

Abstract
Amino acid (aa) polymorphisms in the hepatitis C virus (HCV) genotype 1b core protein have been reported to be a potent predictor for poor response to interferon (IFN)-based therapy and a risk factor for hepatocarcinogenesis. We investigated the effects of these polymorphisms with genotype 1b/2a chimeric viruses that contained polymorphisms of Arg/Gln at aa 70 and Leu/Met at aa 91. We found that infectious virus production was reduced in cells transfected with chimeric virus RNA that had Gln at aa 70 (aa70Q) compared with RNA with Arg at aa 70 (aa70R). Using flow cytometry analysis, we confirmed that HCV core protein accumulated in aa70Q clone transfected cells, and it caused a reduction in cell-surface expression of major histocompatibility complex (MHC) class I molecules induced by IFN treatment through enhanced protein kinase R phosphorylation. We could not detect any effects due to the polymorphism at aa 91. In conclusion, the polymorphism at aa 70 was associated with efficiency of infectious virus production, and this deteriorated virus production in strains with aa70Q resulted in the intracellular accumulation of HCV proteins and attenuation of MHC class I molecule expression. These observations may explain the strain-associated resistance to IFN-based therapy and hepatocarcinogenesis of HCV.
More than 170 million people worldwide have been infected with hepatitis C virus (HCV). Of these, 60% develop chronic hepatitis, and 5–20% develop cirrhosis and hepatocellular carcinoma1,2. More than 350,000 people die from these HCV-related diseases annually. However, the mechanisms of how HCV evades host immunity and maintains chronic infection status are not fully understood. To eradicate the virus from infected patients, interferon (IFN)-based treatments have commonly been used, but they are lacking in efficacy3,4,5. Therefore, predictive factors for outcomes of these treatments have been investigated extensively, and several such factors have been identified, including viral load, genotypes, and polymorphisms in the virus genome. Akuta et al. first reported that amino acid (aa) polymorphisms (Arg/Gln at aa 70 and Leu/Met at aa 91) in the core of the genotype 1b strain were potent predictors of poor response to IFN-based therapy6. Subsequently, several studies revealed that the polymorphism at aa 70 is more potent than the polymorphism at aa 91 and is associated with disease progression and development of hepatocellular carcinoma7,8,9,10,11,12,13,14,15,16,17,18. Notably, this is the only polymorphism in the HCV genome that is associated with both IFN treatment failure and the establishment of hepatocellular carcinoma.
HCV is a positive-stranded RNA virus that possesses an open reading frame encoding a large polyprotein19. This polyprotein is cleaved by cellular and viral proteases into 10 structural and non-structural (NS) proteins20. Core, E1 and E2 are structural proteins and components of virus particles. The others (p7, NS2, NS3, NS4A, NS4B, NS5A and NS5B) are NS proteins that are associated with viral replication. Core forms the capsid shell to house and protect genomic RNA. It has been reported that both the interaction between core and NS5A and the localization of this structure into lipid droplets are indispensable to virus particle assembly21,22. Therefore, polymorphisms in core would affect the efficiency of various steps of the viral life cycle, particularly the viral particle assembly step. In addition to the formational role in viral particles, core has been reported to have pleiotropic effects on various host cell functions, including signal transduction, gene expression, cell cycle, hepatic steatosis and tumorigenesis23,24,25,26,27,28,29,30,31,32,33,34,35,36.
HCV evades host immune responses in several ways. In infected cells, HCV NS3 protein directly cleaves adaptor proteins that are important for IFN production37,38. In addition, HCV blocks the translation of IFN-stimulated genes by inducing the phosphorylation of protein kinase R (PKR) and eukaryotic translation initiation factor 2α39. Recently, we observed HCV-associated attenuation of the IFN-induced cell surface expression of major histocompatibility complex (MHC) class I molecule via PKR phosphorylation40.
In this study, we investigated the effects of polymorphisms in core protein (Arg/Gln at aa 70 and Leu/Met at aa 91) on the steps in the HCV life cycle and on the host immune system, particularly MHC class I molecule expression, in order to elucidate polymorphism-related resistance to IFN-based therapy, immune evasion and hepatocarcinogenesis.
Results
Effects of Polymorphisms in Core on Virus Phenotypes
To investigate the effects of aa polymorphisms in the core of genotype 1b, we generated genotype 1b/2a chimeric HCV with polymorphisms Arg/Gln at aa 70 and Leu/Met at aa 91 (designated TH/JFH1-RL, -RM, -QL, and -QM; Fig. 1a). Three days after the in vitro synthesized RNA of these viral strains was electroporated into Huh-7.5.1 cells, the amounts of extra- and intra-cellular HCV core antigen (Ag) were measured in culture medium and cell lysate, respectively. Among these strains, the intracellular HCV core Ag levels were comparable (Fig. 1b). However, the extracellular core Ag levels of TH/JFH1-RL and -RM were 8.99 × 104 ± 7.40 × 103 and 1.02 × 105 ± 7.18 × 103 fmol/L, respectively, and were 4- to 7- fold higher than those of TH/JFH1-QL and -QM (1.46 × 104 ± 1.32 × 103 and 2.21 × 104 ± 3.64 × 103 fmol/L, respectively). To assess the influence of these polymorphisms on the direct anti-viral effects of IFN, we treated HCV RNA-transfected cells with 10 IU/mL (38.4 pg/mL) of IFN-α2b and measured the intra- and extra-cellular HCV core Ag levels (Fig. 1c and Supplementary Table S1). Despite clinical evidence for polymorphism-associated susceptibility to IFN treatment, the intra- and extra-cellular HCV core Ag levels of chimeric viruses with polymorphism-transfected cells were similarly reduced by IFN-α2b treatment.
Figure 1. Virus production and IFN sensitivity of chimeric viruses with polymorphisms.

(a) Schematic representation of chimeric virus (TH/JFH-1) used in this study. The virus comprises sequences of the TH strain (genotype 1b) and JFH-1 strain (genotype 2a). The junction site (indicated by dotted line) is immediately after the first trans-membrane domain of NS2. Polymorphisms R/Q at aa 70 and L/M at aa 91 are indicated by closed triangles. (b) Huh-7.5.1 cells were transfected with in vitro-transcribed RNA of TH/JFH1-RL, -RM, -QL, and -QM. HCV core Ag levels in cell lysates and medium were measured at 3 days after transfection. (c) To assess IFN sensitivity, transfected cells were given 10 IU/mL of IFN-α 2b at 4 h after transfection. Culture medium and cell lysates were harvested at 3 days after transfection, and the HCV core Ag levels were measured. Data are given in terms of percentage vs. untreated cells. (d,e) Single cycle virus production assay was performed in Huh7-25 cells transfected with in vitro-transcribed RNA of TH/JFH1-RL, -RM, -QL, and -QM. Culture medium and cell lysates were harvested on day 3. HCV core Ag levels (d) and infectivity titers (e) in culture medium and cell lysates were measured. Assays were performed in triplicate, and data represent means ± SD. *p < 0.05 compared with aa70R strains.
Single-cycle Virus Production Assay
For further analysis of the effects of these polymorphisms on different stages of the virus life cycle, we used a single-cycle virus production assay with Huh7-25 cells that lacked the cell surface expression of CD81. Three days after the transfection of full-length RNA of TH/JFH1-RL, -RM, -QL, and -QM, the HCV core Ag levels and infectivity titers were measured. In agreement with our observations in Huh-7.5.1 cells, the HCV core Ag levels in the culture medium were 4.8- to 6.9- fold higher in TH/JFH1-RL and -RM transfected cells than they were in TH/JFH1-QL and -QM. However, the amounts of intracellular HCV core Ag in cell lysates were similar among all strains (Fig. 1d). Both intra- and extra-cellular infectivity titers were 60- to 100- fold higher in TH/JFH1-RL and -RM than in TH/JFH1-QL and -QM, which indicated a lower efficiency for intracellular infectious virus production in strains with Gln at aa 70 (Fig. 1e, p < 0.05).
Detection of Core Protein in HCV Replicating Cells
In experiments with Huh-7.5.1 and Huh7-25 cells, we found that aa polymorphisms in core were associated with efficiency of intracellular infectious virus production. Thus, as a next step, we examined the effects of this polymorphism-associated inefficient virus production in host cells. After transfection of the in vitro transcribed RNA of TH/JFH1-RL, -RM, -QL, and -QM into Huh-7.5.1 cells, the percentages of core protein-positive cells were counted by flow cytometry, and the intracellular levels of core protein within core-positive cells were evaluated by calculating the mean fluorescence intensity (MFI). The percentages of core protein-positive cells at day 1 after transfection were similar among the strains of transfected cells, which suggested comparable levels of transfection efficiency (Supplementary Fig. S1 and Supplementary Table S2). The percentages of HCV core-positive cells were higher in cells transfected with TH/JFH1-RL and -RM than in cells transfected with TH/JFH1-QL and -QM at day 3 after transfection, thus suggesting re-infection by progeny viruses (Fig. 2 and Table 1). However, TH/JFH1-QL and -QM transfection resulted in a higher MFI of HCV core within core-positive cells than TH/JFH1-RL and -RM transfection (Fig. 2 and Table 1). In this experiment, we obtained data concerning the intracellular core levels independent from the percentage of core-positive cells by analyzing the gating of core-positive cells in flow cytometry. Taken together, these data suggest that Gln at aa 70 decreases the efficiency of intracellular infectious virus production and thus induces the accumulation of HCV core proteins in replicating cells.
Figure 2. Population of HCV core-positive cells detected by flow cytometry.

Huh-7.5.1 cells were transfected with RNA of TH/JFH1-RL, -RM, -QL, and -QM. Transfected cells were harvested and stained with anti-HCV core antibody. The percentages of HCV core-positive cells were counted. The shaded areas indicate mock transfections used as controls. The MFI of HCV core in the gate of core-positive cells was calculated and presented in Table 1.
Table 1. Percentage of HCV-positive cells and amounts of core protein.
| Strains | Positive Cells (%) | MFI |
|---|---|---|
| TH/JFH1-RL | 56.4 ± 1.2 | 178.1 ± 1.0 |
| TH/JFH1-RM | 60.6 ± 0.6 | 203.9 ± 1.6 |
| TH/JFH1-QL | 33.4 ± 0.3* | 252.6 ± 1.3* |
| TH/JFH1-QM | 31.6 ± 0.9* | 396.6 ± 1.3* |
MFI, mean fluorescence intensity. *p < 0.05 compared with aa70R strains.
Effects of HCV Core Protein Accumulation on MHC Class I Molecule Induction
To assess the effects of accumulated HCV core protein on the host immune system and the elimination of HCV-infected cells, we evaluated the cell surface expression of MHC class I molecules by flow cytometry. Following administration of type I and II IFNs, we determined that IFN-γ most efficiently induced the cell surface expression of MHC class I molecules in Huh-7.5.1 cells (Supplementary Fig. S2). Thus, we used IFN-γ to assess the effect of core polymorphisms on MHC class I molecule expression. RNA of TH/JFH1-RL, -RM, -QL, and -QM was transfected into Huh-7.5.1 cells by using DEMRIE-C reagent. Two days after transfection, the transfected cells were treated with IFN-γ. The cell surface expression of MHC class I molecules was detected by staining with Alexa Fluor 488 conjugated anti-HLA-ABC before permeabilization. For multicolour staining, HCV core protein was detected using anti-HCV core antibody labelled with Alexa Fluor 647 after permeabilization.
In HCV core-negative cells, MHC class I molecules were largely induced (Fig. 3). In contrast, in HCV core-positive cells, the induction of MHC class I molecules was suppressed. To compare the effects of aa polymorphisms in HCV core, the reduction rate of MHC class I molecule expression was calculated by using the following formula: [(MFI of MHC class I molecules in HCV-negative cells - MFI of MHC class I molecules in HCV-positive cells)/MFI of MHC class I molecules in HCV-negative cells]. The reduction rate in TH/JFH1-QL and -QM was substantially higher than that in TH/JFH1-RL and -RM (p < 0.05, Table 2).
Figure 3. Attenuation of cell surface expression of MHC class I molecules by HCV replication.
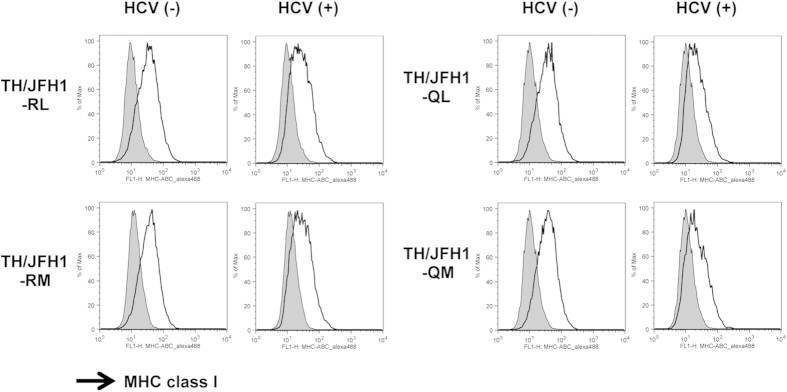
Huh-7.5.1 cells were transfected with RNA of TH/JFH1-RL, -RM, -QL, and -QM. Transfected cells were treated with 200 pg/mL of IFN-γ at day 2 and harvested at day 3 after transfection. Harvested cells were stained with anti-HLA-ABC antibody, permeabilized, and then stained with anti-HCV core antibody. The shaded areas indicate IFN-γ untreated controls. HCV core-negative and -positive populations were gated separately, and cell surface expression of MHC class I molecules was presented in HCV core-negative and -positive populations.
Table 2. Effects of hepatitis C virus replication on MHC class I molecule expression.
| MHC Class I Molecule Expression (MFI) |
Reduction Rate of MHC Class I Molecule Expression (%) | ||
|---|---|---|---|
| HCV (+) | HCV (−) | ||
| TH/JFH1-RL | 34.6 ± 1.0 | 42.0 ± 1.6 | 17.7 ± 0.7 |
| TH/JFH1-RM | 38.4 ± 1.0 | 43.7 ± 1.0 | 12.1 ± 1.2 |
| TH/JFH1-QL | 29.9 ± 2.0 | 43.9 ± 1.4 | 32.0 ± 2.5* |
| TH/JFH1-QM | 31.0 ± 0.6 | 44.9 ± 1.1 | 30.8 ± 0.5* |
MFI, mean fluorescence intensity; MHC, major histocompatibility complex. *p < 0.05 compared with aa70R strains.
Effects of HCV Core Protein Accumulation on PKR Phosphorylation
We previously reported that activation of the PKR pathway is responsible for attenuated expression of MHC class I molecules40. Therefore, we assessed the effects of aa polymorphisms on PKR phosphorylation. Huh-7.5.1 cells were transfected with RNA of TH/JFH1-RL, -RM, -QL, and -QM using DEMRIE-C reagent and subsequently treated with IFN-γ. Cell lysates of these treated cells were analyzed by immunoblotting. The expression levels of PKR were similar among the TH/JFH1 strains that had aa polymorphisms. However, the amounts of phosphorylated PKR were markedly higher in TH/JFH1-QL and -QM transfected cells than they were in TH/JFH1-RL- and -RM transfected cells (Fig. 4). These results indicate that the PKR phosphorylation associated with aa polymorphisms is responsible for the attenuated cell-surface expression of MHC class I molecules.
Figure 4. PKR phosphorylation in full-length HCV RNA transfected cells.
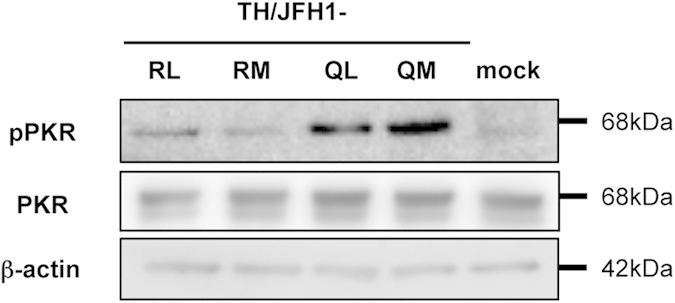
Huh-7.5.1 cells were transfected with RNA of TH/JFH1-RL, -RM, -QL, and -QM. Transfected cells were treated with 200 pg/mL of IFN-γ 2 days after transfection and harvested at day 3. Mock transfection was used as a control. Cell lysates were subjected to immunoblotting with the indicated antibodies.
Discussion
HCV is known to cause chronic hepatitis followed by cirrhosis and hepatocellular carcinoma. Thus, eradicating HCV is a priority for chronically infected patients. For this purpose, IFN-based therapy has played a pivotal role. However, because the efficacy of this therapy is unsatisfactory, much effort has been devoted to elucidating the mechanisms responsible for IFN resistance in this virus. The information obtained by these investigations may contribute to clarifying the strategies employed by HCV in evading the host immune system and identifying predictors of outcome in IFN-based therapy.
The aa polymorphisms in core are among the genetic factors that influence the outcome of IFN-based therapy6,9,41. Although the evidence accumulated from clinical studies supports a correlation between these polymorphisms and responses to IFN-based therapy, the underlying molecular mechanisms are not fully understood. This insufficient progress is partly attributable to a lack of cell-culture systems for HCV genotype1b strains. Clinical studies into these aa polymorphisms have been performed in patients infected with HCV genotype 1b strains. However, efficient cell-culture models for HCV genotype 1b strains without adaptive mutations are not currently available. JFH-1, genotype 2a, is the only strain that can efficiently replicate in cultured cells without mutations42, and aa polymorphisms in core are rarely observed in clinical samples of genotype 2a strains41.
In this study, we used a cell-culture system with JFH-1-based chimeric viruses of genotype 1b/2a to assess the function of aa polymorphisms in core of genotype 1b. By using this system, we confirmed decreased infectious virus production by chimeric viruses with aa70Q. By transfection with full-length RNA, we were able to observe higher levels of HCV core Ag in the culture medium of strains with aa70R-transfected cells compared with aa70Q. In a single-cycle virus production assay with CD81-negative cell lines, intra-cellular infectious virus production was lower in strains with aa70Q compared with aa70R. In flow cytometry, MFI of core protein staining in each core-positive cell was higher after transfection of strains with aa70Q compared with aa70R. These data suggest that the decreased virus production of strains with aa70Q is related to the intracellular accumulation of HCV proteins. Despite the apparent phenotypic association with the polymorphism at aa 70, we were unable to detect any significant differences in comparisons of the polymorphism at aa 91. Although significant differences have been found in clinical observations between patients with the polymorphism at aa 91 in several reports, other reports have been contradictory7,9,12,13,14,16,17.
Although we were able to detect the affected step in the HCV life cycle by studying the polymorphism at aa 70 using a system with genotype 1b/2a chimeric virus, we failed to demonstrate any differences in the direct anti-viral effects of IFN on strains with the polymorphism. The reduction rates for the extra- and intra-cellular levels of HCV core Ag were comparable between the strains with aa70R and aa70Q. These polymorphism-associated differences in susceptibility to IFN treatment have previously been reported43; interleukin-6 and suppressor of cytokine signalling (SOCS3)-mediated suppression of IFN signalling by strains with aa70Q or aa91M was observed in the cell culture system with JFH-1. Possible reasons for this inconsistent observation include the culture system, HCV strain (1b/2a chimeric virus vs JFH-1), and cell line (Huh-7.5.1 cells vs HuH-7 cells), although such conflicting observations have also been reported by two other groups that used genotype 1b strains44,45. In a study with the HCV genotype 1b strain infection system using human hepatocyte-transplanted mice, differences in IFN resistance were not observed among strains with aa polymorphisms in core45.
Instead of direct effects on susceptibility to IFN, we detected another role for aa polymorphisms in the host immune system. We previously reported that HCV replication suppresses cell-surface expression of MHC class I molecules through PKR phosphorylation40. Phosphorylated PKR induces phosphorylation of eIF2α to inhibit translation initiation. In the current study, we demonstrated that strains with aa70Q showed an accumulation of the HCV core protein in replicating cells because of low-efficiency virus particle production, and we showed that this accumulation enhanced PKR phosphorylation. As a result, the cell-surface expression of MHC class I molecules by IFN treatment was suppressed. HCV-specific CD8 + T cell responses are essential for the clearance of HCV infection, and MHC class I molecules play a key role in the recognition of virus-infected cells by CD8 + T cells. Reductions in MHC class I molecule expression by HCV infection may contribute to circumventing the antiviral adaptive immune responses associated with CD8 + T cells. Thus, this observation may explain one of the mechanisms for aa polymorphism-associated IFN resistance. During IFN therapy, HCV genotype 1b strains with aa70Q may remain in cells and survive by avoiding attack by CD8 + T cells. After treatment ends, surviving viruses start to replicate and propagate into other cells. HCV-positive cells evading the elimination of infected cells by T cells may be a causative factor in hepatocellular carcinoma.
In conclusion, we showed that IFN-γ-induced MHC class I molecule expression was suppressed in HCV core aa 70 variants. These findings may explain the mechanisms responsible for clinical resistance to IFN therapy and hepatocarcinogenesis.
Methods
Cell Culture
The HuH-7-derived cell lines Huh-7.5.146 (provided by Francis Chisari; Scripps Research Institute, La Jolla, CA) and Huh7-25 lacking CD81 expression47 were used in this study. Culture conditions for these cell lines were as described previously48.
Plasmid Construction and RNA Transfection
The genotype 1b/2a chimeric virus construct was generated using strains TH (Genotype 1b, accession number is AB985268) and JFH-149 (Genotype 2a, accession number is AB047639) by replacing the sequences for core to the first trans-membrane domain of NS2 of JFH-1 with those of TH (Fig. 1a). The original aa residues at aa 70 and aa 91 of TH were Arg and Met, respectively. Chimeric viruses with the polymorphisms Arg/Gln at aa 70 and Leu/Met at aa 91 were generated by site-directed mutagenesis using appropriate primers (Fig. 1a).
To prepare the template for in vitro RNA synthesis, plasmids of these constructs were linearized at the 3′-end by XbaI digestion. HCV full-length RNA was synthesized using a MEGAscript T7 kit (Life Technologies, Carlsbad, CA) and used for RNA transfection48. Trypsinized Huh-7.5.1 or Huh7–25 cells were washed with Opti-MEM I reduced-serum medium (Life Technologies) and were resuspended at 3 × 105 cells/mL or 1 × 105/mL cells with Cytomix buffer, respectively. Ten micrograms of RNA was mixed with 400 μL of cell suspension and was transferred to an electroporation cuvette (Gene Pulser Cuvette 0.4 cm; Bio-Rad, Hercules, CA). Cells were then pulsed at 260 V and 950 μF with the Gene Pulser II (Bio-Rad). Transfected cells were immediately transferred to 6-well or 12-well plates (Corning, Corning, NY). To assess susceptibility to IFN, IFN-α2b (Intron A, MSD K.K., Tokyo, Japan) was used.
Quantification of HCV Core Antigen
The concentration of HCV core Ag in filtered culture medium and cell lysates was measured by a chemiluminescent enzyme immunoassay (Lumipulse Ortho HCV Ag kit; Ortho Clinical Diagnostics, Tokyo, Japan) in accordance with the manufacturer’s instructions50,51.
Titration of HCV Infectivity
Culture medium was serially diluted in 5-fold increments in complete DMEM and used to infect naïve Huh-7.5.1 cells seeded 24 h earlier in poly-D-lysine coated flat-bottom 96-well plates (Corning, Corning, NY) at a density of 1 × 104 cells per well. Three days after infection, cells were stained with mouse monoclonal HCV core protein antibody (clone 2H9) for 1 h and then washed three times with PBS. Cells were then incubated with Alexa Fluor 488 anti-mouse secondary antibody (Life Technologies) for 1 h. Infectivity titer was expressed as focus-forming units per mL (ffu/mL), expressing the mean number of HCV core-positive foci detected at the highest dilutions48. Intracellular infectivity titer was determined as described previously52,53.
Detection of HCV and MHC Class I Molecule Expression
To detect HCV-positive cells, we used flow cytometry (FACSCalibur; BD Biosciences, San Jose, CA, USA). The antibody used was mouse anti-HCV core 1b (Clone C7–50; Abcam, Cambridge, UK). To label this antibody, Zenon Mouse IgG Labeling Kits (Alexa Fluor 647; Life Technologies) were used. The amount of intracellular core protein was estimated independently from the percentage of core-positive cells by calculating the MFI in the gate of core-positive cells in flow cytometry.
The effects of polymorphisms on MHC class I molecule induction were then assessed. Huh-7.5.1 cells were seeded in 6-well plates at a concentration of 1 × 105/mL, and 10 μg of in vitro-transcribed RNA was transfected with DEMRIE-C reagent (Life Technologies). To induce MHC class I molecule expression, cells were treated with 10 IU/mL (38.4 pg/mL) of IFN-α2b (Intron A), 10 IU/mL (500 pg/mL) of IFN-β (Feron, Toray Medical Co., Ltd., Tokyo, Japan), or 200 pg/mL of IFN-γ (Imunomax-γ; Shionogi & Co., Ltd., Osaka, Japan). Cell surface expression of MHC class I molecules was assessed by staining with Alexa Fluor 488 conjugated anti-HLA-ABC (clone DX17, BD Biosciences).
Immunoblotting
Cell lysates of Huh-7.5.1 cells transfected with in vitro-transcribed RNA were applied to sodium dodecyl sulfate polyacrylamide gel electrophoresis and analyzed by immunoblotting. To detect PKR and its phosphorylated form, rabbit polyclonal anti-PKR (Clone E120; Santa Cruz Biotechnology, Santa Cruz, CA) and rabbit monoclonal anti-phospho-PKR, T446 (Clone E120; Epitomics, Burlingame, CA) were used. As a control for the applied proteins, β-actin was also detected with mouse monoclonal anti-β-actin antibody (Sigma-Aldrich, St. Louis, MO). To detect signals, horseradish peroxidase-conjugated anti-rabbit secondary antibody (Cell Signaling Technology, Danvers, MA) was used.
Statistical Analysis
Assays were performed at least in triplicate. Data from repeated experiments are expressed as the means ± standard deviation (SD). Statistical analysis was performed using Student’s t-test, and p values less than 0.05 were considered statistically significant.
Additional Information
How to cite this article: Tasaka-Fujita, M. et al. Amino Acid Polymorphisms in Hepatitis C Virus Core Affect Infectious Virus Production and Major Histocompatibility Complex Class I Molecule Expression. Sci. Rep. 5, 13994; doi: 10.1038/srep13994 (2015).
Supplementary Material
Acknowledgments
The authors thank Dr. Francis V. Chisari (Scripps Research Institute, La Jolla, CA) for providing the Huh-7.5.1 cell line. This work was supported in part by Grants-in-Aid for Scientific Research from the JSPS KAKENHI Grant Number 24591000; from the Ministry of Health, Labour and Welfare of Japan; from the Ministry of Education, Culture, Sports, Science and Technology; and from the Miyakawa Memorial Research Foundation. W.K. and E-C. S. were supported by the Korea Advanced Institute of Science and Technology (KAIST) Future Systems Healthcare Project from the Ministry of Science, ICT & Future Planning of Korea.
Footnotes
Author Contributions M.T.-F., N.S., T.M., A.M., N.Y., R.S., S.T. and T.K. performed the experiments. W.K., K.W., Y.A., N.S., T.W., E.-C.S. and T.K. supervised various aspects of the study. M.T.-F. and T.K. wrote the manuscript. All authors reviewed the manuscript.
References
- Liang T. J., Rehermann B., Seeff L. B. & Hoofnagle J. H. Pathogenesis, natural history, treatment, and prevention of hepatitis C. Ann. Intern. Med. 132, 296–305 (2000). [DOI] [PubMed] [Google Scholar]
- Shepard C. W., Finelli L. & Alter M. J. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 5, 558–567, 10.1016/s1473-3099(05)70216-4 (2005). [DOI] [PubMed] [Google Scholar]
- Hoofnagle J. H. et al. Treatment of chronic non-A,non-B hepatitis with recombinant human alpha interferon. A preliminary report. N. Engl. J. Med. 315, 1575–1578, 10.1056/NEJM198612183152503 (1986). [DOI] [PubMed] [Google Scholar]
- Ghany M. G., Strader D. B., Thomas D. L., Seeff L. B. & American Association for the Study of Liver, D. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology 49, 1335–1374, 10.1002/hep.22759 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeuzem S. et al. Expert opinion on the treatment of patients with chronic hepatitis C. J. Viral Hepat. 16, 75–90, 10.1111/j.1365-2893.2008.01012.x (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akuta N. et al. Association of amino acid substitution pattern in core protein of hepatitis C virus genotype 1b high viral load and non-virological response to interferon-ribavirin combination therapy. Intervirology 48, 372–380, 10.1159/000086064 (2005). [DOI] [PubMed] [Google Scholar]
- Okanoue T. et al. Predictive values of amino acid sequences of the core and NS5A regions in antiviral therapy for hepatitis C: a Japanese multi-center study. J. Gastroenterol. 44, 952–-963., 10.1007/s00535-009-0087-x (2009). [DOI] [PubMed] [Google Scholar]
- Mori N. et al. Randomized trial of high-dose interferon-alpha-2b combined with ribavirin in patients with chronic hepatitis C: Correlation between amino acid substitutions in the core/NS5A region and virological response to interferon therapy. J. Med. Virol. 81, 640–649, 10.1002/jmv.21438 (2009). [DOI] [PubMed] [Google Scholar]
- Enomoto N. & Maekawa S. HCV genetic elements determining the early response to peginterferon and ribavirin therapy. Intervirology 53, 66–69, 10.1159/000252787 (2010). [DOI] [PubMed] [Google Scholar]
- Nakamoto S. et al. Association between mutations in the core region of hepatitis C virus genotype 1 and hepatocellular carcinoma development. J. Hepatol. 52, 72–78, 10.1016/j.jhep.2009.10.001 (2010). [DOI] [PubMed] [Google Scholar]
- Akuta N. et al. Amino acid substitutions in hepatitis C virus core region predict hepatocarcinogenesis following eradication of HCV RNA by antiviral therapy. J. Med. Virol. 83, 1016–1022, 10.1002/jmv.22094 (2011). [DOI] [PubMed] [Google Scholar]
- El-Shamy A. et al. Sequence heterogeneity of NS5A and core proteins of hepatitis C virus and virological responses to pegylated-interferon/ribavirin combination therapy. Microbiol. Immunol. 55, 418–426, 10.1111/j.1348-0421.2011.00331.x (2011). [DOI] [PubMed] [Google Scholar]
- Hayashi K. et al. Mutations in the core and NS5A region of hepatitis C virus genotype 1b and correlation with response to pegylated-interferon-alpha 2b and ribavirin combination therapy. J. Viral Hepat. 18, 280–286, 10.1111/j.1365-2893.2010.01305.x (2011). [DOI] [PubMed] [Google Scholar]
- El-Shamy A. et al. Polymorphisms of hepatitis C virus non-structural protein 5A and core protein and clinical outcome of pegylated-interferon/ribavirin combination therapy. Intervirology 55, 1–11, doi: 10.1159/000322219 (2012). [DOI] [PubMed] [Google Scholar]
- Akuta N. et al. Complicated relationships of amino acid substitution in hepatitis C virus core region and IL28B genotype influencing hepatocarcinogenesis. Hepatology 56, 2134–2141, 10.1002/hep.25949 (2012). [DOI] [PubMed] [Google Scholar]
- Seko Y. et al. Amino acid substitutions in the hepatitis C Virus core region and lipid metabolism are associated with hepatocarcinogenesis in nonresponders to interferon plus ribavirin combination therapy. Intervirology 56, 13–21, doi: 10.1159/000339993 (2013). [DOI] [PubMed] [Google Scholar]
- El-Shamy A. et al. Polymorphisms of the core, NS3, and NS5A proteins of hepatitis C virus genotype 1b associate with development of hepatocellular carcinoma. Hepatology 58, 555–563, 10.1002/hep.26205 (2013). [DOI] [PubMed] [Google Scholar]
- Miura M. et al. Deep-sequencing analysis of the association between the quasispecies nature of the hepatitis C virus core region and disease progression. J. Virol. 87, 12541–12551, 10.1128/JVI.00826-13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijikata M. et al. Proteolytic processing and membrane association of putative nonstructural proteins of hepatitis C virus. Proc. Natl. Acad. Sci. USA. 90, 10773–10777 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grakoui A., Wychowski C., Lin C., Feinstone S. M. & Rice C. M. Expression and identification of hepatitis C virus polyprotein cleavage products. J. Virol. 67, 1385–1395 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyanari Y. et al. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 9, 1089–1097, 10.1038/ncb1631 (2007). [DOI] [PubMed] [Google Scholar]
- Masaki T. et al. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J. Virol. 82, 7964–7976, 10.1128/JVI.00826-08 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray R. B., Lagging L. M., Meyer K. & Ray R. Hepatitis C virus core protein cooperates with ras and transforms primary rat embryo fibroblasts to tumorigenic phenotype. J. Virol. 70, 4438–4443 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya K. et al. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J. Gen. Virol. 78 (Pt 7), 1527–1531 (1997). [DOI] [PubMed] [Google Scholar]
- Moriya K. et al. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 4, 1065–1067, doi: 10.1038/2053 (1998). [DOI] [PubMed] [Google Scholar]
- Aoki H., Hayashi J., Moriyama M., Arakawa Y. & Hino O. Hepatitis C virus core protein interacts with 14-3-3 protein and activates the kinase Raf-1. J. Virol. 74, 1736–1741 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi J. et al. Hepatitis C virus core protein activates the MAPK/ERK cascade synergistically with tumor promoter TPA, but not with epidermal growth factor or transforming growth factor alpha. Hepatology 32, 958–961, 10.1053/jhep.2000.19343 (2000). [DOI] [PubMed] [Google Scholar]
- Honda M. et al. Hepatitis C virus core protein induces apoptosis and impairs cell-cycle regulation in stably transformed Chinese hamster ovary cells. Hepatology 31, 1351-1359, 10.1053/jhep.2000.7985 (2000). [DOI] [PubMed] [Google Scholar]
- Moriishi K. et al. Proteasome activator PA28gamma-dependent nuclear retention and degradation of hepatitis C virus core protein. J. Virol. 77, 10237–10249 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsumi T. et al. Hepatitis C virus core protein activates ERK and p38 MAPK in cooperation with ethanol in transgenic mice. Hepatology 38, 820–828, 10.1053/jhep.2003.50399 (2003). [DOI] [PubMed] [Google Scholar]
- Miyoshi H. et al. Hepatitis C virus core protein exerts an inhibitory effect on suppressor of cytokine signaling (SOCS)-1 gene expression. J. Hepatol. 43, 757–763, 10.1016/j.jhep.2005.03.028 (2005). [DOI] [PubMed] [Google Scholar]
- Kanda T., Steele R., Ray R. & Ray R. B. Hepatitis C virus core protein augments androgen receptor-mediated signaling. J. Virol. 82, 11066–11072, 10.1128/JVI.01300-08 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N., Moriya K., Kiyosawa K., Koike K. & Aoyama T. Hepatitis C virus core protein induces spontaneous and persistent activation of peroxisome proliferator-activated receptor alpha in transgenic mice: implications for HCV-associated hepatocarcinogenesis. Int. J. Cancer 122, 124–131, 10.1002/ijc.23056 (2008). [DOI] [PubMed] [Google Scholar]
- Angus A. G. et al. Requirement of cellular DDX3 for hepatitis C virus replication is unrelated to its interaction with the viral core protein. J. Gen. Virol. 91, 122–132, 10.1099/vir.0.015909-0 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benzoubir N. et al. HCV core-mediated activation of latent TGF-beta via thrombospondin drives the crosstalk between hepatocytes and stromal environment. J. Hepatol. 59, 1160–1168, 10.1016/j.jhep.2013.07.036 (2013). [DOI] [PubMed] [Google Scholar]
- Stone A. E. et al. Hepatitis C virus core protein inhibits interferon production by a human plasmacytoid dendritic cell line and dysregulates interferon regulatory factor-7 and signal transducer and activator of transcription (STAT) 1 protein expression. PLoS One 9, e95627, 10.1371/journal.pone.0095627 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. H. & Rehermann B. Immune responses to HCV and other hepatitis viruses. Immunity 40, 13–24, 10.1016/j.immuni.2013.12.010 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner S. M. & Gale M. Jr. Regulation of hepatic innate immunity by hepatitis C virus. Nat. Med. 19, 879–888, 10.1038/nm.3253 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaigorta U. & Chisari F. V. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe 6, 513–522, 10.1016/j.chom.2009.11.004 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang W. et al. Hepatitis C Virus Attenuates Interferon-Induced MHC Class I Expression and Decreases CD8 T-Cell Effector Functions. Gastroenterology 146, 1351–1360, 10.1053/j.gastro.2014.01.054 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Shamy A. & Hotta H. Impact of hepatitis C virus heterogeneity on interferon sensitivity: an overview. World J. Gastroenterol. 20, 7555–7569, 10.3748/wjg.v20.i24.7555 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakita T. et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11, 791–796, 10.1038/nm1268 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funaoka Y. et al. Analysis of interferon signaling by infectious hepatitis C virus clones with substitutions of core amino acids 70 and 91. J. Virol. 85, 5986–5994, 10.1128/jvi.02583-10 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda F. et al. Amino acid substitutions of hepatitis C virus core protein are not associated with intracellular antiviral response to interferon-alpha in vitro. Liver Int 30, 1324–1331, 10.1111/j.1478-3231.2010.02299.x (2010). [DOI] [PubMed] [Google Scholar]
- Hiraga N. et al. Impact of viral amino acid substitutions and host interleukin-28b polymorphism on replication and susceptibility to interferon of hepatitis C virus. Hepatology 54, 764–771, 10.1002/hep.24453 (2011). [DOI] [PubMed] [Google Scholar]
- Zhong J. et al. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA. 102, 9294–9299, 10.1073/pnas.0503596102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akazawa D. et al. CD81 expression is important for the permissiveness of Huh7 cell clones for heterogeneous hepatitis C virus infection. J. Virol. 81, 5036–5045, 10.1128/jvi.01573-06 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T. et al. Cell culture and infection system for hepatitis C virus. Nat. Protoc. 1, 2334–2339, 10.1038/nprot.2006.395 (2006). [DOI] [PubMed] [Google Scholar]
- Kato T. et al. Sequence analysis of hepatitis C virus isolated from a fulminant hepatitis patient. J. Med. Virol. 64, 334–339 (2001). [DOI] [PubMed] [Google Scholar]
- Aoyagi K. et al. Development of a simple and highly sensitive enzyme immunoassay for hepatitis C virus core antigen. J. Clin. Microbiol. 37, 1802–1808 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama A. et al. Japanese reference panel of blood specimens for evaluation of hepatitis C virus RNA and core antigen quantitative assays. J. Clin. Microbiol. 50, 1943–1949, 10.1128/JCM.00487-12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T. et al. Hepatitis C virus JFH-1 strain infection in chimpanzees is associated with low pathogenicity and emergence of an adaptive mutation. Hepatology 48, 732–740, 10.1002/hep.22422 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed M. et al. In vivo adaptation of hepatitis C virus in chimpanzees for efficient virus production and evasion of apoptosis. Hepatology 54, 425–433, 10.1002/hep.24399 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.