

Abstract
Osteosarcoma (OS), a highly aggressive primary bone tumor, belongs to the most common solid tumors in growing children. Since specific molecular targets for OS treatment remain to be identified, surgical resection combined with multimodal (neo-)adjuvant chemotherapy is still the only way to help respective individuals. We have previously identified the protein tyrosine phosphatase Rptpζ as a marker of terminally differentiated osteoblasts, which negatively regulates their proliferation in vitro. Here we have addressed the question if Rptpζ can function as a tumor suppressor protein inhibiting OS development in vivo. We therefore analyzed the skeletal phenotype of mice lacking Ptprz1, the gene encoding Rptpζ on a tumor-prone genetic background, i.e. Trp53-heterozygosity. By screening a large number of 52 week old Trp53-heterozygous mice by contact radiography we found that Ptprz1-deficiency significantly enhanced OS development with 19% of the mice being affected. The tumors in Ptprz1-deficient Trp53-heterozygous mice were present in different locations (spine, long bones, ribs), and their OS nature was confirmed by undecalcified histology. Likewise, cell lines derived from the tumors were able to undergo osteogenic differentiation ex vivo. A comparison between Ptprz1-heterozygous and Ptprz1-deficient cultures further revealed that the latter ones displayed increased proliferation, a higher abundance of tyrosine-phosphorylated proteins and resistance towards the influence of the growth factor Midkine. Our findings underscore the relevance of Rptpζ as an attenuator of proliferation in differentiated osteoblasts and raise the possibility that activating Rptpζ-dependent signaling could specifically target osteoblastic tumor cells.
Introduction
Although OS represents the most prevalent primary bone tumor, its incidence is rather low, with less than 1:100.000 [1–3]. Nevertheless, since children are most commonly affected, OS belongs to the leading causes of cancer-related death within the pediatric age group, especially since the tumors are typically very aggressive with the ability to metastasize. This explains why a molecular understanding of OS development is highly relevant, also because conventional chemotherapy following surgical resection only leads to a 5-year survival rate below 70% [4–6]. The general concept of tumorigenesis, i.e. an accumulation of gene mutations affecting oncogenes and tumor suppressor genes, is most likely also valid for OS development, where the mesenchymal osteoprogenitors transform into a state of unlimited proliferation and subsequent bone formation [7,8]. Therefore, many attempts have been made to identify OS-relevant genes by genome-wide expression or sequencing analysis of OS tissue samples [9–18]. Although several candidates have been identified by these approaches, the establishment of a definite gene panel is still difficult, mostly explained by the large heterogeneity and the enhanced genomic instability of OS compared to many other tumors [19–21]. With respect to OS susceptibility, some loci were only recently identified by candidate or genome-wide association studies in large cohorts [22–25]. Moreover, the previously performed genetic analysis of individuals with inherited disorders characterized by high OS incidence has demonstrated that mutations of common tumor suppressor genes (TP53, RB1, RECQL4) also predispose to OS development [8]. One of these disorders is Li-Fraumeni syndrome, which can be caused by heterozygous germline mutations of TP53 and which is characterized by increased cancer risk with OS development in more than 10% of the affected individuals [26–28]. Consistent with the tumor suppressor function of TP53 in OS, somatic TP53 mutations were also identified in the majority of human OS tumors or cell lines [29–31]. Likewise, OS development was observed in mouse models with heterozygous or osteoblast-specific inactivation of Trp53, the murine homologue of TP53 [32–37].
In an attempt to identify markers for terminally differentiated osteoblasts we have previously performed genome-wide expression analysis comparing murine primary calvarial osteoblasts at a non-mineralized and a mineralized stage [38]. In addition to the known markers of osteoblast differentiation, that were all regulated as expected, we identified several genes with increased expression in mineralized cultures that were not previously analyzed in the context of bone remodeling. Of note, the gene displaying the strongest level of induction in this particular experiment was Ptprz1, encoding the transmembrane protein tyrosine phosphatase Rptpζ [39]. Since many growth factors activate cellular proliferation by inducing tyrosine phosphorylation of intracellular signaling proteins, this finding suggested that Rptpζ could negatively regulate osteoblast differentiation, thus being responsible to induce a postmitotic state in terminally differentiated mineralizing osteoblast cultures [40]. This hypothesis was supported by ex vivo experiments, where we took advantage of a previously established Ptprz1-deficient mouse model, which does not display an obvious phenotype in a non-challenged situation [41]. Of note, although Ptprz1-deficient mice only displayed a moderate osteopenia at 1 year of age, primary osteoblasts derived from newborn Ptprz1-deficient mice had an increased proliferation rate compared to wildtype cultures [38]. Likewise, transfection of an Rptpζ expression plasmid into MC3T3-E1 osteoblasts significantly reduced their proliferation rate in a dose-dependent manner.
Although these experiments demonstrated that Rptpζ negatively regulates osteoblast proliferation, we did not observe spontaneous OS development in Ptprz1-deficient mice until the age of 18 months. Since it is well established however that tumor development requires an accumulation of several gene mutations, we hypothesized that a potential OS suppressor function of Rptpζ can only be uncovered in the context of an already existing mutation of a common tumor suppressor gene [42]. We therefore crossed Ptprz1-deficient mice into a Trp53-heterozygous background and monitored OS development by screening through contact Xray. Here we found that OS development in 12 month old Trp53-heterozygous mice was significantly enhanced by Ptprz1-deficiency, thereby providing evidence for a tumor suppressor function of Rptpζ.
Materials and Methods
1. Expression analysis
RNA was isolated from various tissues of 6 week old mice and from primary osteoblasts at various stages of differentiation. More specifically, these cells were isolated by sequential collagenase digestion from the calvariae of 5 days old mice as described [43]. After removal of the co-purified macrophage-like cells by CD11b-immunoaffinity [44,45] cells were plated and cultured in α-MEM until they reached 80% confluency (day 0). We then added ascorbic acid (50 μg/ml) and ß-glycerophosphate (10 mM) to the cultures to induce osteogenic differentiation for 4, 7, 10, 15, 20 or 25 days. RNA isolation was performed with the RNeasyMini kit (Qiagen). For osteoclastogenesis bone marrow cells were isolated from 12 week old mice and induced to differentiate into osteoclasts by addition of 1,25-vitamin D3, M-CSF and RANKL as described [45]. We also isolated RNA from human osteoblasts (PromoCell #C-12720) and human OS cell lines (SaOS-2, ATCC #HTB-85; U2-OS, ATCC #HTB-96), all of them differentiated for 0, 7 or 15 days by using the commercially available osteoblast mineralization medium (PromoCell #C27020) or by addition of ascorbic acid (50 μg/ml), ß-glycerophosphate (10 mM) and dexamethasone (100 nM) to the culture medium. Concentration and quality of RNA were measured using a NanoDrop ND-1000 system (NanoDrop Technology). For qRT-PCR expression analysis, 1 μg of RNA was reversed transcribed using SuperScriptIII (Invitrogen) according to manufacturer’s instructions. Reactions were performed using predesigned TaqMan gene expression assays (Applied Biosystems) with Gapdh as internal control. We additionally isolated RNA from CD11b-negative osteoblast cultures at day 5 and day 12 of differentiation to perform genome-wide expression analysis using Affymetrix Gene Chips (Affymetrix MG 430 2.0). The respective data sets have been deposited in GEO under accession code GSE71565.
2. Mouse models
Trp53-deficient mice were purchased from the Jackson Laboratories (#002101). Their genotyping was performed with the primers 5´-ACA GCG TGG TGG TAC CTT AT-3´, 5´-TAT ACT CAG AGC CGG CCT-3´, and 5´-CTA TCA GGA CAT AGC GTT GG-3´, giving rise to a 450 bp and a 650 bp fragment for the wildtype and mutant allele, respectively. Ptprz1-deficient mice have been described previously [38]. Their genotyping was performed with the primers 5´-AGA TCC ATT CGT CTT GCA GCC TCC-3´, 5´-CAC CTG CCT GGA AAA CTT GTA CTG-3´, 5´-GAA AAG CGC CTC CCC TAC CCG GTA GAA TTG AC-3´ and 5´-CCA GAC ATG ACA CCC CAA TGC CTG AAC ATC TC-3´, giving rise to a 400 bp and a 650 bp fragment for the wildtype and mutant allele, respectively. To exclude any possible influence of genetic background, all analyses were performed with littermates obtained from compound heterozygous matings.
3. Skeletal analysis
After sacrifice all skeletons were immediately analyzed by contact radiography using a Faxitron Xray cabinet (Faxitron Xray Corp.). For further analysis the dissected skeletons were fixed in 3.7% PBS-buffered formaldehyde for 18 hours at 4°C, before they were stored in 80% ethanol. μCT analysis was performed with a μCT 40 (Scanco Medical). For histology, specific skeletal elements were dehydrated in ascending alcohol concentrations and then embedded in methylmetacrylate as described previously [43]. Sections of 5 μm thickness were cut in the sagittal plane on a Microtec rotation microtome (Techno-Med GmbH). All sections were stained by von Kossa/van Gieson or toluidine blue staining procedures as described [43].
4. Characterization of murine OS cell lines
For ex vivo OS cell culture, tumor tissues were dissected and cleaned in PBS containing antibiotics. Tumors were then minced with a scissor and placed into a culture dish in the presence of α-MEM (including 10% FBS and 1% Penicillin/Streptomycin). When the outgrowing cells reached 80–90% confluency, cells were trypsinized and plated onto new culture dishes without tissue residues. OS cell lines were established by repeated passaging (more than 12 times) under regular tissue culture conditions. Osteogenic differentiation was induced by adding ascorbic acid (50 μg/ml) and ß-glycerophosphate (10 mM) to the cultures. After 10 and 20 days we assessed matrix mineralization by alizarin red incorporation as described [45]. To monitor the proliferation rate cells were seeded in triplicate cultures at an initial density of 20.000 cells per well of a 6-well-plate and counted every day using a Neubauer chamber. Additionally we measured BrdU incorporation using the Biotrak Cell Proliferation Kit (Amersham) according to the manufacturer's instructions. To assess the proliferative behavior in the presence of Midkine (Mdk) we performed short-term treatment with increasing concentrations of murine recombinant Mdk (Peprotech) in serum-free medium. After 12 hours of incubation BrdU incorporation was determined as described above. The effects of long-term administration were analyzed by monitoring cell growth for seven days in the presence or absence of 100 ng/ml Mdk.
5. Far Western Blotting with SH2-domains
OS cell lines were deprived of serum for 5 hours in α-MEM and re-stimulated with 10% FBS for 5 minutes. For whole cellular protein extraction cells were washed two times with ice-cold PBS and immediately lysed in KLB-buffer (150 mN NaCl, 25 mM TrisHCL (pH 7.4), 5 mM EDTA, 1% Triton X-100, 10% glycerol, 10 mM sodium pyrophosphate, 1 mM sodium ortho-vanadate, 10 mM ß-glycerolphosphate, 1 mM PMSF, 0.2 mg/ml of aprotinin (Sigma), 10 mM NaF, 0.1 mM freshly prepared sodium pervanadate and 1 mM DTT). After incubation on ice for 30 min cellular lysates were cleared by centrifugation at 4°C for 10 min and supernatants were stored at -80°C. SH2 profiling based on far-Western blotting was essentially performed as described previously [46,47]. Briefly, 15 μg of OS lysates were separated by SDS-PAGE (4–12% acrylamide gels) and transferred to PVDF membranes. After blocking (10% skim milk in TBST-buffer), replicate blots were separately probed with 1 μg/ml of biontinylated SH2 domains precomplexed with streptavidin-horseradish peroxidase. Signals were visualized by chemoluminescence using ECL blotting reagent system (Amersham).
6. Site-directed mutagenesis and DNA transfection in human OS cell lines
To determine the function of Rptpζ as a negative regulator of osteoblast proliferation, we introduced two mutations into the previously described Rptpζ expression plasmid [38]. The first mutation (NM_001206838.1:exon21:c.G3218T:p.C1073S) is located in the active site of Rptpζ and should destroy its ability to desphosphorylate specific substrates [48]. The second mutation (NM_001206838.1:exon22:c.G3227T:p.G1076V) was identified by analysis of exome sequencing data sets from 17 human OS samples. Both mutations were introduced using the QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Stratagene) according to manufacturer’s instructions. For transfections, the human SaOS-2 and U2-OS cells were seeded into 96-well-plates at an initial density of 1000 cells per well. Cells were cultured to 50–70% confluence and transfected with 0.5 μg of wildtype and/or mutant Rptpζ expression plasmids using the Lipofectamine™ 2000 reagent (Invitrogen) according to manufacturer’s instructions. 48 h after transfection BrdU incorporation was measured as described above.
7. Statistical analysis
All data presented in the manuscript were obtained from the analysis of littermates (n ≥ 5) and are presented as means ± standard deviations. Statistical analyses were performed by by the method of Kruskal and Wallis followed by Dunn´s post-test, by Fishers`s exact test, by one-way or two-way ANOVA followed by Dunnett’s or Bonferroni’s post-test (after estbalishing normality and homoscedasticity), or by unpaired, two-tailed Student’s t-test, as indicated in the figure legends, using the commercial software GraphPad Prism5 (version 5.02). P-values below 0.05 were considered statistically significant.
8. Ethics Statement
All animal experiments were approved by the animal facility of the University Medical Center Hamburg Eppendorf and by the “Amt für Gesundheit und Verbraucherschutz” (Org529).
Results
Since our previously published genome-wide expression analysis was performed with primary murine osteoblasts still containing the co-purifying macrophage population, we first analyzed, if Ptprz1 is also differentially expressed in primary osteoblast cultures after depletion of the macrophage population by CD11b immunoaffinity [44]. For that purpose a comparative genome-wide expression analysis was performed with CD11b-negative osteoblasts at day 5 and day 12 of differentiation. By sorting all genes according to the logarithmic ratio of signal intensity (SLR, signal log ratio) we found that the expression of well-established osteoblast markers increased during the course of differentiation (Table 1). Importantly, the same was the case for Ptprz1, whereas other genes encoding protein tyrosine phosphatases were not differentially expressed to the same extent (Table 2). We next monitored Ptprz1 expression in various tissues and primary bone cells from wildtype mice by qRT-PCR. Among tissues we detected the highest expression levels in brain, as well as in femoral and calvarial bone (Fig 1A). Ptprz1 expression was also detectable in lung, but not in fat, heart, kidney, liver and spleen. To identify the primary bone cell type expressing Ptprz1 we next analyzed primary calvarial osteoblasts and primary bone marrow-derived osteoclasts at different stages of differentiation. Here we found that Ptprz1 expression progressively increased during the course of osteoblast differentiation, while expression in osteoclast cultures was barely detectable (Fig 1B–1D). These data demonstrate that Ptprz1 represents an osteoblast differentiation marker, whose expression is restricted to few tissues.
Table 1. Genes encoding osteogenesis markers displaying differential expression during primary osteoblast differentiation.
Given are the Affymetrix signal intensities at two stages of differentiation and the signal log ratios (SLR).
| Gene | day 5 | day 12 | SLR |
|---|---|---|---|
| Mepe | 1.5 | 301.8 | 7.0 |
| Dkk1 | 14.5 | 685.1 | 5.6 |
| Ibsp | 157.8 | 5959.9 | 5.0 |
| Dmp1 | 154 | 3103.2 | 4.4 |
| Phex | 35.6 | 554.7 | 3.9 |
| Ifitm5 | 186.0 | 2244.3 | 3.7 |
| Bglap | 646.7 | 6611.7 | 3.5 |
Table 2. Genes encoding PTPs displaying differential expression during primary osteoblast differentiation.
Given are the Affymetrix signal intensities at two stages of differentiation and the signal log ratios (SLR).
| Gene | day 5 | day 12 | SLR |
|---|---|---|---|
| Ptprz1 | 18.7 | 590.3 | 5.1 |
| Ptpro | 15.7 | 54.2 | 1.8 |
| Ptprc | 60.9 | 134.1 | 1.2 |
| Ptpn18 | 47.0 | 14.1 | 1.2 |
| Ptpn20 | 25.9 | 64.6 | 1.2 |
| Ptprt | 5.2 | 10.7 | 1.1 |
| Ptpn6 | 42.2 | 53.4 | 0.9 |
| Ptpn2 | 1219.1 | 1735.0 | 0.4 |
Fig 1. Ptprz1 expression by differentiated osteoblasts.

(A) qRT-PCR monitoring Ptprz1 expression in brain (Br), femur (Fe), calvaria (Ca), fat (Fa), heart (He), kidney (Ki), liver (Li), lung (Lu) and spleen (Sp). (B) qRT-PCR monitoring Ptprz1 expression in primary osteoblasts (Obl) or osteoclasts (Ocl) at different stages of differentiation. Bars represent mean ± SD (n = 3). (C) qRT-PCR monitoring expression of the osteocyte marker Phex. (D) qRT-PCR monitoring expression of the osteoclast marker Acp5 (encoding TRAP). Values represent copy number (cn) relative to Gadph. Bars represent mean ± SD (n = 3). Asterisks indicate statistical significance vs. Obl day 0 (p<0.05, Kruskal-Wallis followed by Dunn’s post-test).
As we have previously established that Rptpζ negatively regulates osteoblast proliferation in vitro [38], we next addressed the question, whether its inactivation would trigger OS development. Since Ptprz1-deficiency alone does not result in the presence of OS (until the age of 18 months), we first generated mice lacking both, Ptprz1 and Trp53, and compared them to Trp53-deficient littermates. Given the early lethality associated with Trp53-deficiency, mostly explained by lymphomas [32], we performed the skeletal analysis at 12 weeks of age. Using contact Xray we did not detect any signs of OS development in Trp53-deficient mice, regardless of the presence or absence of Ptprz1 (S1 Fig). The same was true for undecalcified sections from the spine or the tibia of the corresponding mice.
Since Trp53-heterozygous mice display reduced lymphoma development and therefore increased life span, we went on to generate Ptprz1-deficient Trp53-heterozygous mice (Trp53 +/- /Ptprz1 -/-) and compared them to Trp53-heterozygous mice (Trp53 +/- /Ptprz1 +/+) and to Trp53-heterozygous mice lacking one Ptprz1 allele (Trp53 +/- /Ptprz1 +/-). All animals were analyzed at 52 weeks of age using contact Xray to screen for the presence of OS, unless OS development was observed earlier (S1 Table). While none of the 24 Trp53 +/- /Ptprz1 +/+ mice displayed obvious skeletal tumors, OS development was detected in 7 out of 38 Trp53 +/- /Ptprz1 -/- mice (Fig 2A). In Trp53 +/- /Ptprz1 +/- mice skeletal tumors were visible in 2 out of the 38 mice that were analyzed. The skeletal tumors found in Trp53 +/- /Ptprz1 -/- and Trp53 +/- /Ptprz1 +/- mice were present in three different locations (Fig 2B), i.e. ribs, spine or long bones, but not in craniofacial bones, and μCT analysis demonstrated that they consisted of mineralized tissue (Fig 2C). Taken together, these data provide strong evidence for a tumor suppressor function of Rptpζ, as the OS incidence in 52 week old Trp53-heterozygous mice was raised from 0% to 19% by Ptprz1-deficiency.
Fig 2. Skeletal tumors in Ptprz1-deficient Trp53-heterozygous mice.

(A) OS development assessed by screening of 12 month old Trp53-heterozygous mice with the indicated Ptprz1 genotypes. The left panel shows the number of analyzed mice (white bars) and the percentage of mice with OS (black bars). The asterisk indicates statistical significance vs. Trp53 +/- /Ptprz1 +/+ (p<0.05, two-tailed Fishers`s exact test). The right panel shows the total number of tumors and their location. (B) Representative contact Xrays from Ptprz1-deficient Trp53-heterozygous mice with OS in the three different locations. (C) μCT images from the same tumors.
To confirm that the tumors represented OS we further applied undecalcified histology (Fig 3A). By toluidine blue staining of proteoglycans we were able to rule out that the tumors are chondrogenic (Fig 3B), which was also supported by the fact, that their mineralized matrix contained osteocytes (Fig 3C). We additionally isolated OS cell lines from two different tumors (Trp53 +/- /Ptprz1 +/- and Trp53 +/- /Ptprz1 -/-) and cultured them in the presence of ascorbic acid and ß-glycerophosphate. Here we found that both cell lines were able to form a mineralized matrix ex vivo (Fig 4A). Moreover, they both displayed differential expression of osteoblast differentiation markers (Fig 4B–4D), thereby confirming the OS nature of the tumors.
Fig 3. OS nature of skeletal tumors in Ptprz1-deficient Trp53-heterozygous mice.

(A) Von Kossa/Van Gieson staining of undecalficied sections confirms that the tumors contain mineralized matrix (stained black). (B) Toluidine blue staining demonstrating dark blue staining of cartilage areas and light blue staining of the tumors. (C) Higher magnification images reveal that the tumors represented bony tissue with osteocytes embedded into the mineralized matrix.
Fig 4. Osteogenic differentiation of OS cell lines.

(A) Alizarin red staining of cells derived from Trp53-heterozygous mice with either one Ptprz1 allele (+/-) or with Ptprz1-deficiency (-/-) reveals that both cell lines are able to form a mineralized matrix after 10 and 20 days of differentiation induced by ascorbic acid and ß-glycerophosphate. (B-D) qRT-PCR expression analysis shows that the differentiation is accompanied by increased expression of Bglap (encoding Osteocalcin), Ibsp (encoding Bone sialoprotein) and Phex. Values represent copy number (cn) relative to Gadph. Bars represent mean ± SD (n = 3). Asterisks indicate significant differences towards day 0 of corresponding genotype (p<0.05, Kruskal-Wallis followed by Dunn’s post-test).
We additionally compared the ex vivo behaviour of the Trp53 +/- /Ptprz1 +/- and Trp53 +/- /Ptprz1 -/- OS cell lines with respect to proliferation and tyrosine phosphorylation status. We observed that the cell line deficient in Rptpζ proliferated at higher rate, which was also confirmed by BrdU incorporation assays (Fig 5A). We further assessed differences in the state of tyrosine phosphorylation between the Trp53 +/- /Ptprz1 +/- and Trp53 +/- /Ptprz1 -/- OS cell lines using far-Western Blotting with different SH2-domains [46,47]. Here we observed that several proteins were specifically detected in Trp53 +/- /Ptprz1 -/- cells with the most obvious differences in the case of SH2-domains of ABL2, CRK, NCK2, Pi3KN and SRC (Fig 5B, S2 Fig). In addition, we analyzed the response of both cell lines to the heparin-binding growth factor Mdk, which has been shown to antagonize Rptpζ in other cell types [49,50]. Using BrdU incorporation assays we found that Mdk increased the proliferation in Trp53 +/- /Ptprz1 +/- cells in a dose-dependent manner, whereas Trp53 +/- /Ptprz1 -/- cells did not respond (Fig 6A). Likewise, long-term administration of Mdk for 7 days significantly enhanced cellular growth only in Trp53 +/- /Ptprz1 +/- cells (Fig 6B). These data demonstrate that the pro-proliferative influence of Mdk on OS cell lines depends on Rptpζ interaction, which is potentially relevant, as MDK was found over-expressed in human OS [51,52].
Fig 5. Proliferation and tyrosine phosphorylation in OS cell lines.

(A) Proliferative capacity of tumor cells derived from Trp53-heterozygous mice with either one Ptprz1 allele (+/-) or with Ptprz1-deficiency (-/-). The growth curves (left) and the BrdU incorporation assays (right) demonstrate increased proliferation in the cases of Ptprz1-deficiency. Bars represent mean ± SD (n≥3). Asterisks indicate significant differences between the two genotypes (p<0.05, two-way ANOVA followed by Bonferroni’s post-test (left panel) or two-tailed Student’s t-test (right panel)). (B) SH2 profiling with different SH2 domains reveals differences in tyrosine phosphorylation of specific proteins (indicated by arrowheads). Re-probing of stripped membranes with anti ß-actin mAb served as control for equal loading. Analyses were performed in duplicate with cell extracts harvested at different cell densities.
Fig 6. Effects of Mdk on proliferation of OS cell lines.
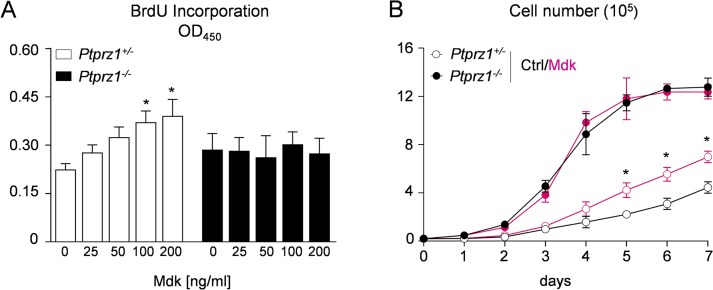
(A) BrdU incorporation assays with the indicated OS cell lines performed in the presence of Mdk at different concentrations. Bars represent mean ± SD (n = 8). Asterisks indicate significant differences towards controls without Mdk (p<0.05, one-way ANOVA followed by Dunnett’s post-test). (B) Growth curves of OS cell lines in the presence or absence of Mdk (100 ng/ml). Asterisks indicate significant differences towards controls of Ptprz1 +/- cells without Mdk (p<0.05, two-way ANOVA followed by Bonferroni’s post-test).
To address the question if Rptpζ would act as a tumor suppressor in human OS we first analyzed, if PTPRZ1 is differentially expressed during differentiation of human osteoblasts. We therefore induced osteogenic differentiation of commercially available primary osteoblasts and of two OS cell lines (SaOS-2 and U2-OS). Here we found that PTPRZ1 expression was increased compared to non-differentiated cells after 15 days in all three cell types, thereby confirming its differential expression in human osteoblasts (Fig 7A). We next analyzed exome sequencing data sets from 17 human OS samples, thereby identifying a heterozygous germline mutation (G3227T) in exon 22 of the PTPRZ1 gene in one of the cases. Since the mutation causes an amino acid substitution (G1076V) within the first protein tyrosine phosphatase domain, we hypothesized that it might interfere with Rptpζ activity. To address this possibility we introduced the corresponding mutation into an Rptpζ expression plasmid [38], and we additionally introduced another mutation (C1073S) within the active site of Rptpζ, serving as a control (Fig 7B). The mutant plasmids were then transfected into non-differentiated SaOS-2 and U2-OS cells, either alone or in combination with intact Rptpζ expression plasmid, before assessing proliferation by BrdU incorporation assays. In both cell lines we found that Rptpζ significantly reduces BrdU incorporation, thereby confirming its role as an attenuator of proliferation also in human osteoblasts (Fig 7C). As expected, the anti-proliferative activity of Rptpζ was fully abolished by the C1073S mutation, which also acted in a dominant negative fashion. Most importantly however, introducing the G1076V mutation did not interfere with the anti-proliferative function of Rptpζ, thereby essentially ruling out its contribution to OS development in the identified individual carrying this mutation in a heterozygous state.
Fig 7. Functional analysis of Rptpζ mutations.

(A) qRT-PCR monitoring expression of BGLAP (left) and PTPRZ1 (right) in human primary osteoblasts (hObl), SaOS-2 or U2-OS cells at different stages of differentiation. Values represent copy number (cn) relative to GAPDH. Bars represent mean ± SD (n = 3). Asterisks indicate statistical significance vs. day 0 (p<0.05, Kruskal-Wallis followed by Dunn’s post-test). (B) Schematic presentation of the PTPRZ1 gene and Rptpζ protein showing the location of the two mutations that have been introduced into a Rptpζ expression plasmid. (C) BrdU incorporation assay with SaOS-2 and U2-OS cells after transfection of wildtype and/or mutant Rptpζ expression plasmids as indicated. Bars represent mean ± SD (n = 6). Asterisks indicate significant differences towards cells transfected with empty vector (ctrl) (p<0.05, one-way ANOVA followed by Dunnett’s post-test).
Discussion
Rptpζ, encoded by the Ptprz1 gene, is primarily expressed in the central nervous system, mostly by astrocytes and oligodendrocytes [53]. Although Ptprz1-deficient mice do not display an obvious phenotype in a non-challenged situation, they were found to have impaired recovery from demyelinating lesions, supporting a role of Rptpζ in oligodendrogenesis [54]. In its full-length form Rptpζ represents a transmembrane protein with two intracellularly located protein tyrosine phosphatase domains [55]. The extracellular part consists of an N-terminal carbonic anhydrase-like domain followed by a fibronectin type III repeat and a large intervening sequence. Importantly, the latter domain is posttranslationally modified by glycoaminglycan attachment, and a soluble form of Rptpζ, also known as phosphacan, lacking the transmembrane and tyrosine phosphatase domains, represents one of the most abundant proteoglycans in the brain [56,57]. With respect to Rptpζ-specific ligands it was found that two heparin-binding growth factors, Pleiotrophin and Mdk, interact with the glycosaminoglycan structures of Rptpζ, thereby inhibiting the tyrosine phoshatase activity of Rptpζ [49,50,58,59]. In terms of OS development, the Mdk/Rptpζ interaction appears particularly interesting, since MDK was found over-expressed in human OS and positively regulated proliferation of human OS cell lines [51,52]. With respect to a possible tumor suppressor function of Rptpζ it was recently shown that its shRNA-mediated repression in prostate cancer cell lines did not only increase the migratory behavior of these cells in vitro, but also enhanced their metastatic potential after injection into nude mice [60]. The putative role of Rptpζ as a suppressor of OS development however, has not been studied so far.
Since we identified Ptprz1 as the only PTP-encoding gene displaying robust differential expression during primary osteoblast differentiation, where it is required to inhibit proliferation, we addressed the question, if Ptprz1-deficiency would increase OS incidence on a tumor-prone genetic background. Since we did not observe OS development in 12 week old mice lacking Trp53 and Ptprz1, we analyzed OS development in Trp53-heterozygous mice, which were monitored by Xray analysis at 12 months of age. One limitation of this screening approach was that we did not necessarily pick up smaller foci with OS characteristics in specific skeletal elements. We therefore cannot fully rule out that such transformation processes occurred in a subset of Trp53-heterozygous mice, as it has been reported previously [45]. On the other hand, the Xray screening allowed us to analyze the full skeletons from a large number of animals, and it was the purpose of our study to detect larger tumors, similar to the previously analyzed mouse model over-expressing the cFos proto-oncogene [61,62]. Especially since our approach immediately identified increased OS susceptibility in Ptprz1-deficient Trp53-heterozygous mice, we did not change our strategy and went on to characterize the tumors by undecalficied histology and ex vivo assays. Using these methods we could demonstrate that Ptprz1-deficiency raised the OS incidence in 52 weeks old Trp53-heterozygous mice from 0% to 19%, thereby providing evidence for a tumor suppressor function of Rptpζ, at least in mice.
To address the question, if Rptpζ would also act as a tumor suppressor in human OS, we first analyzed copy number data sets (CytoScan Arrays) from 160 human OS samples for chromosomal alterations around the PTPRZ1 locus on chromosome 7q31.3, yet we did not detect any specific changes supporting a contribution of PTPRZ1 loss to OS development or severity. We additionally analyzed exome sequencing data sets from 17 human OS samples, thereby identifying only one heterozygous germline mutation in the PTPRZ1 gene, which did however not interfere with the anti-proliferative activity of the Rptpζ protein. We additionally searched the COSMIC database for mutations in PTP-encoding genes previously identified in human OS samples [63]. Consistent with the absence of published data reporting mutations or epigenetic silencing of PTPRZ1 in OS cases, the only identified mutation in a PTP-encoding gene was not affecting PTPRZ1, but PTPRT (COSM1732508AA). Especially this latter observation led us to address the question, if PTPRZ1 is differentially expressed in human osteoblasts, or if Rptpρ, encoded by the PTPRT gene, could be the most relevant phosphatase controlling proliferation of human osteoblasts. We therefore induced osteogenic differentiation of primary osteoblasts, SaOS-2 and U2-OS and found that PTPRZ1 expression followed the same kinetic as observed in primary murine osteoblasts. In contrast, while Ptprt expression was barely and non-differentially expressed in murine cultures, PTPRT transcripts were undetectable in human osteoblasts. These data, together with the reduced proliferation of OS cell lines after transfection of a Rptpζ expression plasmid, imply that Rptpζ has a similar role in human and murine osteoblasts. Nevertheless, the absence of evidence supporting a role of Rptpζ as a human tumor suppressor gene, essentially demonstrates that it is not a major player in human OS development.
On the other hand, since there is evidence for MDK over-expression in human OS, it cannot be fully ruled out Rptpζ, non-mutated and normally expressed, is involved in OS development as a putative Mdk receptor. This is why it is potentially relevant that we were able to provide further evidence for a function of Mdk as an Rptpζ antagonist, by showing that the proliferative influence of Mdk on Trp53 +/- OS cell lines was abrogated by Ptprz1-deficiency. In our opinion, it would additionally be useful to identify osteoblast-specific substrates dephosphorylated by Rptpζ and controlling proliferation. For now we did not succeed to address this question with a candidate approach, since we did not detect specific signals with antibodies against some phosphorylated proteins in Ptprz1-deficient OS cell lines. More specifically, the tested candidates included Fyn, Git-1 (both undetectable in the non-phosphorylated form), Src (pY416), Fak (pY397, pY567/577, pY925), and p42/44-Mapk (pT202/Y204). Therefore, it appears that the identification of osteoblast-specific Rptpζ downstream mediators requires utilization of unbiased approaches, such as substrate trapping. Even though there is no evidence so far supporting a role Rptpζ or its putative substrates in human OS development, it is quite relevant to state that Rptpζ is a transmembrane protein with restricted expression pattern. This implies that it is principally targetable for treatment, for instance by antagonizing its interaction with Mdk.
Supporting Information
Contact Xrays and histological analysis of undecalcified spine or tibia sections from 12 weeks old mice of the indicated genotypes. No OS development was observed in either analysis.
(TIF)
Shown are the profiles after probing the membranes with additional SH2 domains as indicated.
(TIF)
(PDF)
Acknowledgments
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (AM103/10-3).
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (AM103/10-3; www.dfg.de). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Kuijjer ML, Hogendoorn PC, Cleton-Jansen AM. (2013) Genome-wide analyses on high-grade osteosarcoma: making sense of a genomically most unstable tumor. Int J Cancer 133: 2512–2521. 10.1002/ijc.28124 [DOI] [PubMed] [Google Scholar]
- 2. Lietman SA, Joyce MJ. (2010) Bone sarcomas: Overview of management, with a focus on surgical treatment considerations. Cleve Clin J Med 77: S8–S12. 10.3949/ccjm.77.s1.02 [DOI] [PubMed] [Google Scholar]
- 3. Pakos EE, Nearchou AD, Grimer RJ, Koumoullis HD, Abudu A, Bramer JA, et al. (2009) Prognostic factors and outcomes for osteosarcoma: an international collaboration. Eur J Cancer 45: 2367–2375. 10.1016/j.ejca.2009.03.005 [DOI] [PubMed] [Google Scholar]
- 4. Bacci G, Bertoni F, Longhi A, Ferrari S, Forni C, Biagini R, et al. (2003) Neoadjuvant chemotherapy for high-grade central osteosarcoma of the extremity. Histologic response to preoperative chemotherapy correlates with histologic subtype of the tumor. Cancer 97: 3068–3075. [DOI] [PubMed] [Google Scholar]
- 5. Allison DC, Carney SC, Ahlmann ER, Hendifar A, Chawla S, Fedenko A, et al. (2011) A meta-analysis of osteosarcoma outcomes in the modern medical era. Sarcoma 2012: 704872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anninga JK, Gelderblom H, Fiocco M, Kroep JR, Taminiau AH, Hogendoorn PC, et al. (2011) Chemotherapeutic adjuvant treatment for osteosarcoma: where do we stand? Eur J Cancer 47: 2431–2445. 10.1016/j.ejca.2011.05.030 [DOI] [PubMed] [Google Scholar]
- 7. Botter SM, Neri D, Fuchs B. (2014) Recent advances in osteosarcoma. Curr Opin Pharmacol 16C: 15–23. [DOI] [PubMed] [Google Scholar]
- 8. Kansara M, Teng MW, Smyth MJ, Thomas DM. (2014) Translational biology of osteosarcoma. Nat Rev Cancer 14: 722–735. 10.1038/nrc3838 [DOI] [PubMed] [Google Scholar]
- 9. Sadikovic B, Yoshimoto M, Chilton-MacNeill S, Thorner P, Squire JA, Zielenska M. (2009) Identification of interactive networks of gene expression associated with osteosarcoma oncogenesis by integrated molecular profiling. Hum Mol Genet 18: 1962–1975. 10.1093/hmg/ddp117 [DOI] [PubMed] [Google Scholar]
- 10. Kuijjer ML, Rydbeck H, Kresse SH, Buddingh EP, Lid AB, Roelofs H, et al. (2012) Identification of osteosarcoma driver genes by integrative analysis of copy number and gene expression data. Genes Chromosomes Cancer 51: 696–706. 10.1002/gcc.21956 [DOI] [PubMed] [Google Scholar]
- 11. Kansara M, Tsang M, Kodjabachian L, Sims NA, Trivett MK, Ehrich M, et al. (2009) Wnt inhibitory factor 1 is epigenetically silenced in human osteosarcoma, and targeted disruption accelerates osteosarcomagenesis in mice. J Clin Invest 119: 837–851. 10.1172/JCI37175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sadikovic B, Yoshimoto M, Al-Romaih K, Maire G, Zielenska M, Squire JA. (2008) In vitro analysis of integrated global high-resolution DNA methylation profiling with genomic imbalance and gene expression in osteosarcoma. PLoS One 3: e2834 10.1371/journal.pone.0002834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kresse SH, Rydbeck H, Skårn M, Namløs HM, Barragan-Polania AH, Cleton-Jansen AM, et al. (2012) Integrative analysis reveals relationships of genetic and epigenetic alterations in osteosarcoma. PLoS One 7: e48262 10.1371/journal.pone.0048262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Man TK, Lu XY, Jaeweon K, Perlaky L, Harris CP, Shah S, et al. (2004) Genome-wide array comparative genomic hybridization analysis reveals distinct amplifications in osteosarcoma. BMC Cancer 4: 45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang J, Yang D, Sun Y, Sun B, Wang G, Trent JC, et al. (2011) Genetic amplification of the vascular endothelial growth factor (VEGF) pathway genes, including VEGFA, in human osteosarcoma. Cancer 117: 4925–4938. 10.1002/cncr.26116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kresse SH, Ohnstad HO, Paulsen EB, Bjerkehagen B, Szuhai K, Serra M, et al. (2009) LSAMP, a novel candidate tumor suppressor gene in human osteosarcomas, identified by array comparative genomic hybridization. Genes Chromosomes Cancer 48: 679–693. 10.1002/gcc.20675 [DOI] [PubMed] [Google Scholar]
- 17. Lockwood WW, Stack D, Morris T, Grehan D, O'Keane C, Stewart GL, et al. (2011) Cyclin E1 is amplified and overexpressed in osteosarcoma. J Mol Diagn 13: 289–296. 10.1016/j.jmoldx.2010.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pasic I, Shlien A, Durbin AD, Stavropoulos DJ, Baskin B, Ray PN, et al. (2010) Recurrent focal copy-number changes and loss of heterozygosity implicate two noncoding RNAs and one tumor suppressor gene at chromosome 3q13.31 in osteosarcoma. Cancer Res 70: 160–171. 10.1158/0008-5472.CAN-09-1902 [DOI] [PubMed] [Google Scholar]
- 19. Cleton-Jansen AM, Anninga JK, Briaire-de Bruijn IH, Romeo S, Oosting J, Egeler RM, et al. (2009) Profiling of high-grade central osteosarcoma and its putative progenitor cells identifies tumourigenic pathways. Br J Cancer 101: 1909–1918. 10.1038/sj.bjc.6605405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klein MJ, Siegal GP. (2006) Osteosarcoma: anatomic and histologic variants. Am J Clin Pathol 125: 555–581. [DOI] [PubMed] [Google Scholar]
- 21. Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, et al. (2011) Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144: 27–40. 10.1016/j.cell.2010.11.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Savage SA, Mirabello L. (2011) Using epidemiology and genomics to understand osteosarcoma etiology. Sarcoma 2011: 548151 10.1155/2011/548151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Musselman JR, Bergemann TL, Ross JA, Sklar C, Silverstein KA, Langer EK, et al. (2012) Case-parent analysis of variation in pubertal hormone genes and pediatric osteosarcoma: a Children's Oncology Group (COG) study. Int J Mol Epidemiol Genet 3: 286–293. [PMC free article] [PubMed] [Google Scholar]
- 24. Savage SA, Mirabello L, Wang Z, Gastier-Foster JM, Gorlick R, Khanna C, et al. (2013) Genome-wide association study identifies two susceptibility loci for osteosarcoma. Nat Genet 45: 799–803. 10.1038/ng.2645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mirabello L, Yeager M, Mai PL, Gastier-Foster JM, Gorlick R, Khanna C, et al. (2015) Germline TP53 Variants and Susceptibility to Osteosarcoma. J Natl Cancer Inst 107(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Malkin D, Li FP, Strong LC, Fraumeni JF Jr, Nelson CE, Kim DH, et al. (1990) Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250: 1233–1238. [DOI] [PubMed] [Google Scholar]
- 27. Srivastava S, Zou ZQ, Pirollo K, Blattner W, Chang EH. (1990) Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature 348: 747–749. [DOI] [PubMed] [Google Scholar]
- 28. Kleihues P, Schäuble B, zur Hausen A, Estève J, Ohgaki H. (1997) Tumors associated with p53 germline mutations: a synopsis of 91 families. Am J Pathol 150: 1–13. [PMC free article] [PubMed] [Google Scholar]
- 29. Gokgoz N, Wunder JS, Mousses S, Eskandarian S, Bell RS, Andrulis IL. (2001) Comparison of p53 mutations in patients with localized osteosarcoma and metastatic osteosarcoma. Cancer 92: 2181–2189. [DOI] [PubMed] [Google Scholar]
- 30. Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E, et al. (2014) Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep 7: 104–112. 10.1016/j.celrep.2014.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ottaviano L, Schaefer KL, Gajewski M, Huckenbeck W, Baldus S, Rogel U, et al. (2010) Molecular characterization of commonly used cell lines for bone tumor research: a trans-European EuroBoNet effort. Genes Chromosomes Cancer 49: 40–51. 10.1002/gcc.20717 [DOI] [PubMed] [Google Scholar]
- 32. Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, et al. (1994) Tumor spectrum analysis in p53-mutant mice. Curr Biol 4: 1–7. [DOI] [PubMed] [Google Scholar]
- 33. Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. (2004) Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119: 847–860. [DOI] [PubMed] [Google Scholar]
- 34. Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, et al. (2004) Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 119: 861–872. [DOI] [PubMed] [Google Scholar]
- 35. Lengner CJ, Steinman HA, Gagnon J, Smith TW, Henderson JE, Kream BE, et al. (2006) Osteoblast differentiation and skeletal development are regulated by Mdm2-p53 signaling. J Cell Biol 172: 909–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Walkley CR, Qudsi R, Sankaran VG, Perry JA, Gostissa M, Roth SI, et al. (2008) Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev 22: 1662–76. 10.1101/gad.1656808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Berman SD, Calo E, Landman AS, Danielian PS, Miller ES, West JC, et al. (2008) Metastatic osteosarcoma induced by inactivation of Rb and p53 in the osteoblast lineage. Proc Natl Acad Sci USA 105: 11851–11856. 10.1073/pnas.0805462105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schinke T, Gebauer M, Schilling AF, Lamprianou S, Priemel M, Mueldner C, et al. (2008) The protein tyrosine phosphatase Rptpzeta is expressed in differentiated osteoblasts and affects bone formation in mice. Bone 42: 524–534. 10.1016/j.bone.2007.11.009 [DOI] [PubMed] [Google Scholar]
- 39. Peles E, Schlessinger J, Grumet M. (1998) Multi-ligand interactions with receptor-like protein tyrosine phosphatase beta: implications for intercellular signaling. Trends Biochem Sci 23: 121–124. [DOI] [PubMed] [Google Scholar]
- 40. Tonks NK. (2006) Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol 7: 833–846 [DOI] [PubMed] [Google Scholar]
- 41. Harroch S, Palmeri M, Rosenbluth J, Custer A, Okigaki M, Shrager P, et al. (2000) No obvious abnormality in mice deficient in receptor protein tyrosine phosphatase beta. Mol Cell Biol 20: 7706–7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hanahan D, Weinberg RA. (2011) Hallmarks of cancer: the next generation. Cell 144: 646–674. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 43. Albers J, Keller J, Baranowsky A, Beil FT, Catala-Lehnen P, Schulze J, et al. (2013) Canonical Wnt signaling inhibits osteoclastogenesis independent of osteoprotegerin. J Cell Biol 200: 537–549 10.1083/jcb.201207142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chang MK, Raggatt LJ, Alexander KA, Kuliwaba JS, Fazzalari NL, Schroder K, et al. (2008) Osteal tissue macrophages are intercalated throughout human and mouse bone lining tissues and regulate osteoblast function in vitro and in vivo. J Immunol 181: 1232–1244. [DOI] [PubMed] [Google Scholar]
- 45. Schulze J, Bickert T, Beil FT, Zaiss MM, Albers J, Wintges K, et al. (2011) Interleukin-33 is expressed in differentiated osteoblasts and blocks osteoclast formation from bone marrow precursor cells. J Bone Miner Res 26: 704–717. 10.1002/jbmr.269 [DOI] [PubMed] [Google Scholar]
- 46. Schweigel H, Geiger J, Beck F, Buhs S, Gerull H, Walter U, et al. (2013) Deciphering of ADP-induced, phosphotyrosine-dependent signaling networks in human platelets by Src-homology 2 region (SH2)-profiling. Proteomics 13: 1016–1027. 10.1002/pmic.201200353 [DOI] [PubMed] [Google Scholar]
- 47. Machida K, Khenkhar M, Nollau P. (2012) Deciphering Phosphotyrosine-Dependent Signaling Networks in Cancer by SH2 Profiling. Genes Cancer 3: 353–361. 10.1177/1947601912459048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Flint AJ, Tiganis T, Barford D, Tonks NK. (1997) Development of "substrate-trapping" mutants to identify physiological substrates of protein tyrosine phosphatases. Proc Natl Acad Sci USA 94: 1680–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maeda N, Ichihara-Tanaka K, Kimura T, Kadomatsu K, Muramatsu T, Noda M. (1999) A receptor-like protein-tyrosine phosphatase PTPzeta/RPTPbeta binds a heparin-binding growth factor Midkine. Involvement of arginine 78 of Midkine in the high affinity binding to PTPzeta. J Biol Chem 274: 12474–12479. [DOI] [PubMed] [Google Scholar]
- 50. Kadomatsu K, Kishida S, Tsubota S. (2013) The heparin-binding growth factor Midkine: the biological activities and candidate receptors. J Biochem 153: 511–521. 10.1093/jb/mvt035 [DOI] [PubMed] [Google Scholar]
- 51. Sueyoshi T, Jono H, Shinriki S, Ota K, Ota T, Tasaki M, et al. (2012) Therapeutic approaches targeting Midkine suppress tumor growth and lung metastasis in osteosarcoma. Cancer Lett 316: 23–30. 10.1016/j.canlet.2011.10.013 [DOI] [PubMed] [Google Scholar]
- 52. Maehara H, Kaname T, Yanagi K, Hanzawa H, Owan I, Kinjou T, et al. (2007) Midkine as a novel target for antibody therapy in osteosarcoma. Biochem Biophys Res Commun 358: 757–762. [DOI] [PubMed] [Google Scholar]
- 53. Faissner A, Heck N, Dobbertin A, Garwood J. (2006) DSD-1-Proteoglycan/Phosphacan and receptor protein tyrosine phosphatase-beta isoforms during development and regeneration of neural tissues. Adv Exp Med Biol 557: 25–53. [DOI] [PubMed] [Google Scholar]
- 54. Harroch S, Furtado GC, Brueck W, Rosenbluth J, Lafaille J, Chao M, et al. (2002) A critical role for the protein tyrosine phosphatase receptor type Z in functional recovery from demyelinating lesions. Nat Genet 32: 411–414. [DOI] [PubMed] [Google Scholar]
- 55. Barnea G, Grumet M, Sap J, Margolis RU, Schlessinger J. (1994) Close similarity between receptor-linked tyrosine phosphatase and rat brain proteoglycan. Cell 76, 205 [DOI] [PubMed] [Google Scholar]
- 56. Nishiwaki T, Maeda N, Noda M. (1998) Characterization and developmental regulation of proteoglycan-type protein tyrosine phosphatase zeta/RPTPbeta isoforms. J Biochem 123: 458–467. [DOI] [PubMed] [Google Scholar]
- 57. Lamprianou S, Chatzopoulou E, Thomas JL, Bouyain S, Harroch S. (2011) A complex between contactin-1 and the protein tyrosine phosphatase PTPRZ controls the development of oligodendrocyte precursor cells. Proc Natl Acad Sci U S A 108: 17498–174503. 10.1073/pnas.1108774108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pariser H, Perez-Pinera P, Ezquerra L, Herradon G, Deuel TF. (2005) Pleiotrophin stimulates tyrosine phosphorylation of beta-adducin through inactivation of the transmembrane receptor protein tyrosine phosphatase beta/zeta. Biochem Biophys Res Commun 335: 232–239. [DOI] [PubMed] [Google Scholar]
- 59. Meng K, Rodriguez-Peña A, Dimitrov T, Chen W, Yamin M, Noda M, et al. (2000) Pleiotrophin signals increased tyrosine phosphorylation of beta beta-catenin through inactivation of the intrinsic catalytic activity of the receptor-type protein tyrosine phosphatase beta/zeta. Proc Natl Acad Sci U S A 7: 2603–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Diamantopoulou Z, Kitsou P, Menashi S, Courty J, Katsoris P. (2012) Loss of receptor protein tyrosine phosphatase β/ζ (RPTPβ/ζ) promotes prostate cancer metastasis. J Biol Chem 287: 40339–40349. 10.1074/jbc.M112.405852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Grigoriadis AE, Schellander K, Wang ZQ, Wagner EF. (1993) Osteoblasts are target cells for transformation in c-fos transgenic mice. J Cell Biol 122: 685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. David JP, Mehic D, Bakiri L, Schilling AF, Mandic V, Priemel M, et al. (2005) Essential role of RSK2 in c-Fos-dependent osteosarcoma development. J Clin Invest 115: 664–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, et al. (2015) COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 43 (Database issue): D805–811. 10.1093/nar/gku1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Contact Xrays and histological analysis of undecalcified spine or tibia sections from 12 weeks old mice of the indicated genotypes. No OS development was observed in either analysis.
(TIF)
Shown are the profiles after probing the membranes with additional SH2 domains as indicated.
(TIF)
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.