

Abstract
Connexin mutations underlie numerous human genetic diseases. Several connexin genes have been linked to skin diseases, and mechanistic studies have indicated that a gain of abnormal channel function may be responsible for pathology. The topical accessibility of the epidermal connexins, the existence of several mouse models of human skin disease, and the ongoing identification of pharmacological inhibitors targeting connexins provides an opportunity to test new therapeutic approaches.
Keywords: skin, connexin, genetic disease, inhibitor, gap junctions, KID syndrome, inflammation
Graphical abstract
1.1 CONNEXIN CHANNELS & THE EPIDERMIS
The recent decade or so has seen connexins claim heightened attention as key determinants of epidermal homeostasis and, unsurprisingly, also as effectors of a wide spectrum of genetic and acquired cutaneous pathophysiology [1,2]. The connexins are a multi-gene family of highly conserved integral membrane proteins that are dynamically expressed in virtually all vertebrate tissues [3]. Human skin is no exception, containing at least 9 of the 21 human connexin (Cx) isoforms identified to date, including members of both the alpha (Cx37, Cx40, Cx43, Cx45) and beta (Cx26, Cx30, Cx30.3, Cx31, Cx31.1) phylogenetic subgroups [4,5]. Connexin monomers hexamerize to create highly specialized aqueous pores in the plasma membrane, referred to as connexons or hemichannels, and through which messages may pass in the form of ions, small molecules, or cytoplasmic metabolites [6,7]. Hemichannels may associate with a cognate structure on the surface of a contiguous neighbor cell to make intercellular gap junctions (GJs) or may remain ‘unpaired’ to connect the cell interior with the extracellular microenvironment [8–12]. Channels in both configurations exhibit precise spatial and temporal expression patterns according to tissue-specific developmental and functional requirements for electrical and metabolic coupling of cell networks [13,14]. Furthermore, different channel compositions impart unique gating and permeability properties depending on the individual connexin protein constituents and their sensitivities and susceptibilities to factors such as membrane potential and post-translational phosphorylation events [15–18].
1.2 THE CASE FOR EPIDERMAL CONNEXINS AS DRUG TARGETS
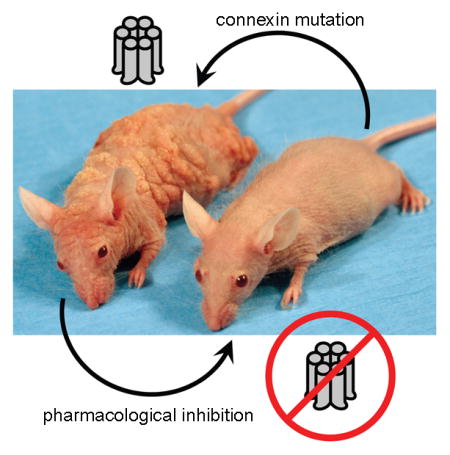
Pharmacological agents have been cleverly implemented in studies to probe for the physiologic relevance of gap junctions and hemichannels in both health and disease. Channel modulators have proven particularly useful as instruments to delineate the cellular consequences of deficient or altered junctional communication in genetic disorders. Mutations of 6 different connexin isoforms have been linked to autosomal dominant hereditary disorders of epidermal cornification, including Vohwinkel syndrome (VS, Cx26), Bart-Pumphrey syndrome (BPS, Cx26), hystrix-like ichthyosis with deafness (HID, Cx26) syndrome, keratitis-ichthyosis-deafness (KID, Cx26) syndrome, erythrokeratoderma variabilis (EKV, Cx30.3/Cx31/Cx31.1), hidrotic ectodermal dysplasia (HED, Cx30), and oculodentaldigital dysplasia (ODDD, Cx43) [2,19]. Interestingly, recent reports indicate that Cx43-related pathology additionally includes erythrokeratodermia variabilis et progressiva (EKVP) [20,21] and keratoderma-hypotrichosis-leukonychia totalis syndrome (KHLS) [22], which share some clinical features with syndromic Cx26 diseases. ODDD manifests with neuropathies, craniofacial and digital anomalies and, occasionally, skin abnormalities such as palmoplantar keratoderma [23]. EKVP results in hyperkeratosis and transient figurate patches of erythema and KHLS is a subtype of palmoplantar keratoderma-congenital alopecia syndrome characterized by profound hyperkeratosis, congenital alopecia, and leukonychia. Generally stated, connexin mutations that cause human disease have been shown to mechanistically proceed through either loss of gap junction and/or hemichannel functionality (i.e. truncation/misfolding/defective oligomerization/defective trafficking) or by dysregulation of controlled channel activity and consequent acquisition of novel pathogenic behaviors [24–26]. Investigators of connexin-mediated cutaneous disease have worked industriously to functionally characterize greater than 30 distinct mutations with numerous reports now suggesting the latter, gain-of-function, scenario to be a common occurrence in the skin [19,27]. For these mutations, inhibitor strategies may hold therapeutic value in addition to serving as research tools to pin down errors in channel gating and permselectivity (Figure 1). This concept is underscored by the topical accessibility of epidermal connexins and the fact that mouse models of Vohwinkel syndrome [28], EKV [29], ODDD [30], HED [31], and KID syndrome [32,33] already exist.
FIGURE 1.

Gap junctions allow for direct intercellular transfer of ions, small molecules, and second messengers including Ca2+, ATP, cAMP, NAD+, IP3, glutamate, and prostaglandins. Connexin hemichannels display controlled exchange of some of these factors with the extracellular space. Mutations that precipitate inflammatory skin diseases show dysregulated Cx26, 30, 31, 43 hemichannels as evidenced by leakage of cytoplasmic ATP and excessive influx of calcium. Small molecule inhibitors capable of suppressing aberrant hemichannel activity, such as mefloquine (pictured), may placate hyperkeratoses in keratitis-ichthyosis-deafness syndrome (Cx26), hidrotic ectodermal dysplasia (Cx30), erythrokeratoderma variabilis (Cx31), and keratoderma-hypotrichosis-leukonychia totalis syndrome (Cx43).
Specialized connexin-inhibitors have proven nontrivial to come by. This is in part due to the challenges of adapting high-throughput drug screens to directly incorporate a readout on membrane biophysics. In addition, the ubiquitous occurrence of connexin-family proteins necessitates inhibitory molecules possessing strict specificity and selectivity properties. Moreover, connexin hemichannels and gap-junctions are increasingly appreciated to operate in independent physiologic niches [6,7], implying a theoretical requirement for inhibitors to be capable of discriminating between the two channel conformations. Indeed, these frustrations have lead to the pursuit of antisense oligonucleotide [34] and siRNA [35,36] approaches as well as the development of the synthetic connexin-mimetic peptides [37–39]. Herein, we provide a perspective on the value of a continued search for pharmacological inhibitors of connexin hemichannels and GJs from among a pool of small molecules recognized to modulate traffic across the plasma membrane and, ideally, also boasting previously verified safety/tolerability profiles. Keratitis-ichthyosis-deafness syndrome will be discussed as an archetypal connexin-driven ectodermal dysplasia for which targeted therapeutic options are conspicuously absent. Within the framework of KID syndrome, our commentary will briefly explore the broader implications of connexin pathology relating to inflammation, wound healing, neoplasia, and innate immunity.
1.3 MOLECULAR GENETICS OF KID SYNDROME
KID syndrome is a rare multisystem genodermatosis caused by dominant mutations of GJB2, the gene that encodes Cx26 [40]. The disorder clinically manifests with coincident ichthyosiform dermatitis, vascularizing keratitis, and sensorineural hearing loss [41]. The incidence of patient cases is fewer than 1 in 100,000 live births, among which there seems to be substantial ‘phenotypic’ heterogeneity [42]. KID syndrome is also appropriately classified among the various forms of syndromic deafness, given the well-established importance of Cx26 in vestibulocochlear organ physiology. In fact, loss-of-function Cx26 mutation is the commonest cause of autosomal recessive prelingual nonsyndromic deafness in the world with greater than 225 sequence variations documented [43]. It is worth highlighting here that the scores of deaf individuals lacking viable Cx26 do not suffer notable skin abnormalities or defective wound healing. By contrast, Cx26 mutations presently appreciated to incite skin disease operate by engendering a gain or aberrance in protein activity and are vanishingly few in comparison [44].
10 distinct germline missense substitutions have been identified in the Cx26 sequence following a clinical diagnosis of KID syndrome [45]. KID mutations are concentrated in the amino-terminus, first transmembrane domain, and first extracellular loop of Cx26 with few exceptions. Electrophysiological functional analyses of several mutant channel forms via cell-based expression assays have suggested that high conductance hemichannels may represent a unifying pathomechanism [46]. Specifically, at least 7 of the Cx26 mutations linked to KID syndrome permit markedly larger hemichannel fluxes than the wild-type homomeric channel under the same experimental conditions [47–55]. Moreover, this phenomenon has been shown to exist without regard to the functional status of the corresponding homomeric-homotypic gap junctions. This is demonstrated clearly in the case of the most frequently detected KID mutation, Cx26-D50N, which forms highly active hemichannels but precludes gap junctional coupling altogether [50,54]. Recent work has elucidated transdominant effects of KID mutations exerted on co-localizing connexins to yield hyperactive Cx26/Cx43 heteromeric hemichannels. Notably, this was shown for the Cx26-S17F mutant that fails to form functioning homomeric Cx26 hemichannels [48]. Though it remains a theory, constitutively active hemichannels in KID syndrome are thought to harm cell viability and tissue integrity by allowing leakage of cytoplasmic contents (i.e. ATP) as well as excessive entry of electrolytes. In particular, corruption of the transepidermal extracellular calcium gradient unhinges signal transduction in differentiating keratinocytes required for successful cornification and turnover [56–60].
1.4 KID SYNDROME CLINICOPATHOLOGY & CURRENT THERAPIES
Patients harboring KID-inducing Cx26 mutations suffer significant morbidity associated with their cutaneous and extracutaneous disease and may encounter life threatening infectious and neoplastic sequelae of the former. Clinical presentation appears to vary in accordance with the specific causative mutation, though genotype-phenotype correlations are weak due to the paucity of patient cases. Two mutations, Cx26-G45E and Cx26-A88V, have been unequivocally linked with a lethal form of the disease [61–66]. Age at diagnosis tends to be during the neonatal period and rarely beyond infancy. Patients are initially recognized to have congenital nonprogressive hearing loss as well as hyperkeratotic and/or erythrodermic skin. Additional abnormalities of the integument may be noted on physical exam including alopecia totalis, palmoplantar keratoderma, dystrophic nails, and hypohydrosis. There have also been reports of trichilemmal and vellus cysts and the follicular occlusion triad, consisting of dissecting folliculitis, hidradenitis suppurativa, and cystic acne [52,67]. Ocular defects may be indicated by corneal opacity and/or photophobia, both of which are signs that warrant prompt ophthalmic evaluation for vascularizing keratitis, keratoconjunctivitis sicca, corneal ulcers, and limbal defects [68].
The primary challenge in the medical management of neonates and infants affected by KID syndrome relates to barrier insufficiency of the inflamed skin that predisposes to persistent cutaneous superinfection. In severe cases, erythroderma and diffuse leathery hyperkeratosis at birth can rapidly progress to reticulated plaques with friable scale and generalized verrucous hyperkeratosis [69]. Fissuring and microwounding of affected skin leads to accelerated transepidermal water loss, maceration of the tissue, and easy excoriation, particularly in the intertriginous folds. Chronic bacterial and mycotic infections are readily established and can overwhelm the naïve infant immune defenses resulting in septicemia. Combinations of emollients, barrier creams, and topical keratolytics (i.e. ureas, α-hydroxy acids, salicylic acid) designed to mitigate hyperkeratoses and preserve the epithelial barrier are the current mainstay of skincare [70–72]. In addition, antiseptic (i.e. bleach) baths are used to prophylax against colonization by pathogenic organisms and subsequent development of serious infections. Nonetheless, topical and systemic antibiotics and antifungal agents are frequently required as life-sustaining measures.
Patients that are able to navigate the hazardous neonatal-infancy period must be closely surveilled for the development of neoplastic complications later in life. Specifically, KID syndrome portends a higher risk of benign and malignant tumors of follicular origin as well as cutaneous and mucosal squamous cell carcinoma [42,52,69,73–78]. Trichilemmal neoplasms arise as well-circumscribed admixed solid and cystic structures formed as a consequence of derailed follicular keratinization, follicular plugging, cyst formation and chronic inflammation [52,69]. A high index of suspicion for malignant transformation is imperative in KID syndrome as there have been reports of metastatic disease appearing as early as the third decade of life [79]. Characteristics of concerns include rapid exophytic growth, ulceration and necrosis, and anchoring to deep subcutaneous tissues that may signal invasion. Squamous cell carcinoma has affected approximately 20% of patients diagnosed with KID syndrome [69,80–82]. Malignant transformation of the epithelium is accelerated by conditions of chronic inflammation and infection, both of which are epitomized by KID syndrome. Complicating matters is that early lesions with malignant potential are difficult to detect among beds of hyperkeratosis, making preemptive biopsy and surgical excision challenging. The systemic retinoids acitretin and alitretinoin have been used in an attempt to advance the stalled desquamation process in hyperkeratotic skin with mixed outcomes [83–87]. A role for acitretin as a chemoprophylactic agent has been proposed with at least one group claiming protective effects against cutaneous malignancy by direct interference with gap junctional communication [88]. However, this study relied on a dye-transfer ‘parachute’ assay to qualitatively assess gap junction activity and did not resolve functional contributions from the individual connexins (i.e. Cx26 and Cx43). Furthermore, as previously mentioned, functional analyses have now demonstrated that several KID-causing mutations produce nonfunctioning Cx26 gap junctions and that rather dysregulated Cx26 hemichannels are predicted to underlie loss of epidermal homeostasis. Paradoxically, retinoic acid has been shown to provoke marked upregulation of Cx26 in human epidermis, at both the transcript and protein level, to an extent not paralleled by other connexins [89]. A discernable danger is that heightened expression of Cx26 may amplify mutation-driven injurious effects. This example emphatically underscores the need for meticulous characterization of molecular pathogenesis and targeted therapies in the genodermatoses.
1.5 NOVEL THERAPEUTIC STRATEGIES IN KID SYNDROME
Efforts toward rational drug discovery for ‘orphan’ genodermatoses ought to concentrate on libraries of existing compounds with recognized safety and tolerability profiles in humans. This is because randomized trials are not feasible in diseases such as KID syndrome from a practical standpoint due to the scarcity of patients and clinical urgency at presentation. Several clinically-approved small molecules belonging to the antimalarial (i.e. mefloquine), glycyrrhetinic acid (i.e. carbenoxolone disodium), and benzopyran (i.e. tonbersat) families have demonstrated avid inhibition of connexin hemichannels and gap junctions [39,90–99]. Because keratinocytes are replete with membrane transport proteins, properties of connexin-specificity and isoform-selectivity are obligatory for therapeutic adaptation. Of particular importance to KID syndrome, molecules should be expressly tested for inhibitory efficacy on nonjunctional hemichannels and furthermore verified to maintain affinity for biochemically modified mutant forms relevant to human disease. For example, mefloquine has displayed dose-dependent selectivity for certain connexin subtypes as well as direct and potent inhibition of mutant Cx26 hemichannels present in KID syndrome [92,100]. Of concern, mefloquine concentrations necessary for Cx26 hemichannel inhibition are nearly 10-fold higher than what is required for its antimalarial action. Although off target effects remain a legitimate worry, risks of systemic toxicity may be assuaged by local topical delivery or combination therapy approaches. Another outstanding dilemma is the failure to differentiate between Cx26 subunits participating in hemichannels and gap junctions. However, this may not impede progress for gain-of-function disorders for which the wild-type protein function is apparently redundant, as is the case in KID syndrome.
The relevance of hemichannel inhibitors in connexin-mediated skin disease is further supported by studies of connexin30 (Cx30, GJB6) and connexin31 (Cx31, GJB3) driven pathology emanating from the stratum granulosum. The EKV mutation Cx31-R42P was shown to form constitutively active hemichannels when expressed in HeLa cells, as evidenced by enhanced dye uptake and necrotic cell death, both of which could be prevented by treatment with a connexin channel inhibitor [101]. Two Cx30 mutations, Cx30-G11R and Cx30-A88V, linked to HED were reported to induce large voltage-activated currents resulting in cell death when expressed in Xenopus oocytes [102,103]. HeLa cells transfected with either Cx30-G11R or Cx30-A88V released ATP into the extracellular milieu, presumably through porous hemichannels. An additional observation of relevance pertains to the KHLS mutation Cx43-G8V, which allows excessive influx of calcium through active hemichannels at the cell membrane [22]. These data further support the notion that pathological connexin hemichannels may represent repeat offenders in hyperproliferative skin disorders.
2.1 CONNEXIN-MEDIATED EVENTS & THE GENERALIZABILITY OF PHARMACOLOGICAL TOOLS
Perhaps most persuasive for the broader scientific community is a rapidly accumulating body of evidence to suggest that connexin-associated cellular events in the skin minimally encompass keratinocyte proliferation [104,105], differentiation [106,107], adhesion [108,109], migration [110–112], inflammation [113–116], and innate immune surveillance [47,117,118]. KID syndrome is a particularly suitable model for discussion, being that these patients live at the intersection of inflammatory, infectious, and neoplastic processes. Connexins do not, however, only hold bearing in rare and esoteric disorders. An expanding impact for connexin modulators is briskly coming into focus as our understanding of interplay between connexins and fundamental cell signaling factors improves.
2.2 CONNEXINS IN THE INFLAMMATORY DIATHESIS & WOUND HEALING
Perturbations in the connexin expression pattern have been well documented in inflammatory and hyperproliferative skin states. For example, Cx26 is highly overexpressed in papilloma lesions [119], experimentally induced wounds [120,121], and psoriatic plaques [122–124]. Overwhelmingly, mining of the psoriatic transcriptome by RNA-seq revealed a greater than 18-fold inflation of GJB2 activity compared to normal skin [125]. Whether Cx26 upregulation in the stratum granulosum in psoriatic lesions contributes to escalating pathology or is instead an irrelevant bystander effect is debated. Of note, connexin hemichannels are a major conduit for ATP release [7,126–128], which may plug into pro-proliferation purinergic signaling in psoriasis [129,130]. An elegant set of experiments in support of this concept was accomplished with transgenic mice designed for ectopic Cx26 overexpression in the granular layer using the involucrin (inv) promotor [131]. These authors demonstrated that Cx26 downregulation is required for barrier acquisition during development and that keratinocytes isolated from adult inv-Cx26 mice displayed enhanced ATP release that correlated with an inflammatory response and psoriasiform phenotypes. Strikingly, the efflux of ATP could be ablated with hemichannel blockers.
Manipulation of connexin expression for enhanced epidermal repair is currently a popular focus of investigation. Connexin remodeling in response to injury appears to orchestrate cellular switches required for mobilization, proliferation, and migration of keratinocytes at the wound periphery [132]. Specifically, the presence of Cx43, Cx26, Cx30, Cx31 and Cx31.1 diminishes at the wound perimeter in the acute phase of healing with basal expression levels restored only after re-epithelialization [120,133,134]. Sustained connexin expression is observed in chronic nonhealing wounds typified by venous stasis ulcers in diabetics [135–137]. Remarkably, Cx43 knockout mice exhibit accelerated wound closure [138,139] and a recent multicenter randomized clinical trial in humans treated with the Cx43 mimetic peptide ACT1 achieved improved healing of refractory ulcers [140–142].
Also noteworthy is a putative pro-inflammatory interplay between paracrine signaling via connexin hemichannels, the cutaneous microflora, and opportunistic pathogens [117]. Bacterial cell wall components are observed to differentially influence expression levels and open probability of Cx26 and Cx43 hemichannels [47,118]. Strikingly, peptidoglycan (PGN) isolated from Staphylococcus Aureus promoted open hemichannel states in HaCaT cells (a human keratinocyte line) transfected with KID-associated Cx26 constructs whereas PGN derived from the commensal skin organism Streptococcus Epidermidis did not. These authors demonstrated that S. Aureus PGN challenge stimulated ATP release into the extracellular milieu and heightened quantities of the inflammatory mediator interleukin-6 (IL-6) in HeLa cells and HaCaT cells expressing KID mutants [47]. This response was not evoked in cells transfected with wild-type Cx26 or positive control non-KID Cx26 mutant variants and could be ablated with the hemichannel blocker carbenoxolone. Although a predisposition to microbe-induced inflammatory states in connexin channelopathies has been suggested, further work is needed to elucidate these interactions.
2.3 CONNEXINS & CANCER
Extending from what is known about cellular proliferation and differentiation, connexin intercellular communication is also under scrutiny for ostensible influences in oncogenesis. Cutaneous neoplasms of non-melanocytic origin show an exaggerated presence of Cx26 and Cx30 without a paralleled increase in Cx43 synthesis [143]. Melanomas have marked upregulation of Cx26 and Cx30 in the epidermis circumferentially adjacent to the tumor [143,144]. This does not occur at the margin of benign melanocytic nevi and has been correlated to malignant potential. The degree of horizontal Cx26 propagation may additionally presage metastasis [144], possibly by linking nests of malignant cells to nearby endothelial cells and thereby facilitating angiogenesis [145].
Immunohistochemical analyses of human malignant melanoma lesions have detected amplified basal levels of Cx43 relative to benign nevi [146]. Surprisingly, ectopic overexpression of Cx43 in the B16-BL6 mouse melanoma cell line curtailed proliferation and anchorage-independent growth in an in vitro model replicating the keratinocyte epithelial microenvironment [147]. Primary tumors generated in a chicken chorioallantoic membrane in vivo system by inoculation with Cx43-expressing BL6 cells were found to have reduced weight compared to those resulting from connexin-deficient or Cx26-expressing malignant cells [147]. Still, purporting Cx43 as a tumor suppressor will require further substantiation, with a conceivable role for pharmacological connexin modulators.
Investigations aimed at exploiting connexin channels to undermine tumorigenesis are ongoing. Gap junctions are recognized as avenues for direct intercellular transfer of microRNAs capable of promulgating cancer signals [148]. Conversely, prominence of gap junctional communication in certain tumor types may enable efficient delivery of nucleic acid based technologies for in situ oncogene silencing. Gap junctions may be further harnessed to potentiate the intended cytotoxic effects of chemotherapeutic agents [149]. If strategies such as these are validated, pharmacological connexin modulators may hypothetically constitute adjuncts in anti-cancer regimens.
2.4 SUMMARY
Hereditary cutaneous disorders involving connexins allude to the clout of these proteins as superintendents of epidermal homeostasis. Much progress has been made with regard to decoding the functional defects engendered by pathogenic mutations along with their physiological upshots. Discrete roles for connexin gap junctions and hemichannels have been dissected apart, in many instances through pharmacological means. However, many outstanding questions remain within connexin pathobiology due to the complexity of the endogenous regulatory mechanisms that direct the gating machinery and impose permselectivity properties. Pathology stemming from an erroneous gain in channel function, such as that in KID syndrome, offers an obvious initial target to catalyze the development of novel therapeutic strategies. Multiple independent investigators have converged on the concept of aberrant hemichannel behavior as permissive of diseased states in the skin. It stands to reason that directly acting connexin inhibitors, if screened for adequate affinity and specificity, may be capable of preserving or restoring epidermal integrity. Particularly exciting are small molecules already known to be free of untoward ancillary effects in human skin. For now the inhibitory paradigm constitutes the lowest hanging fruit, although connexin pharmacology is likely to become highly sophisticated as repercussions for ubiquitous epidermal problems such as inflammation, infection, and hyperplasia are further clarified.
Acknowledgments
Our work was supported by the NIH grant R01 AR59505 (TWW) from the NIAMS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Martin PE, van Steensel M. Connexins and skin disease: Insights into the role of beta connexins in skin homeostasis. Cell Tissue Res. 2015;360:645–658. doi: 10.1007/s00441-014-2094-3. [DOI] [PubMed] [Google Scholar]
- 2.Scott CA, Tattersall D, O’Toole EA, Kelsell DP. Connexins in epidermal homeostasis and skin disease. Biochim Biophys Acta. 2011;1818:1952–1961. doi: 10.1016/j.bbamem.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 3.Goodenough DA, Paul DL. Gap junctions. Cold Spring Harb Perspect Biol. 2009;1:a002576. doi: 10.1101/cshperspect.a002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Richard G. Connexins: A connection with the skin. Exp Dermatol. 2000;9:77–96. doi: 10.1034/j.1600-0625.2000.009002077.x. [DOI] [PubMed] [Google Scholar]
- 5.Salomon D, Masgrau E, Vischer S, Ullrich S, Dupont E, Sappino P, Saurat JH, Meda P. Topography of mammalian connexins in human skin. J Invest Dermatol. 1994;103:240–247. doi: 10.1111/1523-1747.ep12393218. [DOI] [PubMed] [Google Scholar]
- 6.Goodenough DA, Paul DL. Beyond the gap: Functions of unpaired connexon channels. Nat Rev Mol Cell Biol. 2003;4:285–294. doi: 10.1038/nrm1072. [DOI] [PubMed] [Google Scholar]
- 7.Stout C, Goodenough DA, Paul DL. Connexins: Functions without junctions. Current opinion in cell biology. 2004;16:507–512. doi: 10.1016/j.ceb.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 8.Bruzzone R, White TW, Paul DL. Connections with connexins: The molecular basis of direct intercellular signaling. Eur J Biochem. 1996;238:1–27. doi: 10.1111/j.1432-1033.1996.0001q.x. [DOI] [PubMed] [Google Scholar]
- 9.DeVries SH, Schwartz EA. Hemi-gap-junction channels in solitary horizontal cells of the catfish retina. J Physiol. 1992;445:201–230. doi: 10.1113/jphysiol.1992.sp018920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goodenough DA. Bulk isolation of mouse hepatocyte gap junctions. Characterization of the principal protein, connexin. J Cell Biol. 1974;61:557–563. doi: 10.1083/jcb.61.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paul DL, Ebihara L, Takemoto LJ, Swenson KI, Goodenough DA. Connexin46, a novel lens gap junction protein, induces voltage-gated currents in nonjunctional plasma membrane of xenopus oocytes. The Journal of cell biology. 1991;115:1077–1089. doi: 10.1083/jcb.115.4.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Revel JP, Karnovsky MJ. Hexagonal array of subunits in intercellular junctions of the mouse heart and liver. The Journal of cell biology. 1967;33:C7–C12. doi: 10.1083/jcb.33.3.c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sohl G, Willecke K. An update on connexin genes and their nomenclature in mouse and man. Cell Commun Adhes. 2003;10:173–180. doi: 10.1080/cac.10.4-6.173.180. [DOI] [PubMed] [Google Scholar]
- 14.Wiszniewski L, Limat A, Saurat JH, Meda P, Salomon D. Differential expression of connexins during stratification of human keratinocytes. J Invest Dermatol. 2000;115:278–285. doi: 10.1046/j.1523-1747.2000.00043.x. [DOI] [PubMed] [Google Scholar]
- 15.Bedner P, Niessen H, Odermatt B, Kretz M, Willecke K, Harz H. Selective permeability of different connexin channels to the second messenger cyclic amp. J Biol Chem. 2006;281:6673–6681. doi: 10.1074/jbc.M511235200. [DOI] [PubMed] [Google Scholar]
- 16.Goldberg GS, Lampe PD, Nicholson BJ. Selective transfer of endogenous metabolites through gap junctions composed of different connexins. Nature cell biology. 1999;1:457–459. doi: 10.1038/15693. [DOI] [PubMed] [Google Scholar]
- 17.Niessen H, Harz H, Bedner P, Kramer K, Willecke K. Selective permeability of different connexin channels to the second messenger inositol 1,4,5-trisphosphate. J Cell Sci. 2000;113 ( Pt 8):1365–1372. doi: 10.1242/jcs.113.8.1365. [DOI] [PubMed] [Google Scholar]
- 18.Weber PA, Chang HC, Spaeth KE, Nitsche JM, Nicholson BJ. The permeability of gap junction channels to probes of different size is dependent on connexin composition and permeant-pore affinities. Biophysical journal. 2004;87:958–973. doi: 10.1529/biophysj.103.036350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richard G. Connexin disorders of the skin. Clin Dermatol. 2005;23:23–32. doi: 10.1016/j.clindermatol.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 20.Boyden LM, Craiglow BG, Zhou J, Hu R, Loring EC, Morel KD, Lauren CT, Lifton RP, Bilguvar K, Paller AS, Choate KA. Dominant de novo mutations in gja1 cause erythrokeratodermia variabilis et progressiva, without features of oculodentodigital dysplasia. J Invest Dermatol. 2015;135:1540–1547. doi: 10.1038/jid.2014.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duchatelet S, Hovnanian A. Erythrokeratodermia variabilis et progressiva allelic to oculo-dento-digital dysplasia. J Invest Dermatol. 2015;135:1475–1478. doi: 10.1038/jid.2014.535. [DOI] [PubMed] [Google Scholar]
- 22.Wang H, Cao X, Lin Z, Lee M, Jia X, Ren Y, Dai L, Guan L, Zhang J, Lin X, Chen Q, Feng C, Zhou EY, Yin J, Xu G, Yang Y. Exome sequencing reveals mutation in gja1 as a cause of keratoderma-hypotrichosis-leukonychia totalis syndrome. Hum Mol Genet. 2015;24:243–250. doi: 10.1093/hmg/ddu442. [DOI] [PubMed] [Google Scholar]
- 23.Kogame T, Dainichi T, Shimomura Y, Tanioka M, Kabashima K, Miyachi Y. Palmoplantar keratosis in oculodentodigital dysplasia with a gja1 point mutation out of the c-terminal region of connexin 43. The Journal of dermatology. 2014;41:1095–1097. doi: 10.1111/1346-8138.12682. [DOI] [PubMed] [Google Scholar]
- 24.Kelly JJ, Simek J, Laird DW. Mechanisms linking connexin mutations to human diseases. Cell and tissue research. 2015;360:701–721. doi: 10.1007/s00441-014-2024-4. [DOI] [PubMed] [Google Scholar]
- 25.Pfenniger A, Wohlwend A, Kwak BR. Mutations in connexin genes and disease. Eur J Clin Invest. 2011;41:103–116. doi: 10.1111/j.1365-2362.2010.02378.x. [DOI] [PubMed] [Google Scholar]
- 26.Wei CJ, Xu X, Lo CW. Connexins and cell signaling in development and disease. Annu Rev Cell Dev Biol. 2004;20:811–838. doi: 10.1146/annurev.cellbio.19.111301.144309. [DOI] [PubMed] [Google Scholar]
- 27.van Steensel MA. Gap junction diseases of the skin. Am J Med Genet C Semin Med Genet. 2004;131C:12–19. doi: 10.1002/ajmg.c.30030. [DOI] [PubMed] [Google Scholar]
- 28.Bakirtzis G, Choudhry R, Aasen T, Shore L, Brown K, Bryson S, Forrow S, Tetley L, Finbow M, Greenhalgh D, Hodgins M. Targeted epidermal expression of mutant connexin 26(d66h) mimics true vohwinkel syndrome and provides a model for the pathogenesis of dominant connexin disorders. Hum Mol Genet. 2003;12:1737–1744. doi: 10.1093/hmg/ddg183. [DOI] [PubMed] [Google Scholar]
- 29.Schnichels M, Worsdorfer P, Dobrowolski R, Markopoulos C, Kretz M, Schwarz G, Winterhager E, Willecke K. The connexin31 f137l mutant mouse as a model for the human skin disease erythrokeratodermia variabilis (ekv) Hum Mol Genet. 2007;16:1216–1224. doi: 10.1093/hmg/ddm068. [DOI] [PubMed] [Google Scholar]
- 30.Churko JM, Chan J, Shao Q, Laird DW. The g60s connexin43 mutant regulates hair growth and hair fiber morphology in a mouse model of human oculodentodigital dysplasia. J Invest Dermatol. 2011;131:2197–2204. doi: 10.1038/jid.2011.183. [DOI] [PubMed] [Google Scholar]
- 31.Bosen F, Schutz M, Beinhauer A, Strenzke N, Franz T, Willecke K. The clouston syndrome mutation connexin30 a88v leads to hyperproliferation of sebaceous glands and hearing impairments in mice. FEBS letters. 2014;588:1795–1801. doi: 10.1016/j.febslet.2014.03.040. [DOI] [PubMed] [Google Scholar]
- 32.Mese G, Sellitto C, Li L, Wang HZ, Valiunas V, Richard G, Brink PR, White TW. The cx26-g45e mutation displays increased hemichannel activity in a mouse model of the lethal form of keratitis-ichthyosis-deafness syndrome. Mol Biol Cell. 2011;22:4776–4786. doi: 10.1091/mbc.E11-09-0778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schutz M, Auth T, Gehrt A, Bosen F, Korber I, Strenzke N, Moser T, Willecke K. The connexin26 s17f mouse mutant represents a model for the human hereditary keratitis-ichthyosis-deafness syndrome. Hum Mol Genet. 2011;20:28–39. doi: 10.1093/hmg/ddq429. [DOI] [PubMed] [Google Scholar]
- 34.Rogers FA, Hu RH, Milstone LM. Local delivery of gene-modifying triplex-forming molecules to the epidermis. J Invest Dermatol. 2013;133:685–691. doi: 10.1038/jid.2012.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lara MF, Gonzalez-Gonzalez E, Speaker TJ, Hickerson RP, Leake D, Milstone LM, Contag CH, Kaspar RL. Inhibition of cd44 gene expression in human skin models, using self-delivery short interfering rna administered by dissolvable microneedle arrays. Human gene therapy. 2012;23:816–823. doi: 10.1089/hum.2011.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonzalez-Gonzalez E, Kim YC, Speaker TJ, Hickerson RP, Spitler R, Birchall JC, Lara MF, Hu RH, Liang Y, Kirkiles-Smith N, Prausnitz MR, Milstone LM, Contag CH, Kaspar RL. Visualization of plasmid delivery to keratinocytes in mouse and human epidermis. Scientific reports. 2011;1:158. doi: 10.1038/srep00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Desplantez T, Verma V, Leybaert L, Evans WH, Weingart R. Gap26, a connexin mimetic peptide, inhibits currents carried by connexin43 hemichannels and gap junction channels. Pharmacol Res. 2012;65:546–552. doi: 10.1016/j.phrs.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 38.Evans WH, Boitano S. Connexin mimetic peptides: Specific inhibitors of gap-junctional intercellular communication. Biochem Soc Trans. 2001;29:606–612. doi: 10.1042/bst0290606. [DOI] [PubMed] [Google Scholar]
- 39.Wang N, De Bock M, Antoons G, Gadicherla AK, Bol M, Decrock E, Evans WH, Sipido KR, Bukauskas FF, Leybaert L. Connexin mimetic peptides inhibit cx43 hemichannel opening triggered by voltage and intracellular ca2+ elevation. Basic research in cardiology. 2012;107:304. doi: 10.1007/s00395-012-0304-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richard G, Rouan F, Willoughby CE, Brown N, Chung P, Ryynanen M, Jabs EW, Bale SJ, DiGiovanna JJ, Uitto J, Russell L. Missense mutations in gjb2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. Am J Hum Genet. 2002;70:1341–1348. doi: 10.1086/339986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Skinner BA, Greist MC, Norins AL. The keratitis, ichthyosis, and deafness (kid) syndrome. Arch Dermatol. 1981;117:285–289. [PubMed] [Google Scholar]
- 42.Mazereeuw-Hautier J, Bitoun E, Chevrant-Breton J, Man SY, Bodemer C, Prins C, Antille C, Saurat JH, Atherton D, Harper JI, Kelsell DP, Hovnanian A. Keratitis-ichthyosis-deafness syndrome: Disease expression and spectrum of connexin 26 (gjb2) mutations in 14 patients. Br J Dermatol. 2007;156:1015–1019. doi: 10.1111/j.1365-2133.2007.07806.x. [DOI] [PubMed] [Google Scholar]
- 43.Zhao HB, Kikuchi T, Ngezahayo A, White TW. Gap junctions and cochlear homeostasis. J Membr Biol. 2006;209:177–186. doi: 10.1007/s00232-005-0832-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu J, Nicholson BJ. The role of connexins in ear and skin physiology - functional insights from disease-associated mutations. Biochim Biophys Acta. 2013;1828:167–178. doi: 10.1016/j.bbamem.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iossa S, Marciano E, Franze A. Gjb2 gene mutations in syndromic skin diseases with sensorineural hearing loss. Curr Genomics. 2011;12:475–785. doi: 10.2174/138920211797904098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanchez HA, Verselis VK. Aberrant cx26 hemichannels and keratitis-ichthyosis-deafness syndrome: Insights into syndromic hearing loss. Front Cell Neurosci. 2014;8:354. doi: 10.3389/fncel.2014.00354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Donnelly S, English G, de Zwart-Storm EA, Lang S, van Steensel MA, Martin PE. Differential susceptibility of cx26 mutations associated with epidermal dysplasias to peptidoglycan derived from staphylococcus aureus and staphylococcus epidermidis. Exp Dermatol. 2012;21:592–598. doi: 10.1111/j.1600-0625.2012.01521.x. [DOI] [PubMed] [Google Scholar]
- 48.Garcia IE, Maripillan J, Jara O, Ceriani R, Palacios-Munoz A, Ramachandran J, Olivero P, Perez-Acle T, Gonzalez C, Saez JC, Contreras JE, Martinez AD. Keratitis-ichthyosis-deafness syndrome-associated cx26 mutants produce nonfunctional gap junctions but hyperactive hemichannels when co- expressed with wild type cx43. J Invest Dermatol. 2015;135:1338–1347. doi: 10.1038/jid.2015.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gerido DA, DeRosa AM, Richard G, White TW. Aberrant hemichannel properties of cx26 mutations causing skin disease and deafness. Am J Physiol Cell Physiol. 2007;293:C337–345. doi: 10.1152/ajpcell.00626.2006. [DOI] [PubMed] [Google Scholar]
- 50.Lee JR, Derosa AM, White TW. Connexin mutations causing skin disease and deafness increase hemichannel activity and cell death when expressed in xenopus oocytes. J Invest Dermatol. 2009;129:870–878. doi: 10.1038/jid.2008.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mhaske PV, Levit NA, Li L, Wang HZ, Lee JR, Shuja Z, Brink PR, White TW. The human cx26-d50a and cx26-a88v mutations causing keratitis-ichthyosis-deafness syndrome display increased hemichannel activity. Am J Physiol Cell Physiol. 2013;304:C1150–1158. doi: 10.1152/ajpcell.00374.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Montgomery JR, White TW, Martin BL, Turner ML, Holland SM. A novel connexin 26 gene mutation associated with features of the keratitis-ichthyosis-deafness syndrome and the follicular occlusion triad. J Am Acad Dermatol. 2004;51:377–382. doi: 10.1016/j.jaad.2003.12.042. [DOI] [PubMed] [Google Scholar]
- 53.Sanchez HA, Mese G, Srinivas M, White TW, Verselis VK. Differentially altered ca2+ regulation and ca2+ permeability in cx26 hemichannels formed by the a40v and g45e mutations that cause keratitis ichthyosis deafness syndrome. J Gen Physiol. 2010;136:47–62. doi: 10.1085/jgp.201010433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanchez HA, Villone K, Srinivas M, Verselis VK. The d50n mutation and syndromic deafness: Altered cx26 hemichannel properties caused by effects on the pore and intersubunit interactions. J Gen Physiol. 2013;142:3–22. doi: 10.1085/jgp.201310962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stong BC, Chang Q, Ahmad S, Lin X. A novel mechanism for connexin 26 mutation linked deafness: Cell death caused by leaky gap junction hemichannels. Laryngoscope. 2006;116:2205–2210. doi: 10.1097/01.mlg.0000241944.77192.d2. [DOI] [PubMed] [Google Scholar]
- 56.Elias PM, Ahn SK, Denda M, Brown BE, Crumrine D, Kimutai LK, Komuves L, Lee SH, Feingold KR. Modulations in epidermal calcium regulate the expression of differentiation-specific markers. J Invest Dermatol. 2002;119:1128–1136. doi: 10.1046/j.1523-1747.2002.19512.x. [DOI] [PubMed] [Google Scholar]
- 57.Mauro TM, Isseroff RR, Lasarow R, Pappone PA. Ion channels are linked to differentiation in keratinocytes. J Membr Biol. 1993;132:201–209. doi: 10.1007/BF00235738. [DOI] [PubMed] [Google Scholar]
- 58.Menon GK, Price LF, Bommannan B, Elias PM, Feingold KR. Selective obliteration of the epidermal calcium gradient leads to enhanced lamellar body secretion. J Invest Dermatol. 1994;102:789–795. doi: 10.1111/1523-1747.ep12377921. [DOI] [PubMed] [Google Scholar]
- 59.Proksch E, Brandner JM, Jensen JM. The skin: An indispensable barrier. Exp Dermatol. 2008;17:1063–1072. doi: 10.1111/j.1600-0625.2008.00786.x. [DOI] [PubMed] [Google Scholar]
- 60.Reiss M, Lipsey LR, Zhou ZL. Extracellular calcium-dependent regulation of transmembrane calcium fluxes in murine keratinocytes. Journal of cellular physiology. 1991;147:281–291. doi: 10.1002/jcp.1041470213. [DOI] [PubMed] [Google Scholar]
- 61.de Berker D, Branford WA, Soucek S, Michaels L. Fatal keratitis ichthyosis and deafness syndrome (kids). Aural, ocular, and cutaneous histopathology. The American Journal of dermatopathology. 1993;15:64–69. doi: 10.1097/00000372-199302000-00012. [DOI] [PubMed] [Google Scholar]
- 62.Haruna K, Suga Y, Oizumi A, Mizuno Y, Endo H, Shimizu T, Hasegawa T, Ikeda S. Severe form of keratitis-ichthyosis-deafness (kid) syndrome associated with septic complications. J Dermatol. 2010;37:680–682. doi: 10.1111/j.1346-8138.2010.00839.x. [DOI] [PubMed] [Google Scholar]
- 63.Janecke AR, Hennies HC, Gunther B, Gansl G, Smolle J, Messmer EM, Utermann G, Rittinger O. Gjb2 mutations in keratitis-ichthyosis-deafness syndrome including its fatal form. Am J Med Genet A. 2005;133A:128–131. doi: 10.1002/ajmg.a.30515. [DOI] [PubMed] [Google Scholar]
- 64.Jonard L, Feldmann D, Parsy C, Freitag S, Sinico M, Koval C, Grati M, Couderc R, Denoyelle F, Bodemer C, Marlin S, Hadj-Rabia S. A familial case of keratitis-ichthyosis-deafness (kid) syndrome with the gjb2 mutation g45e. Eur J Med Genet. 2008;51:35–43. doi: 10.1016/j.ejmg.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 65.Koppelhus U, Tranebjaerg L, Esberg G, Ramsing M, Lodahl M, Rendtorff ND, Olesen HV, Sommerlund M. A novel mutation in the connexin 26 gene (gjb2) in a child with clinical and histological features of keratitis-ichthyosis-deafness (kid) syndrome. Clin Exp Dermatol. 2011;36:142–148. doi: 10.1111/j.1365-2230.2010.03936.x. [DOI] [PubMed] [Google Scholar]
- 66.Sbidian E, Feldmann D, Bengoa J, Fraitag S, Abadie V, de Prost Y, Bodemer C, Hadj-Rabia S. Germline mosaicism in keratitis-ichthyosis-deafness syndrome: Pre-natal diagnosis in a familial lethal form. Clin Genet. 2010;77:587–592. doi: 10.1111/j.1399-0004.2009.01339.x. [DOI] [PubMed] [Google Scholar]
- 67.Maintz L, Betz RC, Allam JP, Wenzel J, Jaksche A, Friedrichs N, Bieber T, Novak N. Keratitis-ichthyosis-deafness syndrome in association with follicular occlusion triad. Eur J Dermatol. 2005;15:347–352. [PubMed] [Google Scholar]
- 68.Messmer EM, Kenyon KR, Rittinger O, Janecke AR, Kampik A. Ocular manifestations of keratitis-ichthyosis-deafness (kid) syndrome. Ophthalmology. 2005;112:e1–6. doi: 10.1016/j.ophtha.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 69.Coggshall K, Farsani T, Ruben B, McCalmont TH, Berger TG, Fox LP, Shinkai K. Keratitis, ichthyosis, and deafness syndrome: A review of infectious and neoplastic complications. J Am Acad Dermatol. 2013;69:127–134. doi: 10.1016/j.jaad.2012.12.965. [DOI] [PubMed] [Google Scholar]
- 70.Abdollahi A, Hallaji Z, Esmaili N, Valikhani M, Barzegari M, Akhyani M, Toosi S, Miresmaili A. Kid syndrome. Dermatology online journal. 2007;13:11. [PubMed] [Google Scholar]
- 71.Braun-Falco M. Hereditary palmoplantar keratodermas. Journal der Deutschen Dermatologischen Gesellschaft = Journal of the German Society of Dermatology : JDDG. 2009;7:971–984. doi: 10.1111/j.1610-0387.2009.07058.x. quiz 984–975. [DOI] [PubMed] [Google Scholar]
- 72.Patel V, Sun G, Dickman M, Khuu P, Teng JM. Treatment of keratitis-ichthyosis- deafness (kid) syndrome in children: A case report and review of the literature. Dermatol Ther. 2015;28:89–93. doi: 10.1111/dth.12192. [DOI] [PubMed] [Google Scholar]
- 73.Conrado LA, Marques SA, Lastoria JC, Cuce LC, Marques ME, Dillon NL. Keratitis-ichthyosis-deafness (kid) syndrome with squamous cell carcinoma. Int J Dermatol. 2007;46:403–406. doi: 10.1111/j.1365-4632.2007.02977.x. [DOI] [PubMed] [Google Scholar]
- 74.Criscione V, Lachiewicz A, Robinson-Bostom L, Grenier N, Dill SW. Porokeratotic eccrine duct and hair follicle nevus (pehfn) associated with keratitis-ichthyosis-deafness (kid) syndrome. Pediatr Dermatol. 2010;27:514–517. doi: 10.1111/j.1525-1470.2010.01272.x. [DOI] [PubMed] [Google Scholar]
- 75.Grob JJ, Breton A, Bonafe JL, Sauvan-Ferdani M, Bonerandi JJ. Keratitis, ichthyosis, and deafness (kid) syndrome. Vertical transmission and death from multiple squamous cell carcinomas. Arch Dermatol. 1987;123:777–782. [PubMed] [Google Scholar]
- 76.Lancaster L, Jr, Fournet LF. Carcinoma of the tongue in a child: Report of case. J Oral Surg. 1969;27:269–270. [PubMed] [Google Scholar]
- 77.Homeida L, Wiley RT, Fatahzadeh M. Oral squamous cell carcinoma in a patient with keratitis-ichthyosis-deafness syndrome: A rare case. Oral Surg Oral Med Oral Pathol Oral Radiol. 2015;119:e226–232. doi: 10.1016/j.oooo.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 78.Sakabe J, Yoshiki R, Sugita K, Haruyama S, Sawada Y, Kabashima R, Bito T, Nakamura M, Tokura Y. Connexin 26 (gjb2) mutations in keratitis-ichthyosis-deafness syndrome presenting with squamous cell carcinoma. The Journal of dermatology. 2012;39:814–815. doi: 10.1111/j.1346-8138.2011.01414.x. [DOI] [PubMed] [Google Scholar]
- 79.Nyquist GG, Mumm C, Grau R, Crowson AN, Shurman DL, Benedetto P, Allen P, Lovelace K, Smith DW, Frieden I, Hybarger CP, Richard G. Malignant proliferating pilar tumors arising in kid syndrome: A report of two patients. American journal of medical genetics Part A. 2007;143A:734–741. doi: 10.1002/ajmg.a.31635. [DOI] [PubMed] [Google Scholar]
- 80.Hazen PG, Carney P, Lynch WS. Keratitis, ichthyosis, and deafness syndrome with development of multiple cutaneous neoplasms. Int J Dermatol. 1989;28:190–191. doi: 10.1111/j.1365-4362.1989.tb02463.x. [DOI] [PubMed] [Google Scholar]
- 81.Madariaga J, Fromowitz F, Phillips M, Hoover HC., Jr Squamous cell carcinoma in congenital ichthyosis with deafness and keratitis. A case report and review of the literature. Cancer. 1986;57:2026–2029. doi: 10.1002/1097-0142(19860515)57:10<2026::aid-cncr2820571024>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 82.Natsuga K, Akiyama M, Shimizu H. Malignant skin tumours in patients with inherited ichthyosis. The British journal of dermatology. 2011;165:263–268. doi: 10.1111/j.1365-2133.2011.10381.x. [DOI] [PubMed] [Google Scholar]
- 83.Bondeson ML, Nystrom AM, Gunnarsson U, Vahlquist A. Connexin 26 (gjb2) mutations in two swedish patients with atypical vohwinkel (mutilating keratoderma plus deafness) and kid syndrome both extensively treated with acitretin. Acta Derm Venereol. 2006;86:503–508. doi: 10.2340/00015555-0164. [DOI] [PubMed] [Google Scholar]
- 84.Prasad SC, Bygum A. Successful treatment with alitretinoin of dissecting cellulitis of the scalp in keratitis-ichthyosis-deafness syndrome. Acta Derm Venereol. 2013;93:473–474. doi: 10.2340/00015555-1499. [DOI] [PubMed] [Google Scholar]
- 85.Sahoo B, Handa S, Kaur I, Radotra BD, Kumar B. Kid syndrome: Response to acitretin. The Journal of dermatology. 2002;29:499–502. doi: 10.1111/j.1346-8138.2002.tb00315.x. [DOI] [PubMed] [Google Scholar]
- 86.Werchau S, Toberer F, Enk A, Helmbold P. Keratitis-ichthyosis-deafness syndrome: Response to alitretinoin and review of literature. Archives of dermatology. 2011;147:993–995. doi: 10.1001/archdermatol.2011.216. [DOI] [PubMed] [Google Scholar]
- 87.Zhang X, He Y, Zhou H, Luo Q, Li C. Severe ichthyosis-related disorders in children: Response to acitretin. J Dermatolog Treat. 2007;18:118–122. doi: 10.1080/09546630601156348. [DOI] [PubMed] [Google Scholar]
- 88.Rudkin GH, Carlsen BT, Chung CY, Huang W, Ishida K, Anvar B, Yamaguchi DT, Miller TA. Retinoids inhibit squamous cell carcinoma growth and intercellular communication. J Surg Res. 2002;103:183–189. doi: 10.1006/jsre.2001.6346. [DOI] [PubMed] [Google Scholar]
- 89.Masgrau-Peya E, Salomon D, Saurat JH, Meda P. In vivo modulation of connexins 43 and 26 of human epidermis by topical retinoic acid treatment. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 1997;45:1207–1215. doi: 10.1177/002215549704500904. [DOI] [PubMed] [Google Scholar]
- 90.Brokamp C, Todd J, Montemagno C, Wendell D. Electrophysiology of single and aggregate cx43 hemichannels. PloS one. 2012;7:e47775. doi: 10.1371/journal.pone.0047775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cao Y, Zheng OJ. Tonabersat for migraine prophylaxis: A systematic review. Pain physician. 2014;17:1–8. [PubMed] [Google Scholar]
- 92.Cruikshank SJ, Hopperstad M, Younger M, Connors BW, Spray DC, Srinivas M. Potent block of cx36 and cx50 gap junction channels by mefloquine. Proc Natl Acad Sci U S A. 2004;101:12364–12369. doi: 10.1073/pnas.0402044101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Davidson JS, Baumgarten IM, Harley EH. Reversible inhibition of intercellular junctional communication by glycyrrhetinic acid. Biochemical and biophysical research communications. 1986;134:29–36. doi: 10.1016/0006-291x(86)90522-x. [DOI] [PubMed] [Google Scholar]
- 94.Garza I. Tonabersat: A cortical spreading depression inhibitor as potential pharmacologic prophylaxis in migraine with aura. Current neurology and neuroscience reports. 2010;10:7–9. doi: 10.1007/s11910-009-0083-9. [DOI] [PubMed] [Google Scholar]
- 95.Rubinos C, Sanchez HA, Verselis VK, Srinivas M. Mechanism of inhibition of connexin channels by the quinine derivative n-benzylquininium. J Gen Physiol. 2012;139:69–82. doi: 10.1085/jgp.201110678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sarrouilhe D, Dejean C, Mesnil M. Involvement of gap junction channels in the pathophysiology of migraine with aura. Front Physiol. 2014;5:78. doi: 10.3389/fphys.2014.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Spray DC, Rozental R, Srinivas M. Prospects for rational development of pharmacological gap junction channel blockers. Curr Drug Targets. 2002;3:455–464. doi: 10.2174/1389450023347353. [DOI] [PubMed] [Google Scholar]
- 98.Srinivas M, Hopperstad MG, Spray DC. Quinine blocks specific gap junction channel subtypes. Proc Natl Acad Sci U S A. 2001;98:10942–10947. doi: 10.1073/pnas.191206198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Verselis VK, Srinivas M. Connexin channel modulators and their mechanisms of action. Neuropharmacology. 2013;75:517–524. doi: 10.1016/j.neuropharm.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Levit NA, Sellitto C, Wang HZ, Li L, Srinivas M, Brink PR, White TW. Aberrant connexin26 hemichannels underlying keratitis-ichthyosis-deafness syndrome are potently inhibited by mefloquine. J Invest Dermatol. 2015;135:1033–1042. doi: 10.1038/jid.2014.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chi J, Li L, Liu M, Tan J, Tang C, Pan Q, Wang D, Zhang Z. Pathogenic connexin-31 forms constitutively active hemichannels to promote necrotic cell death. PLoS One. 2012;7:e32531. doi: 10.1371/journal.pone.0032531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Common JE, Becker D, Di WL, Leigh IM, O’Toole EA, Kelsell DP. Functional studies of human skin disease- and deafness-associated connexin 30 mutations. Biochemical and biophysical research communications. 2002;298:651–656. doi: 10.1016/s0006-291x(02)02517-2. [DOI] [PubMed] [Google Scholar]
- 103.Essenfelder GM, Bruzzone R, Lamartine J, Charollais A, Blanchet-Bardon C, Barbe MT, Meda P, Waksman G. Connexin30 mutations responsible for hidrotic ectodermal dysplasia cause abnormal hemichannel activity. Hum Mol Genet. 2004;13:1703–1714. doi: 10.1093/hmg/ddh191. [DOI] [PubMed] [Google Scholar]
- 104.Churko JM, Kelly JJ, Macdonald A, Lee J, Sampson J, Bai D, Laird DW. The g60s cx43 mutant enhances keratinocyte proliferation and differentiation. Exp Dermatol. 2012;21:612–618. doi: 10.1111/j.1600-0625.2012.01532.x. [DOI] [PubMed] [Google Scholar]
- 105.Lemaitre G, Sivan V, Lamartine J, Cosset JM, Cavelier-Balloy B, Salomon D, Waksman G, Martin MT. Connexin 30, a new marker of hyperproliferative epidermis. The British journal of dermatology. 2006;155:844–846. doi: 10.1111/j.1365-2133.2006.07439.x. [DOI] [PubMed] [Google Scholar]
- 106.Brissette JL, Kumar NM, Gilula NB, Hall JE, Dotto GP. Switch in gap junction protein expression is associated with selective changes in junctional permeability during keratinocyte differentiation. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:6453–6457. doi: 10.1073/pnas.91.14.6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gibson DF, Bikle DD, Harris J, Goldberg GS. The expression of the gap junctional protein cx43 is restricted to proliferating and non differentiated normal and transformed keratinocytes. Exp Dermatol. 1997;6:167–174. doi: 10.1111/j.1600-0625.1997.tb00201.x. [DOI] [PubMed] [Google Scholar]
- 108.Bao B, Jiang J, Yanase T, Nishi Y, Morgan JR. Connexon-mediated cell adhesion drives microtissue self-assembly. Faseb J. 2011;25:255–264. doi: 10.1096/fj.10-155291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Prochnow N, Dermietzel R. Connexons and cell adhesion: A romantic phase. Histochem Cell Biol. 2008;130:71–77. doi: 10.1007/s00418-008-0434-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kandyba EE, Hodgins MB, Martin PE. A murine living skin equivalent amenable to live-cell imaging: Analysis of the roles of connexins in the epidermis. J Invest Dermatol. 2008;128:1039–1049. doi: 10.1038/sj.jid.5701125. [DOI] [PubMed] [Google Scholar]
- 111.Marquez-Rosado L, Singh D, Rincon-Arano H, Solan JL, Lampe PD. Cask (lin2) interacts with cx43 in wounded skin and their coexpression affects cell migration. Journal of cell science. 2012;125:695–702. doi: 10.1242/jcs.084400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wright CS, van Steensel MA, Hodgins MB, Martin PE. Connexin mimetic peptides improve cell migration rates of human epidermal keratinocytes and dermal fibroblasts in vitro. Wound Repair Regen. 2009;17:240–249. doi: 10.1111/j.1524-475X.2009.00471.x. [DOI] [PubMed] [Google Scholar]
- 113.Calder BW, Matthew Rhett J, Bainbridge H, Fann SA, Gourdie RG, Yost MJ. Inhibition of connexin 43 hemichannel-mediated atp release attenuates early inflammation during the foreign body response. Tissue Eng Part A. 2015;21:1752–1762. doi: 10.1089/ten.tea.2014.0651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chanson M, Derouette JP, Roth I, Foglia B, Scerri I, Dudez T, Kwak BR. Gap junctional communication in tissue inflammation and repair. Biochimica et biophysica acta. 2005;1711:197–207. doi: 10.1016/j.bbamem.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 115.Mori R, Power KT, Wang CM, Martin P, Becker DL. Acute downregulation of connexin43 at wound sites leads to a reduced inflammatory response, enhanced keratinocyte proliferation and wound fibroblast migration. Journal of cell science. 2006;119:5193–5203. doi: 10.1242/jcs.03320. [DOI] [PubMed] [Google Scholar]
- 116.O’Carroll SJ, Becker DL, Davidson JO, Gunn AJ, Nicholson LF, Green CR. The use of connexin-based therapeutic approaches to target inflammatory diseases. Methods Mol Biol. 2013;1037:519–546. doi: 10.1007/978-1-62703-505-7_31. [DOI] [PubMed] [Google Scholar]
- 117.Ceelen L, Haesebrouck F, Vanhaecke T, Rogiers V, Vinken M. Modulation of connexin signaling by bacterial pathogens and their toxins. Cell Mol Life Sci. 2011;68:3047–3064. doi: 10.1007/s00018-011-0737-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Robertson J, Lang S, Lambert PA, Martin PE. Peptidoglycan derived from staphylococcus epidermidis induces connexin43 hemichannel activity with consequences on the innate immune response in endothelial cells. Biochem J. 2010;432:133–143. doi: 10.1042/BJ20091753. [DOI] [PubMed] [Google Scholar]
- 119.Sawey MJ, Goldschmidt MH, Risek B, Gilula NB, Lo CW. Perturbation in connexin 43 and connexin 26 gap-junction expression in mouse skin hyperplasia and neoplasia. Mol Carcinog. 1996;17:49–61. doi: 10.1002/(SICI)1098-2744(199610)17:2<49::AID-MC1>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 120.Goliger JA, Paul DL. Wounding alters epidermal connexin expression and gap junction-mediated intercellular communication. Mol Biol Cell. 1995;6:1491–1501. doi: 10.1091/mbc.6.11.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lucke T, Choudhry R, Thom R, Selmer IS, Burden AD, Hodgins MB. Upregulation of connexin 26 is a feature of keratinocyte differentiation in hyperproliferative epidermis, vaginal epithelium, and buccal epithelium. J Invest Dermatol. 1999;112:354–361. doi: 10.1046/j.1523-1747.1999.00512.x. [DOI] [PubMed] [Google Scholar]
- 122.Hivnor C, Williams N, Singh F, VanVoorhees A, Dzubow L, Baldwin D, Seykora J. Gene expression profiling of porokeratosis demonstrates similarities with psoriasis. J Cutan Pathol. 2004;31:657–664. doi: 10.1111/j.0303-6987.2004.00247.x. [DOI] [PubMed] [Google Scholar]
- 123.Labarthe MP, Bosco D, Saurat JH, Meda P, Salomon D. Upregulation of connexin 26 between keratinocytes of psoriatic lesions. J Invest Dermatol. 1998;111:72–76. doi: 10.1046/j.1523-1747.1998.00248.x. [DOI] [PubMed] [Google Scholar]
- 124.Rivas MV, Jarvis ED, Morisaki S, Carbonaro H, Gottlieb AB, Krueger JG. Identification of aberrantly regulated genes in diseased skin using the cdna differential display technique. J Invest Dermatol. 1997;108:188–194. doi: 10.1111/1523-1747.ep12334217. [DOI] [PubMed] [Google Scholar]
- 125.Li B, Tsoi LC, Swindell WR, Gudjonsson JE, Tejasvi T, Johnston A, Ding J, Stuart PE, Xing X, Kochkodan JJ, Voorhees JJ, Kang HM, Nair RP, Abecasis GR, Elder JT. Transcriptome analysis of psoriasis in a large case-control sample: Rna-seq provides insights into disease mechanisms. J Invest Dermatol. 2014;134:1828–1838. doi: 10.1038/jid.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cotrina ML, Lin JH, Alves-Rodrigues A, Liu S, Li J, Azmi-Ghadimi H, Kang J, Naus CC, Nedergaard M. Connexins regulate calcium signaling by controlling atp release. Proc Natl Acad Sci U S A. 1998;95:15735–15740. doi: 10.1073/pnas.95.26.15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kang J, Kang N, Lovatt D, Torres A, Zhao Z, Lin J, Nedergaard M. Connexin 43 hemichannels are permeable to atp. J Neurosci. 2008;28:4702–4711. doi: 10.1523/JNEUROSCI.5048-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Stout CE, Costantin JL, Naus CC, Charles AC. Intercellular calcium signaling in astrocytes via atp release through connexin hemichannels. The Journal of biological chemistry. 2002;277:10482–10488. doi: 10.1074/jbc.M109902200. [DOI] [PubMed] [Google Scholar]
- 129.Burnstock G, Knight GE, Greig AV. Purinergic signaling in healthy and diseased skin. J Invest Dermatol. 2012;132:526–546. doi: 10.1038/jid.2011.344. [DOI] [PubMed] [Google Scholar]
- 130.Burrell HE, Bowler WB, Gallagher JA, Sharpe GR. Human keratinocytes express multiple p2y-receptors: Evidence for functional p2y1, p2y2, and p2y4 receptors. J Invest Dermatol. 2003;120:440–447. doi: 10.1046/j.1523-1747.2003.12050.x. [DOI] [PubMed] [Google Scholar]
- 131.Djalilian AR, McGaughey D, Patel S, Seo EY, Yang C, Cheng J, Tomic M, Sinha S, Ishida-Yamamoto A, Segre JA. Connexin 26 regulates epidermal barrier and wound remodeling and promotes psoriasiform response. J Clin Invest. 2006;116:1243–1253. doi: 10.1172/JCI27186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Churko JM, Laird DW. Gap junction remodeling in skin repair following wounding and disease. Physiology (Bethesda) 2013;28:190–198. doi: 10.1152/physiol.00058.2012. [DOI] [PubMed] [Google Scholar]
- 133.Coutinho P, Qiu C, Frank S, Tamber K, Becker D. Dynamic changes in connexin expression correlate with key events in the wound healing process. Cell Biol Int. 2003;27:525–541. doi: 10.1016/s1065-6995(03)00077-5. [DOI] [PubMed] [Google Scholar]
- 134.Martin PE, Easton JA, Hodgins MB, Wright CS. Connexins: Sensors of epidermal integrity that are therapeutic targets. FEBS Lett. 2014;588:1304–1314. doi: 10.1016/j.febslet.2014.02.048. [DOI] [PubMed] [Google Scholar]
- 135.Becker DL, Thrasivoulou C, Phillips AR. Connexins in wound healing; perspectives in diabetic patients. Biochimica et biophysica acta. 2012;1818:2068–2075. doi: 10.1016/j.bbamem.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 136.Brandner JM, Houdek P, Husing B, Kaiser C, Moll I. Connexins 26, 30, and 43: Differences among spontaneous, chronic, and accelerated human wound healing. J Invest Dermatol. 2004;122:1310–1320. doi: 10.1111/j.0022-202X.2004.22529.x. [DOI] [PubMed] [Google Scholar]
- 137.Wang CM, Lincoln J, Cook JE, Becker DL. Abnormal connexin expression underlies delayed wound healing in diabetic skin. Diabetes. 2007;56:2809–2817. doi: 10.2337/db07-0613. [DOI] [PubMed] [Google Scholar]
- 138.Cogliati B, Vinken M, Silva TC, Araujo CM, Aloia TP, Chaible LM, Mori CM, Dagli ML. Connexin 43 deficiency accelerates skin wound healing and extracellular matrix remodeling in mice. J Dermatol Sci. 2015;79:50–56. doi: 10.1016/j.jdermsci.2015.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kretz M, Euwens C, Hombach S, Eckardt D, Teubner B, Traub O, Willecke K, Ott T. Altered connexin expression and wound healing in the epidermis of connexin-deficient mice. J Cell Sci. 2003;116:3443–3452. doi: 10.1242/jcs.00638. [DOI] [PubMed] [Google Scholar]
- 140.Ghatnekar GS, Grek CL, Armstrong DG, Desai SC, Gourdie RG. The effect of a connexin43-based peptide on the healing of chronic venous leg ulcers: A multicenter, randomized trial. J Invest Dermatol. 2015;135:289–298. doi: 10.1038/jid.2014.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Grek CL, Prasad GM, Viswanathan V, Armstrong DG, Gourdie RG, Ghatnekar GS. Topical administration of a connexin43-based peptide augments healing of chronic neuropathic diabetic foot ulcers: A multicenter, randomized trial. Wound Repair Regen. 2015;23:203–212. doi: 10.1111/wrr.12275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kirsner RS, Baquerizo Nole KL, Fox JD, Liu SN. Healing refractory venous ulcers: New treatments offer hope. J Invest Dermatol. 2015;135:19–23. doi: 10.1038/jid.2014.444. [DOI] [PubMed] [Google Scholar]
- 143.Haass NK, Wladykowski E, Kief S, Moll I, Brandner JM. Differential induction of connexins 26 and 30 in skin tumors and their adjacent epidermis. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 2006;54:171–182. doi: 10.1369/jhc.5A6719.2005. [DOI] [PubMed] [Google Scholar]
- 144.Haass NK, Ripperger D, Wladykowski E, Dawson P, Gimotty PA, Blome C, Fischer F, Schmage P, Moll I, Brandner JM. Melanoma progression exhibits a significant impact on connexin expression patterns in the epidermal tumor microenvironment. Histochem Cell Biol. 2010;133:113–124. doi: 10.1007/s00418-009-0654-5. [DOI] [PubMed] [Google Scholar]
- 145.Saito-Katsuragi M, Asada H, Niizeki H, Katoh F, Masuzawa M, Tsutsumi M, Kuniyasu H, Ito A, Nojima H, Miyagawa S. Role for connexin 26 in metastasis of human malignant melanoma: Communication between melanoma and endothelial cells via connexin 26. Cancer. 2007;110:1162–1172. doi: 10.1002/cncr.22894. [DOI] [PubMed] [Google Scholar]
- 146.Sargen MR, Gormley RH, Pasha TL, Yum S, Acs G, Xu X, Zhang PJ. Melanocytic tumors express connexin 43 but not 26: Immunohistochemical analysis with potential significance in melanocytic oncogenesis. The American Journal of dermatopathology. 2013;35:813–817. doi: 10.1097/DAD.0b013e318278d401. [DOI] [PubMed] [Google Scholar]
- 147.Ableser MJ, Penuela S, Lee J, Shao Q, Laird DW. Connexin43 reduces melanoma growth within a keratinocyte microenvironment and during tumorigenesis in vivo. J Biol Chem. 2014;289:1592–1603. doi: 10.1074/jbc.M113.507228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hong X, Sin WC, Harris AL, Naus CC. Gap junctions modulate glioma invasion by direct transfer of microrna. Oncotarget. 2015 doi: 10.18632/oncotarget.3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Uzu M, Sato H, Yamada R, Kashiba T, Shibata Y, Yamaura K, Ueno K. Effect of enhanced expression of connexin 43 on sunitinib-induced cytotoxicity in mesothelioma cells. J Pharmacol Sci. 2015;128:17–26. doi: 10.1016/j.jphs.2015.04.002. [DOI] [PubMed] [Google Scholar]