

Abstract
Coxsackieviruses (CVs) are relatively common viruses associated with a number of serious human diseases, including myocarditis and meningo-encephalitis. These viruses are considered cytolytic yet can persist for extended periods of time within certain host tissues requiring evasion from the host immune response and a greatly reduced rate of replication. A member of Picornaviridae family, CVs have been historically considered non-enveloped viruses – although recent evidence suggest that CV and other picornaviruses hijack host membranes and acquire an envelope. Acquisition of an envelope might provide distinct benefits to CV virions, such as resistance to neutralizing antibodies and efficient nonlytic viral spread. CV exhibits a unique tropism for progenitor cells in the host which may help to explain the susceptibility of the young host to infection and the establishment of chronic disease in adults. CVs have also been shown to exploit autophagy to maximize viral replication and assist in unconventional release from target cells. In this article, we review recent progress in clarifying virus replication and dissemination within the host cell, identifying determinants of tropism, and defining strategies utilized by the virus to evade the host immune response. Also, we will highlight unanswered questions and provide future perspectives regarding the potential mechanisms of CV pathogenesis.
Introduction
Enteroviruses (EVs) are widely distributed in nature and frequently cause heart and central nervous system (CNS) diseases (Whitton, Cornell et al., 2005) (Muir & van Loon, 1997). EVs are members of the Picornaviridae family which include notable members such as foot-and-mouth disease virus, poliovirus (PV), rhinovirus and hepatitis A. Some EVs, particularly enterovirus-71 (EV71) in Asia, are considered to be serious emerging CNS pathogens (Shih, Stollar et al., 2011). The EV genus includes an important member, coxsackievirus (CV), which cause severe morbidity and mortality in the newborn and young host (Tebruegge & Curtis, 2009) (Romero, 2008). These viruses have a small, positive-sense single stranded RNA genome, and infection occurs primarily through the fecal-oral route (Whitton, Cornell et al., 2005) (Feng, Langereis et al., 2014b). Approximately 15 million diagnosed cases of EV infections occurred in the US in 1996, revealing that EV remains a substantial problematic viral infection (Sawyer, 2002). The original classification of EVs included the four groups: Coxsackie A viruses, Coxsackie B (CVB) viruses, ECHO (Enteric Cytopathic Human Orphan) viruses and PVs. A new classification system was devised utilizing consecutive numbers for each new isolate (such as EV71, EV72, etc.) due to significant overlap between the historically-named EVs (Oberste, Maher et al., 2002).
In utero and childhood infection is under-recognized but carries long-term consequences whereby intellectual and cognitive abilities of the patient might be compromised (Chiriboga-Klein, Oberfield et al., 1989) (Euscher, Davis et al., 2001;Chang, Huang et al., 2007) (Chamberlain, Christie et al., 1983). A relatively common pediatric virus, CV typically causes mild infections ranging from subclinical to flu-like symptoms and mild gastroenteritis (Weller, Simpson et al., 1989). CV has been shown to infect the heart, pancreas, and CNS (Arnesjo, Eden et al., 1976) (Rhoades, Tabor-Godwin et al., 2011). In rare cases CVs cause severe systemic inflammatory diseases such meningo-encephalitis, pancreatitis, and myocarditis, all of which can be fatal or result in lasting organ dysfunction, including dilated cardiomyopathy and encephalomyelitis (David, Baleriaux et al., 1993) (Hyypia, Kallajoki et al., 1993). The remarkable distribution of CV infections can be appreciated by the high seroprevalence in many countries around the world. In one study, IgG antibodies against CV were detected in 6.7 to 21.6% of individuals throughout various regions of Greece (Mavrouli, Spanakis et al., 2007). An analysis of a French-Canadian population in Montreal showed a seroprevalence as high as 60-80% for some strains of CV (Payment P., 1991). In a region of China, the seroprevalence for a single serotype of CV was shown to be greater than 50% in groups aged 15 years or more (Tao, Li et al., 2013). The wide distribution of CV, their genetic variability, and ability to persist in the human host make it challenging for epidemiologists to link previous viral infection and subsequent pathology, suggesting a potential role for these viruses in chronic human idiopathies (Victoria, Kapoor et al., 2009) in addition to recognized illnesses. Vaccine design against CVs and EVs remain challenging for a number of reasons which include their remarkable genetic variability and inconsistent pathology in humans.
Spontaneous abortions, fetal myocarditis, and neurodevelopmental delays in the newborn remain serious outcomes if CV infection occurs during pregnancy (Ornoy & Tenenbaum, 2006) (Euscher, Davis et al., 2001). Infants infected with CV have a higher likelihood of developing myocarditis, meningitis and encephalitis; and the mortality rate may be as high as 10%. Also, many chronic diseases may be the end result of a previous CV infection. These chronic diseases include chronic myocarditis (Chapman & Kim, 2008), schizophrenia (Rantakallio, Jones et al., 1997), encephalitis lethargica (Cree, Bernardini et al., 2003), and amyotrophic lateral sclerosis (Woodall, Riding et al., 1994) (Woodall & Graham, 2004). The molecular mechanisms determining the tropism of CVs and their ability to persist in the host remain unclear. The lasting consequences of CV infection upon surviving individuals remain largely unknown despite clear dangers associated with infection and the cytolytic nature of the virus.
Many publications have suggested a link between early CV infection and insulin-dependent diabetes (IDDM) (Laitinen, Honkanen et al., 2014) (Jaidane & Hober, 2008) (Christen, Bender et al., 2012), although additional data is needed to support these correlative studies. In addition, a mouse model has shown the development of insulin-dependent diabetes (IDDM) to be associated with CV-induced pancreatitis and replication efficiency (Drescher, Kono et al., 2004), although the factors determining viral tropism and mechanism of disease are not well understood (Tracy, Drescher et al., 2011) (Kanno, Kim et al., 2006).
Type B coxsackieviruses (CVB) include six serotypes, each being associated with acute disease in humans, including acute viral myocarditis and pancreatitis. While CVB is generally regarded as a lytic virus, emerging evidence suggests that persistent infection can be established which may be responsible for chronic inflammation within target organs. Moreover, latency and episodic reactivation could also contribute to the disease process (Feuer, Mena et al., 2002) (Ruller, Tabor-Godwin et al., 2012) (Feuer & Whitton, 2008). Previously, we described the nature of the CVB viral genome in quiescent cells whereby a viral state similar to latency was established (Feuer, Mena et al., 2002) (Feuer, Mena et al., 2004). Following stimulation of quiescent cells by injury, or by the addition of growth factors, viral protein expression was detected and infectious virus was produced, suggesting that latent CVB may be reactivated in response to cellular activation. In parallel, CVB has evolved to modulate cell-signaling networks to evade host antiviral immunity, enter cells, and undergo replication even as the host cell suffers the consequences of a cytolytic viral infection (Esfandiarei, Luo et al., 2004) (Jensen, Garmaroudi et al., 2013) (Esfandiarei & McManus, 2008).
Our review will cover recent progress specifically in CVB research, while acknowledging advances in other areas of EV investigation which have contributed to a greater understanding of CVB replication and pathogenesis.
Molecular Biology of CVB
CVBs, and EVs in general, are non-enveloped viruses which have the ability to survive harsh environments. Infection proceeds via the fecal/oral route, and hence virion stability in the acidic environment of the stomach becomes a necessity for efficient transmission. The virion structure exhibits an icosahedral symmetry with a diameter size of approximately 30nm (Jiang, Liu et al., 2014). Four capsid proteins (VP1-VP4) comprise the virion structure, and these viral proteins are major antigenic determinants following the activation of the host humoral response. The positive-strand viral RNA genome ranges in size ~7-8 kb and is covalently linked at the 5’ end with a viral protein called VPg (the Viral Protein of the genome). VPg, one of the viral proteins 3B, plays an essential role in both positive and negative-strand RNA synthesis by covalently attaching to the 5’ end of the viral genome and acting as a primer for RNA synthesis. It remains unclear how relatively greater amounts of positive-strand genome are produced for every negative-strand genome during an EV infection (Novak & Kirkegaard, 1991). Nevertheless, the ratio of positive-strand genome to negative-strand genome decreases upon the establishment of CVB persistence within the CNS (Feuer, Ruller et al., 2009) (Tam & Messner, 1999). These results suggest that a double-stranded RNA structure might assist in stabilizing the viral genome and contributing to persistent infection.
CVB includes several cis-acting RNA elements (CREs) which are required for efficient viral replication (Steil & Barton, 2009). CREs contribute to the conversion of VPg into VPgpUpUOH which acts as a primer for the viral RNA polymerase to initiate replication. As with other EVs, the CVB RNA genome contains an internal ribosome entry site (IRES) in the 5’ non-translated region (NTR). CVB RNA lacks a 7-methyl guanosine cap structure, yet host cell ribosomes can directly interact with the IRES of CVB RNA to initiate translation of the viral genome (Pelletier & Sonenberg, 1988). The viral genome is translated into a long polyprotein which undergoes a series of cleavages by the viral proteases 2Apro and 3CDpro to generate the mature viral proteins. These viral proteins include VP1-VP4 capsid proteins and seven non-structural proteins (2A-C, 3A-D, and 3DPol - RNA polymerase) (Kitamura, Semler et al., 1981). Following viral protein translation, negative-strand replication begins (Gamarnik & Andino, 1998), and a viral replication complex forms to produce both positive- and negative-strand synthesis at the 5’ cloverleaf structure of the 5’ NTR (Vogt & Andino, 2010). The poly(A) binding protein 1 (PABP1) interacts with the poly(A) tail of the virus leading to circularization of the viral genome during negative-strand synthesis (Herold & Andino, 2001). The virus 2A protein cleaves the cell protein eIF-4G, a host factor necessary for cap-dependent translation (Etchison, Milburn et al., 1982). In this way, CVB maximizes replication by commandeering nearly all of the available resources of the host cell. The viral RNA polymerase 3DPol interacts with viral protein 3AB after attachment to viral membrane vesicles that form after infection (Fujita, Krishnakumar et al., 2007).
CVB Replication Complexes and Remodeling of Intracellular Membranes
In general, positive strand RNA viruses require cellular membranes for genome replication and actively modify intracellular membranes to construct their own replication organelles (Belov & van Kuppeveld, 2012). CVB redirects a number of cell host factors to remodel intracellular membranes for efficient viral replication (Wessels, Duijsings et al., 2007) (Hsu, Ilnytska et al., 2010) (Lanke, van der Schaar et al., 2009). These host proteins include phosphatidylinositol-4-kinase (PI4KIII ), guanine nucleotide exchange factor - GBF1, and ARF1 which help to assemble the membrane replication complex supporting CVB and PV infection (Belov, Altan-Bonnet et al., 2007) (Lanke, van der Schaar et al., 2009). The current model for the initiation of CVB replication organelles in the host cell involves the recruitment of PI4KIIIβ following viral protein 3A binding to GBF1/Arf1 as COPI, a protein that regulates membrane budding, is displaced. PI4KIIIβ catalyzes the production of a phosphatidylinositol-4-phosphate (PI4P) lipid micro-environment leading to the recruitment of viral protein 3Dpol and the synthesis of viral RNA (Hsu, Ilnytska et al., 2010). Cholesterol is actively trafficked from the plasma membrane to viral replication organelles via clathrin-mediated endocytosis, indicating that cholesterol modulation is necessary to support elevated viral replication levels (Ilnytska, Santiana et al., 2013). The detailed examination of host proteins and lipids contributing to the formation of viral replication organelles provides a means of combatting infection by designing a new class of therapeutic small molecules that target these host proteins (such as PI4 kinase inhibitors) (Hsu, Ilnytska et al., 2010), or by blocking cholesterol uptake. Nevertheless as with any antiviral drug candidates, the proclivity of RNA viruses to develop resistance remains clear (van der Schaar, van der Linden et al., 2012). Also, antiviral drugs acting on host proteins might be expected to contribute to cellular anti-proliferative effects and cytotoxicity (Lamarche, Borawski et al., 2012). In addition, therapeutic small molecules shown to reduce viral replication in culture by inhibiting host proteins may not be as effective in vivo. Recent results have shown that kinase inhibitors utilized in a murine model of infection may delay rather than prevent disease, although administration of the drug was abridged due to toxicity (Ford Siltz, Viktorova et al., 2014).
CVB Entry into Target Cells
Two cell receptors have been identified which contribute to CVB entry into target cells. CVB utilizes a transmembrane protein found within the tight junctions of polarized cells called the coxsackievirus and adenovirus receptor (CAR) (Bergelson, Cunningham et al., 1997). Also, human decay accelerating factor (DAF) has been shown to function as a co-receptor for CVB entry for some viral isolates (Bergelson, Chan et al., 1994). In polarized cells, CVB binds to DAF at the apical surface of the cell, and binding stimulates intracellular signaling which assists in virion movement across the cell membrane to the tight junctions (Coyne & Bergelson, 2006). Subsequently, virion binding to CAR within the tight junctions leads to CAR-dependent entry in a caveolin-dependent, dynamin-independent manner. In contrast, CVB entry into non-polarized cells requires CAR expression in a dynamin-dependent, caveolin-independent manner (Patel, Coyne et al., 2009). Although CVB can readily infect via the intraperitoneal route in mice, the gastrointestinal route acts as a barrier to infection (Loria, Shadoff et al., 1976). These findings illuminate clear differences between the natural human host, and the murine model of infection. Expression of the human form of DAF on the intestinal epithelium in transgenic mice failed to facilitate infection by the enteral route (Pan, Zhang et al., 2015), suggesting that other obstacles such as the type I interferon response (Ohka, Igarashi et al., 2007) and interactions with the intestinal flora (Kuss, Etheredge et al., 2008) may limit CVB infection in the murine model.
Receptor expression is necessary for virion entry into possible target cells (Kallewaard, Zhang et al., 2009). For example, mice lacking CAR expression in cardiac tissue completely abolished viral replication in the heart and prevented myocarditis (Shi, Chen et al., 2009). These results show the in vivo importance of CAR expression for CVB infection and disease. Also, decreased CAR expression as primary neurons differentiate in culture correlated with a reduction in CVB infection (Ahn, Jee et al., 2008). The authors concluded that susceptibility to infection is dependent upon the level of CAR expression, and changes in CAR expression during development may limit the number of target cells for CVB. Also, CAR downregulation can occur during a carrier-state infection in cells grown in culture leading to the prevalence of resistant cells over time (Pinkert, Klingel et al., 2011). Since CAR is widely expressed in the host yet distribution of infection is much more limited, additional factors may ultimately control tropism. For example, CAR expression is found at high levels in the murine neonatal CNS (Venkatraman, Behrens et al., 2005) (Honda, Saitoh et al., 2000) (Hotta, Honda et al., 2003). Yet during early infection, CVB is largely restricted to neural progenitor cells (NPCs) or infiltrating myeloid cells (Feuer, Pagarigan et al., 2005). Preferential targeting of these cells may be related to their proliferative status (Feuer, Mena et al., 2002) (Feuer, Mena et al., 2003) (Feuer & Whitton, 2008) (Feuer, Pagarigan et al., 2005) and increased autophagic activity (Tabor-Godwin, Tsueng et al., 2012). While CVB and adenovirus (a DNA virus) both utilize the identical host receptor (CAR), the tropism for each virus appear quite distinct in vivo - indicating other factors at play, such as the tissue-specific type I interferon response, in controlling tissue tropism (Wessely, Klingel et al., 2001).
The Role of Autophagy during CVB Replication
Autophagy is a process by which cells breaks down long-lived, decaying, or damaged organelles and proteins. Several investigators have identified autophagy as a crucial component for the replication and survival of various EVs - including CVB which subverts host autophagy upon infection (Alirezaei, Flynn et al., 2012a). This view is demonstrated in the host by a report showing reduced pancreatic pathology and lower viral titers in mice lacking ATG5 expression in the pancreas after CVB infection (Alirezaei, Flynn et al., 2012b). Autophagy is initiated with the activation of class III phosphatidylinositol 3 kinase (PtdIns3K) signaling. PtdIns3K signaling allows for the nucleation of the phagophore - a cup-shaped double membrane which elongates around cellular components to be degraded. The phagophore structure is commonly believed to originate from the endoplasmic reticulum (ER) (Hamasaki, Furuta et al., 2013). ATG4 truncates microtubule-associated protein 1A/1B-light chain 3 (LC3-I) to expose the C-terminal glycine which becomes conjugated to phophatidylethanolamine to form LC3-II. LC3-II is then recruited to the elongating phagophore, and because of this, LC3-II is a common marker used in the detection of autophagy via western blot and immunostaining (Gustafsson & Gottlieb, 2008) (Klionsky, Abdalla et al., 2012). Once the autophagosome is complete, the structure fuses with a lysosome to form the autolysosome, and the cargo within are degraded by acidic hydrolases.
Previous work has shown that several EVs such as PV, CVB and EV71 trigger autophagy and hijack the autophagosomal membranes to enhance viral replication (Suhy, Giddings, Jr. et al., 2000) (Wong, Zhang et al., 2008a) (Huang, Chang et al., 2009). Also, cell host factors which suppressed autophagy inhibited CVB replication (Delorme-Axford, Morosky et al., 2014). PV proteins 3A and 2BC had been shown to elongate the ER and induce autophagosome formation, respectively. Many viruses have been thought to assemble replication factories onto autophagosomal membranes in order to enhance replication efficiency (Wileman, 2006). With that in mind, CVB has been shown to prevent autophagic flux causing virus-filled autophagosomes to accumulate and merge into “megaphagosomes” (Kemball, Alirezaei et al., 2010a). Additionally, the induction of autophagy may allow for non-cytolytic escape of EVs from the host cell (Taylor & Kirkegaard, 2008) (Bird, Maynard et al., 2014).
Autophagy is crucial for maintaining cellular homeostasis and has been shown to prevent cellular damage in the CNS following activation via short-term fasting (Simonsen, Cumming et al., 2008) (Alirezaei, Kemball et al., 2010). The induction of autophagy in rat primary neurons was associated with increased CVB replication (Yoon, Ha et al., 2008). Curiously, an inverse correlation between autophagy and apoptosis was observed when rat primary neurons were infected with CVB4 (Yoon, Ha et al., 2009). Therefore, deciphering the role of autophagy during the progression of CVB infection in rapidly cycling cells / progenitor cells and during establishment of viral persistence in the heart and CNS may be critical in controlling disease.
CVB is capable of subverting host autophagy via 2A protease which cleaves sequestosome 1 (SQSTM1)/p62 (Shi, Wong et al., 2013). This protein serves in trafficking ubiquitinated proteins to the autophagosome by joining with LC3-II. The disruption of SQSTM1 results in impaired selective autophagy and allows CVB to circumvent host defense signaling. However, CVB has also been shown to become actively trafficked into autophagosomes possibly to promote viral dissemination via the release of intracellular vesicles. How and why CVB prevents general cargo uptake of cellular components in autophagosomes while still allowing its own engulfment is unclear, but these processes may vary based on specific cell types and environmental conditions. Autophagy inhibitors have been shown to decrease extracellular compared to intracellular poliovirus titers (Jackson, Giddings, Jr. et al., 2005a). Hence, modulation of autophagy by candidate drugs might function as therapeutic antivirals during acute or persistent CVB infection.
CVB Infection of Neural Progenitor Cells (NPCs)
Previously, we generated a pediatric model of CVB infection in the CNS and heart in order to study virus tropism and disease (Feuer, Mena et al., 2003) (Feuer, Pagarigan et al., 2005) (Feuer, Ruller et al., 2009). We demonstrated the ability of CVB to target neural progenitor cells (NPCs) within the neonatal CNS (Feuer, Mena et al., 2003), or grown in culture as neurospheres (Tsueng, Tabor-Godwin et al., 2011) (Rhoades, Tabor-Godwin et al., 2011). Neurospheres, or free-floating spheres generated by NPCs in culture, can be differentiated into neurons, astrocytes, and oligodendrocytes (Eriksson, Bjorklund et al., 2003). NPCs were highly susceptible to infection, and cytopathic effects were readily observed following infection. In contrast, differentiated NPCs were less susceptible to CVB infection. We suggested that the reduced susceptibility to CVB infection was due to their decreased proliferative status and cellular changes associated with differentiation (Feuer, Mena et al., 2002). Depletion of NPCs by virus-mediated apoptosis led to CNS developmental defects in the host (Ruller, Tabor-Godwin et al., 2012), and CVB could establish a persistent infection causing chronic inflammation in the brain (Feuer, Ruller et al., 2009). We hypothesized that progenitor cells may survive initial infection and contribute to virus persistence, either indirectly (contributing to virus attenuation) or directly (progenitor cells harboring viral materials) (Rhoades, Tabor-Godwin et al., 2011).
The process of autophagy is thought to be critical for stem cell maintenance and during cell lineage commitment (Guan, Simon et al., 2013) (Vessoni, Muotri et al., 2012). The role of autophagy during CVB infection was shown to be cell-specific, and NPCs supported greater levels of viral infection (Tabor-Godwin, Tsueng et al., 2012). As previous demonstrated by other groups utilizing transformed cell lines (Wong, Zhang et al., 2008b), HL-1 cells (a transformed cardiomyocyte cell line) showed an increase in autophagic signaling following infection with CVB; viral titers increased after autophagy induction and decreased after autophagy inhibition. In contrast, no change in autophagic signaling was seen in NPCs following infection, although basal levels of autophagy were higher compared to HL-1 cells. Furthermore, higher levels of autophagy signaling could be induced in NPCs without increasing viral replication levels. In differentiated NPC precursors, autophagy increased during differentiation. Unexpectedly, a decrease in autophagy was observed in neurons, astrocytes, and oligodendrocytes following CVB infection. These observations were quite surprising since picornaviruses have been shown to consistently activate autophagy during infection (Kirkegaard, 2009), and autophagosomes may be required for optimal picornaviral replication (Taylor & Kirkegaard, 2008).
CVB might be expected to target NPCs for a variety of reasons. The naturally high basal levels of autophagic activity in NPCs may explain their relatively greater susceptibility to CVB infection. NPCs normally undergo rapid expansion and proliferation during development and regeneration of tissue, and CVB has been shown to preferentially replicate in cells undergoing active proliferation (Feuer, Mena et al., 2002) (Feuer, Pagarigan et al., 2005). As NPCs differentiate into neurons, these cells migrate into regions of the hippocampus and the olfactory bulb. Migration of infected NPCs would be expected to assist in virus dissemination, and the olfactory bulb could provide an escape route for the virus through the olfactory neuroepithelium. We hypothesize that viral infection might alter NPCs and neurons derived from infected NPCs, leading to possible behavioral modifications within the host in order to maximize virus transmission. Also, quiescent NPCs may harbor persistent / latent infection until a later point in time when active neurogenesis stimulates virus reactivation (Rhoades, Tabor-Godwin et al., 2011). Finally, immune-privileged regions such as the CNS could limit antiviral responses against CVB providing for a fruitful and protected area of viral replication.
Utilization of Recombinant CVBs Expressing Foreign Proteins
A number of research groups have generated recombinant CVBs (rCVBs) expressing molecular markers, cytokines, and other foreign proteins (Slifka, Pagarigan et al., 2001) (Feuer, Mena et al., 2002) (Henke, Zell et al., 2001). Many of these recombinant viral constructs have proven to be quite stable, although the loss of the foreign insert can appear in the viral population within five passages, depending upon the size of the insert, nucleotide sequence, nature of the gene, and passage conditions. The utilization of these rCVBs in tissue culture and in vivo have clarified questions regarding viral tropism (Feuer, Pagarigan et al., 2005) (Puccini, Ruller et al., 2014), activation of the adaptive immune response against infection (Kemball, Harkins et al., 2009), mechanisms of virulence and disease (Zeng, Chen et al., 2013), and possible uses of CVB as therapeutic/vaccine vectors (Kim, Kim et al., 2012) (Miller, Geng et al., 2009).
The importance of following viral infection temporally using minimally invasive and real time tools may lead better understanding of CVB pathogenesis. Temporal studies for other viruses have demonstrated the role of efficient viral dissemination in enhancing virus replication. For example, vaccinia virus maximizes viral spread by targeting uninfected cells rather than re-infecting nearby cells (Doceul, Hollinshead et al., 2010). A different strategy is utilized by human T-lymphotropic virus-1 (HTLV-1) which creates a specialized area of cell-cell contact – the virological synapse – promoting transmission of virus between cells (Nejmeddine & Bangham, 2010). To inspect the progression of CVB dissemination, we designed a unique rCVB expressing “fluorescent timer” protein (Timer-CVB) to track virus spread in cells grown in culture, and within our animal model of infection (Robinson, Tsueng et al., 2014). “Fluorescent timer” protein (Terskikh, Fradkov et al., 2000) is converted from green fluorescence to red fluorescence over a period of ~48 hours which allows viral spread to be observed in real time based on color changes (Figure 1). By standard plaque assay, Timer-CVB plaques demonstrated a “bull’s-eye” pattern by fluorescence microscopy whereby initial infection was represented by red fluorescence and newly-infected cells via cell-to-cell spread were revealed by green fluorescence. Timer-CVB3 may be of particular benefit in evaluating antiviral compounds in target cells and deciphering their mode of action. For example, antiviral compounds which inhibit the release of infectious virus from the host cell would be represented by infected green cells, followed by yellow, and finally red cells with no new green cells being observed. In contrast, antiviral compounds acting to simply reduce the level of infectious virus production would show new green cells, although at a lower level compared to untreated cultures.
Figure 1. HeLa cells infected with Timer-CVB slowly change fluorescence from green to red.

The gene for “fluorescent timer” protein was inserted into the infectious plasmid clone for CVB. A) Upon infection with recombinant CVB3 expressing “fluorescent timer” protein (Timer-CVB), the slow conversion of the green fluorescing form of timer protein to red occurred over time in cells overlaid with agar. Initial sites of infection fluoresced red, while newly infected cells fluoresced green. B) HeLa cells infected with Timer-CVB (moi = 0.1) initially fluoresced green (recent viral protein) at 24 hours PI as determined by fluorescence microscopy. By 32 hours PI, both green and red fluorescence (matured viral protein) was observed in infected HeLa cells, and by 48 hours PI the majority of cells fluoresced brightly in the red channel. Fewer green and red infected cells were observed by fluorescence microscopy for HeLa cells treated with the antiviral drug ribavirin (100µg/ml) at every time point.
The use of Timer-CVB revealed fascinating aspects of virus replication that we did not expect. First by creating time-lapse videos, we identified extensive intracellular membrane remodeling in infected progenitor cells (Robinson, Tsueng et al., 2014) reminiscent of viral replication organelles previously described by other research groups (Hsu, Ilnytska et al., 2010). These time lapse videos showed viral replication organelles fluorescing in an asynchronous fashion and revealed the dynamics of intracellular membrane reorganization following infection. “Fluorescent timer” protein and viral 3A protein colocalized closely, suggesting that the molecular marker remained trapped within viral replication organelles shortly after translation. Viral 3A protein can bind and modulate host cell factors such as PI4KIII , GBF1 and ARF1, and facilitate intracellular membrane remodeling for efficient viral replication (Wessels, Duijsings et al., 2007) (Hsu, Ilnytska et al., 2010) (Lanke, van der Schaar et al., 2009). Along with participation in the formation of viral replication organelles, viral 3A protein was previously shown to halt protein trafficking and secretion by disrupting the Golgi apparatus (de Jong, Visch et al., 2006) (Cornell, Kiosses et al., 2006) (Cornell, Kiosses et al., 2007). We expect that Timer-CVB may be of value in identifying the formation of viral replication organelles in real time within the cell, and for tracking viral spread in our animal model of infection.
CVB Escapes the Host Cell Through Ejected Autophagosomes
As a non-enveloped, protein encapsulated virus, EVs such as CVB have classically been thought to escape the infected host via cytolysis whereby released virions would rapidly be exposed to host neutralizing antibodies. Interestingly, we observed extracellular microvesicles (EMVs) containing infectious virus released from various progenitor cell types infected with Timer-CVB (Robinson, Tsueng et al., 2014) (Figure 2). Upon further analysis, these structures were shown to be enriched with LC3-II, a common marker for autophagosomes. The presence of LC3-II in virally-induced EMVs, and the lack of LC3-II in basally-released exosomes from uninfected cells suggest that CVB may be actively trafficked into autophagosomes which subsequently promotes their release from the cell. This model would be akin to the previously-described phenomenon of autophagosome-mediated exit without lysis (AWOL) following PV infection (Taylor, Burgon et al., 2009) (Richards & Jackson, 2012). Whereas CVB appeared to escape the cell bound in intact membranes, in the case of PV infection, these microvesicles were hypothesized to be unstable although contributing to the release of viral particles from the host cell in the absence cell lysis. Our results suggest that CVB coordinates a unique method of viral dissemination by utilizing the autophagy pathway, and that these EMVs may be more stable that previously proposed. CVB had been shown to increase the accumulation of autophagosomes in the pancreata of infected mice wherein autophagosomes fused into megaphagosomes (Kemball, Alirezaei et al., 2010a). We previously documented a reduction of autophagosomes in the cytoplasm of partially differentiated NPCs infected with CVB; however upon closer inspection, numerous EMVs appeared to be budding from the surface of these infected cells, possibly as a result of intracellular autophagosomes being expelled into the extracellular space (Tabor-Godwin, Tsueng et al., 2012). More recently, PV, CVB, and rhinovirus particles were shown to be released within phosphatidylserine vesicles by host cells in a non-lytic fashion (Chen, Du et al., 2015). Multiple viral particles were observed within individual vesicles, which may provide for cooperation among viral quasispecies and lead to more efficient infection compared to free viral particles.
Figure 2. Detection of LC3 and CVB viral protein in shed EMVs.

Differentiated NPSCs transduced with adeno-LC3-GFP were infected with dsRED-CVB (moi = 0.1) and observed by fluorescence microscopy at 3 days PI. (A) Abundant shed EMVs (white arrows) expressing viral protein (red) and a marker for autophagosomes (LC3-GFP, green) were readily observed. (E-F) Higher magnification of (C) showed colocalization of viral protein and LC3-GFP in shed EMVs.
The utilization of EMVs could promote viral dissemination and infection in a number of ways (Figure 3). Because EMV-mediated virus dissemination allows CVB to be membrane-bound, this method of release could potentially broaden the range of susceptible target cells by bypassing canonical receptor-mediated endocytosis, and instead fusing with cells that may lack CAR or DAF. Notwithstanding this possibility, CAR expression in some target tissues such as the heart remains a critical determinant of tropism and eventual disease progression (Shi, Chen et al., 2009). Yet, EMVs might still contribute to infection of other cells or tissues prior to eventual dissemination into the heart, or during the establishment virus persistence within cardiac tissue. For example, CVB was shown to efficiently infect and utilize B lymphocytes for dissemination despite barely detectable levels of CAR expression on these important target cells (Mena, Perry et al., 1999). Because exit via EMVs provide a non-cytolytic method for CVB escape, this mechanism could potentially prolong viral replication in the host cell. EMVs could also enhance viral stability in the extracellular space as well as mask the virus from host neutralizing antibodies (Inal & Jorfi, 2013) (Masciopinto, Giovani et al., 2004). Though intact virions were observed in EMVs observed under electron microscopy (Robinson, Tsueng et al., 2014), these structures could presumably contain free viral RNA and still remain infectious. This would allow CVB to infect cells during persistence (Feuer, Ruller et al., 2009) without the need to assemble viral capsids or induce cellular cytopathicity (Taylor, Burgon et al., 2009) (Bird, Maynard et al., 2014). This novel route of dissemination would be analogous to RNA-containing exosomes, a process whereby host RNAs and micro-RNAs are transported for cell-to-cell communication purposes (Meckes, Jr. & Raab-Traub, 2011) (Gallo, Tandon et al., 2012) (Delorme-Axford, Donker et al., 2013).
Figure 3. Model of CVB dissemination in the host by shed EMVs.

High numbers of LC3II+ extracellular microvesicles (EMVs) containing infectious virus were recently observed following infection of progenitor cells in culture. Both the differentiation process and viral infection may enhance shedding of single membrane EMVs derived from the autophagy pathway. A) Virus-associated EMVs may expand the natural tropism of CV to target cells which fail to express canonical virus receptors. B) Neutralizing antibodies may be ineffective against infectious virus sequestered within the protected environment of the extracellular microvesicle. Also, virus-associated EMVs may increase the stability of infectious virus within the host during hematogenous spread. C) EMVs may assist in viral RNA dissemination during the persistent stage of infection whereby the presence of intact virions and/or structural viral proteins may be limited. D) EMVs may help virions travel and enter new target tissues and cross selectively permeable barriers.
CVB has been shown to preferentially target numerous progenitor-like cells such as NPCs and cardiac progenitor cells (CPCs). Because these types of cells often mobilize to areas of needed tissue repair once activated, CVB may be able to “hitchhike” within these cells to access new tissues that the virus would normally be unable to penetrate. Similarly, EMVs may allow CVB to enter selectively permeable tissues such as the CNS by bypassing the blood-brain-barrier or blood-CSF-barrier (Sampey, Meyering et al., 2014). Despite this, host defense mechanisms appear to have co-evolved in response to the release of virus-associated EMVs. Exosome-bound hepatitis C RNA has been shown to activate plasmacytoid dendritic cells, conferring protective innate immunity in the host (Dreux, Garaigorta et al., 2012). Additionally, phagocytic immune cells such as macrophages recognize specific “eat me” markers which may decorate CVB-associated EMVs and target them for degradation (Miyanishi, Tada et al., 2007).
The recent discovery of membrane-stealing picornaviruses, which now include hepatitis A (Feng, Hensley et al., 2013), might point to a two-fold strategy for a family of viruses that can leave the host as a non-enveloped virion, as well as maintain a cloaked, enveloped form - at least after infection of certain host cells. Why might a virus utilize this dual strategy? Perhaps some picornaviruses preserve this duality in order to maximize stability in different environments (Feng & Lemon, 2013). For example, a non-enveloped form of virus may have of greater stability in an inhospitably dry or hypotonic environment. In contrast, an enveloped form of virus may be more advantageous for hematogenic dissemination in the host where circulating neutralizing antibodies might otherwise neutralize virions with exposed antigenic proteins.
“Bus Stop/Trojan Horse” model for CVB entry across the tight junctions
One of the paradoxes regarding receptor usage by a large number of viruses, including CVB, is their dependence upon receptors located in seemingly inaccessible tight junctions of polarized epithelial cells (Bergelson, 2009) (Delorme-Axford & Coyne, 2011). For many viruses including CVB, the epithelial cell layer may be the first barrier encountered for entry into the host. Although polarized epithelial cells grown in culture have been utilized to model CVB entry into target cells (Coyne & Bergelson, 2006) (Coyne, Shen et al., 2007), these cells do not appear to support high levels of CVB replication and virus protein expression in vivo - despite their expression of CAR. Instead, acinar cells of the pancreas, cardiomyocytes, bone marrow and activated lymphocytes within the marginal regions of the spleen, infiltrating nestin+ myeloid cells, progenitor cells in juvenile mice, and immature neurons of the CNS are the major targets of CVB infection in the host.
We suggest that virus binding to intrajunctional proteins might represent a common strategy for viruses to target migratory cells traveling through tight junctions of several tissues (Figure 4). For example, we previously showed that CVB infected nestin+ myeloid cells which entered through the tight junctions of the choroid plexus epithelial cells (Tabor-Godwin, Ruller et al., 2010). The choroid plexus forms the blood-CSF-barrier in the CNS, and entry of activated immune cells is controlled by the tight junctions of the choroid plexus cuboidal epithelium (Ransohoff, Kivisakk et al., 2003). The choroid plexus regulates CSF production, and transthyretin (TTR), a hormone binding protein, is actively transported by the choroid plexus into the CSF (Dickson, Aldred et al., 1986). The choroid plexus also performs unique host functions (Emerich, Skinner et al., 2005), including secretion of growth factors (Shingo, Gregg et al., 2003) and participation in neurogenesis (Falk & Frisen, 2002). The recruitment of these novel myeloid cells was preceded by the induction of CCL12, a chemokine known to attract monocytic cells (Sarafi, Garcia-Zepeda et al., 1997) and fibrocytes (Moore, Murray et al., 2006). Although epithelial cells appeared spared from infection, CVB induced significant acute damage in the choroid plexus (Puccini, Ruller et al., 2014). Also, infected nestin+ myeloid cells were shown to migrate into the CNS and assist in virus dissemination.
Figure 4. “Bus Stop/Trojan Horse” model for CVB entry across the tight junctions of the blood-CSF barrier.

We propose that CVB initially binds to CAR, a tight junction protein, although not entering epithelial cells of the choroid plexus. Upon binding, CCL12 and other chemokines are released by epithelial cells thereby attracting nestin+ myeloid cells which undergo extravasation through tight junctions of choroid plexus epithelial cells. CVB virions enter nestin+ myeloid cells which support infection, and assist with virus entry into the CNS.
Why might CVB target intrajunctional proteins sequestered within highly inaccessible areas of epithelial cells? We propose that CVB, and other viruses, attach to intrajunctional proteins in order to “hitch-hike” on migratory immune cells responding to early infection. The utilization of immune cells for virus spread into primary target organs has been previously described by others (Mena, Perry et al., 1999) (Noda, Aguirre et al., 2006). Infected immune cells might shield the virus from neutralizing antibodies upon migration into secondary tissues. This strategy may not be unique to CVB, although the particular intrajunctional protein utilized by each virus may be distinct (Bergelson, 2009). Other virus families may have evolved to follow a similar method of dispersion via immune target cell “Trojan horses”, although perhaps utilizing a unique signature chemokine profile and matching responding immune cells.
CVB Infection of Cardiac Progenitor Cells (CPCs) AND Pathological Remodeling of the Heart
Though the frequency of CVB exposure in the population is difficult to estimate due to its often asymptomatic nature, an epidemiological study by Petitjean et al found that 39.1% of healthy individuals harbor enteroviral RNA (Petitjean, Kopecka et al., 1992). These data highlight the prevalence of CVB infections which go undetected; however of equal concern was the detection of enteroviral RNA in 66.7% of patients with idiopathic dilated cardiomyopathy. Though a link between acute myocarditis and dilated cardiomyopathy is well-documented (Satoh, Tamura et al., 1994) (Mason, 2002) (Kearney, Cotton et al., 2001) (Cheng, 2006), a causal relationship between mild subclinical infection and subsequent dilated cardiomyopathy - such as in the case of idiopathic cardiomyopathy - is less understood.
Our group has recently published a study which may shed light on the association between mild acute infection and late onset heart failure (Sin, Puccini et al., 2014). We developed a juvenile mouse model of mild CVB infection in which viral RNA and infectious particles could be detected at a high level in the heart immediately following infection, and both c-kit+ and Sca-1+ cardiac progenitor cells (CPCs) were preferential target cells for infection in the heart (Figure 5). Sca-1+ cells in the hearts of infected mice were shown to co-express mature vascular cell markers such as von Willebrand factor (endothelial) and SM22 (smooth muscle). Viral clearance occurred prior to the adult phase, and both mock and CVB-infected animals appeared healthy, suckled normally, and grew at similar rates. Heart weights were normal, and cardiac inflammation could not be detected by hematoxylin and eosin staining up to 11 weeks post-infection. Nevertheless, staining for c-kit antigen in heart sections revealed a 50% reduction in this progenitor cell population at 11 weeks post-infection. After CVB exposure, CPCs showed a strong predisposition to differentiate into vascular cells. CVB has been previously shown to upregulate autophagy, a process which may be essential during cell differentiation (Guan, Simon et al., 2013). Hence in addition to targeting CPCs for infection, CVB may drive their premature differentiation and impair their capacity for self-renewal. This effect ultimately may result in the depletion of the CPC pool seen in juvenile-infected adult mice, in addition to any cytolytic effects.
Figure 5. CVB productively infected progenitor cells in the juvenile heart.
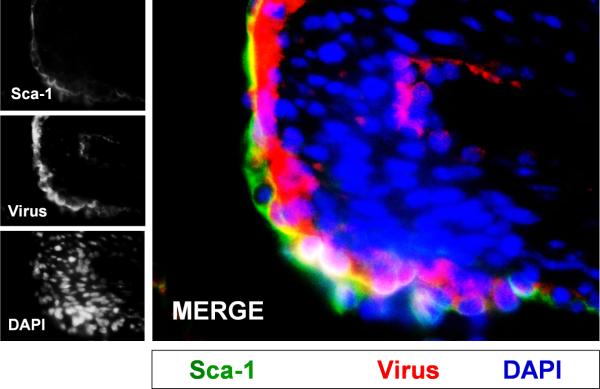
Three day-old mice were infected with eGFP-CVB (105 pfu IP) or mock-infected, and hearts were isolated at 2 days PI. Paraffin-embedded sections of heart tissue were deparaffinized and stained using an antibody against Sca-1 (green) and virus protein (red). Many Sca-1+ cells in heart tissue were shown to be infected with eGFPCVB. DAPI (blue) was utilized to label cell nuclei. Representative images of three infected mice are shown.
What does a compromised CPC population mean for the adult heart? Adult mice given a low inoculum of CVB during the juvenile period appeared healthy and indistinguishable from the mock-infected control mice. Indeed, cardiac hypertrophy and dilation indicative of progression to heart failure was observed only when these juvenile-infected adult mice were subjected to exercise or pharmacologically-induced cardiac stress. Infected animals showed evidence of cardiac hypertrophy and myocardial scarring following swimming exercise or isoproterenol treatment. Further investigation revealed that mock-infected animals were able to augment blood vessel formation following increased cardiac load, whereas CVB-infected animals could not perform this type of vascular remodeling. Stem cells are intimately involved in angiogenesis and neovascularization via paracrine signaling and direct differentiation (Huang, Zhang et al., 2010) (Lu, Pompili et al., 2013) (Leeper, Hunter et al., 2010). We hypothesize that the diminished CPC population in the CVB-infected heart resulted in impaired adaptive vascular remodeling. This impairment in vascular remodeling does not allow for proper perfusion of the myocardium under load, leaving the muscle ischemic and triggering pathological hypertrophy and cardiac damage (Figure 6).
Figure 6. Model of adult heart failure in juvenile CVB-infected mice.

(A) A population of CPCs susceptible to CVB infection resides within the myocardium. Upon augmented cardiac stress, oxygen demand increases within the heart tissue. CPCs are recruited to drive angiogenesis and neovascularization which increases vascular density in the muscle allowing for efficient perfusion of oxygenated blood. (B) When the heart undergoes mild CVB infection, CPCs are preferentially targeted by the virus resulting in a depletion of the CPC population; however the myocardium is otherwise normal. Following cardiac stress, the limited number of remaining CPCs cannot sufficiently stimulate blood vessel formation and the myocardium becomes ischemic. The lack of vascularization causes the heart to become hypertrophic resulting in scar formation and cardiac dysfunction.
Targeting of CPCs by CVB is not entirely unexpected based on the known susceptibility of progenitor cells to infection (Feuer, Pagarigan et al., 2005) (Feuer & Whitton, 2008) (Feuer, Mena et al., 2002) (Willcox, Richardson et al., 2011). CVB may show preferential tropism for progenitor cells, and infection may alter cell lineage commitment or diminish their restorative capacity (Feuer, Mena et al., 2003) (Feuer, Pagarigan et al., 2005) (Tabor-Godwin, Tsueng et al., 2012) (Feuer & Whitton, 2008) (Rhoades, Tabor-Godwin et al., 2011) (Ruller, Tabor-Godwin et al., 2012) (Tsueng, Tabor-Godwin et al., 2011) (Althof & Whitton, 2012). Infection of progenitor cells may also lead to augmented virus dispersal via autophagosome-mediated exit without lysis (AWOL) (Robinson, Tsueng et al., 2014) (Jackson, Giddings, Jr. et al., 2005b). Given that the heart is comprised primarily of post-mitotic myocytes, a pool of cycling CPCs would provide optimal targets for infection. The ultimate fate of infected progenitor cells is unknown, although infection caused premature differentiation of CPCs towards a vascular lineage.
Escape from the Innate Antiviral Immune Response
CVBs and other EVs have evolved many unique mechanisms to evade the host immune response (Harris & Coyne, 2013) (Feng, Langereis et al., 2014b) (Kemball, Alirezaei et al., 2010b). For example, CVB viral proteases which include 3C and 2A proteinases have been shown to attenuate the Type I IFN response by cleaving focal adhesion kinase, MDA5, RIG-1, and MAVS host proteins (Bozym, Delorme-Axford et al., 2012) (Mukherjee, Morosky et al., 2011) (Feng, Langereis et al., 2014a). A common feature following any infection of the host cell includes the induction of the stress response which acts against viral infection by inhibiting protein synthesis (Lloyd, 2012). Viral infection can trigger stress granules (SG) which comprise translationally silenced messenger ribonucleoproteins thereby inhibiting the viral genome from being translated (Onomoto, Yoneyama et al., 2014) (Lloyd, 2013). Poliovirus 3C proteinase can cleave RasGAP-SH3-binding protein (G3BP), a necessary component of SG formation (Reineke & Lloyd, 2015). CVB, similar to poliovirus, disrupted of processing bodies (P bodies) involved in decapping, decay, and translational silencing of mRNA (Dougherty, White et al., 2011). P bodies contain Xrn1, Dcp1a, and Pan3 proteins which play a role in 5’-end mRNA decapping and degradation. These proteins were found to be degraded in target cells following infection with CVB. The degradation of key components of P bodies may provide a mechanism for CVB to replicate to high levels in the host cell despite the initiation of the stress response following infection.
CVB also can antagonize the apoptotic pathway in cells, allowing viral replication to proceed for a longer amount of time necessary to maximize progeny (Harris & Coyne, 2014). For example, CVB can cleave cell components of the pro-apoptotic family, including TRIF (Mukherjee, Morosky et al., 2011), and viral 2B protein can act as viroporin disrupting Ca2+ gradients necessary to initiate apoptosis (Campanella, de Jong et al., 2004). Evasion of the host antiviral response maximizes viral replication during acute infection and may also be critical for the establishment of viral persistence. Recently, in vivo ablation of type I interferon receptor in cardiomyocytes was shown to accelerate myocarditis, although infection in cardiac tissue remained highly focal (Althof, Harkins et al., 2014). These results indicate that other unidentified antiviral factors may prevent more widespread dissemination within the heart.
A recent study has clarified the role of matrix metalloproteinase-12 (MMP-12) in antiviral immunity (Marchant, Bellac et al., 2014) against CVB. Although interferon-α (IFN-α) is essential for antiviral immunity, activated iκBα (encoded by NFKBIA) is necessary for the export of IFN-α from virus-infected cells. MMP-12 mediates NFKBIA transcription which induces IFN-α secretion and protection from CVB infection. Simultaneously, MMP-12 limits the antiviral immune response by cleaving the IFN-α receptor 2 binding site. However, a membrane-impermeable MMP-12 inhibitor was shown to increase IFN-α levels and reduce CVB titers in the pancreas. These studies suggest that modulation of the antiviral response with inhibitors against MMP-12 may assist in controlling CVB infection in the host.
Few studies have inspected the ability of progenitor cells to mount effective antiviral responses and be protected from microbial infection. Utilizing our in vivo model of CNS infection, unique host immune gene expression changes were observed for CVB compared to a different neurotropic virus - LCMV - an arenavirus considered to activate the prototypical immune response in the host (Puccini, Ruller et al., 2014). CCL12, CCL7, CCL4, CXCL4, and CCL2 were upregulated at early time points following CVB infection. In contrast, MHC class I gene expression, several developmental-related Hox genes, and TTR were specifically downregulated after CVB infection. Intriguingly, toll-like receptors have been found to modulate adult hippocampal neurogenesis via MyD88 activation (Rolls, Shechter et al., 2007), and CVB infection in the developing CNS might alter normal development both by targeting progenitor cells (Ruller, Tabor-Godwin et al., 2012), and by inducing a local inflammatory antiviral response.
Adaptive immune response following CVB infection
The significant contribution of a neutralizing antibody response in controlling EV infections can be seen in reports of individuals suffering from agammaglobulimia who develop chronic neuropathies following CVB infection (Misbah, Spickett et al., 1992). Antibodies were shown to be vital for clearing infectious virus in mice lacking B cells (Mena, Perry et al., 1999). B cells also contribute to virus dissemination via the “Trojan horse” dissemination. High levels of viral RNA were observed within the marginal zone of the spleen suggesting active infection of proliferating lymphocytes. Activated microglia and macrophages also contribute to the host response against infection by actively engulfed virally-infected cells (Feuer, Ruller et al., 2009).
Activation of the T cell response in the host may be dependent on the EV genus (Slifka, Pagarigan et al., 2001). Although T cells are critical to controlling viral infections, CVB has evolved numerous strategies to escape CD8+ T cells, for example, by inhibiting MHC class I antigen presentation (Kemball, Harkins et al., 2009). CVB has also been shown to alter the stimulatory capacity of dendritic cells which may impair the host’s ability to induce protective antiviral T cell responses (Kemball, Flynn et al., 2012). Also, CVB can infect the bone marrow and erythroid and lymphoid progenitor cell populations, further impacting host immune responses (Althof & Whitton, 2012).
CVB Vaccines and Antiviral Candidates
Many researchers have developed effective vaccines against CVB using various vaccines strategies including DNA plasmids expressing viral proteins, inactivated virus, or live attenuated forms of virus - although no clinically available vaccine currently exists. A safe and effective vaccine based on RNA was shown to protect mice against virus challenge, although no neutralizing antibodies were detected (Hunziker, Harkins et al., 2004). RNA-based vaccines may be safer than DNA vaccines based on their inability to integrate into cellular DNA. Attenuated viruses have also been shown to be protective against lethal re-challenge in mice (Dan & Chantler, 2005). Recombinant plasmids expressing capsid proteins following in vivo electroporation can induce protective virus-specific antibodies (Park, Kim et al., 2009). Novel virus receptor traps have been designed based on soluble virus receptor fusion proteins which were able to reduce myocardial inflammation, fibrosis, and viral titers in CVB-infected mice (Lim, Choi et al., 2006).
Neonatal patients infected with EVs have been treated with immunoglobulin, although few studies have shown clinical efficacy which remains controversial based on low antibody titers and intratypic variation against particular serotypes circulating within a community (Abzug, 2004) (Galama, Vogels et al., 1997). The method of immunoglobulin preparation involves pooling plasma products from normal donors (Cheng, Chen et al., 2008), and non-specific anti-inflammatory, yet protective responses may be induced following intravenous immunoglobulin treatment (Ooi, Wong et al., 2010).
RNA interference (RNAi)-based strategies have been used to limit CVB replication in culture, and in vivo. The potential to utilize RNAi as an effective antiviral drug against RNA viruses was first shown in 2003 (Gitlin & Andino, 2003). RNAi-based immunity against viral infection is dependent upon Dicer recognition of the viral dsRNA formed during viral replication (Aliyari & Ding, 2009). Small interfering RNAs (siRNAs) directed against protease 2A were shown to inhibit CVB infection in HeLa cells and murine cardiomyocytes (Yuan, Cheung et al., 2005). siRNA molecules designed to target the CVB viral 2A region successfully reduced viral titers and prolonged survival in susceptible mice (Merl, Michaelis et al., 2005). Also, siRNA molecules designed to target the viral 3CPro region of CVB reduced viral replication without showing signs of toxicity (Tan, Wong et al., 2010). Based on the ability of RNA viruses to quickly evolve and become resistant to siRNA molecules, other researchers have combined three different antiviral siRNA molecules to limit the appearance of CVB escape mutants (Merl & Wessely, 2007). In contrast, targeting host host-specific proteins may circumvent the appearance of CVB escape mutants yet reduce viral replication, for example, in cells treated with CAR-specific siRNA molecules (Werk, Schubert et al., 2005).
Ribavirin (1-(_-d-ribofuranosyl)-1H-1,2,4-triazole-3-carboxamide) is a broad-spectrum antiviral compound initially proposed as a nucleoside inhibitor, although more recently shown to work by acting as a mutagen and inducing ‘error catastrophe’ during EV replication (Crotty, Maag et al., 2000) (Crotty & Andino, 2002) (Crotty, Cameron et al., 2001) (Vignuzzi, Stone et al., 2006). We have shown previously that ribavirin could and improve brain wet weight recovery during persistent infection in the CNS (Ruller, Tabor-Godwin et al., 2012). Pleconaril,3-(3,5-dimethyl-4((3-(3-methyl-5-isoxazolyl)propyl]oly)phenyl)-5-(trifluoromethyl)-1,2,4-oxadiazole was originally developed as an anti-picornaviral drug which works by preventing virions from attaching to host cells (Pevear, Tull et al., 1999) (Chen, Weng et al., 2008). Pleconaril was found to be an effective antiviral compound against CVB strains having isoleucine or valine at position 1092 in the VP1 region, whereas leucine at this position was associated with resistance (Schmidtke, Hammerschmidt et al., 2005).
More recently, novel antiviral candidates have been designed against CVB, and EVs in general, by taking advantage of new discoveries regarding virion morphogenesis. Glutathione (GSH), the most prevalent non-protein thiol in the animal cell, was identified as an essential stabilizing cofactor during virion particle formation (Thibaut, van der Linden et al., 2014). A newly discovered inhibitor, TP219, binds GSH and depletes intracellular stores, thereby interfering with virus assembly but not RNA replication. Additional antiviral candidates, such as 1, 2-fluoro-4-(2-methyl-8-(3-(methylsulfonyl) benzylamino) imidazo(1,2-a)pyrazin-3-yl)phenol, have been developed which directly inhibit phosphatidylinositol 4-kinase, an enzyme essential for EV replication (van der Schaar, Leyssen et al., 2013). Also, Itraconazole has been identified as a broad-spectrum inhibitor of EVs by interfering with oxysterol-binding protein (OSBP) and OSBP-related protein 4 functions (Strating, van der Linden et al., 2015). Knockdown of these proteins has been shown to inhibit EV replication by preventing the shuttling of cholesterol and phosphatidylinositol-4-phoshpate between membranes during the formation of viral replication organelles.
Fluoxetine, a selective serotonin reuptake inhibitor, was identified through screening of small molecule libraries as an effective inhibitor of EV replication (Zuo, Quinn et al., 2012). Also, Fluoxetine may show efficacy during a persistent infection, and this antiviral molecule was recently shown to “cure” human pancreatic cells infected in culture with CVB (Alidjinou, Sane et al., 2015). The mechanism of action remains unclear, and utilizing fluoxetine as an antiviral may be problematic based on its neurological effects on serotonin uptake and involvement with an increased risk of bleeding when given with inhibitors of platelet function (Alderman, Moritz et al., 1992). We can expect some antiviral drugs to have greater efficacy or side effects based on genetic differences between individuals. The concept of host molecular profiling and personalized medicine to treat medical illnesses, including viral infections, will be a critical field in the future (Law, Korth et al., 2013). With the advent of induced pluripotent stem cells (iPSCs) and the generation of cardiomyocytes derived from iPSCs, the efficacy of antiviral compounds and CVB-induced viral myocarditis can be quantitatively assessed on patient-derived cardiomyocytes (Sharma, Marceau et al., 2014).
Cleavage of Host Proteins by CVB Proteases
In addition to host proteins required for autophagy and the host antiviral response, CVB has been shown to cleave dystrophin, a vital protein in the heart which supports muscle fiber strength (Badorff, Berkely et al., 2000). Cleavage of dystrophin by CVB viral 2A protease following infection is thought to contribute to dilated cardiomyopathy following infection (Badorff, Lee et al., 1999). Transgenic mice were generated replacing the dystrophin gene with a variant gene no longer containing a CVB viral 2A protease cleavage site. These transgenic mice showed reduced cardiac virus titers following infection and did not suffer from dilated cardiomyopathy normally induced by cardiomyocyte-restricted expression of the CVB viral 2A protease (Lim, Peter et al., 2013).
The targeting of multiple host proteins by CVB viral proteases demonstrates the incredible ability of RNA viruses of limited size to engineer proteins serving multiple functions during replication. Nevertheless, the precise targeting of host proteins and evasion of the host antiviral immune response by viral proteases necessary to maximize viral replication and dissemination might also restrict the ability of CVB, and other EVs, to form a diverse quasispecies cloud to quickly adapt to new environments. The design of inhibitors against CVB proteases might provide an opportunity to limit viral replication and reduce virus-associated pathology. A soluble inhibitor of CVB 3C protease was shown to prevent cardiomyopathy following infection (Lim, Yun et al., 2014).
CVB RNA persistence and chronic disease
Intriguingly, persistent CVB infections have been linked to autoimmune-type diseases such as chronic myocarditis (Chapman & Kim, 2008), diabetes (Sane, Moumna et al., 2011), and chronic inflammatory myopathy (Tam, Fontana et al., 2003). Virus persistence in target tissues is associated with chronic disease, although the mechanism of persistence is not clear and may involve the continued presence of viral RNA rather than active virus replication. Nevertheless, the presence of replication-restricted viral RNA has been shown to contribute to the disease process possibly following the production of viral proteinases or induction of innate immunity against the viral genome (Wessely, Klingel et al., 1998). Numerous studies suggest that persistent viruses, especially those such as CVB, may provide chronic inflammatory events whereby autoreactive T cells become stimulated and secrete inflammatory cytokines through a variety of potential mechanisms, including molecular mimicry and bystander activation (Oldstone, 1998) (Horwitz, Bradley et al., 1998). It is not clear what effect persistent infection and the associated inflammatory events might have on resident progenitor cells, either during development, or in the adult.
Some studies have suggested that the lack of infectious virus during the persistent stage of infection indicates that CVB-associated diseases, which include myocarditis, occur through autoimmune mechanisms. However in a recent study, myocarditis failed to appear in mice lacking CAR expression specifically on cardiomyocytes - suggesting myocarditis requires that cardiomyocytes become infected (Shi, Chen et al., 2009). Since other tissues, such as the pancreas, were readily infected in their model, putative autoreactive T cells against cardiac proteins could have been produced yet failed to appear. These results cast doubt on the notion that CVB infection induces a cross-reactive immune response against cardiac proteins. Rather direct viral infection causing cellular damage and the accompanying virus-mediated immune response greatly contribute to the disease process in infected cardiac tissue.
Few have considered the possibility that RNA viruses may establish a “latent” infection with periodic reactivation – more commonly observed for retroviruses or for DNA viruses in the Herpesviridae family (Feuer, Ruller et al., 2009) (Feuer & Whitton, 2008). We previously published studies suggesting that CVB remains in a “latent” state in quiescent tissue culture cells (Feuer, Mena et al., 2004) (Feuer, Mena et al., 2002). Also, CVB readily establishes a carrier-state infection in cells grown in culture, including HL-1 cells and NPCs continuously passaged in culture (Pinkert, Klingel et al., 2011) (Tsueng, Tabor-Godwin et al., 2011) (Tsueng, Rhoades et al., 2014). We hypothesize that neurogenic regions of the CNS may support CVB persistent infection, and virus reactivation may result upon intermittent progenitor cell expansion and proliferation (Rhoades, Tabor-Godwin et al., 2011).
Viruses with RNA-based genomes tend to be less stable, although some RNA viruses may have developed sophisticated stabilization strategies by limiting RNA decay (Iwakawa, Mizumoto et al., 2008) or forming double-stranded RNA complexes (Tam & Messner, 1999). The molecular mechanism of CVB persistence with restricted viral replication in the heart and pancreas may involve the generation of noncytolytic variants harboring 5’ terminal mutations and deletions (Lee, Kono et al., 2005) (Kim, Tracy et al., 2005) (Chapman & Kim, 2008) (Chapman, Kim et al., 2008) (Tracy, Smithee et al., 2015). The sporadic expression of viral proteins during CVB persistence in the absence of significant viral replication may nevertheless lead to a chronic immune response and immuno-pathology (Whitton & Feuer, 2004).
Recent studies have shown the clinical dangers of suppressing the humoral immune response with anti-CD20 monoclonal antibodies such as rituximab. Treatment with rituximab is routinely given to patients suffering from lymphomas, leukemias, transplant rejection and some autoimmune disorders. However, case reports describing EV meningoencephalitis following treatment with rituximab have been increasing in number (Servais, Caers et al., 2010;Schilthuizen, Berenschot et al., 2010). B cell-dependent immunosuppression following the administration rituximab as a therapy for lymphomas or leukemias would naturally reduce the level of circulating anti-CVB antibodies. If these protective antibodies suppress CVB replication in target tissues such as the CNS harboring persistent viral RNA, meningoencephalitis might be the outcome for some patients (Kiani-Alikhan, Skoulidis et al., 2009).
Conclusions and Future Perspectives
Although recent discoveries have been made regarding determinants of CVB tropism, host proteins involved in CVB replication in target cells, and mechanisms of CVB pathogenesis; many questions remain unanswered. Also, newer and more specific antiviral therapies need to be pursued in order to provide a catalogue of treatment strategies to control both acute and persistent infection. With the advent of stem cell therapy, questions remain if the administration of progenitor cells in tissues harboring persistent infectious agents such as CVB might provide new target cells thereby limiting potential success of tissue regeneration, including in the compromised heart. In addition, can new treatment strategies be devised to control chronic inflammatory response or reactivation during persistent CVB infection? What are the lasting effects of CVB infection on the host, particularly following the infection and recovery of progenitor cells? CVB clearly utilizes autophagy to replicate, but also simultaneously commandeers host proteins such as GBF1, Arf1, and PI4KIIIβ to construct viral replication organelles. How can studies describing induction of autophagy and the formation of virus replication organelles be unified in order to fully comprehend CVB replication within the host cell (Jackson, 2014)?
Is there an inherent need for CVB to access the autophagy pathway to complete the necessary steps of viral replication - such as for virion maturation? Although CVB titers are greatly reduced in culture when inhibiting the autophagy pathway (Wong, Zhang et al., 2008b), or upon infection of mice lacking ATG5 (Alirezaei, Flynn et al., 2012b), viral replication can still proceed – suggesting that a strict requirement for autophagy is not an absolute necessity. Alternatively, perhaps CVB evolved to utilize autophagy for a greater benefit – such as for the fabrication of camouflage vesicles engineered to remain within host cells for a longer period, and for the construction of escape pods to eventually leave host cells (Richards & Jackson, 2013). Can new therapies against CVB be designed based on personalized medicine and having limited toxicity/side effects? Can novel antiviral drugs be identified that target the formation of viral replication complexes or hinder virus-induced autophagy activation? What are the molecular factors that might assist CVB-associated EMVs to enter new target cells? Do CVB-associated EMVs broaden viral tropism within the host? Hence, more research is needed to better understand the mechanisms of CVB-mediated disease in the host and devising the best treatment strategies for patients.
Acknowledgements
This work was supported by National Institutes of Health (NIH) R01 Award NS054108 (to R.F.), and NIH R01 Award HL092136 (to R.A.G.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. No conflicts of interest exist between the subject matter and the authors included in the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Abzug MJ. Presentation, diagnosis, and management of enterovirus infections in neonates. Paediatr.Drugs. 2004;6:1–10. doi: 10.2165/00148581-200406010-00001. [DOI] [PubMed] [Google Scholar]
- Ahn J, Jee Y, Seo I, Yoon SY, Kim D, Kim YK, Lee H. Primary neurons become less susceptible to coxsackievirus B5 following maturation: the correlation with the decreased level of CAR expression on cell surface. J.Med.Virol. 2008;80(3):434–440. doi: 10.1002/jmv.21100. [DOI] [PubMed] [Google Scholar]
- Alderman CP, Moritz CK, Ben-Tovim DI. Abnormal platelet aggregation associated with fluoxetine therapy. Ann.Pharmacother. 1992;26:1517–1519. doi: 10.1177/106002809202601205. [DOI] [PubMed] [Google Scholar]
- Alidjinou EK, Sane F, Bertin A, Caloone D, Hober D. Persistent infection of human pancreatic cells with Coxsackievirus B4 is cured by fluoxetine. Antiviral Res. 2015:10. doi: 10.1016/j.antiviral.2015.01.010. [DOI] [PubMed] [Google Scholar]
- Alirezaei M, Flynn CT, Whitton JL. Interactions between enteroviruses and autophagy in vivo. Autophagy. 2012a;8:973–975. doi: 10.4161/auto.20160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alirezaei M, Flynn CT, Wood MR, Whitton JL. Pancreatic acinar cell-specific autophagy disruption reduces coxsackievirus replication and pathogenesis in vivo. Cell Host.Microbe. 2012b;11:298–305. doi: 10.1016/j.chom.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alirezaei M, Kemball CC, Flynn CT, Wood MR, Whitton JL, Kiosses WB. Short-term fasting induces profound neuronal autophagy. Autophagy. 2010;6(6):702–710. doi: 10.4161/auto.6.6.12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliyari R, Ding SW. RNA-based viral immunity initiated by the Dicer family of host immune receptors. Immunol.Rev. 2009;227:176–188. doi: 10.1111/j.1600-065X.2008.00722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Althof N, Harkins S, Kemball CC, Flynn CT, Alirezaei M, Whitton JL. In vivo ablation of type I interferon receptor from cardiomyocytes delays coxsackieviral clearance and accelerates myocardial disease. J.Virol. 2014;88:5087–5099. doi: 10.1128/JVI.00184-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Althof N, Whitton JL. Coxsackievirus B3 infects the bone marrow and diminishes the restorative capacity of erythroid and lymphoid progenitors. J.Virol. 2012;87:2823–2834. doi: 10.1128/JVI.03004-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnesjo B, Eden T, Ihse I, Nordenfelt E, Ursing B. Enterovirus infections in acute pancreatitis - a possible etiological connection. Scand.J.Gastroenterol. 1976;11:645–649. [PubMed] [Google Scholar]
- Badorff C, Berkely N, Mehrotra S, Talhouk JW, Rhoads RE, Knowlton KU. Enteroviral protease 2A directly cleaves dystrophin and is inhibited by a dystrophin-based substrate analogue. J.Biol.Chem. 2000;275:11191–11197. doi: 10.1074/jbc.275.15.11191. [DOI] [PubMed] [Google Scholar]
- Badorff C, Lee GH, Lamphear BJ, Martone ME, Campbell KP, Rhoads RE, Knowlton KU. Enteroviral protease 2A cleaves dystrophin: evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat.Med. 1999;5:320–326. doi: 10.1038/6543. [DOI] [PubMed] [Google Scholar]
- Belov GA, Altan-Bonnet N, Kovtunovych G, Jackson CL, Lippincott-Schwartz J, Ehrenfeld E. Hijacking components of the cellular secretory pathway for replication of poliovirus RNA. J.Virol. 2007;81:558–567. doi: 10.1128/JVI.01820-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov GA, van Kuppeveld FJ. (+)RNA viruses rewire cellular pathways to build replication organelles. Curr.Opin.Virol. 2012;2:740–747. doi: 10.1016/j.coviro.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergelson JM. Intercellular junctional proteins as receptors and barriers to virus infection and spread. Cell Host.Microbe. 2009;5:517–521. doi: 10.1016/j.chom.2009.05.009. [DOI] [PubMed] [Google Scholar]
- Bergelson JM, Chan M, Solomon KR, St John NF, Lin H, Finberg RW. Decay-accelerating factor (CD55), a glycosylphosphatidylinositol-anchored complement regulatory protein, is a receptor for several echoviruses. Proc.Natl.Acad.Sci.U.S.A. 1994;91:6245–6248. doi: 10.1073/pnas.91.13.6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science. 1997;275:1320–1323. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- Bird SW, Maynard ND, Covert MW, Kirkegaard K. Nonlytic viral spread enhanced by autophagy components. Proc.Natl.Acad.Sci.U.S.A. 2014;111:13081–13086. doi: 10.1073/pnas.1401437111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozym RA, Delorme-Axford E, Harris K, Morosky S, Ikizler M, Dermody TS, Sarkar SN, Coyne CB. Focal Adhesion Kinase Is a Component of Antiviral RIG-I-like Receptor Signaling. Cell Host.Microbe. 2012;11:153–166. doi: 10.1016/j.chom.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanella M, de Jong AS, Lanke KW, Melchers WJ, Willems PH, Pinton P, Rizzuto R, van Kuppeveld FJ. The coxsackievirus 2B protein suppresses apoptotic host cell responses by manipulating intracellular Ca2+ homeostasis. J.Biol.Chem. 2004;279:18440–18450. doi: 10.1074/jbc.M309494200. [DOI] [PubMed] [Google Scholar]
- Chamberlain RN, Christie PN, Holt KS, Huntley RM, Pollard R, Roche MC. A study of school children who had identified virus infections of the central nervous system during infancy. Child Care Health Dev. 1983;9:29–47. doi: 10.1111/j.1365-2214.1983.tb00301.x. [DOI] [PubMed] [Google Scholar]
- Chang LY, Huang LM, Gau SS, Wu YY, Hsia SH, Fan TY, Lin KL, Huang YC, Lu CY, Lin TY. Neurodevelopment and cognition in children after enterovirus 71 infection. N.Engl.J.Med. 2007;356:1226–1234. doi: 10.1056/NEJMoa065954. [DOI] [PubMed] [Google Scholar]
- Chapman NM, Kim KS. Persistent coxsackievirus infection: enterovirus persistence in chronic myocarditis and dilated cardiomyopathy. Curr.Top.Microbiol.Immunol. 2008;323:275–292. doi: 10.1007/978-3-540-75546-3_13. 275-92. [DOI] [PubMed] [Google Scholar]
- Chapman NM, Kim KS, Drescher KM, Oka K, Tracy S. 5' terminal deletions in the genome of a coxsackievirus B2 strain occurred naturally in human heart. Virology. 2008;375:480–491. doi: 10.1016/j.virol.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TC, Weng KF, Chang SC, Lin JY, Huang PN, Shih SR. Development of antiviral agents for enteroviruses. J.Antimicrob.Chemother. 2008;62:1169–1173. doi: 10.1093/jac/dkn424. [DOI] [PubMed] [Google Scholar]
- Chen YH, Du W, Hagemeijer MC, Takvorian PM, Pau C, Cali A, Brantner CA, Stempinski ES, Connelly PS, Ma HC, Jiang P, Wimmer E, Altan-Bonnet G, Altan-Bonnet N. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell. 2015;160:619–630. doi: 10.1016/j.cell.2015.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng MF, Chen BC, Huang TS, Hsieh KS, Chen SN, Liu YC. Clinical application of reverse-transcription polymerase chain reaction and intravenous immunoglobulin for enterovirus encephalitis. Jpn.J.Infect.Dis. 2008;61:18–24. [PubMed] [Google Scholar]
- Cheng TO. Viral myocarditis is a frequent cause of idiopathic dilated cardiomyopathy. Int.J.Cardiol. 2006;109:270. doi: 10.1016/j.ijcard.2005.04.019. [DOI] [PubMed] [Google Scholar]
- Chiriboga-Klein S, Oberfield SE, Casullo AM, Holahan N, Fedun B, Cooper LZ, Levine LS. Growth in congenital rubella syndrome and correlation with clinical manifestations. J.Pediatr. 1989;115:251–255. doi: 10.1016/s0022-3476(89)80073-3. [DOI] [PubMed] [Google Scholar]
- Christen U, Bender C, von Herrath MG. Infection as a cause of type 1 diabetes? Curr.Opin.Rheumatol. 2012;24:417–423. doi: 10.1097/BOR.0b013e3283533719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell CT, Kiosses WB, Harkins S, Whitton JL. Inhibition of protein trafficking by coxsackievirus b3: multiple viral proteins target a single organelle. J.Virol. 2006;80:6637–6647. doi: 10.1128/JVI.02572-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell CT, Kiosses WB, Harkins S, Whitton JL. Coxsackievirus B3 proteins directionally complement each other to downregulate surface major histocompatibility complex class I. J.Virol. 2007;81:6785–6797. doi: 10.1128/JVI.00198-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne CB, Bergelson JM. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell. 2006;124(1):119–131. doi: 10.1016/j.cell.2005.10.035. [DOI] [PubMed] [Google Scholar]
- Coyne CB, Shen L, Turner JR, Bergelson JM. Coxsackievirus entry across epithelial tight junctions requires occludin and the small GTPases Rab34 and Rab5. Cell Host.Microbe. 2007;2:181–192. doi: 10.1016/j.chom.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cree BC, Bernardini GL, Hays AP, Lowe G. A fatal case of coxsackievirus B4 meningoencephalitis. Arch.Neurol. 2003;60:107–112. doi: 10.1001/archneur.60.1.107. [DOI] [PubMed] [Google Scholar]
- Crotty S, Andino R. Implications of high RNA virus mutation rates: lethal mutagenesis and the antiviral drug ribavirin. Microbes.Infect. 2002;4:1301–1307. doi: 10.1016/s1286-4579(02)00008-4. [DOI] [PubMed] [Google Scholar]
- Crotty S, Cameron CE, Andino R. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc.Natl.Acad.Sci.U.S.A. 2001;98:6895–6900. doi: 10.1073/pnas.111085598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S, Maag D, Arnold JJ, Zhong W, Lau JY, Hong Z, Andino R, Cameron CE. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat.Med. 2000;6:1375–1379. doi: 10.1038/82191. [DOI] [PubMed] [Google Scholar]
- Dan M, Chantler JK. A genetically engineered attenuated coxsackievirus B3 strain protects mice against lethal infection. J.Virol. 2005;79(14):9285–9295. doi: 10.1128/JVI.79.14.9285-9295.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David P, Baleriaux D, Bank WO, Amrom D, De TD, Babusiaux C, Matos C, Van SC, Lloret-Pastor C, Szliwowski HB. MRI of acute disseminated encephalomyelitis after coxsackie B infection. J.Neuroradiol. 1993;20:258–265. [PubMed] [Google Scholar]
- de Jong AS, Visch HJ, de MF, van Dommelen MM, Swarts HG, Luyten T, Callewaert G, Melchers WJ, Willems PH, van Kuppeveld FJ. The coxsackievirus 2B protein increases efflux of ions from the endoplasmic reticulum and Golgi, thereby inhibiting protein trafficking through the Golgi. J.Biol.Chem. 2006;281:14144–14150. doi: 10.1074/jbc.M511766200. %19. [DOI] [PubMed] [Google Scholar]
- Delorme-Axford E, Coyne CB. The actin cytoskeleton as a barrier to virus infection of polarized epithelial cells. Viruses. 2011;3:2462–2477. doi: 10.3390/v3122462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorme-Axford E, Donker RB, Mouillet JF, Chu T, Bayer A, Ouyang Y, Wang T, Stolz DB, Sarkar SN, Morelli AE, Sadovsky Y, Coyne CB. Human placental trophoblasts confer viral resistance to recipient cells. Proc.Natl.Acad.Sci.U.S.A. 2013;110:12048–12053. doi: 10.1073/pnas.1304718110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorme-Axford E, Morosky S, Bomberger J, Stolz DB, Jackson WT, Coyne CB. BPIFB3 regulates autophagy and coxsackievirus B replication through a noncanonical pathway independent of the core initiation machinery. MBio. 2014;5:e02147–14. doi: 10.1128/mBio.02147-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson PW, Aldred AR, Marley PD, Bannister D, Schreiber G. Rat choroid plexus specializes in the synthesis and the secretion of transthyretin (prealbumin). Regulation of transthyretin synthesis in choroid plexus is independent from that in liver. J.Biol.Chem. 1986;261:3475–3478. [PubMed] [Google Scholar]
- Doceul V, Hollinshead M, van der Linden L, Smith GL. Repulsion of superinfecting virions: a mechanism for rapid virus spread. Science. 2010;327:873–876. doi: 10.1126/science.1183173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty JD, White JP, Lloyd RE. Poliovirus-mediated disruption of cytoplasmic processing bodies. J.Virol. 2011;85:64–75. doi: 10.1128/JVI.01657-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drescher KM, Kono K, Bopegamage S, Carson SD, Tracy S. Coxsackievirus B3 infection and type 1 diabetes development in NOD mice: insulitis determines susceptibility of pancreatic islets to virus infection. Virology. 2004;329:381–394. doi: 10.1016/j.virol.2004.06.049. [DOI] [PubMed] [Google Scholar]
- Dreux M, Garaigorta U, Boyd B, Decembre E, Chung J, Whitten-Bauer C, Wieland S, Chisari FV. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host.Microbe. 2012;12:558–570. doi: 10.1016/j.chom.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerich DF, Skinner SJ, Borlongan CV, Vasconcellos AV, Thanos CG. The choroid plexus in the rise, fall and repair of the brain. Bioessays. 2005;27:262–274. doi: 10.1002/bies.20193. [DOI] [PubMed] [Google Scholar]
- Eriksson C, Bjorklund A, Wictorin K. Neuronal differentiation following transplantation of expanded mouse neurosphere cultures derived from different embryonic forebrain regions. Exp.Neurol. 2003;184:615–635. doi: 10.1016/S0014-4886(03)00271-1. [DOI] [PubMed] [Google Scholar]
- Esfandiarei M, Luo H, Yanagawa B, Suarez A, Dabiri D, Zhang J, McManus BM. Protein kinase B/Akt regulates coxsackievirus B3 replication through a mechanism which is not caspase dependent. J.Virol. 2004;78:4289–4298. doi: 10.1128/JVI.78.8.4289-4298.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esfandiarei M, McManus BM. Molecular biology and pathogenesis of viral myocarditis. Annu.Rev.Pathol. 2008;3:127–155. doi: 10.1146/annurev.pathmechdis.3.121806.151534. 127-55. [DOI] [PubMed] [Google Scholar]
- Etchison D, Milburn SC, Edery I, Sonenberg N, Hershey JW. Inhibition of HeLa cell protein synthesis following poliovirus infection correlates with the proteolysis of a 220,000-dalton polypeptide associated with eucaryotic initiation factor 3 and a cap binding protein complex. J.Biol.Chem. 1982;257(24):14806–14810. [PubMed] [Google Scholar]
- Euscher E, Davis J, Holzman I, Nuovo GJ. Coxsackie virus infection of the placenta associated with neurodevelopmental delays in the newborn. Obstet.Gynecol. 2001;98:1019–1026. doi: 10.1016/s0029-7844(01)01625-8. [DOI] [PubMed] [Google Scholar]
- Falk A, Frisen J. Amphiregulin is a mitogen for adult neural stem cells. J.Neurosci.Res. 2002;69:757–762. doi: 10.1002/jnr.10410. [DOI] [PubMed] [Google Scholar]
- Feng Q, Langereis MA, Lork M, Nguyen M, Hato SV, Lanke K, Emdad L, Bhoopathi P, Fisher PB, Lloyd RE, van Kuppeveld FJ. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J.Virol. 2014a;88:3369–3378. doi: 10.1128/JVI.02712-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Langereis MA, van Kuppeveld FJ. Induction and suppression of innate antiviral responses by picornaviruses. Cytokine Growth Factor Rev. 2014b;25:577–585. doi: 10.1016/j.cytogfr.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Hensley L, McKnight KL, Hu F, Madden V, Ping L, Jeong SH, Walker C, Lanford RE, Lemon SM. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature. 2013:10. doi: 10.1038/nature12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Lemon SM. Peek-a-boo: membrane hijacking and the pathogenesis of viral hepatitis. Trends Microbiol. 2013 doi: 10.1016/j.tim.2013.10.005. %19. pii: S0966-842X, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuer R, Mena I, Pagarigan R, Slifka MK, Whitton JL. Cell cycle status affects coxsackievirus replication, persistence, and reactivation in vitro. J.Virol. 2002;76:4430–4440. doi: 10.1128/JVI.76.9.4430-4440.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuer R, Mena I, Pagarigan RR, Harkins S, Hassett DE, Whitton JL. Coxsackievirus B3 and the neonatal CNS: the roles of stem cells, developing neurons, and apoptosis in infection, viral dissemination, and disease. Am.J.Pathol. 2003;163:1379–1393. doi: 10.1016/S0002-9440(10)63496-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuer R, Mena I, Pagarigan RR, Hassett DE, Whitton JL. Coxsackievirus replication and the cell cycle: a potential regulatory mechanism for viral persistence/latency. Med.Microbiol.Immunol.(Berl) 2004;193:83–90. doi: 10.1007/s00430-003-0192-z. [DOI] [PubMed] [Google Scholar]
- Feuer R, Pagarigan RR, Harkins S, Liu F, Hunziker IP, Whitton JL. Coxsackievirus targets proliferating neuronal progenitor cells in the neonatal CNS. J.Neurosci. 2005;25:2434–2444. doi: 10.1523/JNEUROSCI.4517-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuer R, Ruller CM, An N, Tabor-Godwin JM, Rhoades RE, Maciejewski S, Pagarigan RR, Cornell CT, Crocker SJ, Kiosses WB, Pham-Mitchell N, Campbell IL, Whitton JL. Viral persistence and chronic immunopathology in the adult central nervous system following Coxsackievirus infection during the neonatal period. J.Virol. 2009;83:9356–9369. doi: 10.1128/JVI.02382-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuer R, Whitton JL. Preferential coxsackievirus replication in proliferating/activated cells: implications for virus tropism, persistence, and pathogenesis. Curr.Top.Microbiol.Immunol. 2008;323:149–173. doi: 10.1007/978-3-540-75546-3_7. [DOI] [PubMed] [Google Scholar]
- Ford Siltz LA, Viktorova EG, Zhang B, Kouiavskaia D, Dragunsky E, Chumakov K, Isaacs L, Belov GA. New small-molecule inhibitors effectively blocking picornavirus replication. J.Virol. 2014;88:11091–11107. doi: 10.1128/JVI.01877-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita K, Krishnakumar SS, Franco D, Paul AV, London E, Wimmer E. Membrane topography of the hydrophobic anchor sequence of poliovirus 3A and 3AB proteins and the functional effect of 3A/3AB membrane association upon RNA replication. Biochemistry. 2007;46(17):5185–5199. doi: 10.1021/bi6024758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galama JM, Vogels MT, Jansen GH, Gielen M, Heessen FW. Antibodies against enteroviruses in intravenous Ig preparations: great variation in titres and poor correlation with the incidence of circulating serotypes. J.Med.Virol. 1997;53:273–276. [PubMed] [Google Scholar]
- Gallo A, Tandon M, Alevizos I, Illei GG. The majority of microRNAs detectable in serum and saliva is concentrated in exosomes. PLoS.One. 2012;7:e30679. doi: 10.1371/journal.pone.0030679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamarnik AV, Andino R. Switch from translation to RNA replication in a positive-stranded RNA virus. Genes Dev. 1998;12(15):2293–2304. doi: 10.1101/gad.12.15.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitlin L, Andino R. Nucleic acid-based immune system: the antiviral potential of mammalian RNA silencing. J.Virol. 2003;77:7159–7165. doi: 10.1128/JVI.77.13.7159-7165.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JL, Simon AK, Prescott M, Menendez JA, Liu F, Wang F, Wang C, Wolvetang E, Vazquez-Martin A, Zhang J. Autophagy in stem cells. Autophagy. 2013:9. doi: 10.4161/auto.24132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson AB, Gottlieb RA. Recycle or die: the role of autophagy in cardioprotection. J.Mol.Cell Cardiol. 2008;44:654–661. doi: 10.1016/j.yjmcc.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, Amano A, Yoshimori T. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389–393. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- Harris KG, Coyne CB. Enter at your own risk: how enteroviruses navigate the dangerous world of pattern recognition receptor signaling. Cytokine. 2013;63:230–236. doi: 10.1016/j.cyto.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KG, Coyne CB. Death waits for no man--does it wait for a virus? How enteroviruses induce and control cell death. Cytokine Growth Factor Rev. 2014;25:587–596. doi: 10.1016/j.cytogfr.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henke A, Zell R, Ehrlich G, Stelzner A. Expression of immunoregulatory cytokines by recombinant coxsackievirus B3 variants confers protection against virus-caused myocarditis. J.Virol. 2001;75:8187–8194. doi: 10.1128/JVI.75.17.8187-8194.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold J, Andino R. Poliovirus RNA replication requires genome circularization through a protein-protein bridge. Mol.Cell. 2001;7(3):581–591. doi: 10.1016/S1097-2765(01)00205-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda T, Saitoh H, Masuko M, Katagiri-Abe T, Tominaga K, Kozakai I, Kobayashi K, Kumanishi T, Watanabe YG, Odani S, Kuwano R. The coxsackievirus-adenovirus receptor protein as a cell adhesion molecule in the developing mouse brain. Brain Res.Mol.Brain Res. 2000;77:19–28. doi: 10.1016/s0169-328x(00)00036-x. [DOI] [PubMed] [Google Scholar]
- Horwitz MS, Bradley LM, Harbertson J, Krahl T, Lee J, Sarvetnick N. Diabetes induced by Coxsackie virus: initiation by bystander damage and not molecular mimicry. Nat.Med. 1998;4:781–785. doi: 10.1038/nm0798-781. [DOI] [PubMed] [Google Scholar]
- Hotta Y, Honda T, Naito M, Kuwano R. Developmental distribution of coxsackie virus and adenovirus receptor localized in the nervous system. Brain Res.Dev.Brain Res. 2003;143:1–13. doi: 10.1016/s0165-3806(03)00035-x. [DOI] [PubMed] [Google Scholar]
- Hsu NY, Ilnytska O, Belov G, Santiana M, Chen YH, Takvorian PM, Pau C, van der Schaar H, Kaushik-Basu N, Balla T, Cameron CE, Ehrenfeld E, van Kuppeveld FJ, Altan-Bonnet N. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell. 2010;141:799–811. doi: 10.1016/j.cell.2010.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Zhang X, Ramil JM, Rikka S, Kim L, Lee Y, Gude NA, Thistlethwaite PA, Sussman MA, Gottlieb RA, Gustafsson AB. Juvenile exposure to anthracyclines impairs cardiac progenitor cell function and vascularization resulting in greater susceptibility to stress-induced myocardial injury in adult mice. Circulation. 2010;121:675–683. doi: 10.1161/CIRCULATIONAHA.109.902221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SC, Chang CL, Wang PS, Tsai Y, Liu HS. Enterovirus 71-induced autophagy detected in vitro and in vivo promotes viral replication. J.Med.Virol. 2009;81(7):1241–1252. doi: 10.1002/jmv.21502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunziker IP, Harkins S, Feuer R, Cornell CT, Whitton JL. Generation and analysis of an RNA vaccine that protects against coxsackievirus B3 challenge. Virology. 2004;330:196–208. doi: 10.1016/j.virol.2004.09.035. [DOI] [PubMed] [Google Scholar]
- Hyypia T, Kallajoki M, Maaronen M, Stanway G, Kandolf R, Auvinen P, Kalimo H. Pathogenetic differences between coxsackie A and B virus infections in newborn mice. Virus Res. 1993;27:71–78. doi: 10.1016/0168-1702(93)90113-2. [DOI] [PubMed] [Google Scholar]
- Ilnytska O, Santiana M, Hsu NY, Du WL, Chen YH, Viktorova EG, Belov G, Brinker A, Storch J, Moore C, Dixon JL, Altan-Bonnet N. Enteroviruses harness the cellular endocytic machinery to remodel the host cell cholesterol landscape for effective viral replication. Cell Host.Microbe. 2013;14:281–293. doi: 10.1016/j.chom.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inal JM, Jorfi S. Coxsackievirus B transmission and possible new roles for extracellular vesicles. Biochem.Soc.Trans. 2013;41:299–302. doi: 10.1042/BST20120272. [DOI] [PubMed] [Google Scholar]
- Iwakawa HO, Mizumoto H, Nagano H, Imoto Y, Takigawa K, Sarawaneeyaruk S, Kaido M, Mise K, Okuno T. A viral noncoding RNA generated by cis-element-mediated protection against 5'->3' RNA decay represses both cap-independent and cap-dependent translation. J.Virol. 2008;82:10162–10174. doi: 10.1128/JVI.01027-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson WT. Poliovirus-induced changes in cellular membranes throughout infection. Curr.Opin.Virol. 2014;9:67–73. doi: 10.1016/j.coviro.2014.09.007. doi: 10.1016/j.coviro.2014.09.007. Epub@2014 Oct 11., 67-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson WT, Giddings TH, Jr., Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS.Biol. 2005a;3(5):e156. doi: 10.1371/journal.pbio.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson WT, Giddings TH, Jr., Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS.Biol. 2005b;3(5):e156. doi: 10.1371/journal.pbio.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaidane H, Hober D. Role of coxsackievirus B4 in the pathogenesis of type 1 diabetes. Diabetes Metab. 2008;34:537–548. doi: 10.1016/j.diabet.2008.05.008. [DOI] [PubMed] [Google Scholar]
- Jensen KJ, Garmaroudi FS, Zhang J, Lin J, Boroomand S, Zhang M, Luo Z, Yang D, Luo H, McManus BM, Janes KA. An ERK-p38 subnetwork coordinates host cell apoptosis and necrosis during coxsackievirus B3 infection. Cell Host.Microbe. 2013;13:67–76. doi: 10.1016/j.chom.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Liu Y, Ma HC, Paul AV, Wimmer E. Picornavirus morphogenesis. Microbiol.Mol.Biol.Rev. 2014;78:418–437. doi: 10.1128/MMBR.00012-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallewaard NL, Zhang L, Chen JW, Guttenberg M, Sanchez MD, Bergelson JM. Tissue-specific deletion of the coxsackievirus and adenovirus receptor protects mice from virus-induced pancreatitis and myocarditis. Cell Host.Microbe. 2009;6:91–98. doi: 10.1016/j.chom.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno T, Kim K, Kono K, Drescher KM, Chapman NM, Tracy S. Group B coxsackievirus diabetogenic phenotype correlates with replication efficiency. J.Virol. 2006;80:5637–5643. doi: 10.1128/JVI.02361-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney MT, Cotton JM, Richardson PJ, Shah AM. Viral myocarditis and dilated cardiomyopathy: mechanisms, manifestations, and management. Postgrad.Med.J. 2001;77:4–10. doi: 10.1136/pmj.77.903.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemball CC, Alirezaei M, Flynn CT, Wood MR, Harkins S, Kiosses WB, Whitton JL. Coxsackievirus infection induces autophagy-like vesicles and megaphagosomes in pancreatic acinar cells in vivo. J.Virol. 2010a;84:12110–12124. doi: 10.1128/JVI.01417-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemball CC, Alirezaei M, Whitton JL. Type B coxsackieviruses and their interactions with the innate and adaptive immune systems. Future.Microbiol. 2010b;5:1329–1347. doi: 10.2217/fmb.10.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemball CC, Flynn CT, Hosking MP, Botten J, Whitton JL. Wild-type coxsackievirus infection dramatically alters the abundance, heterogeneity, and immunostimulatory capacity of conventional dendritic cells in vivo. Virology. 2012;429:74–90. doi: 10.1016/j.virol.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemball CC, Harkins S, Whitmire JK, Flynn CT, Feuer R, Whitton JL. Coxsackievirus B3 inhibits antigen presentation in vivo, exerting a profound and selective effect on the MHC class I pathway. PLoS.Pathog. 2009;5:e1000618. doi: 10.1371/journal.ppat.1000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiani-Alikhan S, Skoulidis F, Barroso A, Nuovo G, Ushiro-Lumb I, Breuer J, Lister A, Mattes F. Enterovirus infection of neuronal cells post-Rituximab. Br.J.Haematol. 2009;146:333–335. doi: 10.1111/j.1365-2141.2009.07748.x. [DOI] [PubMed] [Google Scholar]
- Kim DS, Kim H, Shim SH, Kim C, Song M, Kim YH, Jung YW, Nam JH. Coxsackievirus B3 used as a gene therapy vector to express functional FGF2. Gene Ther. 2012;19:1159–1165. doi: 10.1038/gt.2011.201. [DOI] [PubMed] [Google Scholar]
- Kim KS, Tracy S, Tapprich W, Bailey J, Lee CK, Kim K, Barry WH, Chapman NM. 5'-Terminal deletions occur in coxsackievirus B3 during replication in murine hearts and cardiac myocyte cultures and correlate with encapsidation of negative-strand viral RNA. J.Virol. 2005;79:7024–7041. doi: 10.1128/JVI.79.11.7024-7041.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkegaard K. Subversion of the cellular autophagy pathway by viruses. Curr.Top.Microbiol.Immunol. 2009;335:323–333. doi: 10.1007/978-3-642-00302-8_16. 323-33. [DOI] [PubMed] [Google Scholar]
- Kitamura N, Semler BL, Rothberg PG, Larsen GR, Adler CJ, Dorner AJ, Emini EA, Hanecak R, Lee JJ, van der Werf S, Anderson CW, Wimmer E. Primary structure, gene organization and polypeptide expression of poliovirus RNA. Nature. 1981;291(5816):547–553. doi: 10.1038/291547a0. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, Al-Zeer MA, Albert ML, Albin RL, Alegre-Abarrategui J, Aleo MF, Alirezaei M, Almasan A, Almonte-Becerril M, Amano A, Amaravadi R, Amarnath S, Amer AO, Andrieu-Abadie N, Anantharam V, Ann DK, Anoopkumar-Dukie S, Aoki H, Apostolova N, Auberger P, Baba M, Backues SK, Baehrecke EH, Bahr BA, Bai XY, Bailly Y, Baiocchi R, Baldini G, Balduini W, Ballabio A, Bamber BA, Bampton ET, Banhegyi G, Bartholomew CR, Bassham DC, Bast RC, Jr., Batoko H, Bay BH, Beau I, Bechet DM, Begley TJ, Behl C, Behrends C, Bekri S, Bellaire B, Bendall LJ, Benetti L, Berliocchi L, Bernardi H, Bernassola F, Besteiro S, Bhatia-Kissova I, Bi X, Biard-Piechaczyk M, Blum JS, Boise LH, Bonaldo P, Boone DL, Bornhauser BC, Bortoluci KR, Bossis I, Bost F, Bourquin JP, Boya P, Boyer-Guittaut M, Bozhkov PV, Brady NR, Brancolini C, Brech A, Brenman JE, Brennand A, Bresnick EH, Brest P, Bridges D, Bristol ML, Brookes PS, Brown EJ, Brumell JH, Brunetti-Pierri N, Brunk UT, Bulman DE, Bultman SJ, Bultynck G, Burbulla LF, Bursch W, Butchar JP, Buzgariu W, Bydlowski SP, Cadwell K, Cahova M, Cai D, Cai J, Cai Q, Calabretta B, Calvo-Garrido J, Camougrand N, Campanella M, Campos-Salinas J, Candi E, Cao L, Caplan AB, Carding SR, Cardoso SM, Carew JS, Carlin CR, Carmignac V, Carneiro LA, Carra S, Caruso RA, Casari G, Casas C, Castino R, Cebollero E, Cecconi F, Celli J, Chaachouay H, Chae HJ, Chai CY, Chan DC, Chan EY, Chang RC, Che CM, Chen CC, Chen GC, Chen GQ, Chen M, Chen Q, Chen SS, Chen W, Chen X, Chen X, Chen X, Chen YG, Chen Y, Chen Y, Chen YJ, Chen Z, Cheng A, Cheng CH, Cheng Y, Cheong H, Cheong JH, Cherry S, Chess-Williams R, Cheung ZH, Chevet E, Chiang HL, Chiarelli R, Chiba T, Chin LS, Chiou SH, Chisari FV, Cho CH, Cho DH, Choi AM, Choi D, Choi KS, Choi ME, Chouaib S, Choubey D, Choubey V, Chu CT, Chuang TH, Chueh SH, Chun T, Chwae YJ, Chye ML, Ciarcia R, Ciriolo MR, Clague MJ, Clark RS, Clarke PG, Clarke R, Codogno P, Coller HA, Colombo MI, Comincini S, Condello M, Condorelli F, Cookson MR, Coombs GH, Coppens I, Corbalan R, Cossart P, Costelli P, Costes S, Coto-Montes A, Couve E, Coxon FP, Cregg JM, Crespo JL, Cronje MJ, Cuervo AM, Cullen JJ, Czaja MJ, D'Amelio M, Darfeuille-Michaud A, Davids LM, Davies FE, De FM, de Groot JF, de Haan CA, De ML, De MA, De T, V, Debnath J, Degterev A, Dehay B, Delbridge LM, Demarchi F, Deng YZ, Dengjel J, Dent P, Denton D, Deretic V, Desai SD, Devenish RJ, Di GM, Di PG, Di PC, Diaz-Araya G, Diaz-Laviada I, Diaz-Meco MT, Diaz-Nido J, Dikic I, Dinesh-Kumar SP, Ding WX, Distelhorst CW, Diwan A, Djavaheri-Mergny M, Dokudovskaya S, Dong Z, Dorsey FC, Dosenko V, Dowling JJ, Doxsey S, Dreux M, Drew ME, Duan Q, Duchosal MA. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuss SK, Etheredge CA, Pfeiffer JK. Multiple host barriers restrict poliovirus trafficking in mice. PLoS.Pathog. 2008;4(6):e1000082. doi: 10.1371/journal.ppat.1000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laitinen OH, Honkanen H, Pakkanen O, Oikarinen S, Hankaniemi MM, Huhtala H, Ruokoranta T, Lecouturier V, Andre P, Harju R, Virtanen SM, Lehtonen J, Almond JW, Simell T, Simell O, Ilonen J, Veijola R, Knip M, Hyoty H. Coxsackievirus B1 is associated with induction of beta-cell autoimmunity that portends type 1 diabetes. Diabetes. 2014;63:446–455. doi: 10.2337/db13-0619. [DOI] [PubMed] [Google Scholar]
- Lamarche MJ, Borawski J, Bose A, Capacci-Daniel C, Colvin R, Dennehy M, Ding J, Dobler M, Drumm J, Gaither LA, Gao J, Jiang X, Lin K, McKeever U, Puyang X, Raman P, Thohan S, Tommasi R, Wagner K, Xiong X, Zabawa T, Zhu S, Wiedmann B. Anti-hepatitis C virus activity and toxicity of type III phosphatidylinositol-4-kinase beta inhibitors. Antimicrob.Agents Chemother. 2012;56:5149–5156. doi: 10.1128/AAC.00946-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanke KH, van der Schaar HM, Belov GA, Feng Q, Duijsings D, Jackson CL, Ehrenfeld E, van Kuppeveld FJ. GBF1, a guanine nucleotide exchange factor for Arf, is crucial for coxsackievirus B3 RNA replication. J.Virol. 2009;83:11940–11949. doi: 10.1128/JVI.01244-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law GL, Korth MJ, Benecke AG, Katze MG. Systems virology: host-directed approaches to viral pathogenesis and drug targeting. Nat.Rev.Microbiol. 2013;11:455–466. doi: 10.1038/nrmicro3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CK, Kono K, Haas E, Kim KS, Drescher KM, Chapman NM, Tracy S. Characterization of an infectious cDNA copy of the genome of a naturally occurring, avirulent coxsackievirus B3 clinical isolate. J.Gen.Virol. 2005;86:197–210. doi: 10.1099/vir.0.80424-0. [DOI] [PubMed] [Google Scholar]
- Leeper NJ, Hunter AL, Cooke JP. Stem cell therapy for vascular regeneration: adult, embryonic, and induced pluripotent stem cells. Circulation. 2010;122:517–526. doi: 10.1161/CIRCULATIONAHA.109.881441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim B, Yun S, Ju E, Kim B, Lee Y, Yoo D, Kim Y, Jeon E. Soluble Coxsackievirus B3 3C protease inhibitor prevents cardiomyopathy in an experimental chronic myocarditis murine model. Virus Res. 2014:10. doi: 10.1016/j.virusres.2014.11.030. [DOI] [PubMed] [Google Scholar]
- Lim BK, Choi JH, Nam JH, Gil CO, Shin JO, Yun SH, Kim DK, Jeon ES. Virus receptor trap neutralizes coxsackievirus in experimental murine viral myocarditis. Cardiovasc.Res. 2006;71:517–526. doi: 10.1016/j.cardiores.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Lim BK, Peter AK, Xiong D, Narezkina A, Yung A, Dalton ND, Hwang KK, Yajima T, Chen J, Knowlton KU. Inhibition of Coxsackievirus-associated dystrophin cleavage prevents cardiomyopathy. J.Clin.Invest. 2013;123:5146–5151. doi: 10.1172/JCI66271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RE. How do viruses interact with stress-associated RNA granules? PLoS Pathog. 2012;8:e1002741. doi: 10.1371/journal.ppat.1002741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RE. Regulation of stress granules and P-bodies during RNA virus infection. Wiley.Interdiscip.Rev.RNA. 2013;4:317–331. doi: 10.1002/wrna.1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loria RM, Shadoff N, Kibrick S, Broitman S. Maturation of intestinal defenses against peroral infection with group B coxsackievirus in mice. Infect.Immun. 1976;13:1397–1401. doi: 10.1128/iai.13.5.1397-1401.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Pompili VJ, Das H. Neovascularization and hematopoietic stem cells. Cell Biochem.Biophys. 2013;67:235–245. doi: 10.1007/s12013-011-9298-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchant DJ, Bellac CL, Moraes TJ, Wadsworth SJ, Dufour A, Butler GS, Bilawchuk LM, Hendry RG, Robertson AG, Cheung CT, Ng J, Ang L, Luo Z, Heilbron K, Norris MJ, Duan W, Bucyk T, Karpov A, Devel L, Georgiadis D, Hegele RG, Luo H, Granville DJ, Dive V, McManus BM, Overall CM. A new transcriptional role for matrix metalloproteinase-12 in antiviral immunity. Nat.Med. 2014;20:493–502. doi: 10.1038/nm.3508. [DOI] [PubMed] [Google Scholar]
- Masciopinto F, Giovani C, Campagnoli S, Galli-Stampino L, Colombatto P, Brunetto M, Yen TS, Houghton M, Pileri P, Abrignani S. Association of hepatitis C virus envelope proteins with exosomes. Eur.J.Immunol. 2004;34:2834–2842. doi: 10.1002/eji.200424887. [DOI] [PubMed] [Google Scholar]
- Mason JW. Viral latency: a link between myocarditis and dilated cardiomyopathy? J.Mol.Cell Cardiol. 2002;34:695–698. doi: 10.1006/jmcc.2002.2026. [DOI] [PubMed] [Google Scholar]
- Mavrouli MD, Spanakis N, Levidiotou S, Politi C, Alexiou S, Tseliou P, Hatzitaki M, Foundouli K, Tsakris A, Legakis NJ, Routsias JG. Serologic prevalence of coxsackievirus group B in Greece. Viral Immunol. 2007;20:11–18. doi: 10.1089/vim.2006.0085. [DOI] [PubMed] [Google Scholar]
- Meckes DG, Jr., Raab-Traub N. Microvesicles and viral infection. J.Virol. 2011;85:12844–12854. doi: 10.1128/JVI.05853-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mena I, Perry CM, Harkins S, Rodriguez F, Gebhard J, Whitton JL. The role of B lymphocytes in coxsackievirus B3 infection. Am.J.Pathol. 1999;155:1205–1215. doi: 10.1016/S0002-9440(10)65223-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merl S, Michaelis C, Jaschke B, Vorpahl M, Seidl S, Wessely R. Targeting 2A protease by RNA interference attenuates coxsackieviral cytopathogenicity and promotes survival in highly susceptible mice. Circulation. 2005;111:1583–1592. doi: 10.1161/01.CIR.0000160360.02040.AB. [DOI] [PubMed] [Google Scholar]
- Merl S, Wessely R. Anti-coxsackieviral efficacy of RNA interference is highly dependent on genomic target selection and emergence of escape mutants. Oligonucleotides. 2007;17:44–53. doi: 10.1089/oli.2007.0057. [DOI] [PubMed] [Google Scholar]
- Miller JP, Geng Y, Ng HL, Yang OO, Krogstad P. Packaging limits and stability of HIV-1 sequences in a coxsackievirus B vector. Vaccine. 2009;27:3992–4000. doi: 10.1016/j.vaccine.2009.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misbah SA, Spickett GP, Ryba PC, Hockaday JM, Kroll JS, Sherwood C, Kurtz JB, Moxon ER, Chapel HM. Chronic enteroviral meningoencephalitis in agammaglobulinemia: case report and literature review. J.Clin.Immunol. 1992;12:266–270. doi: 10.1007/BF00918150. [DOI] [PubMed] [Google Scholar]
- Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450:435–439. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- Moore BB, Murray L, Das A, Wilke CA, Herrygers AB, Toews GB. The role of CCL12 in the recruitment of fibrocytes and lung fibrosis. Am.J.Respir.Cell Mol.Biol. 2006;35:175–181. doi: 10.1165/rcmb.2005-0239OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir P, van Loon AM. Enterovirus infections of the central nervous system. Intervirology. 1997;40:153–166. doi: 10.1159/000150542. [DOI] [PubMed] [Google Scholar]
- Mukherjee A, Morosky SA, Delorme-Axford E, Dybdahl-Sissoko N, Oberste MS, Wang T, Coyne CB. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS.Pathog. 2011;7:e1001311. doi: 10.1371/journal.ppat.1001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nejmeddine M, Bangham CR. The HTLV-1 Virological Synapse. Viruses. 2010;2:1427–1447. doi: 10.3390/v2071427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda S, Aguirre SA, Bitmansour A, Brown JM, Sparer TE, Huang J, Mocarski ES. Cytomegalovirus MCK-2 controls mobilization and recruitment of myeloid progenitor cells to facilitate dissemination. Blood. 2006;107:30–38. doi: 10.1182/blood-2005-05-1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak JE, Kirkegaard K. Improved method for detecting poliovirus negative strands used to demonstrate specificity of positive-strand encapsidation and the ratio of positive to negative strands in infected cells. J.Virol. 1991;65(6):3384–3387. doi: 10.1128/jvi.65.6.3384-3387.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberste MS, Maher K, Pallansch MA. Molecular phylogeny and proposed classification of the simian picornaviruses. J.Virol. 2002;76:1244–1251. doi: 10.1128/JVI.76.3.1244-1251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohka S, Igarashi H, Nagata N, Sakai M, Koike S, Nochi T, Kiyono H, Nomoto A. Establishment of a poliovirus oral infection system in human poliovirus receptor-expressing transgenic mice that are deficient in alpha/beta interferon receptor. J.Virol. 2007;81(15):7902–7912. doi: 10.1128/JVI.02675-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldstone MB. Molecular mimicry and immune-mediated diseases. FASEB J. 1998;12:1255–1265. doi: 10.1096/fasebj.12.13.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onomoto K, Yoneyama M, Fung G, Kato H, Fujita T. Antiviral innate immunity and stress granule responses. Trends Immunol. 2014;35:420–428. doi: 10.1016/j.it.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi MH, Wong SC, Lewthwaite P, Cardosa MJ, Solomon T. Clinical features, diagnosis, and management of enterovirus 71. Lancet Neurol. 2010;9:1097–1105. doi: 10.1016/S1474-4422(10)70209-X. [DOI] [PubMed] [Google Scholar]
- Ornoy A, Tenenbaum A. Pregnancy outcome following infections by coxsackie, echo, measles, mumps, hepatitis, polio and encephalitis viruses. Reprod.Toxicol. 2006;21:446–457. doi: 10.1016/j.reprotox.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Pan J, Zhang L, Odenwald MA, Shen L, Turner JR, Bergelson JM. Expression of human decay-accelerating factor on intestinal epithelium of transgenic mice does not facilitate infection by the enteral route. J.Virol. 2015:JVI–14. doi: 10.1128/JVI.03468-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Kim DS, Cho YJ, Kim YJ, Jeong SY, Lee SM, Cho SJ, Yun CW, Jo I, Nam JH. Attenuation of coxsackievirus B3 by VP2 mutation and its application as a vaccine against virus-induced myocarditis and pancreatitis. Vaccine. 2009;27:1974–1983. doi: 10.1016/j.vaccine.2009.01.008. [DOI] [PubMed] [Google Scholar]
- Patel KP, Coyne CB, Bergelson JM. Dynamin- and lipid raft-dependent entry of decay-accelerating factor (DAF)-binding and non-DAF-binding coxsackieviruses into nonpolarized cells. J.Virol. 2009;83(21):11064–11077. doi: 10.1128/JVI.01016-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payment P. Antibody levels to selected enteric viruses in a normal randomly selected Canadian population. Immunology and Infectious Disease. 1991;1:317–322. [Google Scholar]
- Pelletier J, Sonenberg N. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature. 1988;334(6180):320–325. doi: 10.1038/334320a0. [DOI] [PubMed] [Google Scholar]
- Petitjean J, Kopecka H, Freymuth F, Langlard JM, Scanu P, Galateau F, Bouhour JB, Ferriere M, Charbonneau P, Komajda M. Detection of enteroviruses in endomyocardial biopsy by molecular approach. J.Med.Virol. 1992;37:76–82. doi: 10.1002/jmv.1890370114. [DOI] [PubMed] [Google Scholar]
- Pevear DC, Tull TM, Seipel ME, Groarke JM. Activity of pleconaril against enteroviruses. Antimicrob.Agents Chemother. 1999;43:2109–2115. doi: 10.1128/aac.43.9.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkert S, Klingel K, Lindig V, Dorner A, Zeichhardt H, Spiller OB, Fechner H. Virus-host coevolution in a persistently coxsackievirus B3-infected cardiomyocyte cell line. J.Virol. 2011;85:13409–13419. doi: 10.1128/JVI.00621-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puccini JM, Ruller CM, Robinson SM, Knopp KA, Buchmeier MJ, Doran KS, Feuer R. Distinct neural stem cell tropism, early immune activation, and choroid plexus pathology following coxsackievirus infection in the neonatal central nervous system. Lab Invest. 2014;94:161–181. doi: 10.1038/labinvest.2013.138. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Kivisakk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat.Rev.Immunol. 2003;3:569–581. doi: 10.1038/nri1130. [DOI] [PubMed] [Google Scholar]
- Rantakallio P, Jones P, Moring J, Von WL. Association between central nervous system infections during childhood and adult onset schizophrenia and other psychoses: a 28-year follow-up. Int.J.Epidemiol. 1997;26:837–843. doi: 10.1093/ije/26.4.837. [DOI] [PubMed] [Google Scholar]
- Reineke LC, Lloyd RE. The Stress Granule Protein G3BP1 Recruits Protein Kinase R To Promote Multiple Innate Immune Antiviral Responses. J.Virol. 2015;89:2575–2589. doi: 10.1128/JVI.02791-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoades RE, Tabor-Godwin JM, Tsueng G, Feuer R. Enterovirus infections of the central nervous system. Virology. 2011;411:288–305. doi: 10.1016/j.virol.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards AL, Jackson WT. Intracellular vesicle acidification promotes maturation of infectious poliovirus particles. PLoS.Pathog. 2012;8:e1003046. doi: 10.1371/journal.ppat.1003046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards AL, Jackson WT. How positive-strand RNA viruses benefit from autophagosome maturation. J.Virol. 2013;87:9966–9972. doi: 10.1128/JVI.00460-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson SM, Tsueng G, Sin J, Mangale V, Rahawi S, McIntyre LL, Williams W, Kha N, Cruz C, Hancock BM, Nguyen DP, Sayen MR, Hilton BJ, Doran KS, Segall AM, Wolkowicz R, Cornell CT, Whitton JL, Gottlieb RA, Feuer R. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS.Pathog. 2014;10:e1004045. doi: 10.1371/journal.ppat.1004045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls A, Shechter R, London A, Ziv Y, Ronen A, Levy R, Schwartz M. Toll-like receptors modulate adult hippocampal neurogenesis. Nat.Cell Biol. 2007;9:1081–1088. doi: 10.1038/ncb1629. [DOI] [PubMed] [Google Scholar]
- Romero JR. Pediatric group B coxsackievirus infections. Curr.Top.Microbiol.Immunol. 2008;323:223–239. doi: 10.1007/978-3-540-75546-3_10. [DOI] [PubMed] [Google Scholar]
- Ruller CM, Tabor-Godwin JM, Van Deren DAJ, Robinson SM, Maciejewski S, Gluhm S, Gilbert PE, An N, Gude NA, Sussman MA, Whitton JL, Feuer R. Neural stem cell depletion and CNS developmental defects after enteroviral infection. Am.J.Pathol. 2012;180:1107–1120. doi: 10.1016/j.ajpath.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampey GC, Meyering SS, Asad ZM, Saifuddin M, Hakami RM, Kashanchi F. Exosomes and their role in CNS viral infections. J.Neurovirol. 2014;20:199–208. doi: 10.1007/s13365-014-0238-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sane F, Moumna I, Hober D. Group B coxsackieviruses and autoimmunity: focus on Type 1 diabetes. Expert.Rev.Clin.Immunol. 2011;7:357–366. doi: 10.1586/eci.11.11. [DOI] [PubMed] [Google Scholar]
- Sarafi MN, Garcia-Zepeda EA, MacLean JA, Charo IF, Luster AD. Murine monocyte chemoattractant protein (MCP)-5: a novel CC chemokine that is a structural and functional homologue of human MCP-1. J.Exp.Med. 1997;185:99–109. doi: 10.1084/jem.185.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh M, Tamura G, Segawa I. Enteroviral RNA in endomyocardial biopsy tissues of myocarditis and dilated cardiomyopathy. Pathol.Int. 1994;44:345–351. doi: 10.1111/j.1440-1827.1994.tb02934.x. [DOI] [PubMed] [Google Scholar]
- Sawyer MH. Enterovirus infections: diagnosis and treatment. Semin.Pediatr.Infect.Dis. 2002;13:40–47. doi: 10.1053/spid.2002.29756. [DOI] [PubMed] [Google Scholar]
- Schilthuizen C, Berenschot HW, Levin MD. Enteroviral encephalitis in a patient with a marginal zone lymphomatreated with rituximab. Neth.J.Med. 2010;68:221–223. [PubMed] [Google Scholar]
- Schmidtke M, Hammerschmidt E, Schuler S, Zell R, Birch-Hirschfeld E, Makarov VA, Riabova OB, Wutzler P. Susceptibility of coxsackievirus B3 laboratory strains and clinical isolates to the capsid function inhibitor pleconaril: antiviral studies with virus chimeras demonstrate the crucial role of amino acid 1092 in treatment. J.Antimicrob.Chemother. 2005;56:648–656. doi: 10.1093/jac/dki263. [DOI] [PubMed] [Google Scholar]
- Servais S, Caers J, Warling O, Frusch N, Baron F, De PB, Beguin Y. Enteroviral meningoencephalitis as complication of Rituximab therapy in a patient treated for diffuse large B-cell lymphoma. Br.J.Haematol. 2010;150:379–381. doi: 10.1111/j.1365-2141.2010.08202.x. [DOI] [PubMed] [Google Scholar]
- Sharma A, Marceau C, Hamaguchi R, Burridge PW, Rajarajan K, Churko JM, Wu H, Sallam KI, Matsa E, Sturzu AC, Che Y, Ebert A, Diecke S, Liang P, Red-Horse K, Carette JE, Wu SM, Wu JC. Human induced pluripotent stem cell-derived cardiomyocytes as an in vitro model for coxsackievirus B3-induced myocarditis and antiviral drug screening platform. Circ.Res. 2014;115:556–566. doi: 10.1161/CIRCRESAHA.115.303810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Wong J, Piesik P, Fung G, Zhang J, Jagdeo J, Li X, Jan E, Luo H. Cleavage of sequestosome 1/p62 by an enteroviral protease results in disrupted selective autophagy and impaired NFKB signaling. Autophagy. 2013;9:1591–1603. doi: 10.4161/auto.26059. [DOI] [PubMed] [Google Scholar]
- Shi Y, Chen C, Lisewski U, Wrackmeyer U, Radke M, Westermann D, Sauter M, Tschope C, Poller W, Klingel K, Gotthardt M. Cardiac deletion of the Coxsackievirus-adenovirus receptor abolishes Coxsackievirus B3 infection and prevents myocarditis in vivo. J.Am.Coll.Cardiol. 2009;53:1219–1226. doi: 10.1016/j.jacc.2008.10.064. [DOI] [PubMed] [Google Scholar]
- Shih SR, Stollar V, Li ML. Host factors in enterovirus 71 replication. J.Virol. 2011;85:9658–9666. doi: 10.1128/JVI.05063-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shingo T, Gregg C, Enwere E, Fujikawa H, Hassam R, Geary C, Cross JC, Weiss S. Pregnancy-stimulated neurogenesis in the adult female forebrain mediated by prolactin. Science. 2003;299:117–120. doi: 10.1126/science.1076647. [DOI] [PubMed] [Google Scholar]
- Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy. 2008;4(2):176–184. doi: 10.4161/auto.5269. [DOI] [PubMed] [Google Scholar]
- Sin J, Puccini JM, Huang C, Konstandin MH, Gilbert PE, Sussman MA, Gottlieb RA, Feuer R. The impact of juvenile coxsackievirus infection on cardiac progenitor cells and postnatal heart development. PLoS Pathog. 2014;10:e1004249. doi: 10.1371/journal.ppat.1004249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slifka MK, Pagarigan R, Mena I, Feuer R, Whitton JL. Using recombinant coxsackievirus B3 to evaluate the induction and protective efficacy of CD8+ T cells during picornavirus infection. J.Virol. 2001;75:2377–2387. doi: 10.1128/JVI.75.5.2377-2387.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steil BP, Barton DJ. Cis-active RNA elements (CREs) and picornavirus RNA replication. Virus Res. 2009;139(2):240–252. doi: 10.1016/j.virusres.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strating JR, van der Linden L, Albulescu L, Bigay J, Arita M, Delang L, Leyssen P, van der Schaar HM, Lanke KH, Thibaut HJ, Ulferts R, Drin G, Schlinck N, Wubbolts RW, Sever N, Head SA, Liu JO, Beachy PA, De Matteis MA, Shair MD, Olkkonen VM, Neyts J, van Kuppeveld FJ. Itraconazole Inhibits Enterovirus Replication by Targeting the Oxysterol-Binding Protein. Cell Rep. 2015:10. doi: 10.1016/j.celrep.2014.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhy DA, Giddings TH, Jr., Kirkegaard K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J.Virol. 2000;74(19):8953–8965. doi: 10.1128/jvi.74.19.8953-8965.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabor-Godwin JM, Ruller CM, Bagalso N, An N, Pagarigan RR, Harkins S, Gilbert PE, Kiosses WB, Gude NA, Cornell CT, Doran KS, Sussman MA, Whitton JL, Feuer R. A novel population of myeloid cells responding to coxsackievirus infection assists in the dissemination of virus within the neonatal CNS. J.Neurosci. 2010;30:8676–8691. doi: 10.1523/JNEUROSCI.1860-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabor-Godwin JM, Tsueng G, Sayen MR, Gottlieb RA, Feuer R. The role of autophagy during coxsackievirus infection of neural progenitor and stem cells. Autophagy. 2012;8:938–953. doi: 10.4161/auto.19781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam PE, Fontana DR, Messner RP. Coxsackievirus B1-induced chronic inflammatory myopathy: differences in induction of autoantibodies to muscle and nuclear antigens by cloned myopathic and amyopathic viruses. J.Lab Clin.Med. 2003;142:196–204. doi: 10.1016/S0022-2143(03)00108-2. [DOI] [PubMed] [Google Scholar]
- Tam PE, Messner RP. Molecular mechanisms of coxsackievirus persistence in chronic inflammatory myopathy: viral RNA persists through formation of a double-stranded complex without associated genomic mutations or evolution. J.Virol. 1999;73:10113–10121. doi: 10.1128/jvi.73.12.10113-10121.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan EL, Wong AP, Poh CL. Development of potential antiviral strategy against coxsackievirus B4. Virus Res. 2010;150:85–92. doi: 10.1016/j.virusres.2010.02.017. [DOI] [PubMed] [Google Scholar]
- Tao Z, Li B, Xu A, Liu Y, Song L, Wang S, Xiong P, Lin X, Song Y. Seroprevalence of coxsackievirus B3 in Yantai, China. Jpn.J.Infect.Dis. 2013;66:537–538. doi: 10.7883/yoken.66.537. [DOI] [PubMed] [Google Scholar]
- Taylor MP, Burgon TB, Kirkegaard K, Jackson WT. Role of microtubules in extracellular release of poliovirus. J.Virol. 2009;83:6599–6609. doi: 10.1128/JVI.01819-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MP, Kirkegaard K. Potential subversion of autophagosomal pathway by picornaviruses. Autophagy. 2008;4:286–289. doi: 10.4161/auto.5377. [DOI] [PubMed] [Google Scholar]
- Tebruegge M, Curtis N. Enterovirus infections in neonates. Semin.Fetal Neonatal Med. 2009;14:222–227. doi: 10.1016/j.siny.2009.02.002. [DOI] [PubMed] [Google Scholar]
- Terskikh A, Fradkov A, Ermakova G, Zaraisky A, Tan P, Kajava AV, Zhao X, Lukyanov S, Matz M, Kim S, Weissman I, Siebert P. “Fluorescent timer”: protein that changes color with time. Science. 2000;290:1585–1588. doi: 10.1126/science.290.5496.1585. [DOI] [PubMed] [Google Scholar]
- Thibaut HJ, van der Linden L, Jiang P, Thys B, Canela MD, Aguado L, Rombaut B, Wimmer E, Paul A, Perez-Perez MJ, van Kuppeveld FJ, Neyts J. Binding of glutathione to enterovirus capsids is essential for virion morphogenesis. PLoS Pathog. 2014;10:e1004039. doi: 10.1371/journal.ppat.1004039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracy S, Drescher KM, Chapman NM. Enteroviruses and type 1 diabetes. Diabetes Metab Res.Rev. 2011;27:820–823. doi: 10.1002/dmrr.1255. [DOI] [PubMed] [Google Scholar]
- Tracy S, Smithee S, Alhazmi A, Chapman N. Coxsackievirus can persist in murine pancreas by deletion of 5' terminal genomic sequences. J.Med.Virol. 2015;87:240–247. doi: 10.1002/jmv.24039. [DOI] [PubMed] [Google Scholar]
- Tsueng G, Rhoades RE, Nguyen DP, Deline S, Zamudio Montes de Oca AV, Gurney M, Gottlieb RA, Feuer R. Lasting alterations in autophagic flux and increased coxsackievirus virulence during the establishment of a carrier-state infection in neural progenitor cells. 2014. Manuscript In Preparation.
- Tsueng G, Tabor-Godwin JM, Gopal A, Ruller CM, Deline S, An N, Frausto RF, Milner R, Crocker SJ, Whitton JL, Feuer R. Coxsackievirus preferentially replicates and induces cytopathic effects in undifferentiated neural progenitor cells. J.Virol. 2011;85:5718–5732. doi: 10.1128/JVI.02261-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Schaar HM, Leyssen P, Thibaut HJ, de PA, van der Linden L, Lanke KH, Lacroix C, Verbeken E, Conrath K, Macleod AM, Mitchell DR, Palmer NJ, van de Poel H, Andrews M, Neyts J, van Kuppeveld FJ. A novel, broad-spectrum inhibitor of enterovirus replication that targets host cell factor phosphatidylinositol 4-kinase IIIbeta. Antimicrob.Agents Chemother. 2013;57:4971–4981. doi: 10.1128/AAC.01175-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Schaar HM, van der Linden L, Lanke KH, Strating JR, Purstinger G, de VE, de Haan CA, Neyts J, van Kuppeveld FJ. Coxsackievirus mutants that can bypass host factor PI4KIIIbeta and the need for high levels of PI4P lipids for replication. Cell Res. 2012;22:1576–1592. doi: 10.1038/cr.2012.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatraman G, Behrens M, Pyrski M, Margolis FL. Expression of Coxsackie-Adenovirus receptor (CAR) in the developing mouse olfactory system. J.Neurocytol. 2005;34:295–305. doi: 10.1007/s11068-005-8359-8. [DOI] [PubMed] [Google Scholar]
- Vessoni AT, Muotri AR, Okamoto OK. Autophagy in stem cell maintenance and differentiation. Stem Cells Dev. 2012;21:513–520. doi: 10.1089/scd.2011.0526. [DOI] [PubMed] [Google Scholar]
- Victoria JG, Kapoor A, Li L, Blinkova O, Slikas B, Wang C, Naeem A, Zaidi S, Delwart E. Metagenomic analyses of viruses in stool samples from children with acute flaccid paralysis. J.Virol. 2009;83:4642–4651. doi: 10.1128/JVI.02301-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature. 2006;439:344–348. doi: 10.1038/nature04388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt DA, Andino R. An RNA element at the 5'-end of the poliovirus genome functions as a general promoter for RNA synthesis. PLoS.Pathog. 2010;6(6):e1000936. doi: 10.1371/journal.ppat.1000936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller AH, Simpson K, Herzum M, Van HN, Huber SA. Coxsackievirus-B3-induced myocarditis: virus receptor antibodies modulate myocarditis. J.Immunol. 1989;143:1843–1850. [PubMed] [Google Scholar]
- Werk D, Schubert S, Lindig V, Grunert HP, Zeichhardt H, Erdmann VA, Kurreck J. Developing an effective RNA interference strategy against a plus-strand RNA virus: silencing of coxsackievirus B3 and its cognate coxsackievirus-adenovirus receptor. Biol.Chem. 2005;386:857–863. doi: 10.1515/BC.2005.100. [DOI] [PubMed] [Google Scholar]
- Wessels E, Duijsings D, Lanke KH, Melchers WJ, Jackson CL, van Kuppeveld FJ. Molecular determinants of the interaction between coxsackievirus protein 3A and guanine nucleotide exchange factor GBF1. J.Virol. 2007;81:5238–5245. doi: 10.1128/JVI.02680-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessely R, Klingel K, Knowlton KU, Kandolf R. Cardioselective infection with coxsackievirus B3 requires intact type I interferon signaling: implications for mortality and early viral replication. Circulation. 2001;103:756–761. doi: 10.1161/01.cir.103.5.756. [DOI] [PubMed] [Google Scholar]
- Wessely R, Klingel K, Santana LF, Dalton N, Hongo M, Jonathan LW, Kandolf R, Knowlton KU. Transgenic expression of replication-restricted enteroviral genomes in heart muscle induces defective excitation-contraction coupling and dilated cardiomyopathy. J.Clin.Invest. 1998;102:1444–1453. doi: 10.1172/JCI1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitton JL, Cornell CT, Feuer R. Host and virus determinants of picornavirus pathogenesis and tropism. Nat.Rev.Microbiol. 2005;3:765–776. doi: 10.1038/nrmicro1284. [DOI] [PubMed] [Google Scholar]
- Whitton JL, Feuer R. Myocarditis, microbes and autoimmunity. Autoimmunity. 2004;37:375–386. doi: 10.1080/08916930410001713089. [DOI] [PubMed] [Google Scholar]
- Wileman T. Aggresomes and autophagy generate sites for virus replication. Science. 2006;312:875–878. doi: 10.1126/science.1126766. [DOI] [PubMed] [Google Scholar]
- Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Immunohistochemical analysis of the relationship between islet cell proliferation and the production of the enteroviral capsid protein, VP1, in the islets of patients with recent-onset type 1 diabetes. Diabetologia. 2011;54:2417–2420. doi: 10.1007/s00125-011-2192-7. [DOI] [PubMed] [Google Scholar]
- Wong J, Zhang J, Si X, Gao G, Mao I, McManus BM, Luo H. Autophagosome supports coxsackievirus B3 replication in host cells. J.Virol. 2008a;82(18):9143–9153. doi: 10.1128/JVI.00641-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J, Zhang J, Si X, Gao G, Mao I, McManus BM, Luo H. Autophagosome supports coxsackievirus B3 replication in host cells. J.Virol. 2008b;82(18):9143–9153. doi: 10.1128/JVI.00641-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodall CJ, Graham DI. Evidence for neuronal localisation of enteroviral sequences in motor neurone disease/amyotrophic lateral sclerosis by in situ hybridization. Eur.J.Histochem. 2004;48:129–134. doi: 10.4081/877. [DOI] [PubMed] [Google Scholar]
- Woodall CJ, Riding MH, Graham DI, Clements GB. Sequences specific for enterovirus detected in spinal cord from patients with motor neurone disease. BMJ. 1994;308:1541–1543. doi: 10.1136/bmj.308.6943.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SY, Ha YE, Choi JE, Ahn J, Lee H, Kim DH. Autophagy in coxsackievirus-infected neurons. Autophagy. 2009;5(3):388–389. doi: 10.4161/auto.5.3.7723. [DOI] [PubMed] [Google Scholar]
- Yoon SY, Ha YE, Choi JE, Ahn J, Lee H, Kweon HS, Lee JY, Kim DH. Coxsackievirus B4 uses autophagy for replication after calpain activation in rat primary neurons. J.Virol. 2008;82(23):11976–11978. doi: 10.1128/JVI.01028-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Cheung PK, Zhang HM, Chau D, Yang D. Inhibition of coxsackievirus B3 replication by small interfering RNAs requires perfect sequence match in the central region of the viral positive strand. J.Virol. 2005;79:2151–2159. doi: 10.1128/JVI.79.4.2151-2159.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng J, Chen X, Dai J, Zhao X, Xin G, Su Y, Wang G, Li R, Yan Y, Su J, Deng Y, Li K. An attenuated coxsackievirus b3 vector: a potential tool for viral tracking study and gene delivery. PLoS One. 2013;8:e83753. doi: 10.1371/journal.pone.0083753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo J, Quinn KK, Kye S, Cooper P, Damoiseaux R, Krogstad P. Fluoxetine is a potent inhibitor of coxsackievirus replication. Antimicrob.Agents Chemother. 2012;56:4838–4844. doi: 10.1128/AAC.00983-12. [DOI] [PMC free article] [PubMed] [Google Scholar]