

Abstract
Beryllium exposure results in beryllium hypersensitivity in a subset of exposed individuals, leading to granulomatous inflammation and fibrosis in the lung. In addition to its antigenic properties, beryllium has potent adjuvant activity that contributes to sensitization via unknown pathways. Here, we show that beryllium induces cellular death and release of IL-1α and DNA into the lung. Release of IL-1α was inflammasome-independent and required for beryllium-induced neutrophil recruitment into the lung. Beryllium enhanced classical dendritic cell (cDC) migration from the lung to draining LNs in an IL-1R-independent manner, and the accumulation of activated cDCs in the LN was associated with increased priming of CD4+ T cells. DC migration was reduced in TLR9KO mice; however, cDCs in the LNs of TLR9-deficient mice were highly activated, suggesting a role for more than one innate receptor in the effects on DCs. The adjuvant effects of beryllium on CD4+ T cell priming were similar in WT, IL-1R, caspase-1, TLR2, TLR4, TLR7, and TLR9 deficient mice. In contrast, DC migration, activation and the adjuvant effects of beryllium were significantly reduced in MyD88KO mice. Collectively, these data suggest that beryllium exposure results in the release of DAMPs that engage MyD88-dependent receptors to enhance pulmonary DC function.
Keywords: danger-associated molecular pattern (DAMP), dendritic cells, environmental exposure, beryllium, granulomatous lung disease
Introduction
Metal-induced T cell hypersensitivities are common and occur in response to a wide range of metals1. The clinical manifestations of these hypersensitivities range from minor contact allergies to pulmonary fibrosis. Among the more serious hypersensitivities are those induced by beryllium and cobalt, which cause debilitating and life-threatening lung diseases. Chronic beryllium disease (CBD) occurs in genetically susceptible individuals who are exposed to beryllium in the workplace2,3. Beryllium is used in a variety of industries and despite the implementation of strict exposure limits, CBD continues to occur in up to 16% of workers4. The use of beryllium is increasing globally and greater than one million workers worldwide have been exposed3. In addition, the cost of treating a single CBD patient has been estimated to exceed $1 million5. Thus, CBD remains a costly and significant public health concern.
CBD is characterized by the presence of large numbers of effector memory Th1 polarized CD4+ T cells in the lung that produce IFNγ, IL-2 and TNFα upon restimulation with beryllium in vitro6. In contrast to sarcoidosis and other granulomatous lung disorders, there is no evidence for Th17- or Th2-polarized T cells in CBD7,8. In the majority of CBD patients, genetic susceptibility is related to the expression of specific HLA-DP alleles that express a glutamic acid at the 69th position of the β-chain9. This polymorphic amino acid participates in coordination of the Be2+ cation within the MHC class II (MHCII) structure resulting in an altered conformation of the HLA-DP molecule loaded with particular self-peptides (HLA-DP/peptide complex)10,11. Although beryllium does not directly contact the TCR, the modified HLA-DP2/peptide complex is recognized as a novel neoantigen by beryllium-specific TCRs11. Once activated, beryllium-specific effector CD4+ T cells drive the development of granulomatous inflammation in the lung, which can progress to fibrosis, respiratory insufficiency and death12.
Initiation of an effector CD4+ T cell response is a complex process that involves not only the presence of a stimulatory MHCII/peptide complex, but also requires additional signals from mature dendritic cells (DCs). DCs express innate pattern recognition receptors (PRRs) that broadly recognize pathogen-associated molecular patterns (PAMPs) encountered during infection and alarmins or damage-associated molecular patterns (DAMPs) released following cell death13. PRRs drive the migration and maturation of DCs from peripheral tissues to lymph nodes (LNs)14. Naive CD4+ T cells circulating through LNs in search of their cognate antigen/MHCII ligands require additional costimulatory signals, delivered by mature DCs, to proliferate and survive as effector cells15. In the absence of DC maturation, T cells proliferate to the presented antigen but either die or become anergic leading to antigen-specific tolerance16. Some metals, including the commonly used vaccine adjuvant aluminum hydroxide, have adjuvant properties that boost CD4+ T cell responses via effects on DCs17,18. Beryllium has also been shown to have adjuvant properties that drive lymphocyte expansion and Th1 cytokine production19-21. Thus, beryllium may operate not only as an antigenic stimulus but as its own pathogenic adjuvant. We hypothesize that pulmonary exposure to beryllium leads to the release of DAMPs and activation of innate PRRs that promote pulmonary DC maturation and expansion of effector CD4+ T cells.
Here, we show that pulmonary exposure to beryllium induces rapid release of IL-1α and DNA, DAMPs associated with necrotic cell death22. IL-1α induces neutrophil infiltration, and neutrophils partially contribute to the release of DNA. Beryllium-induced release of DAMPs into the lung is associated with accumulation of mature migratory classical DCs (cDCs) in the lung-draining LNs (LDLNs) and enhanced priming of CD4+ T cells to a model antigen. The accumulation of cDCs in the LDLN following beryllium exposure is IL-1R-independent. Although cDC migration is impaired in TLR9KO mice, increased CD80 and CD86 expression on cDCs occurs, and the adjuvant effects of beryllium on CD4+ T cells are intact in these mice. cDC migration, expression of costimulatory molecules and CD4+ T cell priming are all impaired in MyD88KO mice, suggesting that other MyD88-dependent receptors in concert with DNA/TLR9 play a role in the effects on cDCs. Together, these data show that pulmonary beryllium exposure leads to release of DNA and other DAMPs that drive MyD88-dependent maturation and migration of cDCs from the lung to the LDLNs, leading to enhanced priming of naive CD4+ T cell responses.
Results
Pulmonary exposure to beryllium induces cellular death, release of DNA and IL-1α and recruitment of neutrophils into the lung
To determine whether IL-1 or DNA was released following exposure to beryllium in the lung, we performed intratracheal (i.t.) instillations of saline or beryllium hydroxide (Be(OH)2) in WT C57Bl/6 (B6) mice and performed bronchoalveolar lavage (BAL) (Fig. 1). Compared to untreated mice (time 0), IL-1α and extracellular DNA were increased in the BAL fluid (BALF) at 6 and 18hr after beryllium exposure (Fig. 1A, B). This increase was associated with an increased percentage of cells that stained positive with an eFluor506-labeled fixable viability dye (FV-e506) 6 hrs after treatment with Be(OH)2 compared to saline-treated controls (Fig. 1C and Fig S1A). This dye stains necrotic, necroptotic and late apoptotic cells, but not cells undergoing early apoptosis. Analysis of FV-e506- cells for early apoptosis using annexin V indicated an increase in early apoptotic cells in beryllium-exposed mice (Fig. S1B,C) and as previously reported23. However, release of IL-1α occurs following necrosis or necroptosis (a regulated form of necrosis) and not apoptosis22,24. Together, our data suggest that both apoptotic and necrotic/necroptotic cell death occur in the lung in response to beryllium, the latter contributing to the release of DAMPs.
Figure 1. Beryllium exposure induces release of DNA and IL-1α and recruitment of neutrophils to the lung.
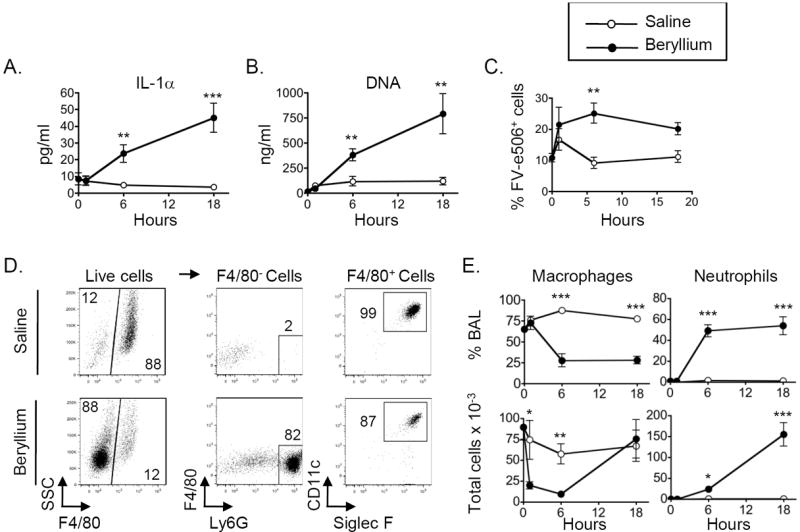
Untreated (0hrs) and mice treated with saline or Be(OH)2 i.t. were sacrificed, and BAL was harvested 1, 6 and 18hrs later. A. IL-1α levels as detected by ELISA in concentrated BALF. B. Concentration of extracellular DNA in BALF. C. Percentage (mean ± SEM) of BAL cells that are FV-e506+. D. A representative example of the gating strategy used to identify live alveolar macrophages and neutrophils after exposure to saline or Be(OH)2 for 6hrs. Gated live cells (FV-e506- as shown if Fig S1A) were analyzed to identify alveolar macrophages and neutrophils. BAL cells were gated on F4/80- and F4/80+ populations (left column). F4/80- cells that were Ly6Ghi were included in the neutrophil gate (middle column). F4/80+ cells that expressed CD11c and Siglec F were included in the alveolar macrophage gate (right column). Numbers on graphs indicate percentage of the gated population falling within each gate. E. The percentage (upper panels) and total cell numbers (lower panels) of BAL alveolar macrophages and neutrophils are shown for each time point compared to baseline (untreated mice, time 0). Total number of cells × 10-3 = number indicated on the axis. Data in A-C and E are combined from two independent experiments (n = 4 mice per group in each experiment). Values on graphs indicate mean ± SEM. A t-test was used to detect differences between treatment groups at each time point. Significance levels are indicated by asterisks (* p < 0.05, ** p < 0.01, *** p < 0.001).
Surprisingly, we did not detect IL-1β in the BALF of mice exposed to beryllium at these time-points (data not shown). We analyzed live cells for the presence of alveolar macrophages (F4/80+ CD11c+ SiglecF+) and neutrophils (F4/80- Ly6G+) in the BAL using flow cytometry (Fig. 1D, E). Beryllium exposure resulted in a drop in the percentage of macrophages and an increase in the percentage of neutrophils in the BAL 6 and 18hr after instillation (Fig. 1D, E). The total number of BAL neutrophils increased 6 and 18hr after beryllium exposure while the number of alveolar macrophages dropped 1hr after exposure and gradually recovered by 18hr (Fig. 1D, E). Together, these data show that beryllium induces rapid release of DAMPs, including DNA and IL-1α, followed by recruitment of neutrophils into the lung.
Beryllium-induced recruitment of neutrophils is dependent on IL-1α
IL-1α and IL-1β bind to the IL-1R, which induces the release of chemokines and recruitment of neutrophils22,25. The release of biologically active IL-1β requires activation of caspase-1 by the inflammasome26,27. The Nlrp3 inflammasome has been shown to be activated in response to a number of particles including silica, asbestos, and aluminum hydroxide28,29. In contrast, IL-1α is released along with DNA from necrotic cells during sterile inflammation (inflammation induced in the absence of microbial components) and does not require caspase-122. In accordance with this, IL-1α release was not impaired in beryllium-treated caspase-1KO mice (Fig. 2A). Both forms of IL-1 can bind to and activate the IL-1R and drive neutrophil recruitment; however, release of IL-1α from necrotic cells drives neutrophil recruitment in the absence of inflammasome activation25. To determine whether the recruitment of neutrophils required IL-1R or caspase-1, we exposed WT, IL-1RKO, MyD88KO and caspase-1KO mice to i.t. Be(OH)2 and analyzed neutrophil recruitment in the lung 18hrs later. Neutrophil recruitment was significantly reduced in beryllium-exposed IL-1RKO and MyD88KO mice, but was intact in similarly treated caspase-1KO mice (Fig. 2B), confirming that IL-1α, and not IL-1β, released after beryllium exposure was critical for neutrophil recruitment to the lung.
Figure 2. Beryllium-induced recruitment of neutrophils is IL-1α dependent.
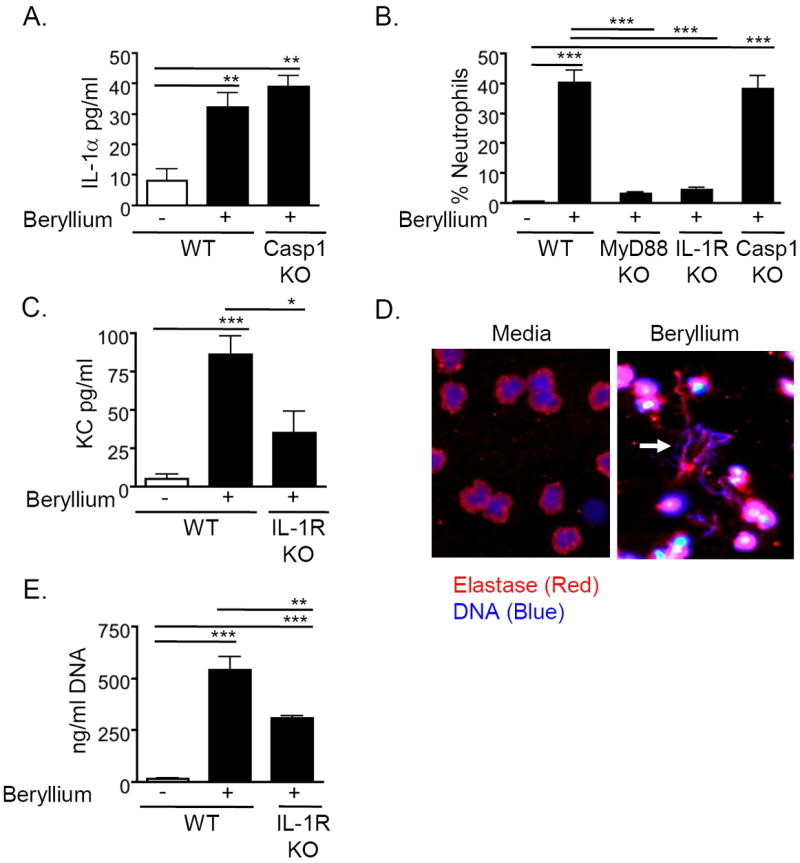
A. IL-1α in concentrated BALF from WT B6 and Caspase-1KO mice (Casp1 KO) treated i.t. with PBS or Be(OH)2 and sacrificed 18hr later. Data are representative of 2 independent experiments (n = 4 mice per group per experiment). B. Percentage of BAL cells that are neutrophils in WT B6, MyD88KO, IL-1RKO and Casp1KO mice treated i.t. with saline or Be(OH)2 and sacrificed 18hr later. Data are combined from two independent experiments (n=4 mice per group in each experiment). C. KC in concentrated BALF of WT B6 and IL-1RKO mice treated i.t. with either saline or Be(OH)2 and sacrificed 18hr later. Data are combined from 2 separate experiments (n = 4 mice per group in each experiment). D. Human neutrophils were stimulated for 4hr in the presence of media or 200 μg/ml Be(OH)2. Arrow indicates the presence of extracellular DNA (blue) colocalized with neutrophil elastase (red). Data are representative of 2 separate experiments. E. DNA in BALF of WT B6 and IL-1RKO mice treated i.t. with saline or Be(OH)2 and sacrificed 18hr later. Data are representative of 2 independent experiments (n = 4 mice per group per experiment). Values on graphs indicate mean ± SEM. A one-way ANOVA was used to determine differences between groups. Significance levels are indicated by asterisks (* p < 0.05, ** p < 0.01, *** p < 0.001).
IL-1α engages IL-1R in the lung and drives expression of the neutrophil chemokine, KC22,30. We observed that KC was increased in the BALF of WT mice exposed to beryllium and was attenuated in beryllium-exposed IL-1RKO mice (Fig. 2C). Thus, our data suggest that release of IL-1α is required for neutrophil recruitment to the lung through induction of KC.
Neutrophils contribute but are not the only source of beryllium-induced release of extracellular DNA
In mice treated with aluminum hydroxide, cellular death leads to the accumulation of DNA having characteristics of NETs31. To determine if beryllium exposure leads to NETosis, we purified human neutrophils and incubated them with media or Be(OH)2 for 4hr. Beryllium induced the release of extracellular DNA that colocalized with the neutrophil granule protein elastase, a situation that only occurs in neutrophils undergoing NETosis (Fig. 2D). However, DNA was released into the BAL of beryllium-treated mice with similar kinetics as IL-1α, suggesting that neutrophils are not required for release of DAMPs. To test this, we compared levels of extracellular DNA in the BALF of beryllium-treated WT mice with those in beryllium-treated IL-1RKO mice, which do not recruit neutrophils to the lung. We observed a reduction in the total quantity of DNA in the BALF of treated IL-1RKO mice compared to WT control mice, but the levels of DNA remained increased above saline-treated control mice, suggesting that neutrophils contribute to the release of extracellular DNA but are not the sole source (Fig. 2E). Thus, other cells such as epithelial cells or macrophages, contribute to DAMPs released following beryllium exposure.
Beryllium exposure drives migration of pulmonary DCs into the draining LN
Our data indicate that release of DAMPs occurs rapidly after exposure to beryllium in the lung. Innate PRRs and IL-1Rs engaged by DAMPs could impact DC function and the development and maintenance of beryllium-specific CD4+ T cells in CBD. To examine this, we determined whether beryllium exposure was associated with increased migration of DCs from the lung to the draining LNs. To track pulmonary DCs, we exposed mice i.t. to AF647-labeled ovalbumin (AF647-ova) in the absence or presence of beryllium and followed the arrival of AF647-ova+ DCs in the LDLNs. The percentage of cDCs (MHCII+CD11c+ cells) in the LDLNs was increased in mice treated with AF647-ova + beryllium compared to mice treated with AF647-ova alone (Fig. 3A). The percentage of LDLN cDCs that were AF647-ova+ was also increased in beryllium-exposed mice (Fig. 3B). As expected, AF-647-ova+ cDCs in the lung expressed high levels of MHCII, a phenotype characteristic of migratory cDCs32 (Fig. 3C).
Figure 3. Beryllium exposure drives migration of pulmonary cDCs into LDLNs.
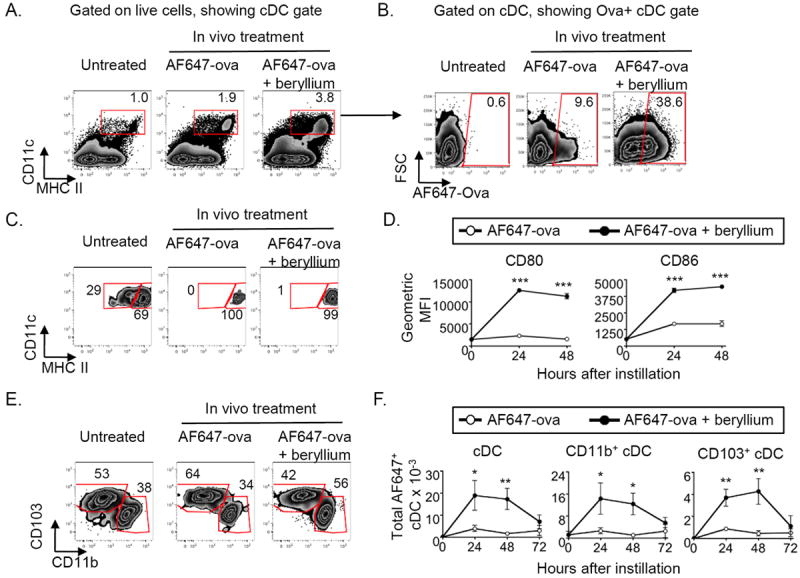
WT B6 mice were treated i.t. with PBS containing AF647-ova alone or mixed with Be(OH)2 (AF647-ova + beryllium) and sacrificed 24-72hrs later. cDCs in the LDLNs were analyzed by flow cytometry. Gating strategy for cDC analysis is shown in panels A-C and E for representative examples of untreated mice or mice treated with AF647-ova ± Be(OH)2 for 24hrs. A. LDLN cells were analyzed for expression of CD11c and MHCII. The percentage of LDLN cells that were CD11c+MHCII+ classical DCs (cDCs) is indicated on each dot plot. This cDC gate was used to analyze other parameters in B-C. B. Gated cDCs were analyzed for the presence of AF647-ova+ cDCs, and the percentage of cDCs that were AF647-Ova+ is indicated on each dot plot. C. Gated cDCs from untreated mice (left panel) and AF647-ova+ cDCs from mice treated with AF647-ova or AF647-ova + beryllium (middle and right panels) were analyzed for MHCII expression. Gates on each plot indicate resident (MHCIIlo) and migratory (MHCIIhi) populations. Gates were based upon the resident and migratory populations of cDCs in untreated mice (left panel). D. Geometric mean fluorescence intensity (GMFI ± SEM) of staining for CD80 and CD86 expression on gated cDCs from untreated mice (time 0, gate shown in panel A) and AF647-ova+ cDCs (gates shown in panel B) from mice treated with AF647-ova (open circles) and AF647-ova + beryllium (filled circles) for the indicated time points in a representative experiment of 2 independent experiments (n = 4 mice per experiment). E. Gated migratory cDCs from untreated mice or AF647-ova+ migratory cDCs from AF647-ova- or AF647-ova + beryllium-treated mice were analyzed for CD11b+ and CD103+ cDC subsets. F. Total numbers (mean ± SEM) of AF647-ova+ cDCs (left) AF647-ova+ CD11b+ (middle) and CD103+ (right) cDCs in the LDLN following pulmonary exposure to AF647-ova alone (open circles) or AF647-ova + beryllium (filled circles) are shown for the indicated time points compared to untreated controls (0 hrs). Data are combined from two separate experiments (n = 4 mice per group in each experiment). A t-test was used to determine differences between the two groups at each time point. Significance levels are indicated by asterisks (* p < 0.05, ** p < 0.01, *** p < 0.001).
We next determined whether beryllium exposure altered costimulatory molecule expression on the surface of cDCs in the LDLNs. AF647-Ova+ cDCs that had migrated to the LDLN had increased levels of CD80 and CD86 on their surface compared to AF647-Ova+ cDCs from mice exposed to AF647-ova alone or to steady state LDLN cDCs from untreated mice (Fig. 3D). LDLN AF647-ova- cDCs in mice treated with AF647-ova + beryllium also upregulated CD80 and CD86 (Fig. S2), suggesting that beryllium exposure resulted in global maturation of cDCs in the LDLNs.
Migratory pulmonary cDCs consist of CD11b+ and CD103+ DC subsets33. CD103+ DCs are specialized to phagocytize apoptotic cells and cross present exogenous antigen on MHCI molecules; however, both subsets can present antigen to CD4+ T cells33. Analysis of CD11b+ and CD103+ cDC subsets (Fig. 3E) showed an increased percentage of the AF647-ova+ cDCs in AF647-ova + beryllium treated mice of the CD11b+ subset compared to mice treated with AF647-ova alone. However, total numbers of both AF647-ova+ CD11b+ and CD103+ cDCs were increased in the LDLNs after beryllium exposure (Fig. 3F).
Beryllium enhances the ability of DCs to prime CD4+ T cells in vivo
To determine whether the effects of beryllium on cDCs enhanced CD4+ T cell priming, we compared CD4+ T cell responses against a model antigen in the LDLNs of B6 mice in the presence or absence of beryllium. We used a well-characterized peptide-protein conjugate of the 3K peptide (FEAQKAKANKAVDGGGC) covalently linked to ova (3K-ova), because this antigen is presented by IAb MHCII molecules in B6 mice, and we can track endogenous 3K-specific CD4+ T cell responses using MHCII tetramers17,34,35. WT B6 mice were exposed i.t. to 3K-ova in the absence or presence of Be(OH)2, and the 3K-specific CD4+ T cell response in the LDLNs was analyzed over time. Beryllium enhanced the magnitude of the primary CD4+ T cell response, which peaked 14 days after instillation (Fig. 4A). Upon secondary exposure to beryllium and 3K-ova, memory CD4+ T cells expanded to greater numbers compared to the primary response (Fig. 4B). Thus, beryllium-induced enhancement of cDC migration and activation is associated with adjuvant effects on CD4+ T cells.
Figure 4. Beryllium-activated DCs prime CD4+ T cells.
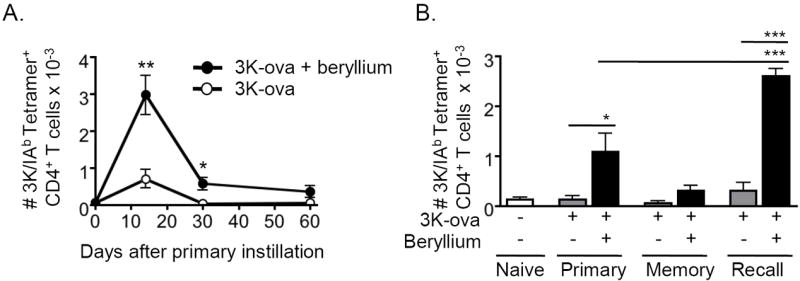
A. WT B6 mice were treated i.t. with nothing (day 0), 3K-ova (open circles) or 3K-ova + beryllium (solid circles) and sacrificed at the indicated time points to analyze the kinetics of the 3K-specific CD4+ T cell response in the LDLNs. LDLNs were harvested and analyzed for the presence of CD4+ T cells that were CD44hi and 3K/IAb tetramer+. The total number of 3K-specific CD4+ T cells (mean ± SEM) in the LDLNs at each indicated time point during the primary response is shown. Data are a representative of 2 independent experiments (n = 4 mice per group). B. Mice were treated with nothing (open bars), 3K-ova (grey bars), or 3K-ova + beryllium (black bars). One cohort of mice (recall) was boosted on day 40 with the same treatment that was used for priming. Mice were sacrificed on day 7 of the primary response (primary), day 47 of the memory response (memory) or 7 days after boosting (recall). Total number (mean ± SEM) of 3K-tetramer+ CD4+ T cells was determined as in A. Data are from a single representative experiment of 2 (n = 4 mice per group). A one-way ANOVA was used to detect differences between groups. Significance levels are indicated by asterisks (* p < 0.05, ** p < 0.01, *** p < 0.001).
Accumulation of pulmonary cDCs in LDLNs after beryllium exposure is IL-1R independent and reduced in TLR9KO mice
To determine whether IL-1R signaling is required for the effects of beryllium on cDC migration and activation, we exposed mice to AF647-ova in the presence or absence of Be(OH)2 and compared the accumulation and activation of AF647-ova+ cDCs in the LDLNs of WT and IL-1RKO mice 48 hrs later. The accumulation of AF647-ova+ CD11b+ and CD103+ cDCs was enhanced following beryllium exposure and was similar in LDLNs of WT and IL-1RKO mice (Fig. 5A). CD80 and CD86 were similarly upregulated on AF647-Ova+ cDCs from beryllium exposed WT and IL-1RKO mice (Fig. 5B). Thus, IL-1R signaling is not required for the observed effects of beryllium on pulmonary cDCs.
Figure 5. Accumulation of beryllium-activated cDCs into the LDLNs is IL-1R independent and reduced in TLR9KO mice.
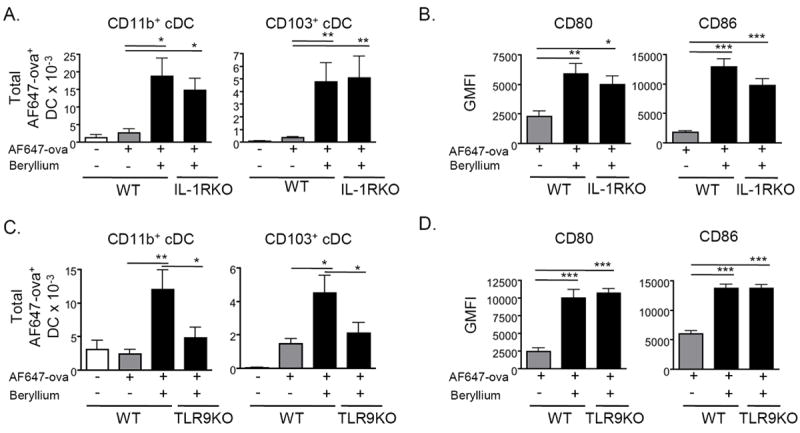
A. Total number (mean ± SEM) of AF647-ova+ CD11b+ and CD103+ cDCs in the LDLNs of WT B6 and IL-1RKO mice treated i.t. with PBS containing AF647-ova ± Be(OH)2 for 48hrs is shown. Gating and analysis was performed as described in Figure 3. B. Expression (GMFI ± SEM) of CD80 and CD86 on AF647-ova+ cDCs derived from WT B6 and IL-1RKO mice treated with AF647-ova ± Be(OH)2 for 48hrs is shown. Data shown in A and B are combined from 2 separate experiments (n = 4 mice per group in each experiment). C. Total number (mean ± SEM) of AF647-ova+ CD11b+ and CD103+ cDCs in the LDLNs of WT B6 and TLR9KO mice treated with AF647-ova ± Be(OH)2 for 48hrs is shown. Data are combined from 3 separate experiments (n = 4-6 mice per group in each experiment). D. Expression (GMFI ± SEM) of CD80 and CD86 on cDCs derived from WT B6 and TLR9KO mice treated with AF647-ova ± Be(OH)2 for 48hrs is shown from a representative experiment of 3 (n = 6 mice per group). A one-way ANOVA was used to determine differences between groups. Significance levels are indicated by asterisks (* p < 0.05, ** p < 0.01, *** p < 0.001).
TLRs are PRRs that can recognize certain DAMPs and promote activation and migration of cDCs to LNs. Self DNA stimulates signaling via TLR9 when delivered to endosomal compartments in DCs36. To determine if TLR9 was involved in the effects of DNA on cDC migration and activation, we treated WT or TLR9KO mice with AF647-ova + beryllium and followed the accumulation of AF647-Ova+ cDCs in the LDLNs 48hrs later. Total numbers of cDCs accumulating in the LDLNs of TLR9KO mice were reduced compared to WT mice (Fig. 5C). However, AF647-Ova+ cDCs in the LDLNs of beryllium-exposed WT and TLR9KO mice expressed similar levels of CD80 and CD86 (Fig. 5D). Thus, TLR9 may recognize DNA and contribute to the observed effects on DC migration, but other pathways contribute to optimal upregulation of costimulatory molecules on cDCs in the draining LN.
Adjuvant effects of beryllium on CD4+ T cell priming are independent of IL-1, TLR4, TLR7, TLR9 and the inflammasome
Although we did not observe significant differences in the accumulation of cDCs in the draining LNs between WT and IL-1RKO mice (Fig. 6A), it was possible that other members of the IL-1 family could contribute to the adjuvant effects of beryllium on CD4+ T cells or that IL-1 could directly impact CD4+ T cell priming. Although we did not detect endotoxin in our Be(OH)2 stock, we also wanted to exclude the possibility that low-level endotoxin contamination was playing a role in our observations. Thus, we exposed WT, IL-1RKO, caspase-1KO and TLR4KO mice to 3K-ova in the presence of beryllium and analyzed the number of 3K-specific CD4+ T cells in the LDLNs 14 days after exposure. We found similar numbers of 3K-specific CD4+ T cells in all groups of beryllium-treated mice (Fig. 6A), suggesting that the adjuvant effects of beryllium on CD4+ T cell priming are independent of IL-1R, the inflammasome, inflammasome-dependent cytokines of the IL-1 family and TLR4.
Figure 6. Adjuvant effects of beryllium on CD4+ T cell priming are independent of IL-1R, TLR2, TLR4, TLR7 and TLR9.
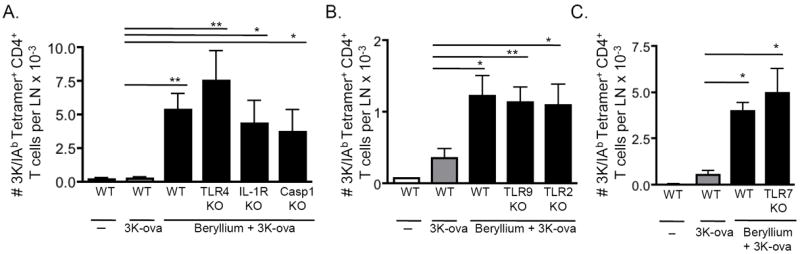
Mice were treated i.t. with 3K-ova ± Be(OH)2 in PBS. A. Total number of 3K-specific CD4+ T cells in the LDLNs of WT B6, TLR4KO, IL-1RKO and Casp1KO mice on day 14 of the primary response. Data are combined from 2 separate experiments (n = 3-4 mice per group in each experiment). B. Total number of 3K-specific CD4+ T cells in the LDLNs of WT B6, TLR9KO and TLR2KO mice on day 12 of the primary response. Data are combined from 3 separate experiments (n = 4-9 mice per group in each experiment). C. Total number of 3K-specific CD4+ T cells in the LDLNs of WT B6 or TLR7KO mice on day 14 of the primary response. Data are combined from 2 separate experiments (n = 3-4 mice per group in each experiment). Values indicate mean ± SEM. A one-way ANOVA was used to determine differences between groups. Significance levels are indicated by asterisks (* p < 0.05, ** p < 0.01, *** p < 0.001).
To determine whether TLR9 or as a control, TLR2, played a role in these adjuvant effects, we treated WT, TLR9KO and TLR2KO mice with 3K-ova in the presence or absence of beryllium. Although accumulation of cDCs in the LDLNs at 48hrs was reduced in TLR9KO mice exposed to beryllium (Fig. 5C), the adjuvant effects of beryllium on CD4+ T cell priming were TLR9 independent (Fig. 6B). TLR7 can respond to nucleic acids; however, the adjuvant effects of beryllium on CD4+ T cell priming were intact in TLR7KO mice (Fig 6C). Collectively, these data suggest that while DNA/TLR9 enhance cDC migration, signaling via other PRRs are sufficient to enhance costimulatory signals on cDCs and the adjuvant effects of beryllium on CD4+ T cell priming.
The adjuvant effects of beryllium are dependent upon MyD88 signaling pathways
Our data suggest that redundant PRRs are involved in beryllium-induced DC migration and CD4+ T cell priming. MyD88 is a signaling adaptor that is downstream of multiple innate PRRs that recognize DAMPs37. Therefore, it is possible that disruption of multiple pathways that rely on MyD88 would eliminate the adjuvant effects of beryllium exposure. To globally test the role of MyD88 dependent receptors on DC migration, we treated WT and MyD88KO mice i.t. with AF647-ova ± Be(OH)2 and analyzed cDCs in the LDLNs at 24 and 48hr. We observed a reduction in the accumulation of AF647-ova+ CD11b+ and CD103+ cDCs in the LDLN after exposure in MyD88KO mice compared to WT mice (Fig. 7A). We also observed that upregulation of CD80 and CD86 on cDCs was impaired in beryllium-exposed MyD88KO mice (Fig. 7B).
Figure 7. MyD88-dependence of the adjuvant effects of pulmonary beryllium exposure.
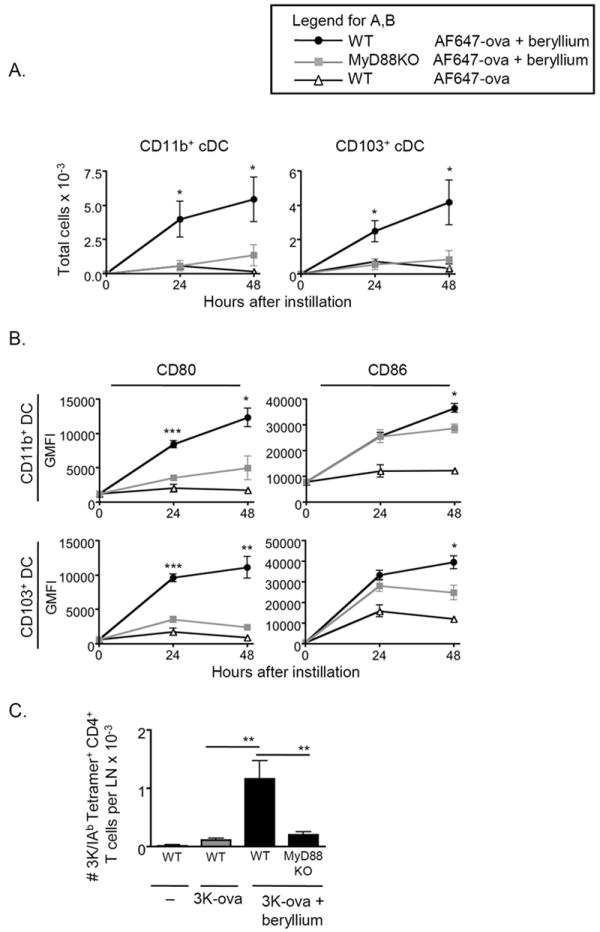
A. Total number (mean ± SEM) of AF647-ova+ cDCs in WT B6 (black circles) or MyD88KO mice (gray squares) treated i.t with AF647-ova + Be(OH)2 (filled symbols) or AF647-ova (open triangles) is shown at indicated time points from a representative experiment of 2 (n = 4 mice per group). B. CD80 (left panels) and CD86 (right panels) expression (geometric MFI) is shown on gated AF647-ova+ CD11b+ and CD103+ cDC subsets derived from AF647-ova + Be(OH)2-treated WT B6 mice (black circles), AF647-ova + Be(OH)2-treated MyD88KO mice (gray squares) and AF647-ova-treated WT B6 mice (open triangles) is shown. Data are from a representative experiment of 2 (n = 4 mice per group). C. Total number (mean ± SEM) of 3K-specific CD4+ T cells in the LDLNs on day 12 of the primary response in WT B6 and MyD88KO mice treated with 3K-ova ± Be(OH)2 is shown. Data is combined from 3 separate experiments (n = 4 mice per group in each experiment). In A and B, a t-test was used to determine differences between WT and MyD88KO mice treated with AF647-ova + Be(OH)2 at each time point, and a one-way ANOVA was used to determine differences between groups in C. Significance levels are indicated by asterisks (* p < 0.05, ** p < 0.01, *** p < 0.001).
To determine if these effects were associated with a reduction in the adjuvant effects of beryllium on CD4+ T cell priming, we treated WT and MyD88KO mice with 3K-ova and beryllium and analyzed the expansion of 3K-specific CD4+ T cells in the LDLN 12 days later. We observed a significant reduction in the total number of 3K-specific CD4+ T cells in MyD88KO mice compared to WT mice (Fig. 7C). Thus, our findings show that the effects of beryllium exposure on DC function and the adjuvant effects of beryllium on CD4+ T cells involve PRRs that signal via MyD88 dependent pathways.
Discussion
CBD is a granulomatous lung disease that is characterized by the accumulation of beryllium-responsive CD4+ T cells in the lung. In the last two decades, much has been learned about beryllium as a disease-inducing MHCII-restricted antigen; however, the development of CBD involves complex interactions between the innate and adaptive arms of the immune system. In this regard, beryllium possesses adjuvant properties that may contribute to CBD pathogenesis; however, the mechanism behind these adjuvant effects is unknown19,20. DCs are critical antigen-presenting cells that determine whether TCR engagement results in a pathogenic or tolerogenic response. However, nothing is known about how beryllium impacts DCs in the target organ. While beryllium-induced apoptosis has been described23, apoptosis is not associated with the release of DAMPs or initiation of inflammation. In addition to apoptosis, our data show that beryllium exposure in the lung induces necrotic cell death and the release of DAMPs, including DNA and IL-1α that drive inflammation. These effects are associated with MyD88-dependent migration of mature cDCs to LDLNs that enhance priming of naive CD4+ T cells, an effect that is IL-1R-independent.
IL-1β has been shown to be important in inducing release of chemokines that drive neutrophils to infiltrate the lung in response to other sterile environmental agents including silica particles and asbestos28,29. However, these studies suggest a critical role for Nlrp3, which becomes activated following phagocytosis of large particles that disrupt lysosomal membranes and induce cellular stress. Activated Nlrp3 promotes assembly of the inflammasome, activation of caspase-1 and release of biologically active IL-1β. In contrast, we did not detect IL-1β in the alveolar space after exposure to beryllium, and neutrophil recruitment was IL-1R-dependent but independent of caspase-1, an enzyme required for the release of biologically active IL-1β. These data suggest that IL-1α plays an essential role in driving neutrophil recruitment after beryllium exposure and are consistent with studies showing a critical role for IL-1α in the recruitment of neutrophils in response to necrotic/necroptotic but not apoptotic cells24,25. It is possible that beryllium induces more necrotic cell death and IL-1α release while other particles induce inflammasome activation following phagocytosis. Thus, the roles of IL-1α and IL-1β may be related to differences in toxicity between different types of sterile particles. Other studies have shown that IL-1α and IL-1β are released at different times and drive recruitment of neutrophils and monocytes, respectively38. Thus, it is possible that IL-1β may be induced later in the response to beryllium.
Our data show that after arrival in the lung, neutrophils contribute to DNA release in vivo, perhaps through NETosis, but are not required for release of DNA. Thus, other cells, including macrophages or epithelial cells, may contribute to the initial DNA release. Our analysis of ex vivo DNA does not discriminate between NETs and extracellular DNA; however, macrophages and epithelial cells have not been described to undergo NETosis. Thus, the DNA detected in the absence of neutrophils likely reflects DNA released by necrosis. Neutrophils have been shown to impact pulmonary DC function during infection39. However, in IL-1RKO mice, which do not recruit neutrophils to the lung after beryllium exposure, cDC migration to LDLNs was intact, suggesting that the effects of beryllium exposure on cDCs are not dependent upon neutrophils or NETosis, but may still be impacted by extracellular DNA.
Introduction of apoptotic cells into the lung has been shown to preferentially induce the migration of CD103+ cDCs into LDLNs33. In contrast, beryllium exposure increases the migration of both cDC subsets. cDCs from beryllium-exposed mice exhibited upregulated levels of the costimulatory molecules CD80 and CD86 and more effectively primed CD4+ T cells to bystander antigen. These effects were not reduced in IL-1RKO or TLR4KO mice. TLR2, TLR9 and TLR7 have been implicated in innate responses to other sterile substances in the lung40. We found no difference in the adjuvant effects of beryllium on CD4+ T cell priming in WT, TLR2, TLR7 or TLR9KO mice. We did observe a reduction in beryllium-induced migration of cDCs in TLR9KO mice; however, cDCs in the draining LN of these mice were highly activated, suggesting that other pathways are involved. It is possible that the high levels of CD80 and CD86 on these cDCs promote enhanced priming of CD4+ T cells, even in the presence of fewer cDCs in the TLR9KO mice. Thus, despite evidence that TLR9 is engaged by DNA, our data suggest that multiple PRRs play a role in optimal cDC function. Together, our data suggest that in addition to TLR9, other PRRs are engaged that activate MyD88 signaling and promote cDC activation and priming of CD4+ T cells. Our findings support a model where DAMPs released in the lung following beryllium exposure impact DC expression of CD80 and CD86 and beryllium does not directly upregulate these costimulatory molecules, consistent with recent studies of human DCs41.
Beryllium and aluminum are both metals with adjuvant properties42. Aluminum hydroxide is a widely used vaccine adjuvant that induces the release of DNA and activation of the inflammasome43,44. In contrast to pulmonary beryllium exposure, aluminum hydroxide injected into the muscle does not enhance DC migration into peripheral LNs, perhaps due to differences in the types of DCs that are mobilized and/or the ability of antigen to be moved without DC help through the lymphatics to the popliteal LN by skeletal muscle contractions17,45,46. In contrast to beryllium which induces Th1 responses and neutrophil recruitment, aluminum hydroxide induces a Th2 type response and eosinophilia47. The adjuvant function of intraperitoneal- or intramuscular-administered aluminum hydroxide on humoral responses is MyD88-independent48. It is possible that the differences between aluminum and beryllium are due to different anatomical locations studied, differences in DC subsets and the PRRs expressed, and/or differences in metal-induced toxicity and DAMPs released at the exposure site.
In the context of CBD, the development of a strong beryllium-specific effector CD4+ T cell response is necessary for the generation of granulomatous inflammation in the lung8. This is limited to individuals expressing particular MHCII molecules capable of presenting beryllium/peptide complexes to CD4+ T cells. We hypothesize that in individuals with this genetic predisposition, beryllium-induced activation of MyD88-dependent pathways may promote the initiation of a beryllium-specific immune response in the lung via effects on DCs and may further impact the development of beryllium-specific effector Th1 and regulatory T cells following beryllium exposure. In addition, these adjuvant properties of beryllium may have a dose-sparing effect, effectively promoting beryllium-induced sensitization in response to low exposures. Although our study is most relevant to understanding the effects of beryllium on the sensitization phase, these pathways may also operate at later stages of disease.
The activation of inflammation and effects on DCs were transient in our study, showing a wave of DC migration into the LDLN that was reduced after 72 hrs. This may be due to fluorescent dye breakdown or to a transient release of DAMPs occurring within a short time after exposure. Further studies are needed to understand how these pathways impact the effects during chronic disease on both DCs and T cells. In reality, workers are exposed to beryllium over long periods of time, and while it is known that beryllium persists long after exposure, the initial events following exposure to DCs are likely most important in driving proliferation and differentiation of naive CD4+ T cells to beryllium (i.e., sensitization). Once primed, effector and memory CD4+ T cells rely less heavily upon costimulation, and antigen-presenting cells in vivo are likely not restricted to DCs alone but include B cells and macrophages. Analysis of the role of these different antigen-presenting cells in CBD and whether DAMP release impacts these cells and granuloma development at later stages of disease remain important unanswered questions.
Taken together, we show for the first time that beryllium drives IL-1-dependent neutrophil accumulation and IL-1-independent migration of activated pulmonary cDCs to LDLNs. Recognition of DNA via TLR9 and engagement of another unknown MyD88-dependent PRR leads to the accumulation of activated DCs in the LDLN and enhancement of CD4+ T cell priming. In addition to its role as a pathogenic antigen in CBD, our data show that beryllium can potently stimulate the innate immune system and enhance DC function, effectively functioning as an adjuvant in CBD.
Methods
Mice
C57Bl/6 WT, MyD88KO, Caspase-1KO, IL-1RKO, TLR2KO, TLR7KO and TLR4KO mice were purchased from Jackson Laboratories. TLR9KO mice were kindly provided by Dr. Jillian Poole (University of Nebraska Medical Center) with permission from Shizuo Akira. Mice were housed and bred at the University of Colorado Biological Resource Center and were used at 6-12 weeks of age. All experiments were approved by the Institutional Animal Care and Use Committee of the University of Colorado Denver, in accordance with the NIH guidelines.
Reagents
Be(OH)2 was generated as described20. AlexaFluor647 was conjugated to low endotoxin ova (Hyglos) using the Pierce AF647 labeling kit according to the manufacturer’s instructions. 3K-ova was generated using the Imject Maleimide Activated Ova kit from Pierce Biotechnology and a cysteine linked 3K peptide49.
Treatments
Mice were anesthetized with isoflurane, and the vocal chords were visualized with a laryngoscope (Penn Technologies) followed by i.t. delivery of 50 μl volume 0.9% sterile saline or PBS with or without sonicated 200 μg Be(OH)2. In some experiments 20 μg AF647-ova or 10 μg 3K-ova were mixed with beryllium.
Early innate responses to beryllium in the lung
Mice were sacrificed after exposure to saline or Be(OH)2, and BAL was performed with 1.5ml of sterile PBS. Cells were separated from the BALF using centrifugation, and viable cells were enumerated using trypan blue. Beryllium crystals have a crystalline morphology that can be clearly distinguished from viable cells. For IL-1 analysis BALF were concentrated 5-fold using Amicon 3kDa filters. IL-1α and IL-1β were quantified using Ready Set Go ELISA (eBioscience).
BAL cells were incubated with FV-eFluor 506 for 30 min on ice (eBioscience), washed, incubated with Fc Block and mAbs directed against Ly6G, Siglec F, F4/80 and Ly6C. Live and dead cells were gated based on unstained controls (Fig S1A). Live cells were analyzed for the presence of neutrophils and macrophages and for apoptotic cells using the FITC Annexin V kit (eBioscience). Data were acquired on a LSRII SORP flow cytometer using FACS Diva Software (BD Biosciences) and analyzed using FlowJo software (Treestar, Inc.).
Quantification of extracellular DNA following in vivo exposure of mice to beryllium
BALF samples were diluted with PBS and added to a 96 well black microplate (Greiner Bio). Mouse genomic DNA (Promega) was used to generate a standard curve. The samples were incubated with a solution of 5μM Sytox Orange (Life Technologies) in PBS for 10 minutes at RT. The plate was analyzed using a Victor3 1420 multilabel plate reader with Wallac 1420 Workstation software (Perkin Elmer) with a 544 nm excitation filter and a 600nm emission filter.
Neutrophil NET release
Neutrophils from healthy human volunteers were isolated as described (see details in supplemental methods)50, analyzed for purity by flow cytometry and stained using fluorescently labeled anti-CD49d and -CD66 mAbs (eBioscience). Cells were resuspended in complete RPMI media (cRPMI) consisting of RPMI 1640 tissue culture medium (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (Hyclone), 10 mM HEPES, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin (all from Invitrogen). Cells were incubated in a 48 well plate for 4hr in media alone or with 1.2 mg Be(OH)2, washed with PBS and fixed for 15 minutes in paraformaldehyde at RT. Cytospins of ~100,000 cells per slide were prepared and stained with PE-labeled anti-human elastase (Calbiochem) and Hoesht to visualize DNA. Cells were washed and coverslipped with VectaMount medium. Slides were analyzed on an Olympus BX-41 fluorescent microscope (CytoViva), and images were taken using a Dage-MTI Excel digital camera XLMCT.
Effects of beryllium on pulmonary DC migration and expression of costimulatory molecules
LDLN were harvested, and single cell suspensions were generated as described (see supplemental methods)17. Cells were incubated with FcBlock and stained with fluorescently-conjugated mAbs to CD11c, IAb, CD11b, CD80, CD103 and CD86.
Effects of beryllium on CD4+ T cell priming
At indicated times after instillation, lungs were flushed of blood by placing an incision in the left atrium of the heart and passing 10ml PBS through the lungs with a syringe and a 25 gauge needle introduced into the left ventricle of the heart. Lungs and LDLNs were harvested into cRPMI media and processed into single cell suspensions as described (details in supplemental methods)8,17. Viable cells were enumerated using Trypan blue exclusion and stained with PE-labeled 3K/IAb tetramers at 37°C for 2hr. Antibodies for CD4, B220, MHC II, F4/80, CD44, and CD8 were added for 20 min at 4°C. 3K/IAb tetramer positive cells were defined after gating on live CD4+CD44hi cells that were CD8, B220, F4/80, and MHCII negative.
Statistical analysis
A one-way ANOVA and unpaired t-test were used to determine significance of differences between groups (Prism 4, GraphPad Software, Inc.). A P value < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
This work is supported by the following grants: American Thoracic Society Unrestricted Grant (to ASM) and NIH grants HL111760 and ES11810 (to APF). We thank Philippa Marrack, Michael Falta, Amanda Goodluck, Nicole Desch, Claudia Jakubzick and Peter Henson for technical feedback on this project. We thank Allison Martin for technical assistance.
Footnotes
Disclosure: The authors have no conflicts of interest to declare.
References
- 1.Budinger L, Hertl M. Immunologic mechanisms in hypersensitivity reactions to metal ions: an overview. Allergy. 2000;55:108–115. doi: 10.1034/j.1398-9995.2000.00107.x. [DOI] [PubMed] [Google Scholar]
- 2.Hardy HL, Tabershaw IR. Delayed chemical pneumonitis occurring in workers exposed to beryllium compounds. J Ind Hyg Toxicol. 1946;28:197–211. [PubMed] [Google Scholar]
- 3.Fontenot AP, Maier LA. Genetic susceptibility and immune-mediated destruction in beryllium-induced disease. Trends in immunology. 2005;26:543–549. doi: 10.1016/j.it.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 4.Henneberger PK, et al. Beryllium sensitization and disease among long-term and short-term workers in a beryllium ceramics plant. Int Arch Occup Environ Health. 2001;74:167–176. doi: 10.1007/s004200100237. [DOI] [PubMed] [Google Scholar]
- 5.Bartell SM, et al. Risk estimation and value-of-information analysis for three proposed genetic screening programs for chronic beryllium disease prevention. Risk Anal. 2000;20:87–99. doi: 10.1111/0272-4332.00009. [DOI] [PubMed] [Google Scholar]
- 6.Pott GB, et al. Frequency of beryllium-specific, TH1-type cytokine-expressing CD4+ T cells in patients with beryllium-induced disease. J Allergy Clin Immunol. 2005;115:1036–1042. doi: 10.1016/j.jaci.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 7.Fontenot AP, Canavera SJ, Gharavi L, Newman LS, Kotzin BL. Target organ localization of memory CD4(+) T cells in patients with chronic beryllium disease. The Journal of clinical investigation. 2002;110:1473–1482. doi: 10.1172/JCI15846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mack DG, et al. Regulatory T cells modulate granulomatous inflammation in an HLA-DP2 transgenic murine model of beryllium-induced disease. Proc Natl Acad Sci U S A. 2014;111:8553–8558. doi: 10.1073/pnas.1408048111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richeldi L, Sorrentino R, Saltini C. HLA-DPB1 glutamate 69: a genetic marker of beryllium disease. Science. 1993;262:242–244. doi: 10.1126/science.8105536. [DOI] [PubMed] [Google Scholar]
- 10.Falta MT, et al. Identification of beryllium-dependent peptides recognized by CD4+ T cells in chronic beryllium disease. J Exp Med. 2013;210:1403–1418. doi: 10.1084/jem.20122426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clayton GM, et al. Structural basis of chronic beryllium disease: linking allergic hypersensitivity and autoimmunity. Cell. 2014;158:132–142. doi: 10.1016/j.cell.2014.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fontenot AP, Falta MT, Freed BM, Newman LS, Kotzin BL. Identification of pathogenic T cells in patients with beryllium-induced lung disease. J Immunol. 1999;163:1019–1026. [PubMed] [Google Scholar]
- 13.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 14.Probst HC, Muth S, Schild H. Regulation of the tolerogenic function of steady-state DCs. Eur J Immunol. 2014;44:927–933. doi: 10.1002/eji.201343862. [DOI] [PubMed] [Google Scholar]
- 15.Banchereau J, et al. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 16.Hawiger D, et al. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med. 2001;194:769–779. doi: 10.1084/jem.194.6.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKee AS, et al. Host DNA released in response to aluminum adjuvant enhances MHC class II-mediated antigen presentation and prolongs CD4 T-cell interactions with dendritic cells. Proc Natl Acad Sci U S A. 2013;110:E1122–1131. doi: 10.1073/pnas.1300392110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kool M, et al. Cutting edge: alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J Immunol. 2008;181:3755–3759. doi: 10.4049/jimmunol.181.6.3755. [DOI] [PubMed] [Google Scholar]
- 19.Behbehani K, Beller DI, Unanue ER. The effects of beryllium and other adjuvants on Ia expression by macrophages. J Immunol. 1985;134:2047–2049. [PubMed] [Google Scholar]
- 20.Hall JG. Studies on the adjuvant action of beryllium. I. Effects on individual lymph nodes. Immunology. 1984;53:105–113. [PMC free article] [PubMed] [Google Scholar]
- 21.Lee JY, et al. Beryllium, an adjuvant that promotes gamma interferon production. Infect Immun. 2000;68:4032–4039. doi: 10.1128/iai.68.7.4032-4039.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eigenbrod T, Park JH, Harder J, Iwakura Y, Nunez G. Cutting edge: critical role for mesothelial cells in necrosis-induced inflammation through the recognition of IL-1 alpha released from dying cells. J Immunol. 2008;181:8194–8198. doi: 10.4049/jimmunol.181.12.8194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sawyer RT, et al. Beryllium-ferritin: lymphocyte proliferation and macrophage apoptosis in chronic beryllium disease. Am J Respir Cell Mol Biol. 2004;31:470–477. doi: 10.1165/rcmb.2004-0090OC. [DOI] [PubMed] [Google Scholar]
- 24.England H, Summersgill HR, Edye ME, Rothwell NJ, Brough D. Release of interleukin-1alpha or interleukin-1beta depends on mechanism of cell death. The Journal of biological chemistry. 2014;289:15942–15950. doi: 10.1074/jbc.M114.557561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen CJ, et al. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–856. doi: 10.1038/nm1603. [DOI] [PubMed] [Google Scholar]
- 26.Kostura MJ, et al. Identification of a monocyte specific pre-interleukin 1 beta convertase activity. Proc Natl Acad Sci U S A. 1989;86:5227–5231. doi: 10.1073/pnas.86.14.5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 28.Hornung V, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dostert C, et al. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hybertson BM, Jepson EK, Clarke JH, Spelts RJ, Repine JB. Interleukin-1 stimulates rapid release of cytokine-induced neutrophil chemoattractant (CINC) in rat lungs. Inflammation. 1996;20:471–483. doi: 10.1007/BF01487040. [DOI] [PubMed] [Google Scholar]
- 31.Munks MW, et al. Aluminum adjuvants elicit fibrin-dependent extracellular traps in vivo. Blood. 2010;116:5191–5199. doi: 10.1182/blood-2010-03-275529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604. doi: 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Desch AN, et al. CD103+ pulmonary dendritic cells preferentially acquire and present apoptotic cell-associated antigen. J Exp Med. 2011;208:1789–1797. doi: 10.1084/jem.20110538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacLeod MK, et al. CD4 memory T cells divide poorly in response to antigen because of their cytokine profile. Proc Natl Acad Sci U S A. 2008;105:14521–14526. doi: 10.1073/pnas.0807449105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Willis RA, Kappler JW, Marrack PC. CD8 T cell competition for dendritic cells in vivo is an early event in activation. Proc Natl Acad Sci U S A. 2006;103:12063–12068. doi: 10.1073/pnas.0605130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yasuda K, et al. Endosomal translocation of vertebrate DNA activates dendritic cells via TLR9-dependent and -independent pathways. J Immunol. 2005;174:6129–6136. doi: 10.4049/jimmunol.174.10.6129. [DOI] [PubMed] [Google Scholar]
- 37.Kawai T, Akira S. Toll-like receptor downstream signaling. Arthritis Res Ther. 2005;7:12–19. doi: 10.1186/ar1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rider P, et al. IL-1alpha and IL-1beta recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol. 2011;187:4835–4843. doi: 10.4049/jimmunol.1102048. [DOI] [PubMed] [Google Scholar]
- 39.Blomgran R, Ernst JD. Lung neutrophils facilitate activation of naive antigen-specific CD4+ T cells during Mycobacterium tuberculosis infection. J Immunol. 2011;186:7110–7119. doi: 10.4049/jimmunol.1100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bauer C, et al. Myeloid differentiation factor 88-dependent signaling is critical for acute organic dust-induced airway inflammation in mice. Am J Respir Cell Mol Biol. 2013;48:781–789. doi: 10.1165/rcmb.2012-0479OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li L, Huang Z, Gillespie M, Mroz PM, Maier LA. p38 Mitogen-Activated Protein Kinase in beryllium-induced dendritic cell activation. Hum Immunol. 2014;75:1155–1162. doi: 10.1016/j.humimm.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fontenot AP, Amicosante M. Metal-induced diffuse lung disease. Semin Respir Crit Care Med. 2008;29:662–669. doi: 10.1055/s-0028-1101276. [DOI] [PubMed] [Google Scholar]
- 43.Marichal T, et al. DNA released from dying host cells mediates aluminum adjuvant activity. Nat Med. 2011;17:996–1002. doi: 10.1038/nm.2403. [DOI] [PubMed] [Google Scholar]
- 44.Li H, Nookala S, Re F. Aluminum hydroxide adjuvants activate caspase-1 and induce IL-1beta and IL-18 release. J Immunol. 2007;178:5271–5276. doi: 10.4049/jimmunol.178.8.5271. [DOI] [PubMed] [Google Scholar]
- 45.Langlet C, et al. CD64 expression distinguishes monocyte-derived and conventional dendritic cells and reveals their distinct role during intramuscular immunization. J Immunol. 2012;188:1751–1760. doi: 10.4049/jimmunol.1102744. [DOI] [PubMed] [Google Scholar]
- 46.Reddy NP. Lymph circulation: physiology, pharmacology, and biomechanics. Crit Rev Biomed Eng. 1986;14:45–91. [PubMed] [Google Scholar]
- 47.Brewer JM, Conacher M, Satoskar A, Bluethmann H, Alexander J. In interleukin-4-deficient mice, alum not only generates T helper 1 responses equivalent to freund’s complete adjuvant, but continues to induce T helper 2 cytokine production. Eur J Immunol. 1996;26:2062–2066. doi: 10.1002/eji.1830260915. [DOI] [PubMed] [Google Scholar]
- 48.Gavin AL, et al. Adjuvant-enhanced antibody responses in the absence of toll-like receptor signaling. Science. 2006;314:1936–1938. doi: 10.1126/science.1135299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dai S, et al. Crossreactive T Cells spotlight the germline rules for alphabeta T cell-receptor interactions with MHC molecules. Immunity. 2008;28:324–334. doi: 10.1016/j.immuni.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu Y. Isolation of human and mouse neutrophils ex vivo and in vitro. Methods Mol Biol. 2012;844:101–113. doi: 10.1007/978-1-61779-527-5_7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.