

Abstract
Background: MicroRNAs are a class of endogenous single strand non-coding RNAs that are involved in many important physiological and pathological processes. The purpose of this study was to investigate the expression levels of miR-29c in human bladder cancer and its potential role in disease pathogenesis. Methods: The expression level of miR-29c was measured in 40 bladder cancer specimens and adjacent normal breast tissues by quantitative polymerase chain reaction (qPCR). Over-expression of miR-29c was established by transfecting mimics into T24.MTT assays, colony formation assays, transwell assays and cell cycle assays were used to explore the potential function of miR-29c inT24 bladder cancer cells. Luciferase reporter assays were performed to analyze the regulation of putative target of miR-29c. The effects of modulating miR-29c on endogenous levels of this target were subsequently confirmed via qRT-PCR and Western blot. Results: The expression of miR-29c in bladder cancer specimens was lower than adjacent normal tissues (P<0.01). Overexpression of miR-29c inhibited cellular growth, suppressed cellular migration and caused an accumulation of cells in the G1 phase of the cell cycle, Dual-luciferase reporter assays showed that miR-29c binds the 3’-untranslated region (3’-UTR) of CDK6, suggesting that CDK6 is a direct target of miR-29c. Furthermore, through qPCR and Western blot assays confirmed that overexpression of miR-29c reduced CDK6 mRNA and protein levels. Conclusions: miR-29c could inhibit the proliferation, migration and invasion of bladder cancer cells via regulating CDK6. in the future, it could be used as a therapeutic target for the treatment of bladder cancer.
Keywords: MiR-29c, proliferation, CDK6, bladder cancer
Introduction
Bladder cancer is one of the most common worldwide malignancies. In developed countries, bladder cancer (BC) is the fifth most commonly diagnosed tumor and the second most common cause of death among genitourinary tumors [1], Bladder cancer is the fourth most common cancer in males and ninth most common in females; it is the ninth most common malignancy overall [2,3]. In middle-aged and elderly men, bladder cancer is the second most prevalent malignancy after prostate cancer [4], So it is urgent to understand the molecular and cellular mechanisms of metastasis for investigating the development of bladder cancer. Downregulation of certain tumor suppressor genes was confirmed to largely contribute to initiation, proliferation, invasion and metastasis of bladder cancer [5]. Therefore, targeted gene therapy has been documented as a reasonable strategy for bladder cancer treatment [6].
MicroRNAs (miRNAs) are a small class of non-coding RNAs which regulate gene expression and may play pivotal roles in the physiological and pathological processes in a variety of eukaryotic organisms [7]. Mature miRNAs negatively regulate their target genes through imperfect complementary sequence pairing to the 3’-untranslated region (UTR) of target genes, resulting to either mRNA degradation or translational repression [8]. Moreover, aberrant miRNA expression has been frequently observed in various types of human tumors. These reports suggest that miRNAs may function as either tumor-suppressor genes or oncogenes [9]. Recent studies indicate that miR-29c is downregulated in rhabdomyosarcoma, cholangiocarcinoma, acute myelogenous leukemia (AML), lung cancer, and nasopharyngeal carcinoma [10,11]. However, the mechanism by which miR-29c contributes to bladder cancer tumorigenesis is still unclear.
CDK6 is a member of a family of serine-threonine kinases and CDK6 is a key regulator during the G1/S cell cycle transition. Aberrant expression of CDK6 protein has been observed in many cancer types [12]. Aberrant CDK6 expression has been reported in pancreatic cancer [13], T-cell lymphoma [14], malignant glioma [15] and medulloblastoma [16], suggesting the involvement of CDK6 in cancer. Furthermore, significant evidence indicates that overexpression of CDK6 in patients with bladder cancer is associated with a worse prognosis [17]. Thus, CDK6 has attracted increasing research interest and understanding the roles of miR-29c in bladder cancer and identifying relevant mRNA targets that mediate its tumor suppressor or oncogenic activities are essential in developing miR-29c as a therapeutic target.
In this study, we report that miR-29c expression is significantly decreased in human bladder cancer, and its overexpression inhibits the proliferation of T24 cells by targeting CDK6. These results indicate that miR-29c functions as a tumor suppressor, whose deregulation may be involved in the initiation and development of human bladder cancer.
Materials and methods
Cell culture and tissue samples
Human T24 bladder cancer cells and human embryonic kidney 293T cells were obtained from the Chinese Science Institute. Cells were culture in DMEM medium supplemented with 10% fetal bovine serum. Bladder tumor tissues and adjacent nontumor bladder tissues were collected from the Department of Urology, Tongji Hospital of Tongji University, Shanghai, China. The samples were immediately snap-frozen in liquid nitrogen. All samples were confirmed as bladder cancer by trained pathologists. No patients received chemotherapy or radiotherapy prior to surgery.
Analysis of miRNA expression using TaqMan RT-PCR
Total RNA from tissue samples and cell lines was harvested using the miRNA Isolation Kit (Ambion, USA). Expression of mature miRNAs was assayed using Taqman MicroRNA Assay (Applied Biosystems) specific for hsa-miR-29c. Briefly, 5 ng of total RNA were reverse transcribed to cDNA with specific stem-loop RT primers. Quantitative real-time PCR was performed by using an Applied Biosystems 7300 Real-time PCR System and a TaqMan Universal PCR Master Mix. All the primers were obtained from the TaqMan miRNA Assays. Small nuclear U6 snRNA (Applied Biosystems) was used as an internal control.
Western blotting
Three groups of monolayer cells were washed with ice-cold PBS for 2 times, added RIPA lysis buffer [1% Triton X-100, 50 mmol/L (Tris pH7.4), 150 mmol/L NaCl, 20 mmol/L iodoacetamide, 1 mmol/L PMSF, 1% aprotinin], lysed on ice for 30 min, scrapped off, transferred into EP tube and centrifuged at 4°C, 12000 xg for 30 min. The supernatants were collected and protein concentration was determined using BCA protein quantitation kit. Each sample with 60 μg protein was added 1X SDS sample buffer [100 mmol/L Tri-HCl (pH6.8), 4% SDS, 0.2% bromophenol blue, 20% glycerol, 200 mmol/L β-mercaptoethanol], and denatured at 95°C for 5 min. Proteins were separated by 10% SDS-PAGE electrophoresis and transferred to 0.45 μm NC membrane. Membrane was blocked with 5% skim milk for 1 h, and incubated with 1:1000 diluted CDK6 monoclonal antibodies at 4°C overnight. The membrane was then incubated with 1:1000 diluted β-actin antibody at room temperature for 1 h, washed with PBST three times, and detected with Odyssey system.
Cell proliferation assay (MTT assay)
Cells (2 × 103) were plated in 96-well plates (BD Biosciences, USA) and incubated at 37°C until the cells reached 30–40% confluence, followed by transfection with 50 nM or 100 nM miR-29c or NC mimics. Cell proliferation was assessed at 24, 48, 72, 96 h as follows: 20 μl (5 mg/ml) MTT solution (Sigma, USA) was added in each well, and after 4 h of incubation at 37°C, the supernatant was discarded and 150 μl of dimethyl sulfoxide (DMSO) were added. After 10 min of low speed shaking (100 rpm) and incubation, the OD at 490 nm was read by a microplate spectrophotometer. Each sample was tested with six replicates. All experiments were performed in biological triplicate.
Colony formation assay
Three hundred cells of each group (miR-29c, mock and NC) were plated in a 6-well plate in complete medium. After incubation at 37°C with 5% CO2 for 7-10 days, or when the colonies were visible by viewing with the eye, the culture was terminated. Complete medium was removed, and the plates were washed twice in phosphate-buffered saline (PBS). The colonies were fixed in 95% ethanol for 10 min, dried and stained with 0.1% crystal violet solution for 10 min. Next, each plate was washed three times with water, and the number of colonies was counted only if the well contained >50 cells. The experiment was performed three times.
Transwell experiment
In accordance with Transwell chamber instructions, DMEM/high glucose medium containing 10% FBS was added to the lower chamber while T24 cell suspension transfected with miR-29c, mock and NC for 48 h was added to the upper chamber. After incubation at 37°C, 5% CO2 for 12-18 h, the lower chamber was observed using inverted microscope. Incubation was terminated when cells passed into lower chamber. Inside of the upper chamber was cleaned with a cotton swab. Lower chamber was immersed and washed with PBS, fixed with 4% paraformaldehyde, stained with 0.1% crystal violet, washed for three times with running water, and then photographed. Membrane-binding crystal violet was dissolved with 300 μl 33% glacial acetic acid, and then absorbance at 573 nm was measured using microplate reader.
Cell cycle assay
miR-29c (100 nM), mock and NC cells were harvested at 48 h after transfection, centrifuged at 1,200 rpm for 10 min and washed three times with cold PBS. Ice-cold 70% ethanol was subsequently added dropwise, and the cells were fixed at 4°C overnight. After a 30-min digestion in RNase (0.1 g/l), a total of 250 μl (0.05 g/l) propidium iodide (PI) staining solution was added to each sample which was then incubated for 30 min at room temperature (RT) in the dark. The cell cycle was then analyzed by a flow cytometer (FACSCanto™ II; BD Biosciences).
Dual-luciferase reporter assay
293T cells were seeded in 12-well plates (BD, USA) and cultured until the cells reached 80–90% confluence. The CDK6 3’-UTR was cloned into the psiCHECK-2 vector, which contains the RL gene, and co-transfected into cells together with miR-29c or NC mimics (100 nM) using lipofectamine, according to the manufacturer’s instructions. Thirty hours after transfection, luciferase activity was measured using the Dual-Luciferase Reporter assay kit (Promega, USA). Briefly, the cells were washed twice with PBS, lysed with passive lysis buffer and incubated at room temperature for 15 min. The supernatants were collected and 20 μl were added into 96-well plates. The FL reporter was measured immediately after adding luciferase assay reagent II (LAR II). After quantifying the firefly luminescence, 100 μl Stop & Glo® Reagent was added to each well to initiate the RL reporter and Renilla luminescence was then measured. The data were analyzed by normalizing RL with FL, and the ratio of FL/RL was calculated to indicate the activity of the reporter.
Statistical methods
Experimental data was showed as mean ± SD. Two groups were compared using t-test comparison and multi-groups were compared using variance analysis by SPSS17.0 statistical software. P<0.05 indicates significant difference.
Results
Lower expression of miR-29c in bladder cancer specimens
Firstly, to examine whether the miR-29c is differentially expressed in cancers, we analyzed levels of miR-29c in 40 paired invasive bladder cancer pecimens and pair-matched adjacent noncancerous bladder tissues by qRT-PCR. Our results demonstrated that the expression of miR-29c was lower in bladder cancer specimens (1.21 ± 0.035) than adjacent noncancerous bladder tissues (10.58 ± 0.63) (P<0.01)(Figure 1).
Figure 1.
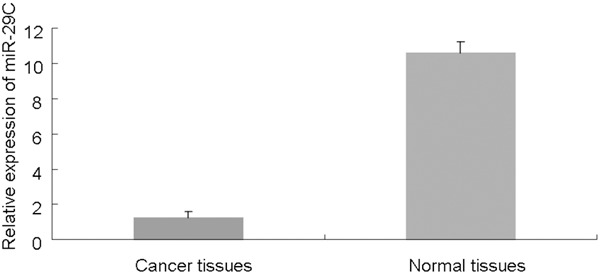
miR-29c levels are significantly decreased in bladder cancer specimens. miRNA was extracted from bladder cancer and adjacent normal tissues. The expression of miR-29c was analyzed by qRT-PCR. The graph represents the 2-ΔΔCt values ± SEM; *P<0.01. SEM, standard error of the mean.
Suppression of bladder cancer proliferation by miR-29c
To explore the effect of miR-29c on bladder cancer cell proliferation, miR-29c mimics were transfected into the human bladder cancer cell line, T24 and proliferation was assessed by MTT assay. As shown in Figure 2, Compared with the mock and the NC group, miR-29c significantly repressed the growth of bladder cancer cells cellular proliferation gradually declined following transfection with miR-29c, in a time- and dose-dependent manner. Thus, 100 nM was used in the following experiments. As shown in Figure 3, Proliferation was also assessed by colony formation assay. The 100 nM miR-29c group exhibited fewer colonies than the mock and NC groups as determined by the colony formation assay. Thus, these data suggest that miR-29c significantly suppresses the proliferation of T24 breast cancer cells.
Figure 2.
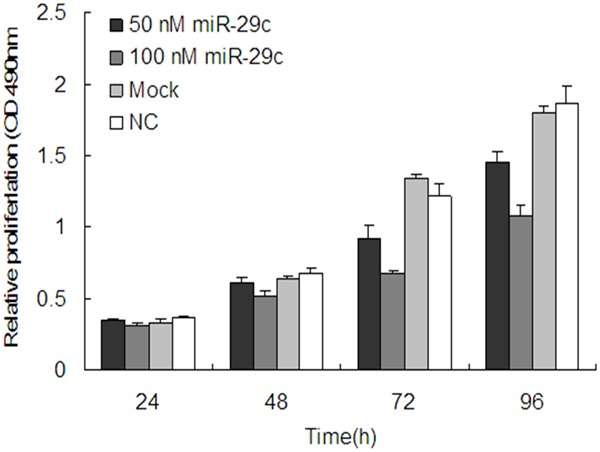
miR-29c inhibits cell proliferation. The proliferation of T24 cells transfected with miR-29c (50 and 100 nM) was measured at the indicated time-points at 490 nm with a microplate spectrophotometer and compared with that of the mock and NC groups. The graph represents OD 490 ± SEM nm; *P<0.05. NC, negative control; SEM, standard error of the mean.
Figure 3.

Colony formation assay. (A-C) Representative images of crystal violet stained colonies in T24 cells. (A) The miR-29c group exhibited fewer colonies than the (B) mock or (C) NC group (D) Quantification of the clone numbers, *p<0.05. NC, negative control.
MiR-29c inhibits migration of T24 cells
The transwell migration assay is a useful method to investigate migratory ability Our results showed that 20 h after transfection, the number of migrating cells in the miR-29c group was significantly less than that in either the mock or NC groups (P<0.05). These data suggest that the migratory ability of T24 cells may be inhibited by miR-29c (Figure 4).
Figure 4.
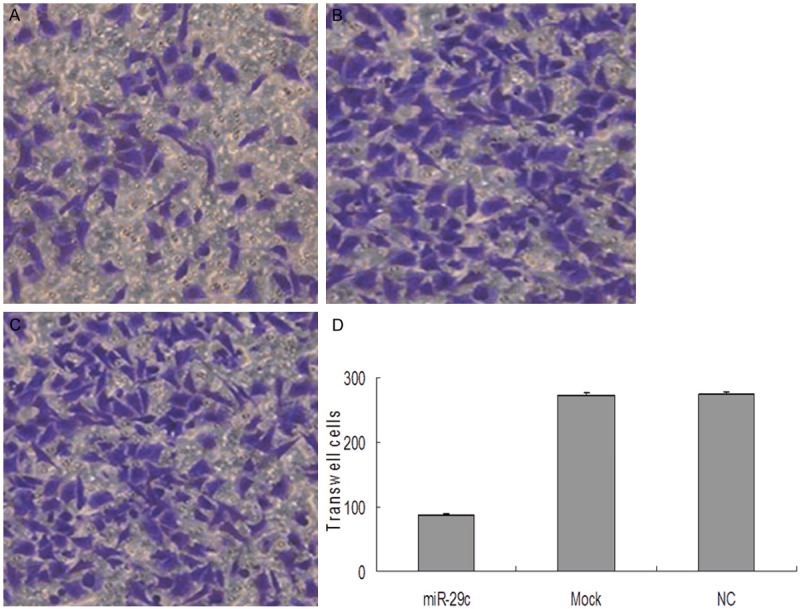
miR-29c inhibited migration of T24 bladder cancer cells. Cell migration ability was analyzed by transwell chamber assay 20 h after miR-29c, mock or NC transfection. A-C: Representative images of crystal violet stained T24 migratory cells transfected with miR-29c, mock or NC. D: Quantification of the number of crystal violet-stained cells. Data represent means ± SEM; *P<0.05. NC, negative control; SEM, standard error of the mean.
MiR-29c disrupts the cell cycle of T24 cells
The cell cycle distribution of the T24 cells with and without transfection of miR-29c mimics was analyzed by flow cytometry. As shown in Figure 5, all these findings demonstrate that miR-29c inhibits the proliferation of T24 bladder cancer cells. Flow cytometer analysis revealed that the percentage of G1 phase T24 cells (59.78 ± 1.58%) dramatically increased in the 100 nmol/L miR-29c mimics-transfected group, which was higher than those in Mock (48.45 ± 0.78%) and NC groups (48.51 ± 0.74%, P<0.05), while the proportion of G2 and S phase cells decreased in the miR-29c group compared with those of the mock and NC groups (P<0.05). These results indicate that the overexpression of miR-29c prevents cells from entering the S phase through initiation of G1 phase arrest in T24 cells.
Figure 5.
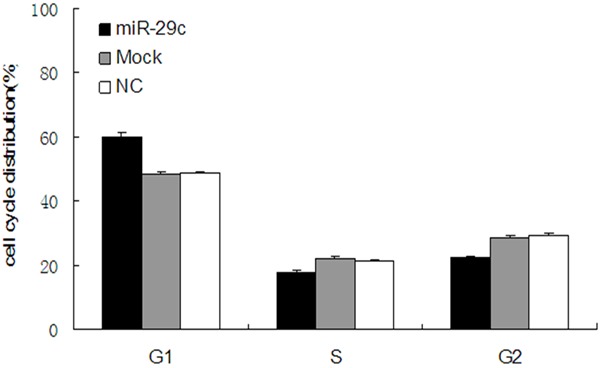
miR-29c disrupts the cell cycle of T24 cells. Cell cycle distribution was analyzed by flow cytometry 48 h after transfection of T24 bladder cancer cells with miR-29c mimics, NC or mock. The respective proportions of G1, S and G2 phase cells are shown, *P<0.05. NC, negative control.
MiR-29c targets CDK6 and regulates its expression in T24 cells
To identify possible miR-29c target genes, we performed a computational screen for genes with complementary sites of miR-29c in their 3’UTR using open-access software. The software included TargetScan, Sanger microRNA target, and Miranda. We focused our attention on CDK6, To examine the possibility that miR-29c targets CDK6, we used dual-luciferase reporter assays, in which the reporter activity is evaluated by normalizing the Renilla luciferase (RL) activity with firefly luciferase (FL) activity the FL/RL ratio indicates the relative activity levels; next we constructed a psiCHECK-2/CDK6 3’-UTR vector, which contained the Renilla luciferase (RL) gene and the 3’-UTR region of CDK6. This construct was transfected into 293T cells together with either miR-29c or NC mimics, and the luciferase activity was analyzed. The ratio of FL/RL was calculated, and showed that the miR-29c group had a ~2-fold higher activity than that of the NC group (P<0.05) (Figure 6B). These results show that miR-29c could directly interact with the CDK6 3’-UTR in the psiCHECK-2 reporter plasmid, leading to the degradation of RL mRNA. Finally, we performed qPCR and western blot analysis of CDK6 expression in T24 cells with and without transfection of miR-29c mimics, or controls. We demonstrated that overexpression of miR-29c significantly decreased CDK6 expression at both the mRNA and protein levels (Figure 7). These data further indicate that CDK6 is a target of miR-29c.
Figure 6.
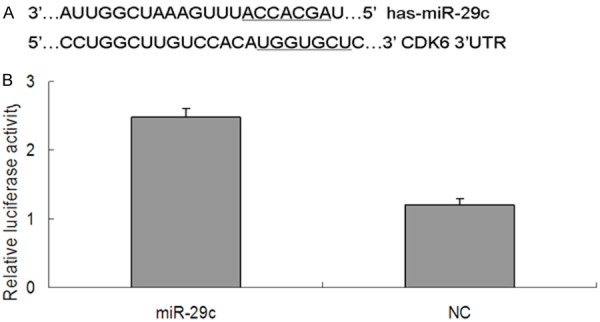
CDK6 is a direct target of miR-29c. A: The binding sites for miR-29c in the 3’-UTR of CDK6 mRNA. B: The relative luciferase activity (FL/RL) was measured in 293T cells after co-transfection of the CDK6 luciferase construct with either miR-29c or NC; *P<0.05. CDK6, Cyclin-dependent kinase 6; 3’-UTR, 3’-untranslated region; NC, negative control.
Figure 7.
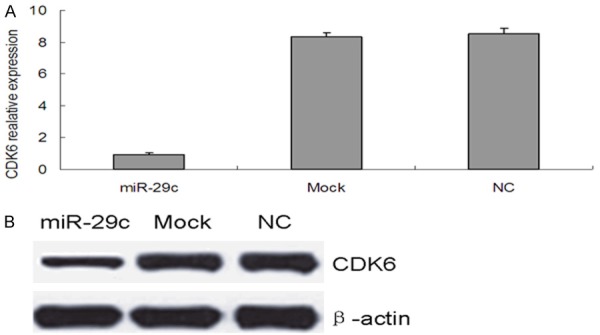
miR-29c inhibits CDK6 expression in T24 cells. (A) qRT-PCR and (B) western blot analysis of CDK6 mRNA and protein levels, respectively. in T24 cells transfected with miR-29c, mock or NC, β-actin was used as a control for loading for western blot analysis.
Discussion
The study on the roles of miRNA in the development of tumors has become a subject of intense investigation. It is estimated that approximately 30% of human genes are miRNA targets [18]. Furthermore, an increasing number of evidences indicate that miRNAs are differentially expressed between normal and malignant tumor tissues, miRNA deregulation is a critical cause of cancer formation. In this study, we examined the expression levels of miR-29c in bladder cancer specimens and adjacent normal tissues. Compared to the normal bladder tissues, miR-29c expression was significantly down-regulated in the bladder cancer specimens. Members of this family have been shown to be silenced or down-regulated in many different types of cancer, such as acute myeloid leukemia [19], chronic lymphocytic leukemia [20], and some solid tumors [21,22], Thus, we hypothesized that miR-29c may function as a tumor suppressor.
MiR-29c has been reported as a critical regulator in carcinogenesis and tumor progression by acting as tumor suppressor gene in various cancers [23,24]. in our study, we transfected miR-29c mimics into T24 cells to induce it overexpression. Exogenous overexpression of miR-29c significantly inhibited the cell growth and colony formation ability of T24 cells as indicated by MTT and colony formation assays. Using the transwell migration assay, we found that the overexpression of miR-29c in bladder cancer cells could suppress their migratory ability. Furthermore, by flow cytometry we found that miR-29c distinctly arrests cancer cells at the G1 phase when compared with the cell cycle of Mock groups and NC groups.
To understand the mechanisms by which miR-29c suppresses tumor cell proliferation in bladder cancer, we searched for potential targets of miR-29c using several online databases, including targetscan, miRanda, and miRGen, and all three databases indicated that the CDK6 mRNA contained miR-29c binding sites. The interaction between miR-29c and CDK6 mRNA has not been previously reported. To confirm whether CDK6 was a real target of miR-29c, we integrated a fragment of the CDK6 3’-UTR containing the target sequence into a luciferase reporter vector. Through luciferase assays, we confirmed that CDK6 was a direct target of miR-29c, Additionally, we found that the mRNA and protein levels of CDK6 were significantly reduced in miR-29c overexpressing cells when compared with those transfected with either mock or NC, thus further indicating that CDK6 is a direct target of miR-29c.
Conclusion
In summary, our study demonstrated that miR-29c is downregulated in bladder cancer specimens compared with normal tissue Overexpression of miR-29c inhibited the proliferation and colony formation ability, suppressed migration and caused G1 phase arrest by targeting CDK6 in T24 bladder cancer cells. These data indicate that miR-29c may serve as a tumor suppressor gene involved in bladder cancer pathogenesis. Therefore, miR-29c may be a potential diagnostic and therapeutic target for bladder cancer.
Acknowledgements
This work is supported by grant 81172426 of the National Natural Science Foundation of China and the Shanghai Education Commission Research and Innovation projects (No. 12ZZ034).
Disclosure of conflict of interest
None.
References
- 1.Parkin DM. The global burden of urinary bladder cancer. Scand J Urol Nephrol Suppl. 2008;218:12–20. doi: 10.1080/03008880802285032. [DOI] [PubMed] [Google Scholar]
- 2.Ploeg M, Aben KK, Kiemeney LA. The present and future burden of urinary bladder cancer in the world. World J Urol. 2009;27:289–293. doi: 10.1007/s00345-009-0383-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crawford JM. The origins of bladder cancer. Lab Invest. 2008;88:686–693. doi: 10.1038/labinvest.2008.48. [DOI] [PubMed] [Google Scholar]
- 4.Micheli A, Francisci S, Krogh V, Rossi AG, Crosignani P. Cancer prevalence in Italian cancer registry areas: the ITAPREVAL study. ITAPREVAL working group. Tumori. 1999;85:309–369. doi: 10.1177/030089169908500502. [DOI] [PubMed] [Google Scholar]
- 5.Voutsinas GE, Stravopodis DJ. Molecular targeting and gene delivery in bladder cancer therapy. J Buon. 2009;14:S69–78. [PubMed] [Google Scholar]
- 6.Malmström PU, Loskog AS, Lindqvist CA, Mangsbo SM, Fransson M, Wanders A, Gårdmark T, Tötterman TH. AdCD40L immunogene therapy for bladder carcinoma the first phase I/IIa trial. Clin Cancer Res. 2010;16:3279–3287. doi: 10.1158/1078-0432.CCR-10-0385. [DOI] [PubMed] [Google Scholar]
- 7.Harris L, Fritsche H, Mennel R, Norton L, Ravdin P, Taube S, Somerfield MR, Hayes DF, Bast RC Jr. Clinical Oncology 2007 update of recommendations for the use of tumor markers in breast cancer. J. Clin. Oncol. 2007;25:5287–5312. doi: 10.1200/JCO.2007.14.2364. [DOI] [PubMed] [Google Scholar]
- 8.Bushati N, Cohen SM. microRNA functions. Annu Rev Cell Dev Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- 9.Ryan BM, Robles AI, Harris CC. Genetic variation in microRNA networks: the implications for cancer research. Nat Rev Cancer. 2010;10:389–402. doi: 10.1038/nrc2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiang Y, Zhu ZQ, Han G, Ye X, Xu B, Peng Z, Ma Y, Yu Y, Lin H, Chen AP, Chen CD. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. PNAS. 2007;104:19226–19231. doi: 10.1073/pnas.0700735104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mo W, Zhang J, Li X, Meng D, Gao Y, Yang S, Wan X, Zhou C, Guo F, Huang Y, Amente S, Avvedimento EV, Xie Y, Li Y. Identification of novel AR-targeted microRNAs mediating androgen signaling through critical pathways to regulate cell viability in prostate cancer. PLoS One. 2013;8:e56592. doi: 10.1371/journal.pone.0056592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malumbres M, Barbacid M. Mammalian cyclindependent kinases. Trends Biochem Sci. 2005;30:630–641. doi: 10.1016/j.tibs.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 13.Lee KH, Lotterman C, Karikari C, Omura N, Feldmann G, Habbe N, Goggins MG, Mendell JT, Maitra A. Epigenetic silencing of MicroRNA miR-107 regulates cyclin-dependent kinase 6 expression in pancreatic cancer. Pancreatology. 2009;9:293–301. doi: 10.1159/000186051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chilosi M, Doglioni C, Yan Z, Lestani M, Menestrina F, Sorio C, Benedetti A, Vinante F, Pizzolo G, Inghirami G. Differential expression of cyclin-dependent kinase 6 in cortical thymocytes and T-cell lymphoblastic lymphoma/leukemia. Am J Pathol. 1998;152:209–217. [PMC free article] [PubMed] [Google Scholar]
- 15.Costello JF, Plass C, Arap W, Chapman VM, Held WA, Berger MS, Su Huang HJ, Cavenee WK. Cyclin-dependent kinase 6 (CDK6) amplification in human gliomas identified using two-dimensional separation of genomic DNA. Cancer Res. 1997;57:1250–1254. [PubMed] [Google Scholar]
- 16.Mendrzyk F, Radlwimmer B, Joos S, Kokocinski F, Benner A, Stange DE, Neben K, Fiegler H, Carter NP, Reifenberger G, Korshunov A, Lichter P. Genomic and protein expression profiling identifies CDK6 as novel independent prognostic marker in medulloblastoma. J. Clin. Oncol. 2005;23:8853–8862. doi: 10.1200/JCO.2005.02.8589. [DOI] [PubMed] [Google Scholar]
- 17.Wang G, Zheng L, Yu Z, Liao G, Lu L, Xu R, Zhao Z, Chen G. Increased cyclin-dependent kinase 6 expression in bladder cancer. Oncol Lett. 2012;4:43–46. doi: 10.3892/ol.2012.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 19.Garzon R, Liu S, Fabbri M, Liu Z, Heaphy CE, Callegari E, Schwind S, Pang J, Yu J, Muthusamy N, Havelange V, Volinia S, Blum W, Rush LJ, Perrotti D, Andreeff M, Bloomfield CD, Byrd JC, Chan K, Wu LC, Croce CM, Marcucci G. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloidleukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113:6411–6418. doi: 10.1182/blood-2008-07-170589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santanam U, Zanesi N, Efanov A, Costinean S, Palamarchuk A, Hagan JP, Volinia S, Alder H, Rassenti L, Kipps T, Croce CM, Pekarsky Y. Chronic lymphocytic leukemia modeled in mouse by targeted miR-29 expression. Proc Natl Acad Sci U S A. 2010;107:12210–12215. doi: 10.1073/pnas.1007186107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sengupta S, den Boon JA, Chen IH, Newton MA, Stanhope SA, Cheng YJ, Chen CJ, Hildesheim A, Sugden B, Ahlquist P. MicroRNA 29c is down-regulated in nasopharyngeal carcinomas, up-regulating mRNAs encoding extracellular matrix proteins. Proc Natl Acad Sci U S A. 2008;105:5874–5878. doi: 10.1073/pnas.0801130105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Ménard S, Palazzo JP, Rosenberg A, Musiani P, Volinia S, Nenci I, Calin GA, Querzoli P, Negrini M, Croce CM. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 23.Wang H, Zhu Y, Zhao M, Wu C, Zhang P, Tang L, Zhang H, Chen X, Yang Y, Liu G. miRNA-29c suppresses lung cancer cell adhesion to extracellular matrix and metastasis by targeting integrin β1 and matrix metalloproteinase2 (MMP2) PLoS One. 2013;8:e70192. doi: 10.1371/journal.pone.0070192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang IP, Tsai HL, Huang CW, Huang MY, Hou MF, Juo SH, Wang JY. The functional significance of microRNA-29c in patients with colorectal cancer: a potential circulating biomarker for predicting early relapse. PLoS One. 2013;8:e66842. doi: 10.1371/journal.pone.0066842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saito Y, Suzuki H, Imaeda H, Matsuzaki J, Hirata K, Tsugawa H, Hibino S, Kanai Y, Saito H, Hibi T. The tumor suppressor microRNA-29c is downregulated and restored by celecoxib in human gastric cancer cells. Int J Cancer. 2013;132:1751–1760. doi: 10.1002/ijc.27862. [DOI] [PubMed] [Google Scholar]
- 26.Bae HJ, Noh JH, Kim JK, Eun JW, Jung KH, Kim MG, Chang YG, Shen Q, Kim SJ, Park WS, Lee JY, Nam SW. MicroRNA-29c functions as a tumor suppressor by direct targeting oncogenic SIRT1 in hepatocellular carcinoma. Oncogene. 2014;33:2557–2567. doi: 10.1038/onc.2013.216. [DOI] [PubMed] [Google Scholar]