

Abstract
The accurate transition from G1 phase of the cell cycle to S phase is crucial for the control of eukaryotic cell proliferation, and its misregulation promotes oncogenesis. During G1 phase, growth-dependent cyclin-dependent kinase (CDK) activity promotes DNA replication and initiates G1-to-S phase transition. CDK activation initiates a positive feedback loop that further increases CDK activity, and this commits the cell to division by inducing genome-wide transcriptional changes. G1–S transcripts encode proteins that regulate downstream cell cycle events. Recent work is beginning to reveal the complex molecular mechanisms that control the temporal order of transcriptional activation and inactivation, determine distinct functional subgroups of genes and link cell cycle-dependent transcription to DNA replication stress in yeast and mammals.
The eukaryotic cell cycle is controlled by a regulatory network, the general features of which are conserved from yeast to humans1. It proceeds through tightly regulated transitions to ensure that specific events take place in an orderly manner. The discovery of cyclins and cyclin-dependent kinases (CDKs), the elucidation of the mechanisms underlying transcriptional control and checkpoint signalling and the characterization of ubiquitin ligase regulatory pathways have revealed that general cell cycle regulatory principles are shared across eukaryotes.
Two crucial aspects of cell cycle regulation are the existence of DNA structure checkpoints, which arrest the cell cycle in response to DNA damage or incomplete replication, and the existence of a ‘commitment point’. This point is known as the ‘restriction point’ in animal cells and ‘start’ in yeast and is defined as the point after which a cell becomes committed to enter the cell cycle and progress through it independently of signals from the environment. The importance of DNA checkpoints and commitment point control for proper cell division is illustrated by the high frequency of mutations found in their constituent regulatory proteins during oncogenesis2. One notable regulatory protein that is often mutated in cancer is the tumour suppressor protein RB3. RB is a potent inhibitor of G1–S transcription (that is, a transcriptional wave that initiates during G1 and is subsequently inactivated during S phase), and its discovery over 20 years ago first suggested the dependency of cell cycle commitment on transcriptional regulation in G1 (REFS 4–6). Subsequent studies showed that the broad mechanisms of eukaryotic G1 cell cycle control are highly conserved7–9,10. Intriguingly, recent work demonstrated that DNA checkpoint control depends on the same transcription factors responsible for commitment point regulation11.
The dynamic changes in gene expression as a function of cell cycle progression are regulated by specific CDK activities. These variations in gene expression levels control the accumulation of several cyclins and thereby regulate CDK activity, thus driving cell cycle progression. Genes regulated during the cell cycle encode several proteins that function in the subsequent phase of the cell cycle. In most eukaryotes, cell cycle-regulated transcription can be grouped into three main waves12. These waves of transcription coincide with the different transition points during the cell cycle, namely G1-to-S, G2-to-M and M-to-G1. Although all three cell cycle transcript waves are well-characterized in yeast, transcription that occurs during the M-to-G1 phase transition in human cells is less well-defined13. Largely on the basis of work carried out in the budding yeast Saccharomyces cerevisiae, it is thought that the subsequent waves of transcription form a continuous regulatory network in which each wave is activated by the previous one and contains activators of the following wave14. Of the cell cycle transcriptional waves, G1–S transcription has been the most studied because of its important role in the tight regulation of G1-to-S phase transition. Derepression of G1–S transcription allows cells to progress into S phase in an unrestrained fashion, a hallmark of cancer. Along with the recently established link of the cell cycle checkpoint response to replication stress, this unrestrained growth illustrates the importance of cell cycle-regulated transcription, which is both driven by and a driving force for cell cycle progression.
Here, we review recent progress in determining the simple but elegant mechanisms by which cells regulate their G1–S phase transcriptional network to control the commitment to cell division and the DNA replication checkpoint response. Although most work has focused on the role of transcriptional activation during cell cycle progression from G1 to S and the genome-wide changes in the transcriptional programme14–16, recent work has uncovered many new insights into the regulation of commitment to cell division, the temporal confinement of G1–S transcription and the response to DNA replication stress (BOX 1). These systems-level properties are conserved across eukaryotes despite frequent lack of protein sequence homology of transcriptional regulators (BOX 1; TABLE 1). We briefly discuss G1–S transcriptional regulation in the context of other cell cycle pathways, such as cyclins and CDKs, checkpoint signalling and the ubiquitin ligase regulatory pathways, but we also refer readers to more comprehensive reviews on these specific topics17–22. Finally, we share our views on how current understanding of the regulation of G1-to-S phase transition may provide a blueprint for future research into the fundamental regulatory mechanisms controlling cellular decision-making processes, dynamic changes in gene expression and checkpoint-dependent rewiring of transcriptional networks.
Box 1. Cell cycle-regulated transcription during the G1 and S phases.
The G1–S transcriptional network is involved in two crucial aspects of cell cycle regulation: cell division cycle control and maintenance of genome stability. Phosphorylation of transcriptional inhibitors by cyclin-dependent kinase (CDK) releases them from transcription factors to activate G1–S genes, including G1 cyclins (see the figure, part a). This reinforces a positive feedback loop, further committing the cell to a new division cycle and activating G1–S transcription. Negative feedback loops subsequently inactivate transcription, which terminates a wave of gene expression (indicated by the red curve) that peaks at the transition from G1 to S phase. A recently identified negative autoregulatory feedback loop involves transcriptional repressors that are G1–S targets themselves. These repressors accumulate and bind to G1–S gene promoters to turn off transcription when cells progress to S phase. In addition, these transcriptional repressors are directly targeted by the DNA replication checkpoint protein kinases to maintain G1–S transcription during a checkpoint arrest. The fundamental regulatory pathways that drive changes in cell cycle-regulated gene expression during the G1 and S phases of the cell cycle are conserved from yeast to humans. Transcriptional regulators involved in this regulation in various eukaryotic systems are listed in TABLE 1. Conserved systems level properties are involved in G1–S transcriptional regulation across eukaryotes (see the figure, part b). Linking a positive feedback mechanism to a negative feedback loop ensures that a switch-like commitment to activation results in timely inactivation via an oscillator. The particular network wiring required for G1-to-S phase transition involves a transcriptional inhibitor and cell cycle-regulated transcription of G1 cyclins and transcriptional repressors (see the figure, part c).

Table 1.
Conservation of cell cycle regulatory proteins*
| Regulator type | Saccharomyces cerevisiae | Schizosaccharomyces pombe | Drosophila melanogaster | Homo sapiens | |
|---|---|---|---|---|---|
| G1–S transcriptional regulators | |||||
| Activators | SBF (Swi6–Swi4) | MBF (Cdc10–Res1–Res2) | E2f1 | E2F1, E2F2, E2F3 | |
| Repressors | MBF (Swi6–Mbp1) | E2f2 | E2F4, E2F5, E2F6, E2F7, E2F8 |
||
| Inhibitors | Whi5 | Possibly Whi5 | Rbf1 | RB | |
| Co-repressors | Nrm1 | Nrm1, Yox1 | Rbf2 | p107, p130 | |
| Cyclin–CDK | |||||
| G1 phase regulator | Cdc28–Cln3 | Cdc2–Puc1 | Cdk4–cyclin D | CDK4–cyclin D, CDK6–cyclin D |
|
| G1–S phase regulator | Cdc28–Cln1, Cdc28–Cln2 | Cdc2–Puc1 Cdc2–Cig1 |
Cdk2–cyclin E | CDK2––cyclin E | |
| S phase regulator | Cdc28– Clb5, Cdc28–Clb6 | Cdc2– Cig1, Cdc2– Cig2 |
Cdk2–cyclin E, Cdk1–cyclin A, Cdk2–cyclin A |
CDK2––cyclin E, CDK1–cyclin A, CDK2–cyclin A |
|
| M phase regulator | Cdc28– Clb1, Cdc28–Clb2, Cdc28–Clb3, Cdc28–Clb4 |
Cdc2–Cdc13 | Cdk1–cyclin B | CDK1–cyclin B | |
| Checkpoint protein kinases | |||||
| Sensor and/or transducer |
Mec1 | Rad3 | ATR | ATR | |
| Tel1 | Tel1 | ATM | ATM | ||
| Effector | Chk1 | Cds1 | Chk1 | CHK1 | |
| Rad53 | Chk1 | Chk2 | CHK2 | ||
ATM, ataxia-telangiectasia mutated; ATR, ataxia-telangiectasia and Rad3-related protein; CDK, cyclin-dependent kinase; CHK, checkpoint kinase; MBF, MCB-binding factor; Mec1, mitosis entry checkpoint 1; Rbf, retinoblastoma family; SBF, SCB-binding factor; Tel1, telomere length regulation 1.
Listed are the functional orthologues between yeast, flies and humans of G1–S phase transcriptional regulators, cyclin-CDKs and checkpoint protein kinases.
Although the functional orthologues of cyclins, CDKs and the checkpoint protein kinases share significant sequence homology, there is a total lack of sequence homology between yeast and the higher eukaryotic G1–S transcriptional regulators.
Activation of G1–S transcription
Cells commit to enter a new cell cycle during G1 by activating cyclin-CDK-dependent transcription (FIG. 1). G1–S transcriptional activation during late G1 promotes entry into S phase after which expression is turned off. This creates a wave of transcription, which peaks at the G1-to-S transition (BOX 1). The mechanism of G1–S transcriptional activation has been well-established and is conserved from yeast to humans.
Figure 1. G1–S transcriptional activation.

a | Schematic showing how the G1–S transcriptional programme, once initiated, is reinforced by a positive feedback loop. b | In mammalian cells, the transcriptional repressors RB, p107 and p130 (collectively known as pocket proteins) are bound to E2F transcription factors to repress expression during early G1. Pocket proteins either prevent activator E2F proteins (such as E2F1, E2F2 and E2F3) to activate transcription or function as co-repressors for repressor E2F proteins (such as E2F4). Phosphorylation of pocket proteins by cyclin D– cyclin-dependent kinase 4 (CDK4) and cyclin D–CDK6 probably releases them from the E2F transcription factors. This induces the transcription of G1–S target gene, including the gene encoding cyclin E. Cyclin E–CDK2 phosphorylates pocket proteins, thereby providing a positive feedback loop. c | Model depicting G1–S transcriptional activation in budding yeast. In early G1, transcription is inhibited by Whi5 binding to the SBF (SCB-binding factor) complex at target promoters. Cln3–Cdk relieves transcriptional inhibition by phosporylating Whi5, which induces its nuclear export and thereby G1–S transcription. Activation of transcription results in the accumulation of Cln1 and Cln2, which in complex with Cdk, further inactivate Whi5 through phosphorylation. This provides positive feedback that results in cell cycle commitment.
E2F family and pocket proteins
In human cells, G1–S transcription depends on the E2F family of transcription factors and their dimerization partner proteins. Misregulation of E2F function is frequently found in cancer, which further supports the role of G1–S transcription in oncogenesis23–28. E2F family members are generally associated with either transcriptional activation (E2F1, E2F2 and E2F3A) or repression (E2F3B, E2F4, E2F5, E2F6, E2F7 and E2F8). However, recent findings revealed a more complex scenario in which activator E2F proteins can act as repressors and repressor E2F proteins can activate transcription29–31. In addition to E2F proteins, confining transcription to the late G1 and S phases of the cell cycle requires regulation by pocket proteins, including RB, p107 and p130, which bind and inhibit the expression of E2F-regulated genes6,32–36.
G1–S transcriptional activation in mammalian cells
E2F family members, their DNA binding partners (dimerization partner proteins) and pocket proteins bind cell cycle gene promoters at different stages of the mitotic cell cycle to ensure the proper temporal expression of target genes4,37 (BOX 2). The association of dimerization partner proteins enhances the DNA-binding affinity of E2F family members so that they can function as transcriptional regulators38. During early G1, activator E2F proteins are bound and inhibited by RB6, whereas E2F4 (and presumably E2F5) bind p130 and p107 at promoters to repress transcription39–42 (FIG. 1b). E2F4 and E2F5 depend on pocket protein binding for nuclear localization. When pocket proteins are phosphorylated by G1 cyclin–CDKs during G1-to-S phase transition, E2F4 and E2F5 are released, shuttled into the cytoplasm4,37,43 and replaced at promoters by activator E2F family members (E2F1, E2F2 and E2F3A) that initiate transcription40,41. This classic paradigm of the role of activator E2F proteins in inducing G1–S gene expression and driving cell cycle entry derives mainly from studies using cultured cells and flies. More recent evidence from in vivo studies in knockout mice has revealed a more complicated picture29,44. The ablation of all activator E2F proteins, E2F1, E2F2 and E2F3, does not prevent normal proliferation of embryonic stem (ES) cells and intestinal and retinal progenitor cells, suggesting that these proteins are dispensable for proliferation in this context. However, an increase in DNA damage and apoptosis is observed in these triple-knockout cells, which suggests a role for transcriptional control by the activator E2F proteins.
Box 2. Mammalian cell cycle transcriptional regulation is dependent on E2F and pocket proteins.
The E2F family of transcription factors and their dimerization partner proteins act as transcriptional regulators of G1– S transcription.
E2F1, E2F2 and E2F3
These proteins are found in complex with RB during G1121,122. They can be detected at E2F target gene promoters by chromatin immunoprecipitation (ChIP) predominantly during G1-to-S transition, which corresponds with transcriptional induction of G1–S cell cycle genes40,41. As they are E2F targets, E2F1, E2F2 and E2F3 accumulate outside of G1 but are detected, to a significantly lesser extent, in G0 and G1 (see the figure).
E2F4 and E2F5
They are found in complex with p130 in G0 and p107 and p130 in G1 (REFS 40–42,123,124). E2F4 can be detected at E2F target promoters by ChIP predominantly during G0, which corresponds with transcriptional repression, but also during G1 (REFS 40,41). E2F4 is shuttled into the cytoplasm during G1-to-S phase transition when pocket proteins disassociate in response to CDK-dependent phosphorylation43,125. Upon return to interphase, dephosphorylated p107 and p130 associate with E2F4, promoting its transport into the nucleus and binding to target promoters41. Like E2F4, E2F5 is found in complex with p130 (REF 126) and p107 (REF 127). The timing at which it binds to target promoters has not been well-established, but it is assumed that binding mirrors that of E2F4 (REF 39).
E2F6, E2F7 and E2F8
This subset of E2F proteins do not require binding to pocket proteins for its repressor function92,96. E2F6 and E2F7 accumulate during the G1-to-S phase transition and bind to target promoters, which coincides with transcriptional inactivation during S phase (REFS. 89, 98, 99, and C.B. and R.A.M. d-B., unpublished data). The timing of target-promoter binding by E2F8 has not been established, but it is assumed that binding is similar to that of E2F7. E2F8 and E2F7 form homo- and heterodimers to repress transcription88,91,128,129.
RB
This protein is found in complex with E2F1, E2F2 and E2F3 in asynchronous cycling cells46. It can be detected by ChIP at target promoters in asynchronous cell cultures and, to a lesser extent, in quiescent cells46. RB accumulates outside of G1.
p107 and p130
These two pocket proteins are found in complex with E2F4 and E2F5 (REFS 35,41,127). p107 accumulates during the G1-to-S phase transition and is detected at target promoters by ChIP during G1 (REF 41). p130 is predominately detected at target promoters by ChIP in G0 and to a lesser extent in G1 (REFS 40,41). High levels of p130 are detected in G0 and low levels throughout the cell cycle130.
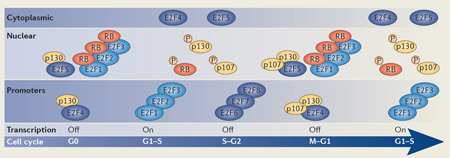
Dimerization partner proteins are omitted for simplicity. Hyperphosphorylation is indicated by ‘P’. Promoter binding for individual E2F family members is only indicated when binding has been established during the cell cycle.
A large number of studies used cultured cells reentering the cell cycle after serum removal to analyse transcriptional activation in G1. After prolonged serum withdrawal, cells enter a condition known as quiescence, which is different from the G1 phase of cycling cells and has been defined as the G0 phase of the cell cycle135,136. Both during G0 and early G1, binding of pocket proteins to E2F proteins prevents transcription. The interaction is disrupted by CDK-dependent phosphorylation in mid-G1, resulting in transcriptional activation. In G0, most E2F-responsive promoters are bound and repressed by p130 together with E2F4 (REFS 40,45), whereas in early G1 of cycling cells, p107 also interacts with DNA-bound E2F4 to repress transcription in a similar manner41. The relative importance of pocket proteins for transcriptional repression during different phases of the cell cycle largely correlates with their protein levels, as pocket proteins function in similar ways to repress transcription4. Although p130 seems to be the most abundant pocket protein during quiescence, its levels are greatly reduced during proliferation4. By contrast, RB and p107 are barely detectable in quiescent cells but are found at higher levels in cycling cells; this is likely to be due to E2F-dependent transcription. Recent chromatin immunoprecipitation (ChIP) data suggests that RB is present at promoters during quiescence and in cycling cells, as was commonly assumed46, as well as in senescent cells, in which it represses G1–S genes. Intriguingly, the permanent exit from the cell cycle in differentiating cells requires activator E2F proteins in complex with RB to repress cell cycle genes29. It is possible that this mechanism of active repression promotes cell cycle exit and is then followed by a more stable repression mediated by E2F4 and E2F5 together with p130 (REF. 29).
Conserved mechanisms govern transcriptional activation
Although there is no conservation at the protein level, a regulatory mechanism for G1–S transcriptional activation similar to that established for mammalian cells was recently found in yeast10,47,48 (FIG. 1c).
In S. cerevisiae many of the genes involved in G1-to-S phase transition are regulated by one of two transcription factor complexes, SBF (SCB-binding factor) or MBF (MCB-binding factor). SBF comprises of a Swi4 DNA-binding component and Swi6 and is required to activate G1–S transcripts during G1. MBF is composed of an Mbp1 DNA-binding component and Swi6 and is required to repress G1–S transcripts outside of G1. Swi4 and Mbp1 bind to specific G1–S target promoters through the SCB (Swi4 cell cycle box) and MCB (MluI cell cycle box) recognition sequences, respectively. In mammalian cells, G1–S genes are regulated by several E2F transcription factor complexes at different stages of the cell cycle. In yeast, G1–S transcripts can be roughly divided into two groups: SBF- or MBF-dependent genes. However, it has been shown that in the absence of the either factor, SBF and MBF may be found at each other’s consensus DNA-binding motif49,50. Although the temporal gene expression pattern induced by either SBF or MBF is similar, the regulatory mechanism is distinct. SBF-regulated genes remain switched off in cells that lack the DNA-binding component Swi4, suggesting that SBF is required for transcriptional induction during G1. Conversely, the repressor complex MBF is required to repress transcription outside of G1, as MBF-regulated genes remain activated in cells lacking DNA-binding component Mbp1. Thus, although SBF acts more like activator E2F proteins, MBF proteins resemble repressor E2F proteins (FIGS 1,2); however, they share no sequence homology with E2F proteins49,51,52. In addition, the S. cerevisiae transcriptional inhibitor Whi5 is functionally orthologous to pocket proteins despite a complete lack of sequence homology10. During G1, binding of Whi5 to SBF inhibits the activity of SBF, much like pocket proteins inhibit E2F activity (FIG. 1). Whi5 is phosphorylated by G1 cyclin–Cdk, leading to its release from SBF at promoters, export from the nucleus53 and inactivation. This ensures SBF-dependent transcriptional activation47,48.
Figure 2. G1–S transcriptional repression.

a | Inactivation of E2F-dependent cell cycle transcription involves multiple negative feedback mechanisms. b | In mammalian cells, G1 cyclin–cyclin-dependent kinase (CDK) (cyclin E–CDK2) together with S phase cyclin–CDK (cyclin A–CDK2) targets the S phase cyclin-specific inhibitor p27 for degradation. The subsequent increase in CDK2 activity results in phosphorylation and release of the activator E2F1, E2F2 and E2F3 transcription factors from gene promoters, thus inactivating transcription. In addition, the E2F targets E2F6, E2F7 and E2F8 accumulate when cells progress to S phase, and they repress transcription when bound to target promoters. The negative feedback mechanism involving the E2F target S phase kinase-associated protein 2 (SKP2), which has a role in targeting E2F1 for degradation via the SCF (SKP2–cullin 1–F-box protein) ubiquitin ligase pathway, has been omitted for simplicity. c | Activation of G1–S transcription in budding yeast results in the accumulation of ~300 gene products, including Nrm1, Cln1, Cln2, Clb5 and Clb6. Some of these proteins are directly or indirectly involved in turning off transcription, thereby forming a negative feedback loop. Cln1–Cdk and Cln2–Cdk prime the Clb–Cdk-specific inhibitor Sic1 for Clb-Cdk phosphorylation, which targets it for degradation (not shown). Clb–Cdk-dependent phosphorylation of SBF (SCB-binding factor) components releases SBF from promoters, and this leads to the inactivation of transcription. MBF (MCB-binding factor)-dependent transcription is inactivated through binding of the MBF-associated co-repressor Nrm1.
A positive feedback switch ensures commitment
Activation of G1–S transcription by a positive feedback loop creates an ‘all-or-none switch’ that results in the commitment of cells to enter the cell cycle54,55 (FIG. 1a). The point at which cells commit to enter a new cell cycle, after which it will progress independently of signals from the environment, is known as the restriction point in mammals and start in yeast. Increased cyclin–CDK activity and the corresponding phosphorylation and inactivation of pocket proteins in mammals, or export and inactivation of Whi5 in S. cerevisiae, allows an initial activation of G1–S transcription factors. The genes encoding the G1 cyclins CLN1 and CLN2 in yeast and cyclin E in mammals are some of the first G1–S genes to be trans-cribed56.Through positive feedback, G1 cyclins increase their own transcription to produce a rapid increase in cyclin–CDK activity that irreversibly leads to cell cycle commitment. In addition to defining the point at which cells commit, the rapid increase in CDK activity driven by this positive feedback results in the timely and coherent activation of the entire G1–S regulon54 (BOX 1; FIG. 1).
Positive feedback loops other than the G1 cyclins loop have also been implicated in mammalian cells, including, but not limited to, the accumulation of activator E2F proteins57–59. It is worth noting that although the molecular link between the commitment point and positive feedback activation has been firmly established in yeast55, it is more contentious in mammalian tissue culture cells, in which the application of temporal high-resolution imaging approaches, to temporally link transcriptional regulation to cell cycle commitment in single cells, has been more limited60. In fact, some data obtained from single-cell measurements suggests a model that places the restriction point well before RB phosphorylation and E2F-dependent transcriptional activation occur61,62.
DNA replication switch and ordering of cell cycle events
The Cdk inhibitor Sic1 causes a delay between the activation of the transcriptional positive feedback loop and the initiation of DNA replication63,64. In a two-step process, G1 cyclin–Cdk activity is required for Sic1 phosphorylation, which primes Sic1 for subsequent Clb-dependent phosphorylation leading to its degradation65. The mutual inhibition of the S phase cyclins and Sic1 form the basis of an ultrasensitive DNA replication switch that depends on the strong binding affinity of Sic1 for the Clb–Cdk complex65. Once activated, the S phase cyclins Clb5 and Clb6 in complex with Cdk phosphorylate Sld2 and Sld3 to initiate the formation of the Sld2–Sld3–Dpb11 complex. This complex mediates the activation of DNA replication66–68.
Cyclin specificity has an important role in ordering cell cycle events18. In particular, the hydrophobic patch on Clb5 binds to RXL motifs, which is important for its specific function in initiating DNA replication69. Similarly, the G1 cyclin Cln2 docks a distinct LP motif, which makes Cln2 highly specific for the transcriptional inhibitor Whi5, and Cln3 binds to another, currently unknown motif70,137. The later B-type cyclin Clb2, although able to initiate replication, lacks the specific docking ability of earlier cyclins. Rather, Clb2 is characterized by a higher intrinsic kinase activity71. Thus, earlier cyclins have weaker intrinsic activity but this is compensated by specific binding motifs, whereas the later mitotic cyclin Clb2 are characterized by a higher intrinsic activity. This yields an updated quantitative model of Cdk activity that progressively targets an increasing number of substrates through a ‘hand-off ‘ from specificity to higher intrinsic activity70,72. Moreover, recent work has demonstrated that a range of complex signal processing functions can be performed by combinatorial multi-site phosphorylation events on individual Cdk substrates65.
Negative feedback turns off transcription
Upon G1–S transcriptional activation, cells progress to S phase, initiate DNA replication and subsequently inactivate transcription. It has become increasingly clear that the mechanism of G1–S transcriptional inactivation in both yeast and mammals involves negative feedback loops (FIG. 2). Several feedback mechanisms target the transcriptional activators that drive the G1-to-S phase transition for inactivation. Moreover, recent work shows that the accumulation of transcriptional repressors during S phase also has an important role in turning off transcription (FIG. 2a).
Inactivation of transcriptional activators
In S. cerevisiae, transcriptional inactivation of SBF targets upon exit from G1 requires the activity of the mitotic cyclin Clb in complex with Cdk1 (also known as Cdc28), which leads to dissociation of SBF from promoters73–76. Clb is initially inhibited during late G1 by the Cdk inhibitor Sic1 (FIG. 2c). The genes encoding G1 cyclins, CLN1 and CLN2, and the Clb cyclins, CLB5 and CLB6, are G1–S target genes, the protein products of which participate in the destruction of Sic1 and the activation of B-type cyclin activity77. Thus, the regulation of Cln and Clb levels by G1–S transcription forms a negative feedback loop through which SBF participates in its own inactivation.
As in yeast, inactivation of G1–S transcription in mammals involves multiple negative feedback loops (FIG. 2b). Similarly to the CDK-dependent inactivation of the transcriptional activator SBF, CDK activity has been proposed to inactivate E2F-mediated transcription during S phase in mammalian cells. This hypothesis was based on the observation that E2F1 is bound and phosphorylated by cyclin A–CDK2 both in vitro and in vivo, and that this promotes the dissociation of E2F1 from DNA and the inactivation of E2F1 target genes78–80. Because the gene encoding cyclin A is itself an E2F target, this constitutes a negative feedback loop. Furthermore, the activity of cyclin A–CDK2 is inhibited by the CDK inhibitor p27, which is targeted for degradation by cyclin E-CDK2 and cyclin A–CDK2; the genes encoding cyclin E and cyclin A are both targets of E2F proteins, thus contributing to the negative feedback loop81,82. In addition, the ubiquitin ligase regulatory SCF (S phase kinase-associated protein 2 (SKP2)– cullin 1–F-box protein) complex has been proposed to regulate the stability of E2F1 during S phase and G2 (REFS 83–85). As the SCF regulatory subunit SKP2 is encoded by an E2F target gene, this also represents a negative feedback loop.
Inactivation by transcriptional repression
The mechanism of inactivation of MBF-dependent transcription is different from that of SBF-dependent transcriptional inactivation and involves a negative feedback loop based on co-repressors. MBF functions as a cell cycle transcriptional repressor in a similar way to the mammalian E2F4, E2F5, E2F6, E2F7 and E2F8. It most closely resembles E2F4 and E2F5, which repress transcription with co-repressor pocket proteins. However, repressor E2F proteins only bind to promoters during specific phases of the cell cycle, whereas MBF is bound to promoters during the entire cell cycle. Temporally confining MBF activity so that transcription is switched on during G1 and switched off in S phase depends on the co-repressor proteins Nrm1 in S. cerevisiae and Nrm1 and Yox1 in the fission yeast Schizosaccharomyces pombe. Because they are both MBF targets, these proteins are involved in a negative feedback loop49,86 (FIG. 2c). Nrm1 is expressed at low levels during early G1, and it is a target of the APC/C (anaphase-promoting complex; also known as the cyclosome) and is degraded at mitotic exit49,87. Thus, co-repressor s accumulate progressively once MBF-regulated gene transcription is activated, leading to binding of the co-repressors to promoter-bound MBF and concomitant repression of G1–S transcription during S phase. Although Nrm1 and Yox1 seem to have a similar function as pocket proteins in mammalian cells, they actually differ in their ability to repress transcription: whereas Nrm1 and Yox1 function as transcriptional co-repressors during S phase, mammalian pocket proteins do not seem to be involved in turning off transcription during S phase when Cdk activity is high.
Finally, additional negative feedback loops that lead to transcriptional repression dependent on E2F6, E2F7 or E2F8, or a combination of these repressors, have been proposed88,89 (FIG. 2b). The genes encoding these E2F proteins are themselves E2F targets and accumulate during the G1-to-S transition90,91. E2F6, E2F7 and E2F8 do not require pocket proteins for their repressor activity88,92–96. As a result, they should be able to repress transcription during S phase and the latter part of the cell cycle when pocket proteins are inhibited by Cdk activity97. Consistent with this view, E2F6 was recently found to repress G1–S transcription in late S phase98 (FIG. 2b). E2F6 accumulates when cells progress to S phase and inactivates transcription in a timely manner by replacing activator E2F proteins at target promoters. Although deletion of E2F6 alone does not affect G1–S transcription in mouse embryonic fibroblasts (MEFs)99, it was shown that, in the absence of E2F6, E2F4 binds to promoters normally bound by E2F6 during S phase, suggesting a compensatory role. Accordingly, depletion of E2F4 in E2F6-knockout MEFs leads to derepression of G1–S genes during S phase. Moreover, E2F7 and E2F8 are likely to be involved in transcriptional inactivation, given that deletion of both E2F7 and E2F8 in MEFs and overexpression of E2F7 in HeLa cells causes activation and inhibition, respectively, of some E2F target genes characteristic of G1-to-S phase transistion88,89. Overall, it seems likely that negative feedback through atypical repressor E2F proteins is required to turn off G1–S transcription.
Subgroups within G1–S transcripts
Single-gene studies in the 1980’s identified subsets of genes differentially regulated during the cell cycle and high-throughput microarray data in the late 1990’s revealed the full extent of cell cycle-dependent gene expression15,100. These and subsequent studies have revealed subgroups of genes, the function and expression timing of which may be correlated.
Subgroups based on function
In S. cerevisiae, over 200 G1–S genes depend on the transcriptional activator SBF and/or the transcriptional repressor MBF. A large body of research has revealed that most of these genes can be grouped into SBF-, MBF-, SBF- and MBF-regulated and ‘switch’ genes (which are regulated by both SBF and MBF at different times during the cell cycle)51,100,101.
Genes that are under the control of either SBF or MBF can also loosely be classified on the basis of their function. Genes involved in driving cell cycle progression, such as the G1 cyclins CLN1 and CLN2, are more likely to be regulated by SBF, whereas MBF-targets are enriched for genes involved in DNA replication, DNA repair and other essential genes.
Controlling essential genes that do not dictate cell cycle timing with a transcriptional repressor such as MBF may constitute a selective advantage in case of MBF absence, as the removal of MBF at any specific promoter results in derepression of those genes rather than lack of expression. Conversely, constitutive derepression of genes involved in cell cycle timing may cause uncontrolled cell proliferation and would be ‘safer’ under the control an activator such as SBF. In addition, the finding that MBF-dependent genes seem to be involved in DNA replication may be a consequence of the transcriptional activation of MBF- but not SBF-dependent genes during replication stress101–103. A detailed description of how MBF-dependent transcription is regulated in response to replication stress is discussed below. Recently, another subgroup of genes regulated by an SBF-to-MBF switch during G1-to-S phase transition was identified103. Interestingly, these switch genes are enriched for G1–S genes that cause a cell cycle progression defect when overexpressed but are upregulated in response to replication stress. The dependency of switch genes on SBF during G1 and MBF outside of G1 prevents them from being constitutively expressed in the event of MBF malfunction, yet renders them responsive to replication stress. Interestingly, whereas only a small number of G1–S targets are regulated by the SBF-to-MBF switch in S. cerevisiae, E2F switching at G1–S target promoters in mammalian cells seems to be the norm. Overall, the use of two distinct transcription factors allows budding yeast to implement combinatorial control of its G1–S transcriptional programme in response to replication stress.
Subgroups based on timing
There is great variation in the expression timing of G1–S genes56. Genes that encode proteins involved in the positive feedback loop, such as those encoding G1 cyclins, are activated before other SBF, MBF and E2F target genes. This results in the decision to divide, which is coincident with the transcriptional activation of positive feedback elements, preceding a genome-wide change in transcription. In other words, the decision to enter a new cell cycle precedes the activation of the genome-wide change in transcription despite the fact that target genes of both processes are regulated by the same transcription factors. In addition, the yeast co-repressor Nrm1, which is involved in a negative feedback, is one of the latest genes to be activated.
The importance of ‘positive feedback first’ and ‘negative feedback last’ was demonstrated when placing CLN2 under control of the NRM1 promoter, which resulted in uncoordinated cell cycle commitment and cell death. Thus, the timing of gene activation involved in feedback regulation to temporally confine G1–S transcription follows a logical order, starting with the robust activation of transcription (dependent on the upregulation of CLN1 and CLN2) in G1 which is finally turned off during S phase (dependent on the upregulation of NRM1). The establishment of this order is not clearly linked to the specific transcription factors that control gene activation and requires further investigation.
Positive feedback first regulation is the most robust feature of transcriptional timing. Even though the timing of expression of nearly all G1–S genes significantly changes when cell cycle synchrony is established by arresting cells either in G1 phase or mitosis, G1 cyclins that affect positive feedback first regulation are among the earliest activated genes in both cases of cell cycle arrest56. The temporal subdivision into blocks of genes across the cell cycle revealed that when cells enter the cell cycle from mitosis, SBF targets are activated before MBF targets. Conversely, when cells re-enter the cell cycle from G1, the order is reversed and MBF targets are activated before SBF targets. Interestingly, genes that encode proteins involved in the positive feedback loop are likely to be under the control of both the SBF and MBF transcription factors. The dually regulated SBF and MBF targets are activated by either of these two factors (the first one activated), which ensures these genes are activated early in the cell cycle. Thus, for the genes that can be activated by either SBF or MBF, the induction of either factor is sufficient to initiate transcription and functions as a logical ‘or’ gate, whereby SBF ‘or’ MBF will activate transcription irrespective of the previous cell cycle. This reveals an important mechanistic aspect of transcriptional activation.
G1–S transcript subgroups in mammalian cells
As in yeast, the activation timing of individual genes within the G1–S wave of transcription varies greatly in mammalian cells with positive feedback-associated genes being activated first16,56. This suggests that the positive feedback first principle is conserved in eukaryotes. By contrast, cell cycle-dependent transcriptional regulation of specific target genes is very poorly conserved across eukaryotes104.
Similarly to SBF and MBF regulating distinct sets of genes in S. cerevisiae, distinct E2F proteins and pocket proteins, perhaps interacting with other co-regulators, might bind to specific promoters to define subgroups. Consistent with this model, many studies suggest that in addition to largely overlapping targets, individual members of the E2F family may display different DNA sequence specificities105,106. However, single-gene analyses indicate similar temporal sequences of E2F and pocket protein binding to the examined genes. It will be interesting to see whether and how the composition of E2F target subgroups emerges.
G1–S transcription and genome stability
When cells become committed to a division cycle, they initiate DNA replication and progress to S phase. Such cells rely on two DNA structure checkpoints (BOX 3) — the DNA damage checkpoint and the DNA replication checkpoint — to protect themselves from irreversible DNA damage. These checkpoints delay mitotic entry and initiate a specific transcriptional programme. The importance of DNA checkpoint signalling is illustrated by the conservation of the subfamily of checkpoint protein kinases (TABLE 1).
Box 3. The DNA structure checkpoints.
To properly replicate the genome and prevent tumorigenesis, cells rely on the DNA structure checkpoints, an evolutionarily conserved set of signalling pathways that monitor DNA damage and the loss of DNA replication fork integrity. These checkpoints delay mitotic entry and initiate a specific transcriptional programme. The signalling pathways involved rely on evolutionarily conserved protein kinases, including the sensor molecules that detect damage or replication stress, that in turn activate transducer proteins that relay the signal to downstream effector proteins required to initiate the full response. DNA structure checkpoints are mediated via the ATM (ataxia-telangiectasia mutated) and ATR (ataxia-telangiectasia and Rad3-related protein) protein kinases and their downstream targets checkpoint kinase 1 (CHK1) and/ or CHK2. The nature of the DNA structure triggering the checkpoint response determines the activity of downstream effector kinases: CHK1 is activated by replication fork arrest during S phase, whereas CHK2 is activated by damaged DNA detected during interphase19–21,107,131,132. On this basis, the DNA structure checkpoint can be divided into the DNA replication checkpoint and the DNA damage checkpoint. The DNA replication checkpoint is essential to prevent DNA damage in response to replication stress during S phase, whereas the DNA damage checkpoint is required to detect and resolve DNA damage during interphase131,133. Both checkpoint signalling cascades arrest cell cycle progression, mostly through the regulation of key regulators of cyclin–cyclin-dependent kinase (CDK) activity, such as the phosphatase CDC25 and the CDK inhibitor p21 (REFS 21,131,132,134). CDC25 removes inhibitory phosphorylation from CDK1 and CDK2 to promote CDK activity and therefore mitotic entry. When the checkpoint is engaged, CDC25 becomes phosphorylated and is subsequently degraded to prevent progression through mitosis. Cell cycle arrest ensures that DNA damage can be avoided or that the detected damage can be repaired before division to limit heritable mutation. Although both checkpoints delay progression of mitosis through largely overlapping mechanisms, they induce distinct responses to specific stresses. Two other important checkpoint responses are the transcriptional response and the initiation of programmed cell death when the damage cannot be resolved. The transcriptional response differs between the replication and damage checkpoints, as, for instance, G1–S transcription is only regulated by the replication checkpoint. Programmed cell death is of particular importance in multicellular organisms and is predominately associated with the DNA damage checkpoint response21.
DNA replication checkpoint
In this Review, we focus on DNA replication checkpoint signalling because recent work has linked this pathway to G1–S transcription. DNA replication stress is defined as inefficient DNA replication that causes DNA replication forks to progress slowly or stall. The DNA replication checkpoint prevents the accumulation of DNA damage as a result of replication stress by stabilizing stalled replication forks, preventing late origins from firing and enabling replication to resume once the stress has been resolved107.
During DNA replication, cells are particularly vulnerable to genomic instability, as replication forks are prone to stall and collapse when encountering replication blocks or damaged DNA templates. The DNA replication checkpoint transcriptional response probably maintains G1–S transcription in order to prevent genomic instability98,101,102,108 (FIG. 3).
Figure 3. G1–S phase transcription and genome stability.

The mechanism by which the DNA replication checkpoint maintains high levels of G1−S transcription in response to replication stress is conserved from yeast to humans. This mechanism involves the inactivation of a transcriptional repressors and/or co-repressors (Nrm1 and Yox1 in yeast and E2F6 in human cells) involved in an autoregulatory negative feedback loop. The downstream effector checkpoint protein kinase (Cds1 in fission yeast, Rad53 in budding yeast and checkpoint kinase 1 (CHK1) in mammals) inactivates the transcriptional repressors Nrm1, Yox1 and E2F6 through phosphorylation to maintain high levels of G1–S transcription. The DNA replication checkpoint protein kinases are conserved from yeast to humans, but the G1–S transcriptional repressors and transcriptional activators are not. MBF, MCB-binding factor.
The replication checkpoint induces G1–S transcription
In both fission and budding yeast, genes that are activated during G1 and that depend on MBF to be inactivated outside of G1 are induced in response to DNA replication stress11,103,109. Activation of these MBF-responsive genes results from checkpoint signalling-dependent inhibition of the negative feedback loop that turns off G1–S transcription during G1-to-S phase transition. The checkpoint effector kinases Rad53 and Cds1 directly target and inhibit the main G1–S transcriptional co-repressor Nrm1 in budding yeast and Nrm1 and Yox1 in fission yeast101,102,110–114 (FIG. 3). Upon phosphorylation, these repressors are no longer able to bind MBF, allowing for continuous expression of MBF-regulated genes.
Maintenance of MBF-regulated gene expression is important for cell survival in response to replication stress because many MBF-targets function in replication, DNA repair and nucleotide synthesis. Consequently, the deletion of Nrm1 and/or Yox1 in fission yeast improves the survival of mutants defective in the DNA replication checkpoint in response to replication stress and, conversely, overexpression of stabilized Nrm1 in budding yeast increases sensitivity to replication stress.
Conservation of the checkpoint transcriptional response
Similarly to the induction of MBF targets by replication stress in yeast, E2F-dependent transcription is induced in mammals by a closely related mechanism98 (FIG. 3). In response to replication stress, checkpoint kinase 1 (CHK1) phosphorylates and inhibits E2F6, which is responsible for inactivating G1–S transcription during the mitotic cell cycle. Inactivation of E2F6 leads to its release from promoters and allows G1–S transcription to persist. The ability of cells to activate G1–S transcription is crucial for survival upon hydroxyurea treatment, probably because E2F targets include proteins that prevent replication fork collapse and DNA damage. This transcriptional response seems to be specific for replication stress, which, in contrast to the DNA damage response, does not induce apoptosis. Interestingly, the DNA damage response can induce pro-apoptotic E2F1 targets115–120. However, these targets are distinct from those normally regulated by E2F1 during the mitotic cell cycle. Despite a distinct lack of conservation of the proteins affecting this regulation, the conservation of the transcriptional response to replication stress underscores the conservation of G1–S control systems-level features across eukaryotes.
Conclusion and perspective
In this Review, we emphasized recent work uncovering simple yet elegant mechanisms of gene expression control during G1 and S phases that regulate commitment to cell division, temporally confine G1–S transcription and respond to replication stress. This work has revealed many new insights into how transcription of different G1–S cell cycle genes is restricted to G1 and how this is regulated by DNA replication checkpoint protein kinases as part of the checkpoint transcriptional response. Despite frequent lack of sequence homology, conservation of systems-level properties across eukaryotes is an emerging theme of cell cycle control. Indeed, the conserved regulation of G1–S transcripts by the replication checkpoint suggests a central role for this transcriptional wave in the maintenance of genome stability.
Genome-wide transcriptional changes are a general feature of cellular transitions. Our knowledge of the transcriptional regulation of the G1-to-S phase transition may be widely applicable to the study of other such transitions. Resulting from decades of concerted effort, our deep understanding of the G1-to-S phase transition provides a blueprint for future research to investigate fundamental regulatory mechanisms controlling cellular decision-making processes, dynamic changes in gene expression during cellular transitions and context-specific rewiring of transcriptional networks.
Acknowledgements
C.B and R.A.M de Bruin were funded by the Medical Research Council (MRC). J.M.S. was funded by the National Institutes of Health (NIH) (GM092925) and the Burroughs Wellcome Fund. The authors thank A. Johnson and C. Schwarz for careful reading of the manuscript. They apologize to colleagues whose work could only be cited indirectly.
Glossary
- Ubiquitin ligase
An enzyme that recognizes Lys residues on a target protein and causes the attachment of ubiquitin to these residues
- RB
A protein that binds activator E2F proteins to inhibit transcription outside of G1–S in animals. RB is an oncoprotein that is dysfunctional in several major cancers
- Pocket proteins
Family of proteins, including RB, p107 and p130, that associates with members of the E2F transcription factor family to inhibit transcription. The pocket domain is essential for tumour suppressing activity
- Whi5
An inhibitor of SBF (SCB-binding factor)-dependent transcription during early G1 in yeast
- Regulon
A collection of genes under the control of the same regulatory protein
- Nrm1 and Yox1
Nrm1 in budding yeast and Nrm1 and Yox1 in fission yeast bind MBF (MCB-binding factor) to inhibit transcription once cells transit into S phase
Footnotes
Competing interests statement
The authors declare no competing financial interests.
FURTHER INFORMATION
Jan M. Skotheim’s homepage: http://skotheimlab.com
Robertus A. M. de Bruin’s homepage: http://www.ucl.ac.uk/lmcb/research-group/rob-de-bruin-research-group
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
Contributor Information
Cosetta Bertoli, Email: c.bertoli@ucl.ac.uk.
Jan M. Skotheim, Email: skotheim@stanford.edu.
Robertus A. M. de Bruin, Email: r.debruin@ucl.ac.uk.
References
- 1.Morgan DO. In: The cell cycle: principles of control. Lawrence E, editor. New Science Press; 2007. pp. 157–173. [Google Scholar]
- 2.Massague J. G1 cell-cycle control and cancer. Nature. 2004;432:298–306. doi: 10.1038/nature03094. [DOI] [PubMed] [Google Scholar]
- 3.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 4.Henley SA, Dick FA. The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div. 2012;7:10. doi: 10.1186/1747-1028-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chinnam M, Goodrich DW. RB1, development, and cancer. Curr. Top. Dev. Biol. 2011;94:129–169. doi: 10.1016/B978-0-12-380916-2.00005-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Helin K. Regulation of cell proliferation by the E2F transcription factors. Curr. Opin. Genet. Dev. 1998;8:28–35. doi: 10.1016/s0959-437x(98)80058-0. [DOI] [PubMed] [Google Scholar]
- 7.Umen JG, Goodenough UW. Control of cell division by a retinoblastoma protein homolog in Chlamydomonas . Genes Dev. 2001;15:1652–1661. doi: 10.1101/gad.892101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cross FR, Buchler NE, Skotheim JM. Evolution of networks and sequences in eukaryotic cell cycle control. Phil. Trans. R. Soc. Lond. B. 2011;366:3532–3544. doi: 10.1098/rstb.2011.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van den Heuvel S, Dyson NJ. Conserved functions of the pRB and E2F families. Nature Rev. Mol. Cell Biol. 2008;9:713–724. doi: 10.1038/nrm2469. [DOI] [PubMed] [Google Scholar]
- 10.Cooper K. Rb, whi it’s not just for metazoans anymore. Oncogene. 2006;25:5228–5232. doi: 10.1038/sj.onc.1209630. [DOI] [PubMed] [Google Scholar]
- 11.de Bruin RA, Wittenberg C. All eukaryotes: before turning off G1–S transcription, please check your DNA. Cell Cycle. 2009;8:214–217. doi: 10.4161/cc.8.2.7412. [DOI] [PubMed] [Google Scholar]
- 12.Bahler J. Cell-cycle control of gene expression in budding and fission yeast. Annu. Rev. Genet. 2005;39:69–94. doi: 10.1146/annurev.genet.39.110304.095808. [DOI] [PubMed] [Google Scholar]
- 13.Fukuoka M, et al. Identification of preferentially reactivated genes during early G1 phase using nascent mRNA as an index of transcriptional activity. Biochem. Biophys. Res. Commun. 2013;430:1005–1010. doi: 10.1016/j.bbrc.2012.12.048. [DOI] [PubMed] [Google Scholar]
- 14.Wittenberg C, Reed SI. Cell cycle-dependent transcription in yeast: promoters, transcription factors, and transcriptomes. Oncogene. 2005;24:2746–2755. doi: 10.1038/sj.onc.1208606. [DOI] [PubMed] [Google Scholar]
- 15.Spellman PT, et al. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whitfield ML, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell. 2002;13:1977–2000. doi: 10.1091/mbc.02-02-0030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Skaar JR, Pagano M. Control of cell growth by the SCF and APC/C ubiquitin ligases. Curr. Opin. Cell Biol. 2009;21:816–824. doi: 10.1016/j.ceb.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bloom J, Cross FR. Multiple levels of cyclin specificity in cell-cycle control. Nature Rev. Mol. Cell Biol. 2007;8:149–160. doi: 10.1038/nrm2105. [DOI] [PubMed] [Google Scholar]
- 19.Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nature Rev. Mol. Cell Biol. 2010;11:208–219. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- 20.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nature Rev. Mol. Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nature Rev. Cancer. 2006;6:369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 23.Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nature Rev. Cancer. 2009;9:785–797. doi: 10.1038/nrc2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson DG, Schneider-Broussard R. Role of E2F in cell cycle control and cancer. Front Biosci. 1998;3:d447–d448. doi: 10.2741/a291. [DOI] [PubMed] [Google Scholar]
- 25.Asano M, Nevins JR, Wharton RP. Ectopic E2F expression induces S phase and apoptosis in Drosophila imaginal discs. Genes Dev. 1996;10:1422–1432. doi: 10.1101/gad.10.11.1422. [DOI] [PubMed] [Google Scholar]
- 26.Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 27.Lukas J, Petersen BO, Holm K, Bartek J, Helin K. Deregulated expression of E2F family members induces S-phase entry and overcomes p16INK4A-mediated growth suppression. Mol. Cell. Biol. 1996;16:1047–1057. doi: 10.1128/mcb.16.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nature Rev. Cancer. 2001;1:222–231. doi: 10.1038/35106065. [DOI] [PubMed] [Google Scholar]
- 29. Chong JL, et al. E2f1-3 switch from activators in progenitor cells to repressors in differentiating cells. Nature. 2009;462:930–934. doi: 10.1038/nature08677. Establishes the in vivo relevance of E2F1, E2F2 and E2F3, indicating an important role for their repressor function in addition to their well-established function in transcriptional activation
- 30.Weijts BG, et al. E2F7 and E2F8 promote angiogenesis through transcriptional activation of VEGFA in cooperation with HIF1. EMBO J. 2012;31:3871–3884. doi: 10.1038/emboj.2012.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee BK, Bhinge AA, Iyer VR. Wide-ranging functions of E2F4 in transcriptional activation and repression revealed by genome-wide analysis. Nucleic Acids Res. 2011;39:3558–3573. doi: 10.1093/nar/gkq1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Helin K, Harlow E, Fattaey A. Inhibition of E2F-1 transactivation by direct binding of the retinoblastoma protein. Mol. Cell. Biol. 1993;13:6501–6508. doi: 10.1128/mcb.13.10.6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hatakeyama M, Weinberg RA. The role of RB in cell cycle control. Prog. Cell Cycle Res. 1995;1:9–19. doi: 10.1007/978-1-4615-1809-9_2. [DOI] [PubMed] [Google Scholar]
- 34.Wirt SE, Sage J. p107 in the public eye: an Rb understudy and more. Cell Div. 2010;5:9. doi: 10.1186/1747-1028-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ginsberg D, et al. E2F-4, a new member of the E2F transcription factor family, interacts with p107. Genes Dev. 1994;8:2665–2679. doi: 10.1101/gad.8.22.2665. [DOI] [PubMed] [Google Scholar]
- 36.Grana X, Garriga J, Mayol X. Role of the retinoblastoma protein family, pRB, p107 and p130 in the negative control of cell growth. Oncogene. 1998;17:3365–3383. doi: 10.1038/sj.onc.1202575. [DOI] [PubMed] [Google Scholar]
- 37.Dimova DK, Dyson NJ. The E2F transcriptional network: old acquaintances with new faces. Oncogene. 2005;24:2810–2826. doi: 10.1038/sj.onc.1208612. [DOI] [PubMed] [Google Scholar]
- 38.Hitchens MR, Robbins PD. The role of the transcription factor DP in apoptosis. Apoptosis. 2003;8:461–468. doi: 10.1023/a:1025586207239. [DOI] [PubMed] [Google Scholar]
- 39.Gaubatz S, et al. E2F4 and E2F5 play an essential role in pocket protein-mediated G1 control. Mol. Cell. 2000;6:729–735. doi: 10.1016/s1097-2765(00)00071-x. [DOI] [PubMed] [Google Scholar]
- 40.Takahashi Y, Rayman JB, Dynlacht BD. Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev. 2000;14:804–816. [PMC free article] [PubMed] [Google Scholar]
- 41.Balciunaite E, et al. Pocket protein complexes are recruited to distinct targets in quiescent and proliferating cells. Mol. Cell. Biol. 2005;25:8166–8178. doi: 10.1128/MCB.25.18.8166-8178.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beijersbergen RL, et al. E2F-4, a new member of the E2F gene family, has oncogenic activity and associates with p107 in vivo . Genes Dev. 1994;8:2680–2690. doi: 10.1101/gad.8.22.2680. [DOI] [PubMed] [Google Scholar]
- 43.Gaubatz S, Lees JA, Lindeman GJ, Livingston DM. E2F4 is exported from the nucleus in a CRM1-dependent manner. Mol. Cell. Biol. 2001;21:1384–1392. doi: 10.1128/MCB.21.4.1384-1392.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen D, et al. Division and apoptosis of E2f-deficient retinal progenitors. Nature. 2009;462:925–929. doi: 10.1038/nature08544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Litovchick L, et al. Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol. Cell. 2007;26:539–551. doi: 10.1016/j.molcel.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 46.Chicas A, et al. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell. 2010;17:376–387. doi: 10.1016/j.ccr.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Bruin RA, et al. Cln3 activates G1-specific transcription via phosphorylation of the SBF bound repressor Whi5. Cell. 2004;117:887–898. doi: 10.1016/j.cell.2004.05.025. [DOI] [PubMed] [Google Scholar]
- 48. Costanzo M, et al. CDK activity antagonizes Whi5, an inhibitor of G1/S transcription in yeast. Cell. 2004;117:899–913. doi: 10.1016/j.cell.2004.05.024. Establishes, together with reference 47, the role of Whi5 in G1 cyclin-Cdk-mediated activation of G1–S transcription in budding yeast.
- 49. de Bruin RA, et al. Constraining G1-specific transcription to late G1 phase: the MBF-associated corepressor Nrm1 acts via negative feedback. Mol. Cell. 2006;23:483–496. doi: 10.1016/j.molcel.2006.06.025. Shows that the transcriptional co-repressor Nrm1 is involved in a negative feedback mechanism to turn off G1–S transcription in budding and fission yeast
- 50.Wijnen H, Landman A, Futcher B. The G1 cyclin Cln3 promotes cell cycle entry via the transcription factor Swi6. Mol. Cell. Biol. 2002;22:4402–4418. doi: 10.1128/MCB.22.12.4402-4418.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ferrezuelo F, Colomina N, Futcher B, Aldea M. The transcriptional network activated by Cln3 cyclin at the G1-to-S transition of the yeast cell cycle. Genome Biol. 2012;11:R67. doi: 10.1186/gb-2010-11-6-r67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bean JM, Siggia ED, Cross FR. High functional overlap between MluI cell-cycle box binding factor and Swi4/6 cell-cycle box binding factor in the G1/S transcriptional program in Saccharomyces cerevisiae . Genetics. 2005;171:49–61. doi: 10.1534/genetics.105.044560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kosugi S, Hasebe M, Tomita M, Yanagawa H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc. Natl Acad. Sci. USA. 2009;106:10171–10176. doi: 10.1073/pnas.0900604106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Skotheim JM, Di Talia S, Siggia ED, Cross FR. Positive feedback of G1 cyclins ensures coherent cell cycle entry. Nature. 2008;454:291–296. doi: 10.1038/nature07118. Establishes the positive feedback mechanism required for robust activation of G1–S transcription
- 55.Doncic A, Falleur-Fettig M, Skotheim JM. Distinct interactions select and maintain a specific cell fate. Mol. Cell. 2011;43:528–539. doi: 10.1016/j.molcel.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Eser U, Falleur-Fettig M, Johnson A, Skotheim JM. Commitment to a cellular transition precedes genome-wide transcriptional change. Mol. Cell. 2011;43:515–527. doi: 10.1016/j.molcel.2011.06.024. Identifies differential timing for the expression of G1–S genes in yeast and humans and shows the importance of feedback first regulation
- 57.Bracken AP, Ciro M, Cocito A, Helin K. E2F target genes: unraveling the biology. Trends Biochem. Sci. 2004;29:409–417. doi: 10.1016/j.tibs.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 58.Yung Y, Walker JL, Roberts JM, Assoian RK. A Skp2 autoinduction loop and restriction point control. J. Cell Biol. 2007;178:741–747. doi: 10.1083/jcb.200703034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johnson DG, Ohtani K, Nevins JR. Autoregulatory control of E2F1 expression in response to positive and negative regulators of cell cycle progression. Genes Dev. 1994;8:1514–1525. doi: 10.1101/gad.8.13.1514. [DOI] [PubMed] [Google Scholar]
- 60.Foster DA, Yellen P, Xu L, Saqcena M. Regulation of G1 cell cycle progression: distinguishing the restriction point from a nutrient-sensing cell growth checkpoint(s) Genes Cancer. 2010;1:1124–1131. doi: 10.1177/1947601910392989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martinsson HS, Starborg M, Erlandsson F, Zetterberg A. Single cell analysis of G1 check points-the relationship between the restriction point and phosphorylation of pRb. Exp. Cell Res. 2005;305:383–391. doi: 10.1016/j.yexcr.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 62.Hitomi M, et al. p27Kip1 and cyclin dependent kinase 2 regulate passage through the restriction point. Cell Cycle. 2006;5:2281–2289. doi: 10.4161/cc.5.19.3318. [DOI] [PubMed] [Google Scholar]
- 63.Schwob E, Bohm T, Mendenhall MD, Nasmyth K. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition inS. cerevisiae. Cell. 1994;79:233–244. doi: 10.1016/0092-8674(94)90193-7. [DOI] [PubMed] [Google Scholar]
- 64.Nash P, et al. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature. 2001;414:514–521. doi: 10.1038/35107009. [DOI] [PubMed] [Google Scholar]
- 65. Koivomagi M, et al. Cascades of multisite phosphorylation control Sic1 destruction at the onset of S phase. Nature. 2012;480:128–131. doi: 10.1038/nature10560. Establishes the role of semi-processive multi-site phosphorylation events in the degradation of the Cdk inhibitor Sic1
- 66.Masumoto H, Muramatsu S, Kamimura Y, Araki H. S-Cdk-dependent phosphorylation of Sld2 essential for chromosomal DNA replication in budding yeast. Nature. 2002;415:651–655. doi: 10.1038/nature713. [DOI] [PubMed] [Google Scholar]
- 67.Zegerman P, Diffley JF. Phosphorylation of Sld2 and Sld3 by cyclin-dependent kinases promotes DNA replication in budding yeast. Nature. 2007;445:281–285. doi: 10.1038/nature05432. [DOI] [PubMed] [Google Scholar]
- 68.Kang YH, Galal WC, Farina A, Tappin I, Hurwitz J. Properties of the human Cdc45/Mcm2-7/ GINS helicase complex and its action with DNA polymerase epsilon in rolling circle DNA synthesis. Proc. Natl Acad. Sci. USA. 2012;109:6042–6047. doi: 10.1073/pnas.1203734109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wilmes GM, et al. Interaction of the S-phase cyclin Clb5 with an ‘RXL’ docking sequence in the initiator protein Orc6 provides an origin-localized replication control switch. Genes Dev. 2004;18:981–991. doi: 10.1101/gad.1202304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koivomagi M, et al. Dynamics of Cdk1 substrate specificity during the cell cycle. Mol. Cell. 2012;42:610–623. doi: 10.1016/j.molcel.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Loog M, Morgan DO. Cyclin specificity in the phosphorylation of cyclin-dependent kinase substrates. Nature. 2005;434:104–108. doi: 10.1038/nature03329. [DOI] [PubMed] [Google Scholar]
- 72.Stern B, Nurse P. A quantitative model for the cdc2 control of S phase and mitosis in fission yeast. Trends Genet. 1996;12:345–350. [PubMed] [Google Scholar]
- 73.Amon A, Tyers M, Futcher B, Nasmyth K. Mechanisms that help the yeast cell cycle clock tick: G2 cyclins transcriptionally activate G2 cyclins and repress G1 cyclins. Cell. 1993;74:993–1007. doi: 10.1016/0092-8674(93)90722-3. [DOI] [PubMed] [Google Scholar]
- 74.de Bruin RA, Kalashnikova TI, Wittenberg C. Stb1 collaborates with other regulators to modulate the G1-specific transcriptional circuit. Mol. Cell. Biol. 2008;28:6919–6928. doi: 10.1128/MCB.00211-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Siegmund RF, Nasmyth KA. The Saccharomyces cerevisiae Start-specific transcription factor Swi4 interacts through the ankyrin repeats with the mitotic Clb2/Cdc28 kinase and through its conserved carboxy terminus with Swi6. Mol. Cell. Biol. 1996;16:2647–2655. doi: 10.1128/mcb.16.6.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koch C, Schleiffer A, Ammerer G, Nasmyth K. Switching transcription on and off during the yeast cell cycle: Cln/Cdc28 kinases activate bound transcription factor SBF (Swi4/Swi6) at start, whereas Clb/Cdc28 kinases displace it from the promoter in G2. Genes Dev. 1996;10:129–141. doi: 10.1101/gad.10.2.129. [DOI] [PubMed] [Google Scholar]
- 77.Moll T, et al. Transcription factors important for starting the cell cycle in yeast. Phil. Trans. R. Soc. Lond. B. 1993;340:351–360. doi: 10.1098/rstb.1993.0078. [DOI] [PubMed] [Google Scholar]
- 78.Xu M, Sheppard KA, Peng CY, Yee AS, Piwnica-Worms H. Cyclin A/CDK2 binds directly to E2F-1 and inhibits the DNA-binding activity of E2F-1/DP-1 by phosphorylation. Mol. Cell. Biol. 1994;14:8420–8431. doi: 10.1128/mcb.14.12.8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dynlacht BD, Flores O, Lees JA, Harlow E. Differential regulation of E2F transactivation by cyclin/cdk2 complexes. Genes Dev. 1994;8:1772–1786. doi: 10.1101/gad.8.15.1772. [DOI] [PubMed] [Google Scholar]
- 80.Krek W, et al. Negative regulation of the growth-promoting transcription factor E2F-1 by a stably bound cyclin A-dependent protein kinase. Cell. 1994;78:161–172. doi: 10.1016/0092-8674(94)90582-7. [DOI] [PubMed] [Google Scholar]
- 81.Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nature Cell Biol. 1999;1:193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 82.Montagnoli A, et al. Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev. 1999;13:1181–1189. doi: 10.1101/gad.13.9.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marti A, Wirbelauer C, Scheffner M, Krek W. Interaction between ubiquitin-protein ligase SCFSKP2 and E2F-1 underlies the regulation of E2F-1 degradation. Nature Cell Biol. 1999;1:14–19. doi: 10.1038/8984. [DOI] [PubMed] [Google Scholar]
- 84.Campanero MR, Flemington EK. Regulation of E2F through ubiquitin-proteasome-dependent degradation: stabilization by the pRB tumor suppressor protein. Proc. Natl Acad. Sci. USA. 1997;94:2221–2226. doi: 10.1073/pnas.94.6.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hateboer G, Kerkhoven RM, Shvarts A, Bernards R, Beijersbergen RL. Degradation of E2F by the ubiquitin-proteasome pathway: regulation by retinoblastoma family proteins and adenovirus transforming proteins. Genes Dev. 1996;10:2960–2970. doi: 10.1101/gad.10.23.2960. [DOI] [PubMed] [Google Scholar]
- 86.Aligianni S, et al. The fission yeast homeodomain protein Yox1p binds to MBF and confines MBF-dependent cell-cycle transcription to G1–S via negative feedback. PLoS Genet. 2009;5:e1000626. doi: 10.1371/journal.pgen.1000626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ostapenko D, Solomon MJ. Anaphase promoting complex-dependent degradation of transcriptional repressors Nrm1 and Yhp1 in Saccharomyces cerevisiae . Mol. Biol. Cell. 2011;22:2175–2184. doi: 10.1091/mbc.E11-01-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li J, et al. Synergistic function of E2F7 and E2F8 is essential for cell survival and embryonic development. Dev. Cell. 2008;14:62–75. doi: 10.1016/j.devcel.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Westendorp B, et al. E2F7 represses a network of oscillating cell cycle genes to control S-phase progression. Nucleic Acids Res. 2012;40:3511–3523. doi: 10.1093/nar/gkr1203. Establishes a role for E2F7 in the repression of G1–S transcription during S phase in mammalian cells.
- 90.Lyons TE, Salih M, Tuana BS. Activating E2Fs mediate transcriptional regulation of human E2F6 repressor. Am. J. Physiol. Cell Physiol. 2006;290:C189–C199. doi: 10.1152/ajpcell.00630.2004. [DOI] [PubMed] [Google Scholar]
- 91.Moon NS, Dyson N. E2F7 and E2F8 keep the E2F family in balance. Dev. Cell. 2008;14:1–3. doi: 10.1016/j.devcel.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 92.Cartwright P, Muller H, Wagener C, Holm K, Helin K. E2F-6: a novel member of the E2F family is an inhibitor of E2F-dependent transcription. Oncogene. 1998;17:611–623. doi: 10.1038/sj.onc.1201975. [DOI] [PubMed] [Google Scholar]
- 93.de Bruin A, et al. Identification and characterization of E2F7, a novel mammalian E2F family member capable of blocking cellular proliferation. J. Biol. Chem. 2003;278:42041–42049. doi: 10.1074/jbc.M308105200. [DOI] [PubMed] [Google Scholar]
- 94.Di Stefano L, Jensen MR, Helin K. E2F7, a novel E2F featuring DP-independent repression of a subset of E2F-regulated genes. EMBO J. 2003;22:6289–6298. doi: 10.1093/emboj/cdg613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Logan N, et al. E2F-8: an E2F family member with a similar organization of DNA-binding domains to E2F-7. Oncogene. 2005;24:5000–5004. doi: 10.1038/sj.onc.1208703. [DOI] [PubMed] [Google Scholar]
- 96.Trimarchi JM, et al. E2F-6, a member of the E2F family that can behave as a transcriptional repressor. Proc. Natl Acad. Sci. USA. 1998;95:2850–2855. doi: 10.1073/pnas.95.6.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lammens T, Li J, Leone G, De Veylder L. Atypical E2Fs: new players in the E2F transcription factor family. Trends Cell Biol. 2009;19:111–118. doi: 10.1016/j.tcb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bertoli C, Klier S, McGowan C, Wittenberg C, de Bruin RAM. Chk1 inhibits E2F6 repressor function in response to replication stress to maintain cell cycle transcription. Curr. Biol. in the press doi: 10.1016/j.cub.2013.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Giangrande PH, et al. A role for E2F6 in distinguishing G1/S- and G2/M-specific transcription. Genes Dev. 2004;18:2941–2951. doi: 10.1101/gad.1239304. Proposes that E2F6 could be involved in a negative feedback loop to repress G1–S transcription when cells progress to S phase
- 100.Powell BL, et al. Leukapheresis induced changes in cell cycle distribution and nucleoside transporters in patients with untreated acute myeloid leukemia. Leukemia. 1991;5:1037–1042. [PubMed] [Google Scholar]
- 101.Bastos de Oliveira FM, Harris MR, Brazauskas P, de Bruin RA, Smolka MB. Linking DNA replication checkpoint to MBF cell-cycle transcription reveals a distinct class of G1/S genes. EMBO J. 2012;31:1798–1810. doi: 10.1038/emboj.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Travesa A, et al. DNA replication stress differentially regulates G1/S genes via Rad53-dependent inactivation of Nrm1. EMBO J. 2012;31:1811–1822. doi: 10.1038/emboj.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Smolka MB, Bastos de Oliveira FM, Harris MR, de Bruin RA. The checkpoint transcriptional response: Make sure to turn it off once you are satisfied. Cell Cycle. 2012;11:3166–3174. doi: 10.4161/cc.21197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jensen LJ, Jensen TS, de Lichtenberg U, Brunak S, Bork P. Co-evolution of transcriptional and post-translational cell-cycle regulation. Nature. 2006;443:594–597. doi: 10.1038/nature05186. [DOI] [PubMed] [Google Scholar]
- 105.Trojer P, et al. L3MBTL2 protein acts in concert with PcG protein-mediated monoubiquitination of H2A to establish a repressive chromatin structure. Mol. Cell. 2011;42:438–450. doi: 10.1016/j.molcel.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xu X, et al. A comprehensive ChIP-chip analysis of E2F1, E2F4, and E2F6 in normal and tumor cells reveals interchangeable roles of E2F family members. Genome Res. 2007;17:1550–1561. doi: 10.1101/gr.6783507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zegerman P, Diffley JF. DNA replication as a target of the DNA damage checkpoint. DNA Repair (Amst.) 2009;8:1077–1088. doi: 10.1016/j.dnarep.2009.04.023. [DOI] [PubMed] [Google Scholar]
- 108.Chu Z, et al. Modulation of cell cycle-specific gene expressions at the onset of S phase arrest contributes to the robust DNA replication checkpoint response in fission yeast. Mol. Biol. Cell. 2007;18:1756–1767. doi: 10.1091/mbc.E06-10-0928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gomez-Escoda B, et al. Yox1 links MBF-dependent transcription to completion of DNA synthesis. EMBO Rep. 2011;12:84–89. doi: 10.1038/embor.2010.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Caetano C, Klier S, de Bruin RA. Phosphorylation of the MBF repressor Yox1p by the DNA replication checkpoint keeps the G1/S cell-cycle transcriptional program active. PLoS ONE. 2011;6:e17211. doi: 10.1371/journal.pone.0017211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. de Bruin RA, et al. DNA replication checkpoint promotes G1–S transcription by inactivating the MBF repressor Nrm1. Proc. Natl Acad. Sci. USA. 2008;105:11230–11235. doi: 10.1073/pnas.0801106105. Establishes, together with reference 98, the mechanism of how the DNA replication checkpoint co-opts the cell cycle transcriptional programme to respond to replication stress in fission yeast and in human cells
- 112.Dutta C, et al. The DNA replication checkpoint directly regulates MBF-dependent G1/S transcription. Mol. Cell. Biol. 2008;28:5977–5985. doi: 10.1128/MCB.00596-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ivanova T, Gomez-Escoda B, Hidalgo E, Ayte J. G1/S transcription and the DNA synthesis checkpoint: common regulatory mechanisms. Cell Cycle. 2011;10:912–915. doi: 10.4161/cc.10.6.14963. [DOI] [PubMed] [Google Scholar]
- 114.Purtill FS, et al. A homeodomain transcription factor regulates the DNA replication checkpoint in yeast. Cell Cycle. 2011;10:664–670. doi: 10.4161/cc.10.4.14824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lin WC, Lin FT, Nevins JR. Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev. 2001;15:1833–1844. [PMC free article] [PubMed] [Google Scholar]
- 116.Pediconi N, et al. Differential regulation of E2F1 apoptotic target genes in response to DNA damage. Nature Cell Biol. 2003;5:552–558. doi: 10.1038/ncb998. [DOI] [PubMed] [Google Scholar]
- 117.Stevens C, La Thangue NB. E2F and cell cycle control: a double-edged sword. Arch. Biochem. Biophys. 2003;412:157–169. doi: 10.1016/s0003-9861(03)00054-7. [DOI] [PubMed] [Google Scholar]
- 118.Stevens C, Smith L, La Thangue NB. Chk2 activates E2F-1 in response to DNA damage. Nature Cell Biol. 2003;5:401–409. doi: 10.1038/ncb974. [DOI] [PubMed] [Google Scholar]
- 119.Urist M, Tanaka T, Poyurovsky MV, Prives C. p73 induction after DNA damage is regulated by checkpoint kinases Chk1 and Chk2. Genes Dev. 2004;18:3041–3054. doi: 10.1101/gad.1221004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang B, Liu K, Lin FT, Lin WC. A role for 14-3-3 tau in E2F1 stabilization and DNA damage-induced apoptosis. J. Biol. Chem. 2004;279:54140–54152. doi: 10.1074/jbc.M410493200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhang HS, Postigo AA, Dean DC. Active transcriptional repression by the Rb-E2F complex mediates G1 arrest triggered by p16INK4a, TGFβ, and contact inhibition. Cell. 1999;97:53–61. doi: 10.1016/s0092-8674(00)80714-x. [DOI] [PubMed] [Google Scholar]
- 122.Hurford RK, Jr, Cobrinik D, Lee MH, Dyson N. pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev. 1997;11:1447–1463. doi: 10.1101/gad.11.11.1447. [DOI] [PubMed] [Google Scholar]
- 123.Vairo G, Livingston DM, Ginsberg D. Functional interaction between E2F-4 and p130: evidence for distinct mechanisms underlying growth suppression by different retinoblastoma protein family members. Genes Dev. 1995;9:869–881. doi: 10.1101/gad.9.7.869. [DOI] [PubMed] [Google Scholar]
- 124.Moberg K, Starz MA, Lees JA. E2F-4 switches from p130 to p107 and pRB in response to cell cycle reentry. Mol. Cell. Biol. 1996;16:1436–1449. doi: 10.1128/mcb.16.4.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Deschenes C, Alvarez L, Lizotte ME, Vezina A, Rivard N. The nucleocytoplasmic shuttling of E2F4 is involved in the regulation of human intestinal epithelial cell proliferation and differentiation. J. Cell. Physiol. 2004;199:262–273. doi: 10.1002/jcp.10455. [DOI] [PubMed] [Google Scholar]
- 126.Hijmans EM, Voorhoeve PM, Beijersbergen RL, van ‘t Veer LJ, Bernards R. E2F-5, a new E2F family member that interacts with p130 in vivo . Mol. Cell. Biol. 1995;15:3082–3089. doi: 10.1128/mcb.15.6.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chen CR, Kang Y, Siegel PM, Massague J. E2F4/5 and p107 as Smad cofactors linking the TGFβ receptor to c-myc repression. Cell. 2002;110:19–32. doi: 10.1016/s0092-8674(02)00801-2. [DOI] [PubMed] [Google Scholar]
- 128.Christensen J, et al. Characterization of E2F8, a novel E2F-like cell-cycle regulated repressor of E2F-activated transcription. Nucleic Acids Res. 2005;33:5458–5470. doi: 10.1093/nar/gki855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Maiti B, et al. Cloning and characterization of mouse E2F8, a novel mammalian E2F family member capable of blocking cellular proliferation. J. Biol. Chem. 2005;280:18211–18220. doi: 10.1074/jbc.M501410200. [DOI] [PubMed] [Google Scholar]
- 130.Bhattacharya S, et al. SKP2 associates with p130 and accelerates p130 ubiquitylation and degradation in human cells. Oncogene. 2003;22:2443–2451. doi: 10.1038/sj.onc.1206339. [DOI] [PubMed] [Google Scholar]
- 131.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 132.Sorensen CS, Syljuasen RG. Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 2012;40:477–486. doi: 10.1093/nar/gkr697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nature Rev. Mol. Cell Biol. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- 134.Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 135.Coller HA, Sang L, Roberts JM. A new description of cellular quiescence. PLoS Biol. 2006;4:e83. doi: 10.1371/journal.pbio.0040083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sang L, Coller HA, Roberts JM. HES1 regulates the reversibility of cellular quiescence. Science. 2008;321:1095–1100. doi: 10.1126/science.1155998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bhaduri S, Pryciak PM. Cyclin-specific docking motifs promote phosphorylation of yeast signaling proteins by G1/S Cdk complexes. Curr. Biol. 2011;21:1615–1623. doi: 10.1016/j.cub.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]