) than the IC50 values measured, and thus competition is negligible according to the Cheng-Prusoff equation (30). Therefore, in the case of CXCR4, we report Kd values that are approximately equal to the IC50 values measured in the binding experiments. For ACKR3, we report IC50 values because the concentration of tracer CXCL12 is similar to the IC50 values in many cases, which necessitates the Cheng-Prusoff correction for the calculation of Kd. Moreover the Kd value for the interaction of ACKR3 with 125I-CXCL12 is not known. CXCR4 Kd values can be directly compared with the IC50 values reported for ACKR3. It is important to note that although HEK293 cells contain endogenous CXCR4, its expression levels are insufficient for detectable ligand binding. To test this, a control binding experiment with untransfected HEK293S cells and wild type CXCL12 was performed identically to the experiment with transfected cells. The untransfected cells did not yield a binding curve, and therefore, we attribute the binding response to ACKR3 in the ACKR3-transfected cells rather than to endogenous CXCR4.
CXCL12 Functional Assays
Calcium Mobilization
Chemokine-dependent increases in cytosolic calcium were tested with a FLIPR Calcium 4 assay kit (Molecular Devices) using the HEK293 cell line transfected with CXCR4. Each well contained 2 × 105 cells in Hanks' balanced salt solution supplemented with 20 mm HEPES, pH 7.4. Increasing amounts of chemokine were added, and the resulting response was recorded. Experiments were performed in triplicate and plotted as the mean peak fluorescence ±S.D.
Migration Assays
CXCL12-dependent cell migration was tested in a Transwell filter assay (Costar) with 5.0-μm membrane as described previously (31). Briefly, Jurkat cells (2.5 × 105 cells) were placed on top of each filter, and migrated cells were counted after 2 h of incubation at 37 °C with a FACSCalibur flow cytometer (BD Biosciences). Assays were performed in triplicate, and data are plotted as the mean ± S.D. of the percentage of migrated cells.
Internalization Assays
MDA-MB-231 cells stably expressing hemagglutinin (HA)-tagged CXCR4 (MDA-HA-CXCR4) were cultured in RPMI 1640 medium supplemented with 10% FBS and 5 μg/ml puromycin and grown at 37 °C with 5% CO2. For the internalization assays, cells were lifted with PBS + 1 mm EDTA, resuspended to 1 × 106 cells/ml in assay buffer (RPMI 1640 medium with 0.5% BSA and 10 mm HEPES), and chilled on ice. Chemokine (WT CXCL12 or N-terminally modified variant) was added to a final concentration of 200 nm. Two hundred microliters of sample was removed for the zero time point prior to incubation of the cells in a 37 °C waterbath. Additional aliquots were removed from the bulk sample at 5, 15, 30, and 45 min, immediately placed on ice, and diluted 1:3 with ice-cold PBS. To analyze the surface levels of CXCR4, cells were spun down at 250 × g for 5 min at 4 °C, resuspended to 1 × 106 cells/ml in FACS buffer (PBS + 0.5% BSA), and stained with anti-HA-phycoerythrin (Miltenyi Biotec) or isotype control for 30 min on ice in the dark. Cells were detected on a Guava EasyCyte 8HT flow cytometer (EMD Millipore). Data were analyzed with FlowJo (Treestar Inc.) and plotted with GraphPad Prism (GraphPad Software). Results are the mean ± S.E. from three independent experiments performed in duplicate.
Murine EAE Experiments
Female SJL/J mice (8–12 weeks of age) were immunized subcutaneously in the hind flanks with 50 μg of proteolipid protein(139–151) emulsified in complete Freund's adjuvant containing 0.5 mg/ml Mycobacterium butyricum (Difco) and 8.33 mg/ml Mycobacterium tuberculosis H37Ra (Difco). On days 0 and 2, 300 ng of pertussis toxin (List Biological Laboratories) in 250 μl of endotoxin-free PBS was injected intravenously. Mice were injected into the peritoneal cavity with 100 μl of endotoxin-free PBS containing 100 μg of LGGG-CXCL12 or the control peptide CCL2Ala every 48 h from day 0 following immunization. CCL2Ala is a variant of the chemokine CCL2 that is misfolded due to replacement of all four cysteine residues by alanine (32). It is well characterized and widely used as an inert control peptide in EAE experiments involving multiple chemokines (21, 32). Animals were monitored daily, and clinical disease was measured by assessment of paralysis using the following criteria: 0.5, slight tail weakness; 1, tail weakness; 2, full tail paralysis; 2.5, tail and some hind limb paralysis; 3, full hind limb paralysis; 3.5, hind limb and some forelimb paralysis; 4, full hind and forelimb paralysis; 5, moribund. Experiments were carried out in accordance with approvals obtained from the University of Adelaide's institutional animal ethics committee. Statistical tests used were analysis of variance and multiple comparison post-tests.
Molecular Modeling and Docking
Models of CXCR4 complexes with CXCL12 variants were built in the ICM software package (33). A hybrid CXC template was first built from the CXCR4 molecule in the CXCR4·vMIP-II structure (Protein Data Bank code 4RWS (34)) and a CXCL12 structure (Protein Data Bank code 3GV3 (35)) after rigidly superimposing the backbone atoms of CXCL12 residues Cys-11, Arg-12, Val-49, and Cys-50 onto the corresponding atoms in vMIP-II (the core of the so-called chemokine recognition site 1 (CRS1)). Residue side chains in the complex models were refined with 5 × 105 steps of Monte Carlo optimization in internal coordinates. Interactions between CXCR4 and chemokine involving regions referred to as CRS1 and CRS1.5 were taken from Qin et al. (34). For the prediction of CRS2 interactions (under “Results” and from Qin et al. (34)), the receptor was converted into a set of three-dimensional grid potential maps representing van der Waals, electrostatic, hydrogen bonding, and apolar surface interactions. The chemokine N terminus (up to the second N-terminal cysteine residue) was built ab initio, its CXC motif was tethered to the positions of the corresponding atoms in the template, and the N terminus was thoroughly sampled in the receptor potential grids. The obtained stack of N-terminal conformations was merged with the full atom model of the receptor, and another 10–20 × 106 steps of Monte Carlo optimization were performed, this time using full atom receptor representation with flexible binding pocket side chains.
Results
Experimental Design of the CXCL12 Phage Libraries and the Selection Strategy
We designed two CXCL12 libraries targeting the N terminus because of its critical role in receptor binding and activation. The N-addition library extends the N terminus with an additional residue, which is simultaneously randomized along with the first three residues of CXCL12 (Table 1). A second library, the N-truncation library, omits the first four residues and randomizes residues 5–8. This library was inspired by the fact that CXCL12 is inactivated in vivo by proteolytic processing of the N terminus, which makes it a natural antagonist (36). Although removal of the first few residues dramatically reduces receptor affinity (by 100-fold) (23), we hoped that exploring a large sequence library would permit the identification of high affinity CXCL12 variants that are naturally resistant to proteolytic inactivation in vivo.
TABLE 1.
Design of CXCL12 sequence libraries
Each variable residue position is denoted with a bold X in the sequence, and the N-terminal addition residue is denoted with a plus symbol (+). The cysteine residues within the CXC motif of the chemokine at residues 9 and 11 are underlined.
| Position index | +123456789012345678 … |
|---|---|
| Wild type | KPVSLSYRCPCRFFESHV … |
| N-addition | XXXXSLSYRCPCRFFESHV … |
| N-truncation | XXXXCPCRFFESHV … |
In addition to selecting for increased receptor affinity, we set out to select CXCL12 variants that do not activate G protein or recruit β-arrestin, i.e. receptor antagonists and inverse agonists. To this end, biopanning on cells was performed at a temperature permissive for receptor internalization. Agonist binding typically results in receptor activation and subsequent β-arrestin-mediated internalization; therefore, this experimental setup was expected to remove receptor agonists but retain antagonists at the cell surface for elution, reamplification, and further selection (Fig. 1). We also attempted to select high affinity internalizing receptor agonists by collecting the cells after removal of the surface-bound phage. This strategy was successful in identifying internalizing CCL5-based agonists of CCR5 by Hartley and co-workers (29, 37) and in generating internalizing antibodies of the epidermal growth factor (EGF) receptor (38).
FIGURE 1.

The steps in the phage display experimental design are shown in the middle panel. Multiple CXCL12 phagemid constructs were tested when optimizing the CXCL12 phage display system (left, magenta box), and the experimental conditions for generating recombinant CXCL12 phage are given (left, green box). Selections were performed on live cells at 37 °C, which is permissive of internalization, to bias the output toward receptor antagonists (right, blue box). MBP, maltose-binding protein; Ubq, ubiquitin; IPTG, isopropyl β-d-thiogalactopyranoside.
CXCL12 Is a Difficult Target to Phage Display and Requires Optimization
Although a powerful technology for high throughput identification of high affinity protein-protein interactions, phage display is not universally applicable to all targets. It requires that the displayed protein is efficiently expressed and properly folded in bacterial hosts, exported to the bacterial periplasm, resistant to proteolysis, and compatible with phage assembly and host infection. Like many mammalian proteins and in part because of their disulfide-bonded topology, chemokines expressed in E. coli are generally insoluble and are targeted into inclusion bodies. Additionally, the N termini of chemokines are prone to proteolysis. These features can make chemokines challenging phage display targets, and thus it may be no coincidence that most of the reported phage display efforts targeting chemokine receptors involved displayed peptides rather than full chemokine sequences (39–41).
In contrast to the successful application of phage display to CCL5 (29, 37), CXCL12 behind the OmpA signal sequence could not be displayed using a similar (and relatively standard) phage display protocol (data not shown). Assuming that inefficient export to the periplasmic space and/or insolubility contributed to the lack of phage display, two approaches were devised to solve this problem: (i) optimization of the CXCL12 expression construct and (ii) optimization of E. coli growth, induction, and infection conditions for the production of CXCL12-displaying phage particles. For the first approach, we generated fusion constructs that in addition to CXCL12 in-frame with the g3p coat protein included maltose-binding protein (MBP-CXCL12-g3p) or ubiquitin (Ubq-CXCL12-g3p) at the N terminus of CXCL12. Both maltose-binding protein and ubiquitin can function as solubilizing chaperones for otherwise insoluble proteins (42), including CXCL12 (43) (Fig. 1). Because the chemokine N terminus is important for receptor interactions, an enterokinase site was included after maltose-binding protein, and ubiquitinase was used to remove ubiquitin before biopanning. However, the resulting phage particles displayed no detectable amount of CXCL12 as determined with an anti-CXCL12 antibody in a sandwich ELISA format (data not shown). We hypothesized that CXCL12 produced in these fusion systems is not well incorporated into phage.
In the second approach, we experimented with conditions for E. coli growth, induction, and infection. In particular, we lowered the growth temperature during protein expression to slow the overall process and thereby increase the solubility of CXCL12 and the efficiency of its periplasmic export. We also initiated protein expression (with isopropyl β-d-thiogalactopyranoside) prior to infection of the cells with helper phage. This allowed the cells to accumulate the CXCL12-g3p fusion in the periplasm before cellular protein production and secretion machinery was overwhelmed with robust expression of phage proteins (Fig. 1). This strategy enabled detection of CXCL12 on the surface of phage and permitted selection of CXCL12 recombinant phage libraries (Fig. 2). Because insolubility of heterologous proteins expressed in E. coli is a common hurdle, we expect that this optimized phage display strategy may be broadly applicable to other clinically relevant but experimentally difficult protein-protein interactions.
FIGURE 2.
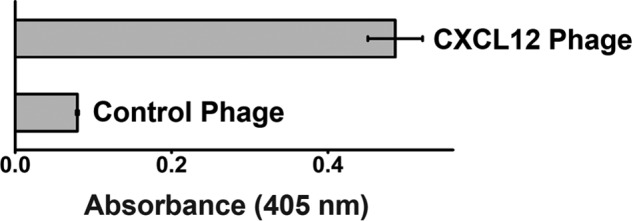
CXCL12 recombinant phage are detected in a sandwich ELISA experiment over control phage. Recombinant CXCL12 phage or control WT phage were captured with an anti-CXCL12 antibody and detected with an anti-phage antibody. Error bars represent S.D.
Enriched Sequences Result from Selection of N-addition CXCL12 Libraries on Cell Surfaces
The N-truncation library was selected against cells expressing CXCR4, whereas the N-addition library was selected against cells expressing CXCR4 or cells expressing ACKR3 in the background of low levels of endogenous CXCR4 (the endogenous CXCR4 expression in these cells can be detected with an anti-CXCR4 antibody, but is not sufficient for a detectable CXCL12 binding response in a scintillation proximity assay). The composition of the output sequences from cell surface selection of both the addition and truncation libraries is similar, with glutamine, tyrosine, phenylalanine, arginine, and leucine significantly enriched (Table 2). In a few cases, the same sequence was identified multiple times from the output population, reflecting substantial sequence refinement. By contrast, sequences resulting from the internalized selection approach were dominated by frameshift mutations and premature stop codons; thus it appears that this experiment did not select for recombinant CXCL12 phage that was available for recovery from cell lysates.
TABLE 2.
Sequence results for the CXCL12 phage library selection experiments
Numbers in parentheses following sequences indicate the number of times the sequence was identified in the output population.
| N-addition vs. CXCR4 | N-addition vs. CXCR4/ACKR3 | N-truncation vs. CXCR4 | N-truncation vs. CXCR4/ACKR3 |
|---|---|---|---|
| AGQS | APQR | AKQS | CQSR |
| CRFF | CDQC | APWQ | CSQC |
| CRQL | DSQF | AVQL | DGQRa |
| CSVC | FNQL | FPLQa | ETLQ |
| FQGD | GVQQ | HQCF | FEQLa |
| GLLS | IKSQ | LIQYa | FPSQ |
| ICAD | LKQV | LQPL | GWPQa |
| LGGG | LLQC | LQRC | ITQL |
| LRYI | LRFQ | PQRV | LSQYa |
| LTQF | LRHQ (5) | QFPLa | LVSQ |
| MLGI | LRSQ (4) | QQSF | QFGS |
| MVGY | MQFV | QSPL | QLSL |
| QFYS | MRHQ | QYPLa | QSGL |
| RGFE | QCIS | RQCF | QSLF |
| RQQF | QFES | SCQP | QYCV |
| SCQS | QFFG | SQPMa | SEVQ |
| SECL | QLPR | TFYQa | SITQ |
| SFMS | QVQF | SLQPa | |
| SQLA | QWVA | VAMQ | |
| SQRV | RHQF | VQLC | |
| TSCV | RYCQ | VQPR | |
| VGEV | SCQY | YQQFa | |
| VPGA (2) | SQFV | ||
| VQFG | SQSQ | ||
| TSQF | |||
| WQLF |
a Sequences were chosen for experimental validation and found to have low affinity for the receptor (Kd ≥1 μm).
With many targets, sequence enrichment can be observed after only a single round of selection, and three to four rounds of panning are sufficient for substantial sequence refinement. In our cell surface experiments, sequence enrichment generally required six rounds of panning with a gradual increase in selection stringency. We hypothesize that the slow sequence enrichment may be due to the properties of the receptor, in particular the structural plasticity of its binding pocket, which is discussed in more detail below and in Kufareva et al. (44).
The Selected CXCL12 Variants Are High Affinity Antagonists of CXCR4
Thirteen CXCL12 variants from the N-addition and 10 from the N-truncation libraries were selected for recombinant expression and in-depth experimental characterization. Sequences with high similarity to conserved patterns were chosen as these were likely to be high affinity binders along with sequences dissimilar to the conserved patterns to sample additional sequence space. Competition binding experiments revealed that six of the 13 characterized CXCL12 variants from the N-addition library have Kd values below 10 nm for CXCR4 with the most potent variant, LGGG-CXCL12, showing a 2-fold improvement in affinity compared with WT CXCL12 (0.95 versus 2.1 nm) (Table 3 and Fig. 3A). Ten of the CXCL12 variants were also characterized for their ability to bind ACKR3-expressing cells (Table 3 and Fig. 3B), and seven were found to have IC50 values below 1 nm for ACKR3. CXCL12 variants from the N-truncation library were found to have low affinity (Kd ≥ 1 μm) for CXCR4 and were not pursued further (Table 2).
TABLE 3.
Affinities of the CXCL12 variants for CXCR4 and ACKR3
Specificity for ACKR3 is calculated as the ratio of the affinity for CXCR4 to affinity for ACKR3 for each variant.
| CXCL12 variant | CXCR4 Kda | ACKR3 IC50 | Specificity for ACKR3 |
|---|---|---|---|
| nm | nm | ||
| Wild type | 2.1 ± 0.7 | 0.014 ± 0.004 | 150 |
| P2G | 4.2 ± 0.1 | 1.1 ± 0.3 | 3.8 |
| FNQL | 12.0 ± 0.5 | 0.8 ± 0.3 | 20 |
| LGGG | 0.9 ± 0.2 | 11 ± 2 | .08 |
| LKQV | 6.8 ± 0.5 | 0.30 ± 0.07 | 23 |
| LRHQ | 2.1 ± 0.5 | 0.06 ± 0.03 | 40 |
| LRSQ | 14 ± 0.6 | 0.5 ± 0.1 | 30 |
| MLGI | 6.0 ± 0.1 | 0.09 ± 0.03 | 70 |
| MRHQ | 2.3 ± 0.1 | 0.16 ± 0.04 | 14 |
| VPGA | 5.0 ± 0.8 | 1.7 ± 0.7 | 3 |
| QWVA | 110 ± 50 | 0.5 ± 0.1 | 200 |
| QFNI | 500 ± 100 | Not determined | Not determined |
| SQCS | 400 ± 100 | Not determined | Not determined |
| SQSQ | 550 ± 50 | 28 ± 2 | 20 |
| SQLA | 400 ± 200 | Not determined | Not determined |
a CXCR4 Kd values are approximately equal to the measured IC50 values.
FIGURE 3.

Characterization of CXCL12 variants. Competition binding curves for CXCL12 proteins against CXCR4 (A) or ACKR3 (B) in a scintillation proximity assay binding assay with 40 pm 125I-CXCL12 as a tracer are shown. A control experiment was performed between untransfected HEK293S cells and wild type CXCL12 (inset). The observed background signal is not competed off by increasing concentrations of CXCL12; thus the endogenous levels of CXCR4 are not sufficient to produce a binding signal. All binding assays were performed in quadruplicate and plotted as mean ± S.D. (error bars). C, the affinity of each CXCL12 variant for ACKR3 (IC50 value in nm) is plotted versus its affinity for CXCR4 (Kd value in nm). D, calcium flux dose-response curve for CXCL12 proteins, including WT CXCL12, LKQV, P2G, LGGG, LRHQ, LRSQ, MLGI, VPGA, QWVA, QFNI, SQCS, SQLA, and SQSQ. Data for all nonfunctional mutants are shown as overlapping cross marks (×), and the line appears as a flat baseline. Data shown are mean ± S.D. (error bars) of triplicates. E, cell migration of Jurkat cells in response to a chemokine gradient for CXCL12 proteins, including WT CXCL12, LKQV, P2G, LGGG, LRHQ, LRSQ, MLGI, VPGA, QWVA, QFNI, SQCS, SQLA, and SQSQ. Data for all nonfunctional mutants are shown as overlapping cross marks (×), and the line appears as a flat baseline. Data shown are mean ± S.D. of triplicates. F, CXCR4 internalization assay for WT CXCL12 and two of the highest affinity variants, LGGG-CXCL12 and LRHQ-CXCL12. For these experiments, MDA-MD-231 cells were transfected with HA epitope-tagged CXCR4, and result shown are the means ± S.E. (error bars) from three independent experiments performed in duplicate.
Binding to and activation by WT CXCL12 result in internalization of the receptor (45); thus biopanning was performed at 37 °C to select for antagonists by virtue of their retention on the cell surface. To test whether the CXCL12 variants identified through this process were indeed antagonists, two functional assays, calcium mobilization and cell migration toward chemokine (Fig. 3, D and E), were performed. All variants failed to elicit a calcium mobilization response with the exception of LKQV-CXCL12, which retained a low level of signaling (<20% of the WT response; Fig. 3D). Similar results were observed with cell migration assays; most variants showed no detectable activity with the exception of LKQV-CXCL12 (Fig. 3E). For the two highest affinity CXCL12 variants (LGGG-CXCL12 and LRHQ-CXCL12), their inability to internalize CXCR4 was directly confirmed (Fig. 3F). Taken together, these results demonstrate that LGGG-CXCL12 and LRHQ-CXCL12 are bona fide antagonists of CXCR4, validating our selection strategy. Unlike functional characterization of CXCR4, studying ligand activation of ACKR3 is challenging because this receptor does not signal through classical G protein-mediated pathways (46). Therefore, the effects of the selected variants on the pharmacology of ACKR3 are subjects of future investigations. However, because ACKR3 scavenges CXCL12 from the cells surface (10, 11), we expect that, similar to CXCR4, our selection strategy has produced antagonists of ACKR3 internalization that would also block the interaction of the receptor with endogenous CXCL12.
LGGG-CXCL12 Reduces the Progression of EAE
EAE is an inflammatory disease with pathology and clinical symptoms similar to multiple sclerosis in humans. In prior work, an N-terminally modified antagonist variant of CXCL12, P2G-CXCL12, was shown to reduce the progression of disease symptoms in an SJL/J mouse model of EAE via effects on the priming phase of the immune response (21). Accordingly, we tested our highest affinity CXCR4 antagonist, LGGG-CXCL12, for therapeutic efficacy in EAE. LGGG-CXCL12 was administered following immunization for EAE and led to a significant delay in disease onset and a decrease in both peak disease score and cumulative disease when compared with mice treated with a control peptide, CCL2Ala, or WT CXCL12 (Fig. 4 and Table 4).
FIGURE 4.
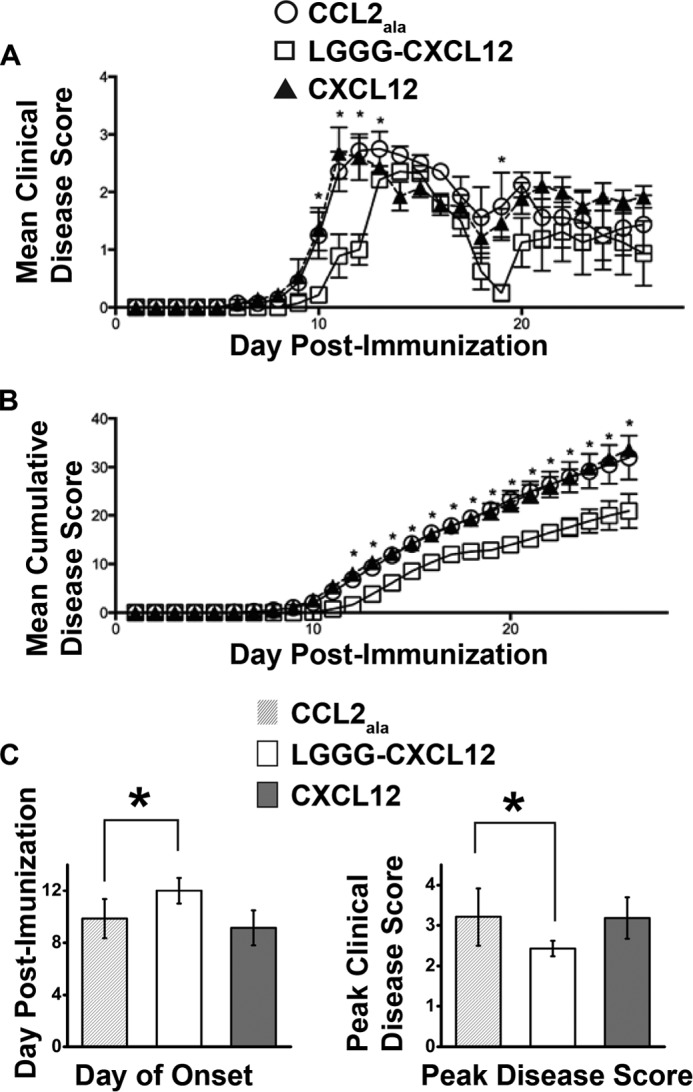
LGGG-CXCL12 slows onset of EAE and reduces peak and cumulative disease. EAE was induced in SJL/J mice as described, and mice were treated with 100 μg of LGGG-CXCL12, CXCL12, or CCL2Ala on even days following immunization up to day 17. A, the mice were scored daily for clinical signs of the disease. Statistically significant differences were observed between the CCL2Ala and LGGG-CXCL12 groups at p < 0.05 on days 9–12 and 19. B, cumulative disease over the disease course. Statistically significant differences were observed between the CCL2Ala and LGGG-CXCL12 groups at p < 0.05 on days 12–26. The data are presented as mean ± S.E. (error bars) (n = 7). C, post-test comparison of the day of disease onset (left) and peak disease score (right) where LGGG-CXCL12 and CCL2Ala were statistically different at p < 0.05 (analysis of variance). In all three cases (CCL2Ala, LGGG-CXCL12, and CXCL12), the incidence of the disease was seven of seven.
TABLE 4.
The effect of antagonist treatment on the development of clinical EAE
SJL/J mice were induced with EAE and then treated with CCL2Ala, LGGG-CXCL12, or CXCL12. Upon disease completion, various parameters of disease were analyzed and are presented below.
| Group | Incidence | Day of onseta | Peak disease scorea |
|---|---|---|---|
| CCL2Ala | 7 of 7 | 9.85 ± 1.5 | 3.21 ± 0.71 |
| LGGG-CXCL12 | 7 of 7 | 12.0 ± 1.0b | 2.43 ± 0.19b |
| CXCL12 | 7 of 7 | 9.14 ± 1.34 | 3.18 ± 0.51 |
a Mean ± S.E.
b Significantly different from CCL2Ala at p < 0.05 by analysis of variance.
Structure-Function Insights into Select CXCL12 Variants by Molecular Modeling
Molecular modeling was used to gain insight into the structural basis of action of select antagonistic variants of CXCL12. The modeling efforts were based on the recently solved crystal structure of CXCR4 in complex with a high affinity viral antagonist chemokine, vMIP-II (34). Chemokine interactions with CXCR4 are mediated by two distinct interfaces: CRS1 involves the binding of the receptor N terminus to the globular core of the chemokine, whereas CRS2 consists of the chemokine N terminus binding in the transmembrane (TM) pocket of the receptor (34, 47). The two interfaces are connected by an intermediate region, CRS1.5, where the conserved cysteine motif of the chemokine interacts with the conserved N-terminal cysteine of the receptor (34).
Variations in the N terminus of the chemokine are not expected to affect binding in CRS1 and CRS1.5; therefore, these modeled interactions were taken from previous work (34, 47) (Fig. 5A), whereas CRS2 interactions were sampled and studied in detail here (Fig. 5, B–E). For the purpose of comparison, WT CXCL12 and the antagonist variants P2G-CXCL12, LGGG-CXCL12, and LRHQ-CXCL12 were docked to CXCR4. A consistent pattern of interactions was observed with all chemokine variants with the N-terminal amine of the chemokine binding to CXCR4 Asp-972.63 (the superscript number denotes the Ballesteros-Weinstein index for helical domain residues (48)) and the CXCL12 arginine residue at position 8 binding to CXCR4 Asp-2626.58 (Fig. 5, B–E). In WT CXCR4 and P2G-CXCL12, the amine group of Lys-1 hydrogen bonds to CXCR4 Glu-2887.39 (Fig. 5, B and C); however, this residue is absent in LGGG and LRHQ (Fig. 5, D and E). Consequently, in the modeled CXCR4·LGGG-CXCL12 complex, CXCR4 Glu-2887.39 makes a hydrogen bond with the amide nitrogen of chemokine Gly-3 (Fig. 5D) in the same way it does with Ala-3 of vMIP-II in the crystal structure (34). This interaction is made possible by glycine, a small and flexible residue, at position 2 of both vMIP-II and LGGG-CXCL12. In several plausible models of the CXCR4·LRHQ-CXCL12 complex, CXCR4 Glu-2887.39 forms hydrogen bonds with the side chain of either Arg-2 (Fig. 5E) or His-3. Residues Ser-4 and Tyr-7 of the chemokine make favorable polar interactions with Asp-187 in the second extracellular loop (ECL2) of CXCR4 (Fig. 5, B–E). Importantly, all of the above residues (Asp-972.63, Glu-2887.39, Asp-187, and Asp-2626.58) were shown to be important for binding and activation of CXCR4 by CXCL12 (49–53).
FIGURE 5.

Predicted binding modes of CXCL12 and its antagonist variants to CXCR4. A, overall architecture of the complexes with the conserved CRS1 interaction shown between the globular core of the chemokine (skin) and the N terminus of the receptor (black ribbon and sticks). Acidic and basic residues in the chemokine are colored red and blue, respectively; they form favorable contacts with the basic (Lys-25) and acidic (Asp-22 and Glu-26) residues in the CXCR4 N terminus. The sulfated N-terminal tyrosine of CXCR4 (sY21) interacts with the backbone amines of Arg-20 and Ala-21 as well as the side chain of Arg-20 in a way that is observed for sulfate groups and free sulfate ions in several x-ray structures of CXCL12 (71–74). The predicted positions of the N termini of four CXCL12 variants are shown in ribbon: orange, WT; green, P2G; purple, LGGG; cyan, LRHQ. B–E, close-up views and important residue contacts for the four CXCL12 variants in the CRS2. The transmembrane helices of CXCR4 are numbered (roman numerals).
Modeling supports the idea that CXCR4-binding chemokines can tolerate (and even benefit from) addition of one N-terminal hydrophobic residue such as leucine (in LGGG-CXCL12 and LRHQ variants) or methionine (in Met-CXCL12, a high affinity agonist variant of CXCL12 (54)). N-terminal hydrophobic residues favorably pack in a small subpocket formed by CXCR4 residues Trp-942.60, Asp-972.63, Trp-102ECL1, Val-1123.28, His-1133.29, and Cys-186ECL1 (Fig. 5, D and E) in a manner similar to Leu-1 of vMIP-II (34) and the cyclohexane ring of the small molecule antagonist IT1t (55). Further extensions of the CXCL12 N terminus may not be possible without loss of affinity, assuming the present mode of binding is conserved.
We previously hypothesized that the agonist action of WT CXCL12 is caused by direct interaction of chemokine residue Pro-2 with Tyr-1163.32 of CXCR4 (34) (Fig. 5B) as residues in this position are involved in activation of many G protein-coupled receptors (56). In P2G-CXCL12, the singular replacement of Pro-2 by glycine eliminates this contact and with it the agonist action of the chemokine (Fig. 5C). Consistent with this hypothesis, direct interaction with CXCR4 Tyr-1163.32 is absent in the modeled complexes of CXCR4 with LGGG-CXCL12 and LRHQ-CXCL12 (Fig. 5, D and E) and, as for P2G-CXCL12, may explain their antagonistic pharmacology.
Discussion
High Affinity CXCR4 Antagonists Result from the CXCL12 N-addition but Not the N-truncation Library
In this work, we set out to select high affinity CXCR4 antagonists from N-terminal CXCL12 sequence libraries. Two libraries were explored: in one library, the chemokine N terminus was extended by one residue, and this residue plus the first three residues (corresponding to WT CXCL12 residues 1KPV3) were varied, whereas in the other, the chemokine N terminus was truncated by four residues, and WT CXCL12 residues 5LSYR8 were varied (Table 1). With respect to the latter, we hypothesized that truncation of CXCL12 might render it less susceptible to proteolysis in vivo and that by exploring a large sequence library (>105 members, the theoretical number of sequences for four randomized positions) a few high affinity antagonists might emerge.
To remove CXCR4 agonists from the population, selection was performed on live cells at a temperature permissive to receptor internalization. Although selection on live cells presents a number of technical challenges, it has the advantage of preserving the target molecule in a biologically relevant context and conformation. As a result of this selection strategy, 12 of 13 characterized CXCL12 N-addition variants from the cell surface selections were full CXCR4 antagonists, and only one showed partial agonism. Most N-addition variants also showed high affinity for the receptor with eight of 12 variants having affinities higher than 20 nm and four variants more potent than 5 nm. By contrast, the N-truncation library did not produce high affinity binders to CXCR4. This suggests that CXCL12 residues 1–4 are critical for high affinity CXCR4·CXCL12 interactions and is in agreement with a prior report that truncated CXCL12 (residues 5–68) has a dramatically reduced affinity compared with WT CXCL12 (23). It is also consistent with our hypothesis that in CXC receptor·chemokine pairs, high affinity binding is dominated by CRS2 interactions in contrast to CC receptor·chemokine complexes that rely more on CRS1 interactions (34). In CRS2 of experimental and predicted CXCR4·chemokine complexes (34), hydrogen bonding between the backbone amines of the N-terminal residues of the chemokine and acidic residues (Asp-972.63 and Glu-2887.39) in CXCR4 plays a critical role; truncation of four N-terminal residues in CXCL12 places its N-terminal amine as far as 5.5–6 Å from these acidic residues and makes these important interactions impossible. As our data show, the resulting loss in affinity is difficult to regain by mutating residues 5–8. However, in line with the above hypothesis, mutations to the CRS1 interaction of CXCL12 may enable the recovery of high affinity.
Selected CXCL12 Variants Show a High Degree of Sequence Enrichment
The N-addition sequences selected by iterative rounds of biopanning against CXCR4- and ACKR3-expressing cells exhibit a high degree of sequence variability but at the same time a strong compositional bias (Table 2 and Fig. 6, A and B). They are significantly enriched in glutamine (as many as 79% of selected sequences have at least one glutamine residue), leucine (39%), phenylalanine (36%), serine (35%), and arginine (31%). At the same time, the sequences are depleted in asparagine, lysine, proline, tryptophan, threonine, and aspartic and glutamic acids. In 29 of 71 selected sequences (41%), the N-terminal addition position is occupied by an aliphatic residue (leucine, isoleucine, methionine, or valine), whereas in 22 additional sequences (31%), this residue is neutral polar (glutamine, serine, threonine, or asparagine). Interestingly, this enrichment was observed in the amino acid composition of the selected N-terminal sequences but not as much in the order of amino acids (with the exception of LR(H/S)Q). This may be due to the flexibility of the chemokine N terminus that, combined with the high plasticity of the receptor pocket (34), allows for similar interactions to be made across scrambled sequences but with structural rearrangements.
FIGURE 6.
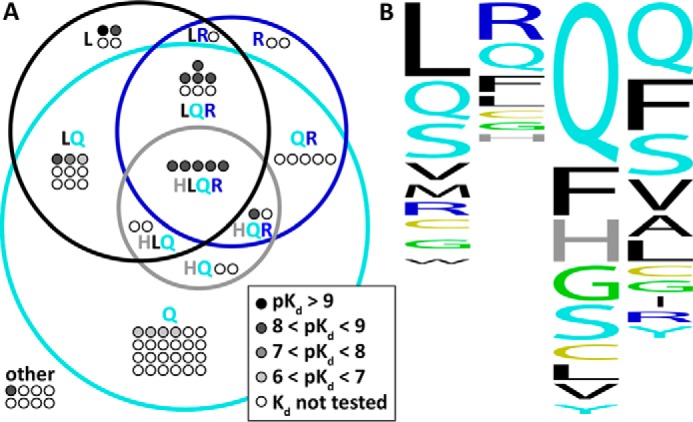
A, a Venn diagram for 71 sequences selected by biopanning of N-addition CXCL12 library against cells expressing CXCR4 and ACKR3. Sequences are shown as dots; each sequence is shown as many times as it appeared in the selected set. Sequences for which Kd values against CXCR4 were determined are colored black (pKd > 9), dark gray (8 < pKd < 9), gray (7 < pKd < 8), and light gray (6 < pKd < 7). 63 of 71 sequences had at least one of glutamine (Q; cyan), leucine (L; black), arginine (R; blue), or histidine (H; gray) and are pictured inside the corresponding Venn diagram regions; the remaining eight sequences are pictured outside and labeled “other.” The diagram describes the amino acid composition of the selected variants regardless of the relative positions of the amino acids within the sequences. B, a sequence profile for the 71 sequences where the relative conservation of amino acids at each of the four N-terminal positions of the chemokine is indicated by height. The graphic was prepared with WebLogo.
The results of biopanning are consistent with the nature of the binding pocket of CXCR4, which is wide open and polar and contains a large fraction of acidic residues (34). Accordingly, very few selected variants contain negatively charged residues. Positively charged arginine is enriched; however, positively charged lysine is depleted. The presence of lysine among the N-terminal residues of the chemokine may be associated with agonistic pharmacology toward CXCR4; WT CXCL12 (a full agonist) has a lysine residue at position 1 that is critical for its signaling properties (23), and the only lysine-containing variant among those tested in functional assays, LKQV-CXCL12, appeared to be a partial agonist. The preference for aliphatic residues in the N-terminal addition position is consistent with the existence of a small hydrophobic subpocket formed by TM2, TM3, and ECL2; this subpocket can favorably accommodate the N-terminal aliphatic residue in our selected variants as well as in virally encoded chemokine antagonist vMIP-II (34) or in Met-CXCL12 (54). Finally, N-terminal glutamine is common in the chemokine family; it is known to spontaneously cyclize to pyroglutamate, which is a rigid bulky polar group with favorable properties for receptor binding (57). This might explain the high frequency of glutamine residues in position 1 of the selected variants (Fig. 6B).
In 20% of the selected sequences, at least one of the four N-terminal residues is a glycine. This amino acid increases the flexibility of the N terminus and enables it to adopt a favorable fold for interactions of the backbone amide groups with CXCR4 Glu-2887.39. The highest affinity CXCR4 antagonist from our selection is LGGG-CXCL12; this sequence enables a unique fold and exceptionally efficient engagement of the polar interactions. Glycine in position 2 plays a similar role in vMIP-II (N-terminal sequence, 1LGAS4) and in P2G-CXCL12 (N-terminal sequence, 1KGVS4).
Given the high level of amino-acid enrichment observed in our experiments, we considered the possibility of bias caused by selective pressure factors rather than the intended receptor-chemokine interactions. First, we considered bias in the initial library; however, direct sequencing of the input population revealed random sequences. Additionally, because the N-terminal libraries are directly adjacent to the secretion signal, we considered amino acid preferences due to interaction with the bacterial protein secretion machinery. The OmpA secretion machinery has a preference for proline, threonine, and histidine, whereas cysteine, arginine, and glycine are disfavored (58). However, these biases do not appear to contribute to the observed sequence enrichment, and we infer that the sequences are enriched primarily due to interaction with the target surface receptor.
Selected CXCL12 Variants Bind Both ACKR3 and CXCR4 with High Affinity Despite Significant Differences in the Receptor Binding Pockets
Mutants and truncation variants of CXCL12 have been studied with respect to their interaction with CXCR4; however, little has been reported for CXCL12 variants binding to ACKR3. To address this void, we included selections against ACKR3-expressing cells and assayed variants for binding to ACKR3. For WT CXCL12, P2G-CXCL12, and nine of 10 selected CXCL12 variants (with the exception of LGGG-CXCL12), binding affinities to ACKR3 were shown to exceed those of CXCR4 but generally follow the same rank order (Fig. 3, B and C). The specificity for ACKR3, which is the ratio of the affinity for CXCR4 to affinity for ACKR3 for each variant, ranges from near WT (100-fold preference for ACKR3) to a reversal of specificity in the variant LGGG-CXCL12 (10-fold preference for CXCR4) (Table 3). Homology modeling demonstrated that the anatomy of the binding pockets is quite different between CXCR4 and ACKR3, and although both receptors possess numerous acidic and other polar residues, their spatial positions inside the pockets are not conserved (Fig. 7, A and B). These structural differences are reflected in the subtly different specificities of the variants even though remarkably both pockets recognize WT CXCL12 and multiple N-terminally modified variants with high affinity.
FIGURE 7.
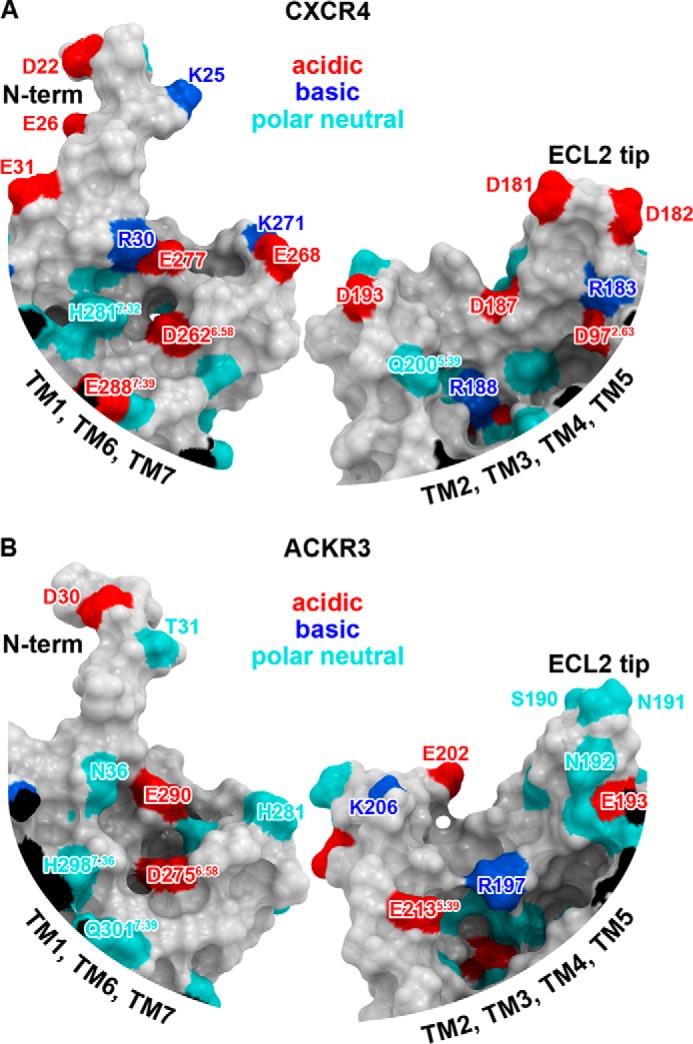
Maps of polar residues in the binding pockets of CXCR4 (A) and ACKR3 (B) built from an x-ray structure and a homology model, respectively. Each pocket is split approximately in half by a plane perpendicular to the membrane, and two sides of each pocket are shown: one comprising the N terminus; TM helices 1, 6, 7; and ECL3 (left), and the other is formed by TMs 2, 3, 4, and 5 and ECLs 1 and 2 (right). The surfaces are colored according to the character of the amino acid side chains forming them: acidic, red; basic blue; polar neutral, cyan. The maps illustrate that the distribution of pocket charged residues is very different between CXCR4 and ACKR3.
Conversely, in addition to chemokines, both pockets are also known to recognize multiple unrelated peptides and small molecules, and at least in the case of CXCR4, this is achieved via substantial structural plasticity involving reshaping and conformational rearrangements of binding determinants inside the pocket (34). Although modeling may provide suggestions about chemokine recognition determinants in ACKR3, only high resolution structure determination of ACKR3 complexes will ultimately and unambiguously reveal the specific interactions that enable these two receptors to recognize similar ligands.
Future Improvements for Challenging Phage Display Targets
In contrast to identifying receptor antagonists by selecting phage that remained on the cell surface, our attempts to select for internalizing variants were not successful. In these experiments, we collected the cells after removal of surface-bound phage, and reamplified the phage released after cell lysis. In previous studies, this strategy yielded agonist variants of CCL5 that promote internalization of CCR5 (29, 37) and internalizing antibodies to the EGF receptor (38). However, in our experiments, the CXCL12 sequences harvested from internalized phage populations were dominated by frameshift mutations and premature stop codons. CXCR4 differs from CCR5 in that it is degraded through lysosomal pathways after agonist stimulation, whereas CCR5 is recycled back to the cell surface (59–61). Because the fate of the internalized phage is largely dictated by the trafficking properties of the displayed protein (62), it is probable that phage particles displaying CXCL12 agonists were efficiently degraded and therefore not available for recovery. In cases where the receptor traffics through degradative pathways, using lysosomal inhibitors may permit selection of internalizing receptor agonists in future phage display experiments. Other improvements are also suggested such as biasing the N-terminal sequence of CXCL12 to avoid proteolysis (63) and thereby improve the potential therapeutic utility of the resulting protein.
Therapeutic Promise of Agents Targeting CXCR4 and ACKR3
Administration of CXCR4 antagonists (P2G-CXCL12 reported previously (21) and LGGG-CXCL12 reported herein)) reduces symptoms and slows the progression of EAE. This is a promising trend supporting the role of CXCR4 antagonists in EAE management. However, in a different study (64), a similar beneficial outcome was observed with a CXCL12-immunoglobulin fusion protein, a CXCR4 agonist. Moreover, in a third study, treatment with the small molecule CXCR4 antagonist AMD3100 in a mild case of EAE produced results opposite to those presented in Kohler et al. (21) and herein and worsened the disease outcome (65). This contradictory evidence complicates establishing the relation between the pharmacology of the CXCR4 ligand and its therapeutic efficacy and highlights the need for reagents with defined pharmacology to study the roles of receptors in disease.
Recently, the second receptor for CXCL12, ACKR3, has also been shown to play a role in EAE, and blockade with an ACKR3-selective antagonist, CCX771, was found to be beneficial (66). ACKR3 does not activate G proteins but instead signals through β-arrestin-mediated ERK1/2 pathways, the outcome of which is unclear (46, 67). As our present study demonstrates, not only CXCL12 but also its numerous N-terminally modified variants have activity against ACKR3. Moreover, AMD3100, which was originally developed and touted as a selective CXCR4 antagonist, has been shown to act as a positive (agonist) allosteric modulator of ACKR3 (68). The involvement of both CXCR4 and ACKR3 in EAE complicates the interpretation of experiments with ligands that may interact with both receptors especially when the pharmacological properties of the ligands are not fully understood. Complicating the issue further, several studies indicate that ACKR3 modulates the activity of CXCR4 by scavenging CXCL12 (69) and through receptor heterodimerization (9, 70).
Although the mechanisms of CXCR4 and ACKR3-specific effects in inflammatory diseases and cancers are difficult to separate and as such the respective benefit of drugs selectively targeting CXCR4 and ACKR3 is not clear, our success with LGGG-CXCL12 in vivo suggests that N-terminally modified CXCL12 variants have therapeutic potential. Well characterized reagents with different specificities may help differentiate the respective roles of CXCR4 and ACKR3 in both normal and pathological situations. Additionally, ligand specificity may play a role in clinical outcomes, and future studies are undoubtedly necessary to delineate the respective and shared contributions of CXCR4 and ACKR3.
Conclusion
Our study is the first report of a CXCL12 phage display strategy that may facilitate the development of high affinity dual or selective CXCL12-based variants with defined pharmacological properties that modulate the activity of CXCR4 or ACKR3. Such variants, including those with diverse pharmacological properties, are needed to more accurately define the roles of the two receptors in various physiological and pathological processes. The sequence space explored in our study leads to valuable structure-activity insights, including the intolerance of the receptor N-terminal truncations of the chemokine, and insight into the requirements for CXCR4 activation. Our optimized phage display strategy may be broadly applicable to other experimentally difficult phage display targets. Future iterations of the library design and selection process should facilitate the development of CXCL12 variants with properties such as proteolytic resistance that make them viable biologics. Because of the role of CXCR4 and ACKR3 in many diseases, including cancer, inflammatory diseases, and AIDS, these reagents have the potential to be broadly useful therapeutics.
Author Contributions
M. S. H., C. L. S., and A. B. C. collected experimental data. I. K. performed computational studies. I. C. and S. R. M. designed and performed the murine experiments. M. S. H., C. L. S., I. K., and T. M. H. wrote the paper. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We are grateful to the members of the Handel laboratory who provided critical discussion throughout the project and also thank Katherine Chan for cloning many of the CXCL12 variants.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 AI037113 and U01 GM094612 (to T. M. H.), R01 GM071872 and U54 GM094618 (to I. K.), and R01 AI118985 (to T. M. H. and I. K.). This work was also supported by National Health and Medical Research Council Grant APP1066781 and Multiple Sclerosis Research Australia Grant 12-001 (to S. R. M. and I. C.). The authors declare that they have no conflicts of interest with the contents of this article.
- CXCR
- CXC-type chemokine receptor
- EAE
- experimental autoimmune encephalomyelitis
- CCR
- CC-type chemokine receptor
- CCL
- CC-type chemokine ligand
- CXCL
- CXC-type chemokine ligand
- ACKR
- atypical chemokine receptor
- RANTES
- regulated on activation normal T cell expressed and secreted
- vMIP-II
- viral macrophage inflammatory protein-II
- CRS
- chemokine recognition site
- TM
- transmembrane
- ECL
- extracellular loop.
References
- 1. Bleul C. C., Farzan M., Choe H., Parolin C., Clark-Lewis I., Sodroski J., Springer T. A. (1996) The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature 382, 829–833 [DOI] [PubMed] [Google Scholar]
- 2. Hori T., Sakaida H., Sato A., Nakajima T., Shida H., Yoshie O., Uchiyama T. (1998) Detection and delineation of CXCR-4 (fusin) as an entry and fusion cofactor for T cell-tropic HIV-1 by three different monoclonal antibodies. J. Immunol. 160, 180–188 [PubMed] [Google Scholar]
- 3. Bleul C. C., Wu L., Hoxie J. A., Springer T. A., Mackay C. R. (1997) The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 94, 1925–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sun Y., Cheng Z., Ma L., Pei G. (2002) β-Arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J. Biol. Chem. 277, 49212–49219 [DOI] [PubMed] [Google Scholar]
- 5. Busillo J. M., Benovic J. L. (2007) Regulation of CXCR4 signaling. Biochim. Biophys. Acta 1768, 952–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ma Q., Jones D., Borghesani P. R., Segal R. A., Nagasawa T., Kishimoto T., Bronson R. T., Springer T. A. (1998) Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 95, 9448–9453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nagasawa T., Tachibana K., Kishimoto T. (1998) A novel CXC chemokine PBSF/SDF-1 and its receptor CXCR4: their functions in development, hematopoiesis and HIV infection. Semin. Immunol. 10, 179–185 [DOI] [PubMed] [Google Scholar]
- 8. Balabanian K., Lagane B., Infantino S., Chow K. Y., Harriague J., Moepps B., Arenzana-Seisdedos F., Thelen M., Bachelerie F. (2005) The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J. Biol. Chem. 280, 35760–35766 [DOI] [PubMed] [Google Scholar]
- 9. Levoye A., Balabanian K., Baleux F., Bachelerie F., Lagane B. (2009) CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood 113, 6085–6093 [DOI] [PubMed] [Google Scholar]
- 10. Naumann U., Cameroni E., Pruenster M., Mahabaleshwar H., Raz E., Zerwes H.-G., Rot A., Thelen M. (2010) CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS One 5, e9175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Berahovich R. D., Zabel B. A., Lewén S., Walters M. J., Ebsworth K., Wang Y., Jaen J. C., Schall T. J. (2014) Endothelial expression of CXCR7 and the regulation of systemic CXCL12 levels. Immunology 141, 111–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Werner L., Guzner-Gur H., Dotan I. (2013) Involvement of CXCR4/CXCR7/CXCL12 interactions in inflammatory bowel disease. Theranostics 3, 40–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hattermann K., Mentlein R. (2013) An infernal trio: the chemokine CXCL12 and its receptors CXCR4 and CXCR7 in tumor biology. Ann. Anat. 195, 103–110 [DOI] [PubMed] [Google Scholar]
- 14. Patrussi L., Baldari C. (2011) The CXCL12/CXCR4 axis as a therapeutic target in cancer and HIV-1 infection. Curr. Med. Chem. 18, 497–512 [DOI] [PubMed] [Google Scholar]
- 15. Xia X.-M., Wang F.-Y., Xu W.-A., Wang Z.-K., Liu J., Lu Y.-K., Jin X.-X., Lu H., Shen Y.-Z. (2010) CXCR4 antagonist AMD3100 attenuates colonic damage in mice with experimental colitis. World J. Gastroenterol. 16, 2873–2880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McDermott D. H., Liu Q., Ulrick J., Kwatemaa N., Anaya-O'Brien S., Penzak S. R., Filho J. O., Priel D. A., Kelly C., Garofalo M., Littel P., Marquesen M. M., Hilligoss D., Decastro R., Fleisher T. A., Kuhns D. B., Malech H. L., Murphy P. M. (2011) The CXCR4 antagonist plerixafor corrects panleukopenia in patients with WHIM syndrome. Blood 118, 4957–4962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sarchio S. N., Scolyer R. A., Beaugie C., McDonald D., Marsh-Wakefield F., Halliday G. M., Byrne S. N. (2014) Pharmacologically antagonizing the CXCR4-CXCL12 chemokine pathway with AMD3100 inhibits sunlight-induced skin cancer. J. Invest. Dermatol. 134, 1091–1100 [DOI] [PubMed] [Google Scholar]
- 18. Steinberg M., Silva M. (2010) Plerixafor: a chemokine receptor-4 antagonist for mobilization of hematopoietic stem cells for transplantation after high-dose chemotherapy for non-Hodgkin's lymphoma or multiple myeloma. Clin. Ther. 32, 821–843 [DOI] [PubMed] [Google Scholar]
- 19. Walters M. J., Ebsworth K., Berahovich R. D., Penfold M. E., Liu S. C., Al Omran R., Kioi M., Chernikova S. B., Tseng D., Mulkearns-Hubert E. E., Sinyuk M., Ransohoff R. M., Lathia J. D., Karamchandani J., Kohrt H. E., Zhang P., Powers J. P., Jaen J. C., Schall T. J., Merchant M., Recht L., Brown J. M. (2014) Inhibition of CXCR7 extends survival following irradiation of brain tumours in mice and rats. Br. J. Cancer 110, 1179–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Veazey R. S., Ling B., Green L. C., Ribka E. P., Lifson J. D., Piatak M. Jr., Lederman M. M., Mosier D., Offord R., Hartley O. (2009) Topically applied recombinant chemokine analogues fully protect macaques from vaginal simian-human immunodeficiency virus challenge. J. Infect. Dis. 199, 1525–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kohler R. E., Comerford I., Townley S., Haylock-Jacobs S., Clark-Lewis I., McColl S. R. (2008) Antagonism of the chemokine receptors CXCR3 and CXCR4 reduces the pathology of experimental autoimmune encephalomyelitis. Brain Pathology 18, 504–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chevigné A., Fievez V., Schmit J.-C., Deroo S. (2011) Engineering and screening the N-terminus of chemokines for drug discovery. Biochem. Pharmacol. 82, 1438–1456 [DOI] [PubMed] [Google Scholar]
- 23. Crump M. P., Gong J. H., Loetscher P., Rajarathnam K., Amara A., Arenzana-Seisdedos F., Virelizier J. L., Baggiolini M., Sykes B. D., Clark-Lewis I. (1997) Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 16, 6996–7007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tan Y., Li Y., Xiao J., Shao H., Ding C., Arteel G. E., Webster K. A., Yan J., Yu H., Cai L., Li X. (2009) A novel CXCR4 antagonist derived from human SDF-1β enhances angiogenesis in ischaemic mice. Cardiovasc. Res. 82, 513–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ravn P., Madhurantakam C., Kunze S., Matthews E., Priest C., O'Brien S., Collinson A., Papworth M., Fritsch-Fredin M., Jermutus L., Benthem L., Gruetter M., Jackson R. H. (2013) Structural and pharmacological characterization of novel potent and selective monoclonal antibody antagonists of glucose-dependent insulinotropic polypeptide receptor. J. Biol. Chem. 288, 19760–19772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jähnichen S., Blanchetot C., Maussang D., Gonzalez-Pajuelo M., Chow K. Y., Bosch L., De Vrieze S., Serruys B., Ulrichts H., Vandevelde W., Saunders M., De Haard H. J., Schols D., Leurs R., Vanlandschoot P., Verrips T., Smit M. J. (2010) CXCR4 nanobodies (VHH-based single variable domains) potently inhibit chemotaxis and HIV-1 replication and mobilize stem cells. Proc. Natl. Acad. Sci. U.S.A. 107, 20565–20570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chevigné A., Fischer A., Mathu J., Counson M., Beaupain N., Plesséria J.-M., Schmit J.-C., Deroo S. (2011) Selection of a CXCR4 antagonist from a human heavy chain CDR3-derived phage library. FEBS J. 278, 2867–2878 [DOI] [PubMed] [Google Scholar]
- 28. Zhao B., Mankowski M. K., Snyder B. A., Ptak R. G., Liwang P. J. (2011) Highly potent chimeric inhibitors targeting two steps of HIV cell entry. J. Biol. Chem. 286, 28370–28381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gaertner H., Cerini F., Escola J.-M., Kuenzi G., Melotti A., Offord R., Rossitto-Borlat I., Nedellec R., Salkowitz J., Gorochov G., Mosier D., Hartley O. (2008) Highly potent, fully recombinant anti-HIV chemokines: reengineering a low-cost microbicide. Proc. Natl. Acad. Sci. U.S.A. 105, 17706–17711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cheng Y., Prusoff W. H. (1973) Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (IC50) of an enzyme reaction. Biochem. Pharmacol. 22, 3099–3108 [DOI] [PubMed] [Google Scholar]
- 31. Kawamura T., Stephens B., Qin L., Yin X., Dores M. R., Smith T. H., Grimsey N., Abagyan R., Trejo J., Kufareva I., Fuster M. M., Salanga C. L., Handel T. M. (2014) A general method for site specific fluorescent labeling of recombinant chemokines. PLoS One 9, e81454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gong J.-H., Ratkay L. G., Waterfield J. D., Clark-Lewis I. (1997) An antagonist of monocyte chemoattractant protein 1 (MCP-1) inhibits arthritis in the MRL-lpr mouse model. J. Exp. Med. 186, 131–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Abagyan R., Totrov M. (1994) Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 235, 983–1002 [DOI] [PubMed] [Google Scholar]
- 34. Qin L., Kufareva I., Holden L. G., Wang C., Zheng Y., Zhao C., Fenalti G., Wu H., Han G. W., Cherezov V., Abagyan R., Stevens R. C., Handel T. M. (2015) Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 347, 1117–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Murphy J. W., Yuan H., Kong Y., Xiong Y., Lolis E. J. (2010) Heterologous quaternary structure of CXCL12 and its relationship to the CC chemokine family. Proteins 78, 1331–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mortier A., Gouwy M., Van Damme J., Proost P. (2011) Effect of posttranslational processing on the in vitro and in vivo activity of chemokines. Exp. Cell Res. 317, 642–654 [DOI] [PubMed] [Google Scholar]
- 37. Hartley O., Dorgham K., Perez-Bercoff D., Cerini F., Heimann A., Gaertner H., Offord R. E., Pancino G., Debré P., Gorochov G. (2003) Human immunodeficiency virus type 1 entry inhibitors selected on living cells from a library of phage chemokines. J. Virol. 77, 6637–6644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Heitner T., Moor A., Garrison J. L., Marks C., Hasan T., Marks J. D. (2001) Selection of cell binding and internalizing epidermal growth factor receptor antibodies from a phage display library. J. Immunol. Methods 248, 17–30 [DOI] [PubMed] [Google Scholar]
- 39. Molek P., Strukelj B., Bratkovic T. (2011) Peptide phage display as a tool for drug discovery: targeting membrane receptors. Molecules 16, 857–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Houimel M., Mazzucchelli L. (2009) Identification of biologically active peptides that inhibit binding of human CXCL8 to its receptors from a random phage-epitope library. J. Leukoc. Biol. 85, 728–738 [DOI] [PubMed] [Google Scholar]
- 41. Houimel M., Mazzucchelli L. (2012) hCXCR1 and hCXCR2 antagonists derived from combinatorial peptide libraries. Cytokine 57, 322–331 [DOI] [PubMed] [Google Scholar]
- 42. Raran-Kurussi S., Waugh D. S. (2012) The ability to enhance the solubility of its fusion partners is an intrinsic property of maltose-binding protein but their folding is either spontaneous or chaperone-mediated. PLoS One 7, e49589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Allen S. J., Hamel D. J., Handel T. M. (2011) A rapid and efficient way to obtain modified chemokines for functional and biophysical studies. Cytokine 55, 168–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kufareva I., Salanga C. L., Handel T. M. (2015) Chemokine and chemokine receptor structure and interactions: implications for therapeutic strategies. Immunol. Cell Biol. 93, 372–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Neel N. F., Schutyser E., Sai J., Fan G.-H., Richmond A. (2005) Chemokine receptor internalization and intracellular trafficking. Cytokine Growth Factor Rev. 16, 637–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rajagopal S., Kim J., Ahn S., Craig S., Lam C. M., Gerard N. P., Gerard C., Lefkowitz R. J. (2010) β-Arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc. Natl. Acad. Sci. U.S.A. 107, 628–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Scholten D. J., Canals M., Maussang D., Roumen L., Smit M. J., Wijtmans M., de Graaf C., Vischer H. F., Leurs R. (2012) Pharmacological modulation of chemokine receptor function. Br. J. Pharmacol. 165, 1617–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ballesteros J. A., Weinstein H. (1995) Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. in Methods in Neurosciences: Receptor Molecular Biology (Sealfon S. C., ed), pp. 366–428, Academic Press, San Diego [Google Scholar]
- 49. Tian S., Choi W.-T., Liu D., Pesavento J., Wang Y., An J., Sodroski J. G., Huang Z. (2005) Distinct functional sites for human immunodeficiency virus type 1 and stromal cell-derived factor 1α on CXCR4 transmembrane helical domains. J. Virol. 79, 12667–12673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brelot A., Heveker N., Montes M., Alizon M. (2000) Identification of residues of CXCR4 critical for human immunodeficiency virus coreceptor and chemokine receptor activities. J. Biol. Chem. 275, 23736–23744 [DOI] [PubMed] [Google Scholar]
- 51. Våbenø J., Nikiforovich G. V., Marshall G. R. (2006) Insight into the binding mode for cyclopentapeptide antagonists of the CXCR4 receptor. Chem. Biol. Drug Des. 67, 346–354 [DOI] [PubMed] [Google Scholar]
- 52. Choi W.-T., Tian S., Dong C.-Z., Kumar S., Liu D., Madani N., An J., Sodroski J. G., Huang Z. (2005) Unique ligand binding sites on CXCR4 probed by a chemical biology approach: implications for the design of selective human immunodeficiency virus type 1 inhibitors. J. Virol. 79, 15398–15404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhou N., Luo Z., Luo J., Liu D., Hall J. W., Pomerantz R. J., Huang Z. (2001) Structural and functional characterization of human CXCR4 as a chemokine receptor and HIV-1 co-receptor by mutagenesis and molecular modeling studies. J. Biol. Chem. 276, 42826–42833 [DOI] [PubMed] [Google Scholar]
- 54. Yang O. O., Swanberg S. L., Lu Z., Dziejman M., McCoy J., Luster A. D., Walker B. D., Herrmann S. H. (1999) Enhanced inhibition of human immunodeficiency virus type 1 by Met-stromal-derived factor 1β correlates with down-modulation of CXCR4. J. Virol. 73, 4582–4589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu B., Chien E. Y., Mol C. D., Fenalti G., Liu W., Katritch V., Abagyan R., Brooun A., Wells P., Bi F. C., Hamel D. J., Kuhn P., Handel T. M., Cherezov V., Stevens R. C. (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Katritch V., Cherezov V., Stevens R. C. (2013) Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 53, 531–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Burg J. S., Ingram J. R., Venkatakrishnan A. J., Jude K. M., Dukkipati A., Feinberg E. N., Angelini A., Waghray D., Dror R. O., Ploegh H. L., Garcia K. C. (2015) Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science 347, 1113–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Duffaud G., Inouye M. (1988) Signal peptidases recognize a structural feature at the cleavage site of secretory proteins. J. Biol. Chem. 263, 10224–10228 [PubMed] [Google Scholar]
- 59. Malik R., Marchese A. (2010) Arrestin-2 interacts with the endosomal sorting complex required for transport machinery to modulate endosomal sorting of CXCR4. Mol. Biol. Cell 21, 2529–2541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Marchese A., Benovic J. L. (2001) Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4 mediates lysosomal sorting. J. Biol. Chem. 276, 45509–45512 [DOI] [PubMed] [Google Scholar]
- 61. Signoret N., Pelchen-Matthews A., Mack M., Proudfoot A. E., Marsh M. (2000) Endocytosis and recycling of the HIV coreceptor Ccr5. J. Cell Biol. 151, 1281–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kim A., Shin T.-H., Shin S.-M., Pham C. D., Choi D.-K., Kwon M.-H., Kim Y.-S. (2012) Cellular internalization mechanism and intracellular trafficking of filamentous M13 phages displaying a cell-penetrating transbody and TAT peptide. PLoS One 7, e51813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Eldridge B., Cooley R. N., Odegrip R., McGregor D. P., Fitzgerald K. J., Ullman C. G. (2009) An in vitro selection strategy for conferring protease resistance to ligand binding peptides. Protein Eng. Des. Sel. 22, 691–698 [DOI] [PubMed] [Google Scholar]
- 64. Meiron M., Zohar Y., Anunu R., Wildbaum G., Karin N. (2008) CXCL12 (SDF-1α) suppresses ongoing experimental autoimmune encephalomyelitis by selecting antigen-specific regulatory T cells. J. Exp. Med. 205, 2643–2655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. McCandless E. E., Wang Q., Woerner B. M., Harper J. M., Klein R. S. (2006) CXCL12 limits inflammation by localizing mononuclear infiltrates to the perivascular space during experimental autoimmune encephalomyelitis. J. Immunol. 177, 8053–8064 [DOI] [PubMed] [Google Scholar]
- 66. Cruz-Orengo L., Holman D. W., Dorsey D., Zhou L., Zhang P., Wright M., McCandless E. E., Patel J. R., Luker G. D., Littman D. R., Russell J. H., Klein R. S. (2011) CXCR7 influences leukocyte entry into the CNS parenchyma by controlling abluminal CXCL12 abundance during autoimmunity. J. Exp. Med. 208, 327–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang Y., Li G., Stanco A., Long J. E., Crawford D., Potter G. B., Pleasure S. J., Behrens T., Rubenstein J. L. (2011) CXCR4 and CXCR7 have distinct functions in regulating interneuron migration. Neuron 69, 61–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kalatskaya I., Berchiche Y. A., Gravel S., Limberg B. J., Rosenbaum J. S., Heveker N. (2009) AMD3100 is a CXCR7 ligand with allosteric agonist properties. Mol. Pharmacol. 75, 1240–1247 [DOI] [PubMed] [Google Scholar]
- 69. Puchert M., Engele J. (2014) The peculiarities of the SDF-1/CXCL12 system: in some cells, CXCR4 and CXCR7 sing solos, in others, they sing duets. Cell Tissue Res. 355, 239–253 [DOI] [PubMed] [Google Scholar]
- 70. Décaillot F. M., Kazmi M. A., Lin Y., Ray-Saha S., Sakmar T. P., Sachdev P. (2011) CXCR7/CXCR4 heterodimer constitutively recruits β-arrestin to enhance cell migration. J. Biol. Chem. 286, 32188–32197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dealwis C., Fernandez E. J., Thompson D. A., Simon R. J., Siani M. A., Lolis E. (1998) Crystal structure of chemically synthesized [N33A] stromal cell-derived factor 1α, a potent ligand for the HIV-1 “fusin” coreceptor. Proc. Natl. Acad. Sci. U.S.A. 95, 6941–6946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ohnishi Y., Senda T., Nandhagopal N., Sugimoto K., Shioda T., Nagal Y., Mitsui Y. (2000) Crystal structure of recombinant native SDF-1α with additional mutagenesis studies: an attempt at a more comprehensive interpretation of accumulated structure-activity relationship data. J. Interferon Cytokine Res. 20, 691–700 [DOI] [PubMed] [Google Scholar]
- 73. Murphy J. W., Cho Y., Sachpatzidis A., Fan C., Hodsdon M. E., Lolis E. (2007) Structural and functional basis of CXCL12 (stromal cell-derived factor-1α) binding to heparin. J. Biol. Chem. 282, 10018–10027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Smith E. W., Liu Y., Getschman A. E., Peterson F. C., Ziarek J. J., Li R., Volkman B. F., Chen Y. (2014) Structural analysis of a novel small molecule ligand bound to the CXCL12 chemokine. J. Med. Chem. 57, 9693–9699 [DOI] [PMC free article] [PubMed] [Google Scholar]