

Abstract
AngII (angiotensin II) may contribute to cardiovascular risk in obesity via adverse effects on insulin sensitivity and endothelial function. In the present study, we examined the effects of ARB (angiotensin receptor blocker) therapy (losartan, 100 mg/day) on insulin sensitivity and endothelial function in 53 subjects with stage I hypertension, abdominal obesity and impaired fasting glucose. The study design was a randomized double-blinded parallel design placebo-controlled multicentre trial of 8 weeks duration. We used the hyperinsulinaemic–euglycaemic clamp technique to measure insulin sensitivity (expressed as the ‘M/I’ value) and RH-PAT (reactive hyperaemia-peripheral arterial tonometry) to measure endothelial function. Additional measures included HOMA (homoeostasis model assessment)-B, an index of pancreatic β-cell function, and markers of inflammation [e.g. CRP (C-reactive protein)] and oxidative stress (e.g. F2-isoprostanes). ARB therapy did not alter insulin sensitivity [5.2 (2.7) pre-treatment and 4.6 (1.6) post-treatment] compared with placebo therapy [6.1 (2.9) pre-treatment and 5.3 (2.7) post-treatment; P value not significant], but did improve the HOMA-B compared with placebo therapy (P = 0.05). ARB therapy also did not change endothelial function [RH-PAT, 2.15 (0.7) pre-treatment and 2.11 (0.7) post-treatment] compared with placebo therapy [RH-PAT, 1.81 (0.5) pre-treatment and 1.76 (0.7) post-treatment; P value not significant]. Markers of inflammation and oxidative stress were not significantly changed by ARB therapy. In conclusion, ARB therapy did not alter peripheral insulin sensitivity or endothelial function in this cohort of patients with essential hypertension, abdominal obesity and impaired fasting glucose, but did improve pancreatic β-cell function.
Keywords: angiotensin II, clinical trial, hypertension, insulin signalling, obesity, vascular function
INTRODUCTION
Epidemic obesity threatens to reverse progress made in reducing the burden of cardiovascular disease in the United States [1]. Two pathophysiological links underlying obesity and its associated cardiovascular risk are insulin resistance and endothelial dysfunction. Insulin resistance, highly prevalent in obesity, predisposes to diabetes and also adversely affects BP (blood pressure) and lipid metabolism. Endothelial dysfunction, also common in obesity, increases susceptibility to vascular injury by permitting a pro-inflammatory and pro-thrombotic state. Although often considered as distinct entities, insulin sensitivity and endothelial function are in fact integrally linked [2]. Insulin stimulates endothelial NO production via a signalling pathway homologous to that of insulin-stimulated glucose transport in skeletal muscle. In turn, NO activity is essential for effective insulin-stimulated glucose disposal.
Overactivity of the RAAS (renin–angiotensin–aldosterone system), and specifically the vasoactive mediator AngII (angiotensin II), appears to be of particular importance in the genesis of insulin resistance and endothelial dysfunction in obesity. AngII is known to interfere with insulin metabolic signalling at multiple levels [3,4]. In addition, AngII limits NO bioavailability by stimulating vascular ROS (reactive oxygen species) generation [5]. In experimental models, targeting of AngII improves both insulin resistance and endothelial function [6,7].
These observations suggest that pharmacological inhibition of AngII activity may uniquely address the pathophysiological mechanisms related to excess cardiovascular risk in obesity. Accordingly, we sought to examine the effect of angiotensin receptor blockade with losartan on insulin sensitivity and endothelial function in hypertensive individuals with abdominal obesity and impaired fasting glucose.
MATERIALS AND METHODS
Subjects
Eligible subjects comprised non-diabetic men or women, 18–75 years of age, with mild hypertension and taking ≤1 antihypertensive medication. Inclusion criteria included a screening visit BMI (body mass index) between 30 and 40 kg/m2, waist circumference >40 inches in men or >35 inches in women, and fasting glucose ≥100 mg/dl and <126 mg/dl. At the screening visit, subjects’ SBP (systolic BP) had to be <160 mmHg, and a DBP (diastolic BP) of <100 mmHg. At the pre-randomization visit, the SBP had to be ≥120 and <160 mmHg, and the DBP ≥80 and <100 mmHg.
Subjects with known sensitivity or intolerance to ARBs (angiotensin receptor blockers) or a history of angio-oedema were not eligible. Other exclusion criteria included the presence or suspicion of a secondary cause of hypertension, a history of malignant hypertension, smoking within the past year, renal impairment, defined as a serum creatinine >1.5 mg/dl or >2 + proteinuria by dipstick, the use of antioxidant supplements, an inability or unwillingness to practice acceptable methods of birth control, the presence of coronary artery disease, congestive heart failure or stroke, or any other medical condition that, in the investigator’s judgment, would alter the conduct or outcome of the study.
Study design
The present study was an investigator-initiated industry-sponsored multi-site double-blinded placebo-controlled randomized parallel design clinical trial. The number of subjects at each site was: four at the University of Texas Southwestern Medical Center at Dallas, Dallas, TX, U.S.A., two at the St. Luke’s Roosevelt Hospital, New York, NY, U.S.A., six at the Brigham and Women’s Hospital, Boston, MA, U.S.A., 19 at the Veterans Administration Health Care System, San Diego, CA, U.S.A., two at the University of Miami Diabetes Research Institute, Miami, FL, U.S.A., 14 at the Indiana University School of Medicine, Indianapolis, IN, U.S.A., two at the Baylor Clinic, Houston, TX, U.S.A., and four at the University of Pennsylvania School of Medicine, Philadelphia, PA, U.S.A.
The study duration was 12 weeks, with 4 weeks of placebo run-in followed by 8 weeks of double-blind medication. Participants were randomized to either losartan (100 mg/day) or placebo in a 1:1 manner. At the screening visit, subjects gave written informed consent. Each study site institutional review board approved the protocol. The study is registered on ClinicalTrials.gov (http://www.ClinicalTrials.gov) under the identifier NCT 00675987.
Antihypertensive medication was tapered and discontinued after the screening visit. Once off medication, subjects began a 4-week single-blind placebo run-in period. BP and adherence were measured midway and at the end of the run-in period. Subjects were excluded for non-compliance at the end of the placebo period (<75 % or >120 % of correct number of pills taken) or if the BP fell outside accepted limits (see above).
Eligible patients then underwent 8 weeks of double-blind treatment. Endothelial function and insulin sensitivity were measured on day 1 of treatment and on the final day (testing visits). Subjects were instructed to maintain a constant diet throughout the study. Before all study visits, subjects fasted, avoided exercise for >12 h, arrived via motorized transportation, did not use sildenafil, tadalafil or vardenafil for 72 h, and did not use cold medication, nutritional supplements and niacin for >24 h. Study medication was not taken on the morning of a scheduled visit. Study visits all occurred before 10.00 hours. For pre-menopausal women, study visits were scheduled to occur during the follicular phase of the menstrual cycle. Efforts were made to maintain a calm environment throughout study visits, and study measurements were made in a temperature-controlled room (37 °C).
Study measurements
Anthropometric measurements
Body weight and height were measured with the subject wearing light clothing and no shoes. Waist circumference was measured at the level of the umbilicus on the bare abdomen.
BP
A study investigator measured BP using a standard aneroid sphygmomanometer. All site investigators underwent standardized training in accord with American Heart Association guidelines [8]. The average of at least three measurements was taken, and the values were rounded to the nearest whole number. If a single DBP reading deviated >5 mmHg from the average, the BP was considered unstable and the measurements were repeated on another occasion.
Endothelial function
Endothelium-dependent vasodilation was assessed by measuring the flow-induced change in digital PVA (pulse volume amplitude) of the first finger. Digital PVA was measured using a peripheral arterial tonometer (EndoPAT; Itamar-Medical). The peripheral arterial tonometer apparatus comprises a finger-mounted probe that surrounds the distal phalanx with an electronically controlled inflatable pressuring air cushion confined within a rigid external case. The pressure changes within the probe that accompany pulsatile volume changes in the finger are fed to a personal computer where the signal is band-pass-filtered (0.3–30 Hz), amplified, displayed and stored. The PVA is a measure of the height of the pulse wave and is proportional to pressure changes accompanying volume changes within the probe.
Investigators at each site were trained and had to demonstrate competence with the technique prior to subject enrolment. The baseline PVA was measured over a period of at least 5 min to ensure stability. Thereafter, a cuff was rapidly inflated to occlusive pressure (at least 50 mmHg >SBP) on the upper arm to induce limb ischaemia. After exactly 5 min, the cuff was rapidly deflated and pulse volume measurements were repeated during this reactive hyperaemic phase. The increase in PVA during RH (reactive hyperaemia) following cuff release is in large part caused by NO [9]. Accordingly, endothelium-dependent vasodilation was calculated from the stored digitized data as the ratio of PVA during RH over a 1-min interval starting 1 min after cuff deflation and divided by the baseline PVA obtained over the 1-min interval immediately preceding cuff inflation. The PVA from the same finger of the contralateral non-ischaemic hand was used to adjust for any drift in the magnitude of the signal due to systemic factors, and the PVA ratio was normalized to the control contralateral finger [RH-PVA (PVA during RH relative to baseline)]. The measurement of endothelial function was repeated once after at least 15 min of rest, and the average of the two measurements was calculated.
Insulin sensitivity
A hyperinsulinaemic–euglycaemic clamp was performed to assess peripheral insulin sensitivity after the measurement of endothelial function. An intravenous catheter was placed in each arm, one for blood withdrawal (the hand) and the other for infusion (the contralateral forearm). The hand of the blood-withdrawal arm was placed in a box heated to 50 °C to ensure arterialization of venous blood. The subject’s weight was measured at each clamp visit, and the BSA (body surface area) was determined by the Gehan and George equation:
A 2.5 h hyperinsulinaemic–euglycaemic clamp was performed using a primed continuous insulin infusion. The priming was 160 m-units/m2 per min for the first 10 min, followed by 80 m-units/m2 per min for 140 min. Plasma glucose was assessed every 5 min at the bedside, and 20 % dextrose was infused at a variable rate to maintain plasma glucose of 90 mg/dl ± 5 % [10]. Samples for insulin measurement were obtained in triplicate at baseline, every 30 min for the first 120 min, and then every 10 min for the last 30 min. The volume remaining from the 20 % dextrose infusion pump was recorded every 15 min. Insulin sensitivity (M) was calculated as the average glucose infusion rate (mg/kg of body weight per min) over the last 30 min of the clamp. The infusion rate is approximately equivalent to the GDR (glucose disposal rate), as endogenous production is suppressed or at least greatly minimized at this dose of insulin administration [11]. Steady-state insulin levels varied among subjects and we therefore calculated the insulin-adjusted insulin sensitivity or insulin sensitivity index (M/I; where I is mean level of insulin), which is the GDR/I ratio during the last 30 min of the clamp multiplied by 200 (arbitrary units). HOMA-IR [HOMA (homoeostasis model assessment) of insulin resistance], an index that correlates inversely with peripheral insulin sensitivity, and HOMA-B (HOMA of β-cell function), an index that correlates directly with pancreatic β-cell function, were calculated as described previously [12].
Additional assays
A fasting venous blood specimen was obtained at the initial visit for standard safety labs, such as blood count (LH750; Beckman Coulter), and hepatic and renal profiles (Modular Biochemical Analyser; Roche Diagnostics), as well as for glucose and insulin levels (ADVIA Centaur; Siemens Healthcare Diagnostics). A urine dipstick pregnancy test was performed at the initial visit as well as the two testing visits. At the two testing visits, blood was withdrawn for measurements of hsCRP (high-sensitivity C-reactive protein) (Modular P analyser; Roche), VCAM-1 (vascular cell-adhesion molecule-1) (ELISA; R&D Systems), MCP-1 (monocyte chemoattractant protein-1) (ELISA; R&D Systems), ox-LDL (oxidized low-density lipoprotein) (ELISA; Mercodia), F2-isoprostane (GC–MS; Vanderbilt University, Nashville, TN, U.S.A.) and E-selectin (ELISA; R&D Systems), and urine was obtained to assess for the presence of microalbuminuria (Modular P Analyser; Roche Diagnostics).
Statistical methods
The primary approach to the statistical analysis of the efficacy data was a modified intention-to-treat approach. This analysis included all patients who received at least one dose of the double-blind study drug and had a valid measurement of insulin sensitivity and/or endothelial function at baseline and a valid measurement after baseline. Patients were analysed based on the treatment group to which they were randomized.
There were two co-primary end points. The first was the change in peripheral insulin sensitivity after losartan therapy compared with after placebo therapy. The second was the change in endothelial function after losartan therapy compared with after placebo therapy. Secondary end points included the changes in microalbuminuria, hsCRP, VCAM-1, MCP-1, ox-LDL, F2-isoprostane and E-selectin after losartan compared with placebo therapy.
It was anticipated that a total enrolment of 60 subjects randomized into the losartan and placebo groups at a 1:1 ratio would yield approximately 25 evaluable subjects in each group. On the basis of two published studies demonstrating a positive effect of losartan on insulin resistance [13,14], the losartan treatment effect was estimated to be 1.38 mg/kg of body weight per min. Assuming an S.D. of 1.5 mg/kg of body weight per min and assuming no change in the GDR in the placebo group, 25 evaluable patients per group would then provide 89 % power to detect a significant difference between placebo and losartan treatment. Losartan was expected to improve endothelial function by 24.5 % with an S.D. of 21 % [15–18]. Assuming a similar treatment effect size, 25 evaluable patients per group would provide 98 % power to detect a significant difference between losartan and placebo.
An ANCOVA (analysis of covariance) model was used to compare the treatments on the primary and secondary efficacy variables. No adjustment for multiple hypothesis testing was made. The ANCOVA included terms for treatment, investigator sites and the corresponding baseline measurement. Investigator sites with low enrolment were combined into one or more groups for analysis. Baseline characteristics are described as means (S.D.) or medians (S.D.) for parametric and non-parametric data respectively. The S.D. of the median was calculated as S.D. = (quartile 3 − quartile 1)/1.075.
RESULTS
A total of 171 potential subjects were screened. Of these, 118 were disqualified for the reasons listed in Figure 1, leaving 53 subjects who were randomized, 26 to losartan therapy and 27 to placebo. Of those enrolled, 51 subjects completed the trial, 25 in the losartan group and 26 in the placebo group. Quality review of the data resulted in three subjects being excluded from the analysis of insulin sensitivity due to poor quality insulin clamps. In one subject, the serum insulin level did not increase from baseline, in one subject the serum insulin level inexplicably increased ~5-fold mid-clamp, and in one subject steady-state was not achieved. No digital endothelial function study was considered inadequate. These determinations were made in a blinded manner prior to locking of the database. Baseline characteristics of the 53 randomized subjects are presented by treatment assignment in Table 1.
Figure 1.
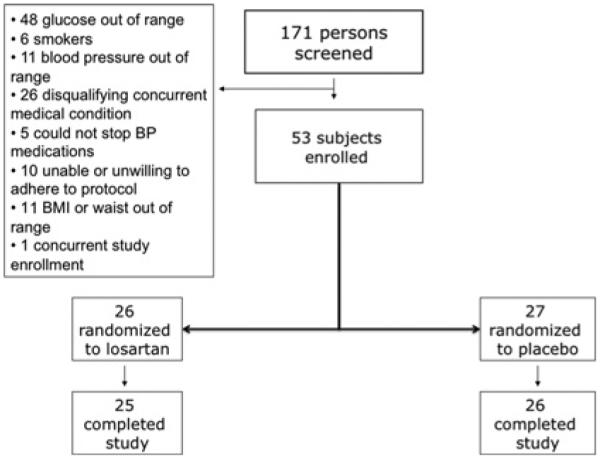
CONSORT flow diagram for the trial
Table 1.
Characteristics of all study participants at screening and by treatment assignment at completion of placebo run-in
| Screening visit | Baseline visit |
||
|---|---|---|---|
| Characteristic | All subjects (n = 53) | Placebo (n = 27) | Losartan (n = 26) |
| Age (years) | 52.5 (9.5) | 53.8 (8.3) | 51.1 (10.5) |
| Sex (female) (n) | 26 (49.1 %) | 12 (44.4 %) | 14 (53.8 %) |
| Race | |||
| White (n) | 40 (75.4 %) | 24 (88.9 %) | 16 (61.5 %) |
| Black (n) | 10 (18.9 %) | 1 (3.7 %) | 9 (34.6 %) |
| Other (n) | 3 (5.7 %) | 2 (7.4 %) | 1 (3.8 %) |
| Taking antihypertensive therapy (n) | 1 (1.9 %) | 0 | 1 (3.8 %) |
| Weight (kg) | 101.2 (13.5) | 101.6 (14.1) | 101.0 (13.7) |
| BMI (kg/m2) | 34.7 (3.8) | 34.7 (3.8) | 34.8 (3.8) |
| Waist circumference (cm)* | 111.2 (9.9) | n/a | n/a |
| SBP (mmHg) | 136.0 (10.8) | 135.0 (9.6) | 136.3 (10.9) |
| DBP (mmHg) | 87.2 (6.4) | 86.6 (5.3) | 88.7 (5.3) |
| Heart rate (beats/min) | 71.5 (10.6) | 74.8 (12.2) | 72.4 (9.2) |
| Potassium (mmol/l) | 4.3 (0.3) | 4.0 (3.3, 4.7) | 4.1 (3.6, 4.9) |
| Glucose (mmol/l) | 104.8 (8.6) | 97.5 (1.7) | 97.5 (1.7) |
| Insulin (μ-units/ml) | 14.3 (8.0) | 10.2 (0.9) | 11.7 (1.2) |
| HOMA-IR | 3.8 (2.3) | 2.5 (0.2) | 2.9 (0.3) |
| Triacyglycerols (mmol/l) | n/a | 136.5 (56.0, 309.0) | 113.0 (51.0, 235.0) |
| HDL-cholesterol (mmol/l) | n/a | 44.0 (22.0, 77.0) | 43.0 (31.0, 83.0) |
| Creatinine (mg/dl) | 1.0 (0.2) | 0.93 (0.03) | 0.88 (0.04) |
| Urine albumin/creatinine ratio (mg/mmol) | n/a | 1.22 (0.35) | 1.02 (0.18) |
Measured at the screening visit only.
Continuous variables as means (S.E.M.) or medians (range). HDL, high-density lipoprotein; n/a, not available.
Insulin sensitivity
At the baseline visit, insulin sensitivity was 5.65 (2.7) mg/kg of body weight per min among all subjects, confirming the presence of insulin resistance in the study population in comparison with historical healthy populations [19]. Insulin sensitivity at baseline was not different between the losartan- and placebo-treated subjects (Table 2). The primary end point, the change in insulin sensitivity, was not different between the losartan-treated and the placebo-treated groups. Of note, HOMA-B tended to increase from baseline in the losartan-treated group { + 26.1 [95 % CI (confidence interval), −2.75, 55.0]} and decrease in the placebo-treated group [ −9.6 (95 % CI, −38.8, 19.5)], a difference on the margin of statistical significance (P = 0.05).
Table 2.
Insulin clamp data in losartan- and placebo-treated patients
| Parameter | Treatment assignment | Pre-treatment | Post-treatment | Change from baseline | P value |
|---|---|---|---|---|---|
| Fasting glucose (mg/dl) | Losartan | 97.5 (9.5) | 98.2 (6.5) | 2.3 (−1.38, 5.92) | 0.69 |
| Placebo | 97.5 (8.6) | 97.9 (10.5) | 1.4 (−2.26, 5.04) | ||
| Fasting insulin (μ-units/ml) | Losartan | 11.7 (6.1) | 14.5 (8.3) | 3.5 (0.56, 6.34) | 0.07 |
| Placebo | 10.2 (4.3) | 10.9 (7.2) | 0.2 (−2.69, 3.04) | ||
| HOMA-IR | Losartan | 2.9 (1.7) | 3.3 (2.1) | 0.7 (−0.02, 1.41) | 0.20 |
| Placebo | 2.5 (1.2) | 2.7 (2.2) | 0.1 (−0.61, 0.85) | ||
| HOMA-B | Losartan | 127.3 (71) | 150.0 (90) | 26.1 (−2.75, 55.0) | 0.05 |
| Placebo | 108.3 (44) | 108.1 (50) | −9.6 (−38.76, 19.52) | ||
| Insulin sensitivity corrected for the steady-state insulin level [mg/kg of body weight per min/(μ-units/ml)] |
Losartan | 5.2 (2.7) | 4.5 (1.7) | −0.8 (−1.62, −0.07) | 0.62 |
| Placebo | 6.1 (2.9) | 5.3 (2.8) | −0.6 (−1.39, 0.19) |
Values are means (S.D.) or least-squares mean (95 % CI). P values are a comparison between the groups for a change from the baseline value adjusted for baseline value and study site.
Endothelial function
At the baseline visit, the RH-PVA was 2.0, a result similar to what has been observed previously in healthy populations [20]. Baseline endothelial function was not different between the losartan- and placebo-treated subjects (Table 3). The change in endothelial function was not different after losartan therapy compared with placebo therapy (P value not significant).
Table 3.
EndoPAT results in losartan- and placebo-treated patients
| EndoPAT RH | Pre-treatment | Post-treatment | Change from baseline | P value |
|---|---|---|---|---|
| Treatment assignment | ||||
| Losartan | 2.15 (0.7) | 2.11 (0.7) | −0.06 (−0.33, 0.21) | 0.31 |
| Placebo | 1.81 (0.5) | 1.76 (0.7) | −0.23 (−0.51, 0.04) |
Values are means (S.D.) or least-squares mean (95 % CI). P values are a comparison between the groups for a change from the baseline value adjusted for baseline value and study site.
Secondary outcomes
In addition to changes in insulin sensitivity and endothelial function, we also examined a series of measures of inflammation, oxidative stress and endothelial injury (Table 4). There were no significant changes in any of these after losartan compared with after placebo therapy.
Table 4.
Blood and urine markers in losartan- and placebo-treated patients
| Marker | Treatment assignment | Pre-treatment | Post-treatment | Change from baseline | P value |
|---|---|---|---|---|---|
| hsCRP (mg/l)* | Losartan | 2.6 | 2.0 | −34 % (−50 %, −13 %) | 0.07 |
| Placebo | 3.3 | 2.9 | −10 % (−32 %, 20 %) | ||
| VCAM-1 (ng/ml) | Losartan | 598 | 585 | −21 (−70, 28) | 0.10 |
| Placebo | 663 | 680 | 29 (−20, 79) | ||
| MCP-1 (pg/ml) | Losartan | 335 | 338 | −24 (−65, 18) | 0.62 |
| Placebo | 373 | 352 | −37 (−80, 6.8) | ||
| oxLDL (units/l) | Losartan | 50.2 | 39.8 | −5.5 (−11.9, 0.9) | 0.37 |
| Placebo | 41.4 | 39.8 | −2.0 (−8.1, 4.1) | ||
| F2-isoprostanes (ng/mg of creatinine) | Losartan | 2.1 | 3.1 | 0.9 (−0.4, 2.2) | 0.51 |
| Placebo | 1.7 | 2.5 | 0.4 (−0.9, 1.6) | ||
| E-selectin (ng/ml) | Losartan | 41.8 | 41.6 | −0.6 (−3.5, 2.3) | 0.55 |
| Placebo | 43.4 | 42.7 | −1.6 (−4.5, 1.3) | ||
| Urine albumin/creatine (mg/mmol) | Losartan | 1.0 | 1.4 | 0.2 (−0.3, 0.8) | 0.69 |
| Placebo | 1.2 | 1.5 | 0.4 (−0.1, 0.9) |
Geometric means and the geometric mean of the percentage change from baseline are reported.
Values are means (S.D.) or least-squares mean (95 % CI), unless otherwise stated. P values are a comparison between the groups for a change from the baseline value adjusted for baseline value and study site.
BP and heart rate
BP was not different between the two treatment groups at baseline (Table 1). The change in BP after losartan therapy was not significantly different than after placebo therapy (Table 5). SBP tended to fall 3.3 (13.0) mmHg in the losartan group and to increase 1.4 (8.4) mmHg in the placebo group. DBP tended to fall 1.5 (8.5) mmHg in the losartan group and to increase 1.2 (7.4) mmHg in the placebo group. There was no difference in the change in heart rate in the losartan-compared with the placebo-treated patients.
Table 5.
Select clinical characteristics in losartan- and placebo-treated patients
| Placebo |
Losartan |
||||||
|---|---|---|---|---|---|---|---|
| Characteristic | Pre-treatment | Post-treatment | Change from baseline | Pre-treatment | Post-treatment | Change from baseline | P value |
| SBP (mmHg) | 135.0 (9.8) | 136.4 (11.3) | 1.4 (8.4) | 136.2 (11.1) | 132.9 (12.3) | −3.3 (13.0) | 0.36 |
| DBP (mmHg) | 86.3 (5.3) | 87.5 (9.9) | 1.2 (7.4) | 88.6 (5.4) | 87.1 (10.2) | −1.5 (8.5) | 0.26 |
| Heart rate (beats/min) | 74.8 (12.2) | 73.2 (10.5) | −1.7 (11.7) | 72.4 (9.2) | 71.4 (9.3) | −1.0 (9.9) | 0.81 |
| Blood urea nitrogen (mg/dl) | 13.5 (6.0, 19.0) | 14.0 (6.0, 21.0) | 0.5 (−7.0, 7.0) | 12.0 (6.0, 20.0) | 12.0 (6.0, 17.0) | 0.0 (−7.0, 4.0) | 0.48 |
| Creatinine (mg/dl) | 0.9 (0.7, 1.3) | 0.9 (0.6, 1.3) | 0.0 (−0.2, 0.1) | 0.9 (0.7, 1.4) | 0.8 (0.7, 1.4) | 0.0 (−0.1, 0.3) | 0.83 |
| Serum potassium (mmol/l) | 3.9 (0.3) | 4.1 (0.3) | 0.2 (0.5) | 4.1 (0.3) | 4.1 (0.3) | 0.0 (0.3) | 0.39 |
Values are means (S.D.) or medians (95 % CI). P values are the comparison of the change after losartan therapy with that after placebo therapy, using ANCOVA with terms for treatment, investigator sites and the corresponding baseline measurement.
Renal function and potassium
Baseline renal function, measured as the serum creatinine level, was within the normal range and did not differ between the two treatment groups (Table 5). There was no significant difference in the change in renal function after losartan compared with placebo therapy. Baseline and post-treatment serum potassium levels were within the normal range and did not differ between the two treatment groups.
DISCUSSION
We conducted an 8-week randomized double-blinded placebo-controlled trial of the ARB losartan (100 mg/day) in 53 patients with stage I hypertension, abdominal obesity and impaired fasting glucose, with co-primary end points of insulin sensitivity and endothelial dysfunction. We did not observe a significant effect of ARB therapy on either peripheral insulin sensitivity or endothelial function compared with placebo therapy. However, we did observe an improvement in pancreatic β-cell function attributable to ARB therapy. In addition, we did not observe an effect of losartan treatment on a panel of biomarkers of inflammation, oxidative stress and endothelial injury. Losartan did tend to lower BP to a similar extent to that observed in a related trial of 261 patients with essential hypertension randomized to either losartan or placebo for 8 weeks; however, the present study was not powered to detect differences in BP [21].
Insulin resistance
There is extensive experimental evidence suggesting that AngII induces insulin resistance. Specifically, AngII directly interferes with insulin metabolic signalling. AngII inhibits IRS (insulin receptor substrate)-1 and IRS-2 associated PI3K (phosphoinositide 3-kinase) activity and reduces the ability of IRS-1 to associate with the activated insulin receptor [4,22,23]. AngII also decreases IRS-1 protein levels in vascular smooth muscle cells and induces SOCS-3 (suppressor of cytokine signalling-3), which further inhibits insulin signalling and insulin-stimulated glucose disposal [3,24]. Thus the biology supports a potential role for ARB therapy to improve insulin sensitivity via improved insulin metabolic signalling.
In the present study, however, losartan did not improve peripheral insulin sensitivity, as measured using a hyperinsulinaemic–euglycaemic clamp. This differs with findings in some, but not all previous, reports. Paolissi et al. [14] reported an ~25 % improvement in GDR with losartan compared with placebo therapy in 16 mild-to-moderate hypertensive subjects, but, of note in that study, an unusually large reduction in BP (16/10 mmHg) was attributed to losartan therapy. Moan et al. [13] described a 27–30 % improvement in GDR and GDR/I with losartan (100 mg/day for 6 weeks), but this was an uncontrolled study of only five subjects with severe (DBP ≥115 mmHg) hypertension. In a subsequent controlled study by the same authors involving 20 mildly hypertensive subjects [25], losartan had no effect on insulin sensitivity. Interestingly, Moan et al. [25] and others [26–28] found that losartan, when compared with calcium channel blocker therapy, improved insulin sensitivity in hypertensive subjects. Laakso et al. [29] found that 12 weeks of treatment with losartan compared with metoprolol in hypertensive patients with hyperinsulinaemia had no effect on insulin-stimulated GDR. Finally, Fogari et al. [30–32] have reported three studies of different populations of hypertensive patients in which the effect of losartan on insulin-stimulated GDR was compared with ACE (angiotensin-converting enzyme) inhibitor therapy; in each trial, losartan failed to improve GDR, whereas the ACE inhibitor did. Our present controlled study of mild hypertensive subjects did not detect an effect of losartan on insulin sensitivity. On balance, these trials fail to support the notion that ARB therapy with losartan in mild hypertension improves insulin resistance, as measured using the euglycaemic clamp technique.
This failure to improve insulin sensitivity seems in conflict with the reported reduction in risk for diabetes attributed to ARB therapy [33]. This discordance may be due to the fact that the GDR during a euglycaemic clamp is mostly determined by skeletal muscle insulin sensitivity, whereas the ARB therapy may be improving glycaemia by beneficial effects on the pancreatic β-cells. Santoro et al. [34], who found that ACE inhibitor therapy improved glycaemia in essential hypertension not by improvement in peripheral insulin sensitivity, but rather by preservation of pancreatic islet β-cell function, suggested this.
In fact, experimental data suggests that AngII contributes to β-cell dysfunction via adverse effects on islet blood flow, and by promoting islet cell fibrosis, inflammation and oxidative stress [35–37]. Indeed, ARB therapy was shown to improve early-phase insulin responses in hypertensive subjects with impaired glucose tolerance [38]. Our present study confirms that ARB therapy may preserve β-cell function in hypertension.
Endothelial function
AngII increases vascular ROS generation by induction of vascular NADPH oxidase [39]. As a consequence of this, the AngII-infused rat and the rat doubly transgenic for human renin and angiotensinogen develop hypertension, vascular oxidative stress and endothelial dysfunction, each of which is remedied by ARB therapy [5,40]. However, in our present study of obese hypertensive patients with impaired fasting glucose, losartan therapy did not improve endothelial function, as assessed by the change in digital PVA during RH. This is consistent with some, but not all, published reports. For example, ACE inhibitor therapy had no effect on forearm resistance vessel endothelial function compared with placebo therapy in 24 subjects with essential hypertension [41]. In contrast, Koh et al. [42] found that each of losartan, candesartan and irbesartan improved FMD (flow-mediated dilation) of the brachial artery compared with placebo in patients with mild-to-moderate essential hypertension. We did not detect an effect of ARB therapy on endothelial function measured by RH-PVA; however, our subjects’ RH-PVA response prior to intervention was similar to that reported previously in healthy subjects [9,20,43]. Although the present study did not have an internal control group of healthy individuals, these historical data suggest that our selection criteria did not effectively identify a group of subjects with endothelial dysfunction.
Inflammation and oxidative stress
As a secondary aim of the present study, we examined the effect of losartan therapy on markers of inflammation, oxidative stress and endothelial damage. We anticipated that abnormalities in these biomarkers would provide insight into the mechanisms of AngII-mediated insulin resistance and endothelial dysfunction had these findings been confirmed. For each of these markers there was biological plausibility [40,44]; however, in the present study, losartan did not cause a significant improvement in any of these markers.
Limitations
The results of the present study should be considered in light of a few study limitations. First, the hyperinsulinaemic–euglycaemic clamp technique, as performed, measures skeletal muscle insulin resistance, but does address β-cell function. Secondly, our sample size may have been insufficient to detect changes in many of the examined biomarkers. Thirdly, both Bonetti et al. [43] and Liu et al. [20] found low interday reproducibility of the RH-PVA measurements. These reports suggest that our power calculation, based upon the repeatability of the brachial artery ultrasound technique, may have overestimated the power of our sample size. In addition, although subjects fulfilled entry criteria for BP and fasting glucose level, average BPs and glucose levels decreased somewhat (mean values, 135–136/88–89 mmHg and 96–98 mg/dl respectively) after the placebo run-in period. Greater baseline abnormalities may be necessary to observe a positive effect of ARBs on insulin resistance or endothelial dysfunction.
Conclusions
In summary, we did not observe an effect of ARB therapy on either insulin sensitivity or endothelial dysfunction in stage I hypertension characterized by abdominal obesity and insulin resistance. Our results do suggest though that ARB therapy improves pancreatic β-cell function in this population.
ACKNOWLEDGEMENTS
We thank Dr Andrew M. Tershakovec and Mr Kevin Gergich for their assistance in the conduct of this trial. The opinions expressed in this paper are those of the authors and do not necessarily represent those of Merck & Co. Inc.
FUNDING
This work was support in part by the Merck-Assisted Studies Program of Merck & Co. Inc. T.S.P. is a recipient of a National Heart, Lung, and Blood Institute (NHLBI) Career Development Award in Vascular Medicine [grant number K12 HL083786); M.A.C. is the Simon C. Fireman Scholar in Cardiovascular Medicine; J.L.P. received support from the Eads Family Research Fund at Baylor College of Medicine.
Abbreviations
- ACE
angiotensin-converting enzyme
- ANCOVA
analysis of covariance
- AngII
angiotensin II
- ARB
angiotensin receptor blocker
- BMI
body mass index
- BP
blood pressure
- BSA
body surface area
- CI
confidence interval
- DBP
diastolic BP
- GDR
glucose disposal rate
- GDR/I ratio
GDR/mean level of insulin ratio
- HOMA
homoeostasis model assessment
- HOMA-B
HOMA of pancreatic β-cell function
- HOMA-IR
HOMA of insulin resistance
- hsCRP
high-sensitivity C-reactive protein
- IRS
insulin receptor substrate
- MCP-1
monocyte chemoattractant protein-1
- ox-LDL
oxidized low-density lipoprotein
- PVA
pulse volume amplitude
- RH
reactive hyperaemia
- RH-PVA
PVA during RH relative to baseline
- ROS
reactive oxygen species
- SBP
systolic BP
- VCAM-1
vascular cell-adhesion molecule-1
Footnotes
AUTHOR CONTRIBUTION
Todd Perlstein performed the study, analysed the data and wrote the paper; Robert Henry, Kieren Mather, Michael Rickels, Nicola Abate, Scott Grundy, Jeanine Albu, Jennifer Marks and James Pool performed the study; Yabing Mai analysed the data; and Mark Creager initiated, designed and performed the study, and wrote the paper. All authors were involved in revising the paper and approving the final version.
REFERENCES
- 1.Olshansky SJ, Passaro DJ, Hershow RC, Layden J, Carnes BA, Brody J, Hayflick L, Butler RN, Allison DB, Ludwig DS. A potential decline in life expectancy in the United States in the 21st century. N. Engl. J. Med. 2005;352:1138–1145. doi: 10.1056/NEJMsr043743. [DOI] [PubMed] [Google Scholar]
- 2.Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–1904. doi: 10.1161/CIRCULATIONAHA.105.563213. [DOI] [PubMed] [Google Scholar]
- 3.Calegari VC, Alves M, Picardi PK, Inoue RY, Franchini KG, Saad MJ, Velloso LA. Suppressor of cytokine signaling-3 provides a novel interface in the cross-talk between angiotensin II and insulin signaling systems. Endocrinology. 2005;146:579–588. doi: 10.1210/en.2004-0466. [DOI] [PubMed] [Google Scholar]
- 4.Andreozzi F, Laratta E, Sciacqua A, Perticone F, Sesti G. Angiotensin II impairs the insulin signaling pathway promoting production of nitric oxide by inducing phosphorylation of insulin receptor substrate-1 on Ser312 and Ser616 in human umbilical vein endothelial cells. Circ. Res. 2004;94:1211–1218. doi: 10.1161/01.RES.0000126501.34994.96. [DOI] [PubMed] [Google Scholar]
- 5.Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J. Clin. Invest. 1996;97:1916–1923. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wassmann S, Czech T, van Eickels M, Fleming I, Bohm M, Nickenig G. Inhibition of diet-induced atherosclerosis and endothelial dysfunction in apolipoprotein E/angiotensin II type 1A receptor double-knockout mice. Circulation. 2004;110:3062–3067. doi: 10.1161/01.CIR.0000137970.47771.AF. [DOI] [PubMed] [Google Scholar]
- 7.Hsieh PS. Reversal of fructose-induced hypertension and insulin resistance by chronic losartan treatment is independent of AT2 receptor activation in rats. J. Hypertens. 2005;23:2209–2217. doi: 10.1097/01.hjh.0000189871.94031.e2. [DOI] [PubMed] [Google Scholar]
- 8.Pickering TG, Hall JE, Appel LJ, Falkner BE, Graves J, Hill MN, Jones DW, Kurtz T, Sheps SG, Roccella EJ. Recommendations for blood pressure measurement in humans and experimental animals: part 1: blood pressure measurement in humans: a statement for professionals from the Subcommittee of Professional and Public Education of the American Heart Association Council on High Blood Pressure Research. Circulation. 2005;111:697–716. doi: 10.1161/01.CIR.0000154900.76284.F6. [DOI] [PubMed] [Google Scholar]
- 9.Nohria A, Gerhard-Herman M, Creager MA, Hurley S, Mitra D, Ganz P. Role of nitric oxide in the regulation of digital pulse volume amplitude in humans. J. Appl. Physiol. 2006;101:545–548. doi: 10.1152/japplphysiol.01285.2005. [DOI] [PubMed] [Google Scholar]
- 10.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am. J. Physiol. 1979;237:E214–E223. doi: 10.1152/ajpendo.1979.237.3.E214. [DOI] [PubMed] [Google Scholar]
- 11.Rizza RA, Mandarino LJ, Gerich JE. Dose-response characteristics for effects of insulin on production and utilization of glucose in man. Am. J. Physiol. 1981;240:E630–E639. doi: 10.1152/ajpendo.1981.240.6.E630. [DOI] [PubMed] [Google Scholar]
- 12.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 13.Moan A, Hoieggen A, Nordby G, Eide IK, Kjeldsen SE. Effects of losartan on insulin sensitivity in severe hypertension: connections through sympathetic nervous system activity? J. Hum. Hypertens. 1995;9(Suppl. 5):S45–S50. [PubMed] [Google Scholar]
- 14.Paolisso G, Tagliamonte MR, Gambardella A, Manzella D, Gualdiero P, Varricchio G, Verza M, Varricchio M. Losartan mediated improvement in insulin action is mainly due to an increase in non-oxidative glucose metabolism and blood flow in insulin-resistant hypertensive patients. J. Hum. Hypertens. 1997;11:307–312. doi: 10.1038/sj.jhh.1000434. [DOI] [PubMed] [Google Scholar]
- 15.Cheetham C, Collis J, O’Driscoll G, Stanton K, Taylor R, Green D. Losartan, an angiotensin type 1 receptor antagonist, improves endothelial function in non-insulin-dependent diabetes. J. Am. Coll. Cardiol. 2000;36:1461–1466. doi: 10.1016/s0735-1097(00)00933-5. [DOI] [PubMed] [Google Scholar]
- 16.Cheetham C, O’Driscoll G, Stanton K, Taylor R, Green D. Losartan, an angiotensin type I receptor antagonist, improves conduit vessel endothelial function in Type II diabetes. Clin. Sci. 2001;100:13–17. [PubMed] [Google Scholar]
- 17.Deng YB, Wang DW, Li CL, Yang HY. Effects of the angiotensin receptor antagonist losartan on the response of the left main coronary artery to cold pressor test in patients with essential hypertension as assessed by echocardiography. Can. J. Cardiol. 2002;18:389–396. [PubMed] [Google Scholar]
- 18.Deng YB, Yang HY, Li CL, Chang Q. Angiotensin receptor antagonist losartan improves endothelial function of epicardial coronary arteries in patients with essential hypertension. Clin. Cardiol. 2002;25:422–428. doi: 10.1002/clc.4960250906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perlstein TS, Gerhard-Herman M, Hollenberg NK, Williams GH, Thomas A. Insulin induces renal vasodilation, increases plasma renin activity, and sensitizes the renal vasculature to angiotensin receptor blockade in healthy subjects. J. Am. Soc. Nephrol. 2007;18:944–951. doi: 10.1681/ASN.2006091026. [DOI] [PubMed] [Google Scholar]
- 20.Liu J, Wang J, Jin Y, Roethig HJ, Unverdorben M. Variability of peripheral arterial tonometry in the measurement of endothelial function in healthy men. Clin. Cardiol. 2009;32:700–704. doi: 10.1002/clc.20668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oparil S, Abate N, Chen E, Creager MA, Galet V, Jia G, Julius S, Lerman A, Lyle PA, Pool J, Tershakovec AM. A double-blind, randomized study evaluating losartan potassium monotherapy or in combination with hydrochlorothiazide versus placebo in obese patients with hypertension. Curr. Med. Res. Opin. 2008;24:1101–1114. doi: 10.1185/030079908x280716. [DOI] [PubMed] [Google Scholar]
- 22.Folli F, Kahn CR, Hansen H, Bouchie JL, Feener EP. Angiotensin II inhibits insulin signaling in aortic smooth muscle cells at multiple levels. A potential role for serine phosphorylation in insulin/angiotensin II crosstalk. J. Clin. Invest. 1997;100:2158–2169. doi: 10.1172/JCI119752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Izawa Y, Yoshizumi M, Fujita Y, Ali N, Kanematsu Y, Ishizawa K, Tsuchiya K, Obata T, Ebina Y, Tomita S, Tamaki T. ERK1/2 activation by angiotensin II inhibits insulin-induced glucose uptake in vascular smooth muscle cells. Exp. Cell Res. 2005;308:291–299. doi: 10.1016/j.yexcr.2005.04.028. [DOI] [PubMed] [Google Scholar]
- 24.Taniyama Y, Hitomi H, Shah A, Alexander RW, Griendling KK. Mechanisms of reactive oxygen species-dependent downregulation of insulin receptor substrate-1 by angiotensin II. Arterioscler. Thromb. Vasc. Biol. 2005;25:1142–1147. doi: 10.1161/01.ATV.0000164313.17167.df. [DOI] [PubMed] [Google Scholar]
- 25.Moan A, Hoieggen A, Seljeflot I, Risanger T, Arnesen H, Kjeldsen SE. The effect of angiotensin II receptor antagonism with losartan on glucose metabolism and insulin sensitivity. J. Hypertens. 1996;14:1093–1097. doi: 10.1097/00004872-199609000-00008. [DOI] [PubMed] [Google Scholar]
- 26.Aksnes TA, Reims HM, Guptha S, Moan A, Os I, Kjeldsen SE. Improved insulin sensitivity with the angiotensin II-receptor blocker losartan in patients with hypertension and other cardiovascular risk factors. J. Hum. Hypertens. 2006;20:860–866. doi: 10.1038/sj.jhh.1002087. [DOI] [PubMed] [Google Scholar]
- 27.Nishimura H, Sanaka T, Tanihata Y, Naito T, Higuchi C, Otsuka K. Losartan elevates the serum high-molecular weight-adiponectin isoform and concurrently improves insulin sensitivity in patients with impaired glucose metabolism. Hypertens. Res. 2008;31:1611–1618. doi: 10.1291/hypres.31.1611. [DOI] [PubMed] [Google Scholar]
- 28.Jin HM, Pan Y. Angiotensin type-1 receptor blockade with losartan increases insulin sensitivity and improves glucose homeostasis in subjects with type 2 diabetes and nephropathy. Nephrol. Dial. Transplant. 2007;22:1943–1949. doi: 10.1093/ndt/gfm049. [DOI] [PubMed] [Google Scholar]
- 29.Laakso M, Karjalainen L, Lempiainen-Kuosa P. Effects of losartan on insulin sensitivity in hypertensive subjects. Hypertension. 1996;28:392–396. doi: 10.1161/01.hyp.28.3.392. [DOI] [PubMed] [Google Scholar]
- 30.Fogari R, Zoppi A, Corradi L, Lazzari P, Mugellini A, Lusardi P. Comparative effects of lisinopril and losartan on insulin sensitivity in the treatment of non diabetic hypertensive patients. Br. J. Clin. Pharmacol. 1998;46:467–471. doi: 10.1046/j.1365-2125.1998.00811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fogari R, Zoppi A, Lazzari P, Preti P, Mugellini A, Corradi L, Lusardi P. ACE inhibition but not angiotensin II antagonism reduces plasma fibrinogen and insulin resistance in overweight hypertensive patients. J. Cardiovasc. Pharmacol. 1998;32:616–620. doi: 10.1097/00005344-199810000-00014. [DOI] [PubMed] [Google Scholar]
- 32.Fogari R, Zoppi A, Preti P, Fogari E, Malamani G, Mugellini A. Differential effects of ACE-inhibition and angiotensin II antagonism on fibrinolysis and insulin sensitivity in hypertensive postmenopausal women. Am. J. Hypertens. 2001;14:921–926. doi: 10.1016/s0895-7061(01)02140-9. [DOI] [PubMed] [Google Scholar]
- 33.Gillespie EL, White CM, Kardas M, Lindberg M, Coleman CI. The impact of ACE inhibitors or angiotensin II type 1 receptor blockers on the development of new-onset type 2 diabetes. Diabetes Care. 2005;28:2261–2266. doi: 10.2337/diacare.28.9.2261. [DOI] [PubMed] [Google Scholar]
- 34.Santoro D, Natali A, Palombo C, Brandi LS, Piatti M, Ghione S, Ferrannini E. Effects of chronic angiotensin converting enzyme inhibition on glucose tolerance and insulin sensitivity in essential hypertension. Hypertension. 1992;20:181–191. doi: 10.1161/01.hyp.20.2.181. [DOI] [PubMed] [Google Scholar]
- 35.Tikellis C, Wookey PJ, Candido R, Andrikopoulos S, Thomas MC, Cooper ME. Improved islet morphology after blockade of the renin–angiotensin system in the ZDF rat. Diabetes. 2004;53:989–997. doi: 10.2337/diabetes.53.4.989. [DOI] [PubMed] [Google Scholar]
- 36.Carlsson PO, Berne C, Jansson L. Angiotensin II and the endocrine pancreas: effects on islet blood flow and insulin secretion in rats. Diabetologia. 1998;41:127–133. doi: 10.1007/s001250050880. [DOI] [PubMed] [Google Scholar]
- 37.Ko SH, Kwon HS, Kim SR, Moon SD, Ahn YB, Song KH, Son HS, Cha BY, Lee KW, Son HY, et al. Ramipril treatment suppresses islet fibrosis in Otsuka Long–Evans Tokushima fatty rats. Biochem. Biophys. Res. Commun. 2004;316:114–122. doi: 10.1016/j.bbrc.2004.02.023. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki K, Nakagawa O, Aizawa Y. Improved early-phase insulin response after candesartan treatment in hypertensive patients with impaired glucose tolerance. Clin. Exp. Hypertens. 2008;30:309–314. doi: 10.1080/10641960802269927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 40.Haas JA, Krier JD, Bolterman RJ, Juncos LA, Romero JC. Low-dose angiotensin II increases free isoprostane levels in plasma. Hypertension. 1999;34:983–986. doi: 10.1161/01.hyp.34.4.983. [DOI] [PubMed] [Google Scholar]
- 41.Creager MA, Roddy MA. Effect of captopril and enalapril on endothelial function in hypertensive patients. Hypertension. 1994;24:499–505. doi: 10.1161/01.hyp.24.4.499. [DOI] [PubMed] [Google Scholar]
- 42.Koh KK, Han SH, Chung WJ, Ahn JY, Jin DK, Kim HS, Park GS, Kang WC, Ahn TH, Shin EK. Comparison of effects of losartan, irbesartan, and candesartan on flow-mediated brachial artery dilation and on inflammatory and thrombolytic markers in patients with systemic hypertension. Am. J. Cardiol. 2004;93:1432–1435. doi: 10.1016/j.amjcard.2004.02.050. [DOI] [PubMed] [Google Scholar]
- 43.Bonetti PO, Barsness GW, Keelan PC, Schnell TI, Pumper GM, Kuvin JT, Schnall RP, Holmes DR, Higano ST, Lerman A. Enhanced external counterpulsation improves endothelial function in patients with symptomatic coronary artery disease. J. Am. Coll. Cardiol. 2003;41:1761–1768. doi: 10.1016/s0735-1097(03)00329-2. [DOI] [PubMed] [Google Scholar]
- 44.Alvarez A, Cerda-Nicolas M, Naim Abu Nabah Y, Mata M, Issekutz AC, Panes J, Lobb RR, Sanz MJ. Direct evidence of leukocyte adhesion in arterioles by angiotensin II. Blood. 2004;104:402–408. doi: 10.1182/blood-2003-08-2974. [DOI] [PubMed] [Google Scholar]