

Assembly of the C1d projection of the central microtubule pair apparatus in mammalian motile cilia requires the ciliary protein CFAP54. Loss of the C1d projection in mice lacking CFAP54 impairs ciliary motility and cilia-driven fluid flow and results in a primary ciliary dyskinesia phenotype.
Abstract
Motile cilia and flagella play critical roles in fluid clearance and cell motility, and dysfunction commonly results in the pediatric syndrome primary ciliary dyskinesia (PCD). CFAP221, also known as PCDP1, is required for ciliary and flagellar function in mice and Chlamydomonas reinhardtii, where it localizes to the C1d projection of the central microtubule apparatus and functions in a complex that regulates flagellar motility in a calcium-dependent manner. We demonstrate that the genes encoding the mouse homologues of the other C. reinhardtii C1d complex members are primarily expressed in motile ciliated tissues, suggesting a conserved function in mammalian motile cilia. The requirement for one of these C1d complex members, CFAP54, was identified in a mouse line with a gene-trapped allele. Homozygous mice have PCD characterized by hydrocephalus, male infertility, and mucus accumulation. The infertility results from defects in spermatogenesis. Motile cilia have a structural defect in the C1d projection, indicating that the C1d assembly mechanism requires CFAP54. This structural defect results in decreased ciliary beat frequency and perturbed cilia-driven flow. This study identifies a critical role for CFAP54 in proper assembly and function of mammalian cilia and flagella and establishes the gene-trapped allele as a new model of PCD.
INTRODUCTION
The motile cilium is an organelle vital to human health. Motile cilia extend from the basal bodies of epithelial cells in the respiratory system and the oviduct and ependymal cells in the brain (Ibanez-Tallon et al., 2003; Satir and Christensen, 2007; Lee, 2011). Unlike the immotile primary cilia, motile cilia beat in a wave-like motion to clear mucus in the respiratory system, enable cerebrospinal fluid (CSF) flow in the ventricular system of the brain, and assist in egg and sperm transport through the oviduct (Ibanez-Tallon et al., 2003; Satir and Christensen, 2007; Berbari et al., 2009; Lee, 2011). The core, also known as the axoneme, of motile cilia consists of a 9+2 microtubule structure with nine doublet microtubules surrounding a central microtubule pair (Ibanez-Tallon et al., 2003; Satir and Christensen, 2007; Lee, 2011). The ciliary motor force is generated by dynein arms associated with the outer doublet microtubules. The central microtubule apparatus is believed to play a crucial role in regulation of the dynein motor force through its association with the radial spokes that connect the outer doublets to the central pair (Smith and Lefebvre, 1997; Mitchell, 2004). Motile 9+0 cilia lacking a central pair are found on the embryonic node and play a role in establishing left–right asymmetry (Ibanez-Tallon et al., 2003; Satir and Christensen, 2007; Lee, 2011). Sperm flagella have a 9+2 axonemal structure similar to motile cilia and are required for sperm motility (Ibanez-Tallon et al., 2003; Satir and Christensen, 2007; Lee, 2011). Proteomic analyses of eukaryotic motile cilia have demonstrated a remarkable complexity (Ostrowski et al., 2002; Pazour et al., 2005), and the molecular mechanisms regulating ciliary motility are not fully understood.
Dysfunction of motile cilia and flagella typically results in primary ciliary dyskinesia (PCD), a pediatric syndrome affecting ∼1 in 16,000 live births (Lee, 2011; Knowles et al., 2013; Horani et al., 2014). Patients commonly suffer from chronic rhinosinusitis, bronchiectasis, chronic otitis media, male infertility, and situs inversus, with female infertility and hydrocephalus also reported in some individuals (Lee, 2011, 2013; Knowles et al., 2013; Horani et al., 2014). PCD is usually inherited in an autosomal recessive manner and is genetically heterogeneous (Lee, 2011; Knowles et al., 2013; Horani et al., 2014). Several mouse models have been described that have enabled a more detailed understanding of the disease’s pathology and identification of additional genes required for proper cilia function (Lee, 2011, 2013).
We previously demonstrated that loss of ciliary and flagellar protein 221 (CFAP221), also known as primary ciliary dyskinesia protein 1 (PCDP1), results in a PCD phenotype in mice (Lee et al., 2008). Mice homozygous for the nm1054 deletion have hydrocephalus, male infertility, accumulation of mucus in the sinus cavity, and an enhanced inflammatory response to pulmonary Streptococcus pneumoniae infection (Lee et al., 2008; McKenzie et al., 2013). The PCD phenotype is rescued by a transgene containing Cfap221 (Lee et al., 2008). Mutant respiratory epithelial cilia appear ultrastructurally normal but have a reduced beat frequency, while the male infertility results from an absence of mature sperm flagella, indicating that CFAP221 is required for both ciliary motility and spermatogenesis (Lee et al., 2008).
CFAP221 localizes to motile cilia and flagella in humans, mice, and the flagellated alga Chlamydomonas reinhardtii (Lee et al., 2008; DiPetrillo and Smith, 2010). The C. reinhardtii homologue FAP221 forms a complex with four additional proteins (FAP74, FAP54, FAP46, and C1d-87/WDR93) that associates with the C1d projection of the central microtubule apparatus (DiPetrillo and Smith, 2010; Brown et al., 2012). In response to changes in intracellular calcium, this C1d complex regulates dynein motor force through an interaction with the calcium-binding protein calmodulin (DiPetrillo and Smith, 2010, 2011). Ciliary dysfunction in mice lacking CFAP221 is consistent with flagellar defects in C. reinhardtii lacking FAP74 or FAP46 (DiPetrillo and Smith, 2010; Brown et al., 2012). Knockdown of FAP74 results in loss of the C1d projection, reduced and uncoordinated flagellar motility, and an inability of the C1d complex to properly assemble or associate with calmodulin (DiPetrillo and Smith, 2010). Similarly, a FAP46 null mutant also lacks the C1d projection, displays reduced flagellar beating, and has a disrupted C1d complex (Brown et al., 2012). These studies indicate that these individual members of the C1d complex are each required for proper function of the complex and suggest that there is little evidence of functional redundancy between complex members.
To further understand the role and requirement for the mammalian C1d complex, we demonstrate that the genes encoding the C1d complex members are primarily expressed in motile ciliated tissues, suggesting that they have a conserved function in motile cilia. In addition, we generated a mouse line with a gene-trapped allele of ciliary and flagellar protein 54 (Cfap54). Mutant mice have phenotypes associated with PCD, including hydrocephalus, male infertility, and accumulation of mucus in the sinuses. The male infertility results from a defect in spermatogenesis, and while respiratory epithelial cilia are present, they possess a defect in the C1d projection and have a reduced beat frequency, which results in perturbed cilia-driven flow. These findings suggest that CFAP54 is required for assembly and function of mammalian cilia and flagella.
RESULTS
The mouse genes encoding C1d complex members are primarily expressed in motile ciliated tissues
To investigate the role of the C1d complex members in mammals, we analyzed the tissue expression pattern of the genes encoding the mouse homologues by quantitative reverse transcription PCR (qRT-PCR). Cfap74, Cfap54, Cfap46, and Wdr93 are all expressed at highest levels in the testis and at a lower level in the lung, and Cfap74 and Cfap54 are also expressed at a low level in the brain (Figure 1). Each of these tissues possesses motile ciliated or flagellated cells. In contrast, there is little to no Cfap74, Cfap54, or Cfap46 expression in heart, kidney, or liver, which have only primary ciliated cells. This pattern is consistent with Cfap221, which was previously shown to express in the testis, respiratory system, and brain (Lee et al., 2008). These data suggest that the gene products have a conserved function in motile cilia. Interestingly, there is also a low level of Wdr93 expression in kidney, suggesting that this protein may serve an additional tissue-specific function.
FIGURE 1:

The C1d complex members are expressed in mammalian motile ciliated tissues. Tissue expression profiles for the mouse homologues of C. reinhardtii C1d complex members Cfap74 (A), Cfap54 (B), Cfap46 (C), and Wdr93 (D) by qRT-PCR analysis. Each gene is primarily expressed in tissues with cell types possessing motile cilia or flagella.
Generation of a Cfap54 gene-trapped mouse line
The Cfap54 gene is located on human chromosome 12 and mouse chromosome 10. The full-length mouse cDNA was sequenced from reverse-transcribed testis cDNA, and that sequence was aligned to the genomic sequence to experimentally validate the gene structure. The mouse Cfap54 gene has 68 coding exons and encodes a predicted 3171–amino acid protein (unpublished data). Based on the NCBI Conserved Domain search tool, the only predicted domain is a domain of unknown function represented by amino acids 103 to 643.
To understand the requirement for Cfap54 in mammalian cilia, we generated a mouse line with a gene-trapped allele of Cfap54 (Cfap54gt/gt). The gene-trapping cassette inserts into the first intron of Cfap54 and is expected to result in truncation of the Cfap54 transcript after exon one (Figure 2A). Following germ-line transmission of the gene-trap insertion, heterozygotes (Cfap54gt/+) were intercrossed to generate homozygous Cfap54gt/gt animals, which were obtained at ∼25%, the expected Mendelian ratio for autosomal recessive inheritance. Genotyping was enabled by PCR using three sets of primers (Figure 2B). Primers WT-F and WT-R flank the insertion site and only amplify the wild-type (WT) allele. Primers WT-F and GT1-R amplify the 5′ end of the gene-trapping cassette, and primers GT2-F and WT-R amplify the 3′ end. Detection of amplicons from all three primer pairs indicates a heterozygous genotype, while the homozygous Cfap54gt/gt allele is detected by only the gene trapping–cassette primer pairs WT1-F/GT1-R and GT2-F/WT-R. qRT-PCR was used to validate the mutation using forward and reverse primers in exons 56 and 57, respectively, which are downstream of the gene-trap insertion site and span an intron to ensure that genomic DNA is not amplified. While there is substantial expression of Cfap54 in WT testis, only a very low level of transcript was detected in the Cfap54gt/gt testis (Figure 2C), suggesting that the Cfap54gt/gt allele is a null.
FIGURE 2:
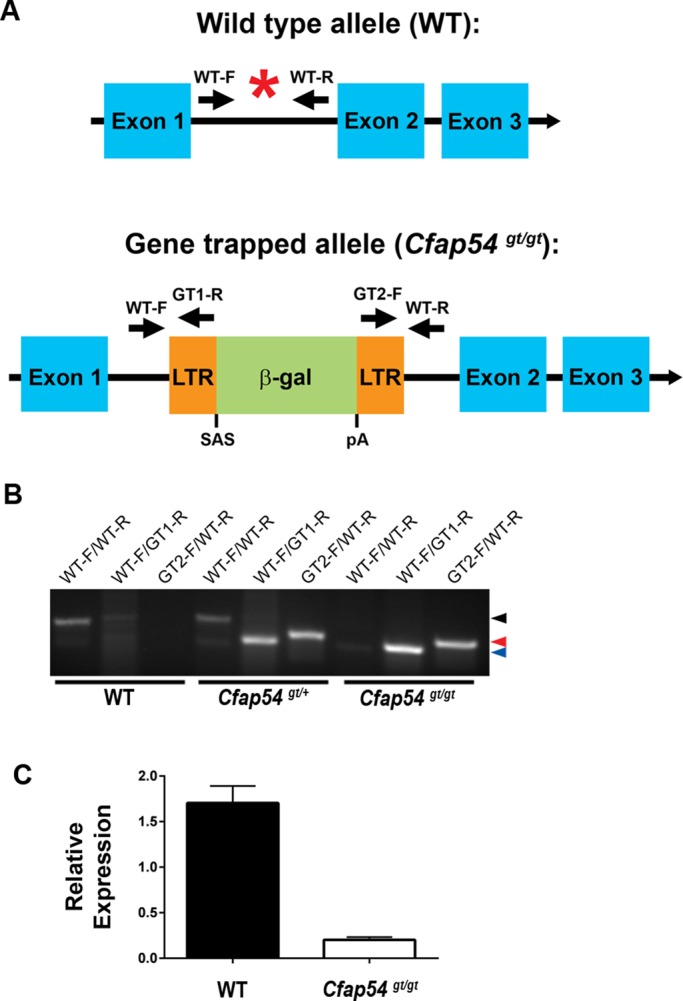
Generation of a Cfap54 gene-trapped mouse line. (A) Schematic diagram of the WT (top) and Cfap54 gene-trapped (bottom) alleles. The insertion site is indicated by a red asterisk in the WT allele. The gene-trapping cassette inserts between exons one and two of the 68-exon Cfap54 gene. The WT allele is detected by PCR primers WT-F and WT-R. The gene-trapped allele is detected by PCR primers WT-F and GT1-R at the 5′ end and GT2-F and WT-R at the 3′ end. β-gal: beta-galactosidase; LTR: long terminal repeat; pA: polyA tail; SAS: splice acceptor site. (B) Genotyping of WT, Cfap54gt/+, and Cfap54gt/gt mice by PCR. Primer pairs for each reaction are shown above the gel, and the genotype of the mouse is shown below. The bands for the WT allele (234 base pairs), the 5′ end of the gene-trapping cassette (186 base pairs), and the 3′ end of the gene-trapping cassette (194 base pairs) are indicated by black, blue, and red arrowheads, respectively. (C) qRT-PCR analysis of Cfap54 expression in WT and Cfap54gt/gt testis. Cfap54 is only detected at a very low level in mutant testis (p = 0.0002).
Mice lacking CFAP54 have hydrocephalus
The Cfap54gt/gt line was maintained on the C57BL/6J (B6) and 129S6/SvEvTac (129) backgrounds. There is known to be strain-dependent severity of hydrocephalus in PCD mouse models, likely due to the presence of genetic modifiers, with models typically exhibiting a more severe hydrocephalic phenotype on the B6 background than 129 (Lee, 2013; Finn et al., 2014). Cfap54gt/gt animals on the B6 background develop severe, gross hydrocephalus, which is indicated by an enlarged, dome-shaped head and results in early mortality (Figure 3A). Of five B6 Cfap54gt/gt animals that died naturally, three succumbed before 5 wk of age, while the other two died at approximately 7 wk of age. All B6 mutants utilized for phenotypic analysis were killed by 5 wk of age due to severe hydrocephalus. In contrast, 129 mutants and intercrossed (B6×129)F1 mutants did not develop severe, gross hydrocephalus and exhibited no signs of early mortality.
FIGURE 3:

Hydrocephalus in Cfap54gt/gt mice. (A) WT (top) and Cfap54gt/gt (bottom) mice. The enlarged cranial vault (arrowhead) observed in mutant mice is suggestive of hydrocephalus. (B–G) Coronal histological sections of B6 WT (B), B6 Cfap54gt/gt (C), 129 WT (D), 129 Cfap54gt/gt (E), (B6×129)F1 WT (F), and (B6×129)F1 Cfap54gt/gt (G) brains through the lateral ventricles showing one hemisphere. There is dramatic dilatation of the lateral ventricles (LV) in the B6 mutant brain, as well as loss of the ciliated ependymal cells, damage to the white matter (WM) and cerebral cortex (Ctx), and hemorrhaging (arrowhead) (C). Only mild dilatation without substantial secondary tissue damage is observed in 129 (E) or (B6×129)F1 (G) Cfap54gt/gt mutant brains. Sections are stained with H&E. Scale bars: 1 mm.
Coronal sections through the lateral ventricles of the B6 WT brain show very narrow ventricles and well-organized white matter and cerebral cortex (Figure 3B). In contrast, in the B6 Cfap54gt/gt brain, there is severe dilatation of the lateral ventricles, denudation of the ciliated ependymal cells that line the ventricles, damage to the underlying white matter and cerebral cortex, and evidence of intraventricular hemorrhaging (Figure 3C). The ventricular enlargement is presumably due to accumulation of CSF, and the tissue damage likely results from pressure exerted by the accumulating CSF against the ventricular walls. Although 129 and (B6×129)F1 Cfap54gt/gt mutants do not develop gross hydrocephalus or exhibit early mortality, there is evidence of mild dilatation of the lateral ventricles without substantial secondary tissue damage in mutants on the 129 (Figure 3, D and E) and (B6×129)F1 (Figure 3, F and G) backgrounds compared with WT.
Male infertility in Cfap54gt/gt mice results from defects in spermatogenesis
Because Cfap54gt/gt mutants die before the age of sexual maturity on the B6 background, fertility was assessed in (B6×129)F1 Cfap54gt/gt males. Four male mutants at 8 wk of age were paired with WT females for at least 3 d. Vaginal plugs were observed, but the females never became pregnant, suggesting that the males were infertile. The cause of the male infertility was investigated through histological analysis. In the WT testis, flagella on the elongating spermatids extend into the lumen of the seminiferous tubule during spermiogenesis (Figure 4A), the final stage of spermatogenesis before mature sperm are released to the epididymis (Hermo et al., 2010a – c). In the Cfap54gt/gt mutant, there is an absence of flagella in the lumen (Figure 4B), indicating a defect in spermatogenesis. Absence of any obvious defects associated with spermatogonia or spermatocytes suggest that loss of CFAP54 primarily affects spermiogenesis.
FIGURE 4:

Defects in Cfap54gt/gt spermatogenesis. (A and B) Histological sections of WT (A) and Cfap54gt/gt (B) testis. There is an absence of flagella extending into the lumen of the seminiferous tubule from the Cfap54gt/gt elongating spermatids. Sections are stained with H&E. (C and D) Transmission electron microscopic analysis of WT (C) and Cfap54gt/gt (D) testis showing disorganization of the axonemal microtubules (arrowheads) in cross-sections of Cfap54gt/gt flagella. (E–G) Light microscopic analysis of epididymal sperm from WT (E) and Cfap54gt/gt (F and G) mice showing shortened flagella (arrowheads) on mutant sperm. Sperm are stained with the Camco differential stain kit. Scale bars: 200 μm (A and B); 200 nm (C and D); 25 μm (E–G).
The absence of mature flagella was confirmed by transmission electron microscopy. Cross-sections of WT flagella show the normal 9+2 axonemal structure (Figure 4C). Mutant flagella were rarely detected, and those that were present were highly disorganized with axonemal structures largely absent (Figure 4D). The presence of only occasional flagellar remnants suggests that spermatogenesis is aborted early in spermiogenesis.
The defects observed in the Cfap54gt/gt testis are consistent with the morphology of mutant epididymal sperm, which were analyzed by light microscopy. WT sperm have a hook-shaped head and a long flagellum (Figure 4E). Very few sperm were found in the Cfap54gt/gt epididymis, and they possessed substantially shortened tails (Figure 4, F and G), confirming a defect in flagellar elongation. As some sperm were present in the mutant epididymis, it is likely that spermiation, the process by which mature spermatozoa are released into the lumen of the seminiferous tubule, is not affected by absence of CFAP54.
Cfap54gt/gt mice have an accumulation of mucus in the sinus cavity
Coronal sections through the maxillary sinus of B6 and (B6×129)F1 Cfap54gt/gt mice were analyzed by histology. While the sinus cavity is clear in WT mice (Figure 5A), there is an accumulation of mucus in the sinuses of Cfap54gt/gt mice (Figure 5B). As mucus is normally cleared by the epithelial cilia lining the airway, the accumulation in mutant animals suggests that loss of CFAP54 results in a defect in the mucociliary clearance mechanism. This condition could predispose the mice to respiratory infection, a phenotype common in PCD patients. In contrast to the severe defects in spermatid flagellar formation, immunohistochemical analysis of the ciliary marker acetylated tubulin shows that the Cfap54gt/gt airway epithelial cilia are present, organized, and appear morphologically indistinguishable from WT cilia (Figure 5, C and D), suggesting that ciliogenesis is unaffected in Cfap54gt/gt mice.
FIGURE 5:

Abnormal airway pathology in Cfap54gt/gt sinuses. (A and B) Coronal histological sections of the WT (A) and Cfap54gt/gt (B) maxillary sinuses. There is an accumulation of mucus (arrowheads) in the sinus cavity of Cfap54gt/gt mice. Sections are stained with H&E. (C and D) Immunohistochemical analysis of the ciliary marker acetylated tubulin in WT (C) and Cfap54gt/gt (D) trachea. Arrowheads indicate acetylated tubulin staining in the epithelial cilia. Scale bars: 100 μm (A and B); 5 μm (C and D).
Defects in ciliary structure and function in Cfap54gt/gt mice
Although Cfap54gt/gt cilia are present and organized (Figure 5D), transmission electron microscopy was used to investigate their axonemal ultrastructure. Unlike mutant sperm flagella, Cfap54gt/gt tracheal epithelial cilia have a normal 9+2 microtubule structure (Figure 6, A and B). However, there is an absence of electron-dense material, indicating a loss of the C1d projection of the central microtubule apparatus that is not observed in WT cilia (Figure 6, C and D). In addition, loss of the C1d projection appears to prevent association between the central apparatus and the radial spoke at this location. The consistency of this defect in mutant cilia is evident in an overlay of 10 axonemal cross-sections of Cfap54gt/gt cilia (Figure 6, E and F, and Supplemental Figure 1). Interestingly, this defect is in contrast to mice lacking CFAP221, which appear to have ultrastructurally normal cilia (Lee et al., 2008).
FIGURE 6:
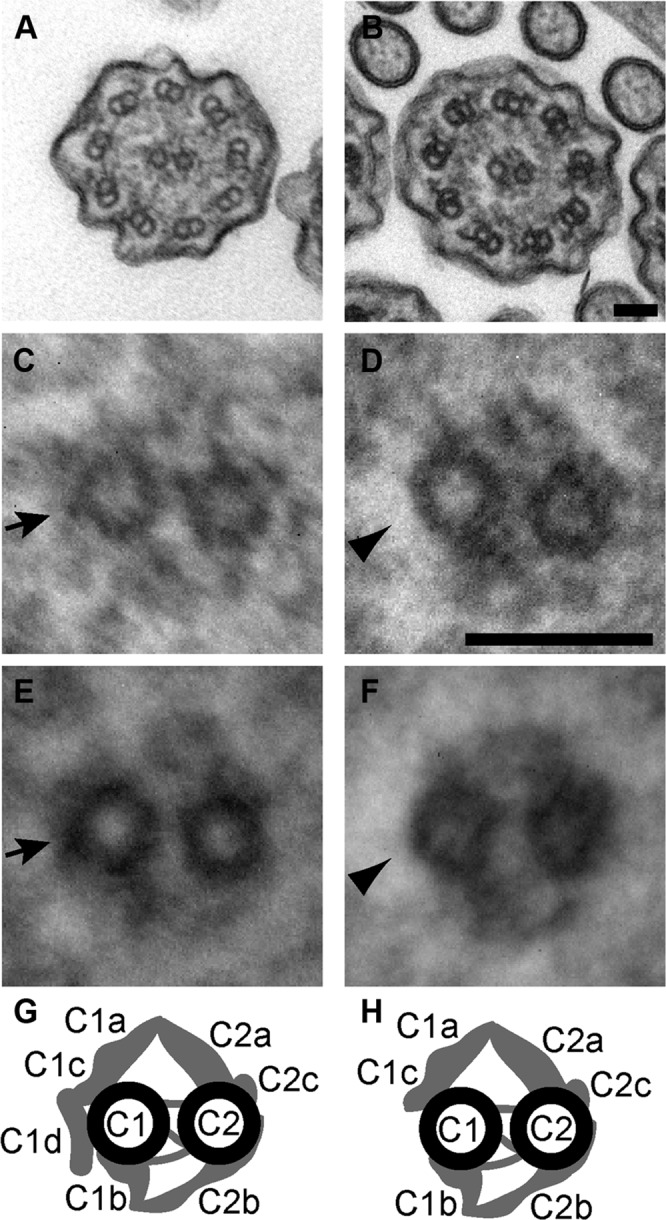
Structural ciliary abnormalities in Cfap54gt/gt mice. (A–F) Transmission electron microscopic analysis of WT (A, C, and E) and Cfap54gt/gt (B, D, and F) tracheal epithelial cilia. An absence of electron-dense material (arrowhead) in cross-sections of Cfap54gt/gt cilia indicates a loss of the C1d projection (arrow) of the central apparatus (A–D). Overlays of 10 WT (E) and Cfap54gt/gt (F) ciliary cross-sections show consistency of the absent C1d projection in mutants (arrowhead) compared with its presence in WT mice (arrow). Scale bars: 50 nm. (G and H) Schematic diagrams of the central pair apparatus indicating the presence (G) and absence (H) of the C1d projection in WT and Cfap54gt/gt mice, respectively (modified with permission from Lechtreck et al., 2008).
Analysis of Cfap54gt/gt tracheal ciliary beat frequency (CBF) shows a statistically significant decrease of ∼14.2% compared with WT animals (Figure 7A), suggesting that loss of the C1d projection results in a functional defect in ciliary motility. To determine whether this defect perturbs ciliary clearance, we analyzed the rate of cilia-driven ink flow over exposed tracheal epithelial cilia and brain ependymal cilia ex vivo. The flow rate over Cfap54gt/gt tracheal epithelial cilia is decreased by ∼46.9% compared with WT (Figure 7B), and the flow rate over Cfap54gt/gt ependymal cilia is decreased by ∼84.0% (Figure 7C). This defect in cilia-driven flow likely accounts for the inability to properly clear mucus in the sinus cavity and may also hinder CSF flow in the brain. Taken together, these data demonstrate that CFAP54 is required for proper assembly and function of mammalian cilia and flagella.
FIGURE 7:
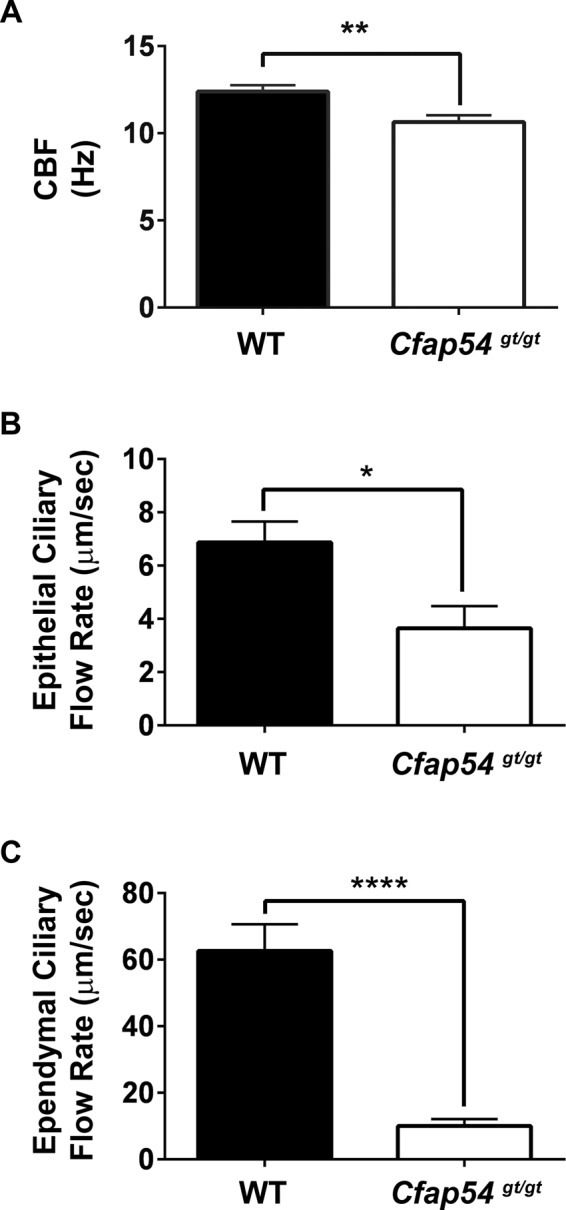
Functional defects in Cfap54gt/gt ciliary motility. (A) Analysis of tracheal epithelial CBF from WT and Cfap54gt/gt mice in beats per second (Hz). The Cfap54gt/gt CBF is ∼14.2% lower than WT (p = 0.003). (B and C) Analysis of ex vivo cilia-driven flow over ciliated tracheal epithelia (B) and brain ependyma (C) from WT and Cfap54gt/gt mice in micrometers per second. The Cfap54gt/gt tracheal epithelial ciliary flow rate is ∼46.9% lower than WT (p = 0.02), and the Cfap54gt/gt ependymal ciliary flow rate is ∼84.0% lower than WT (p = 5 × 10−6).
DISCUSSION
In this study, we have demonstrated that loss of CFAP54, a member of the C. reinhardtii C1d complex, in mice results in phenotypes associated with PCD, including hydrocephalus, male infertility, and accumulation of mucus in the sinus cavity. The C1d projection of the central microtubule apparatus is absent from tracheal epithelial cilia, resulting in decreases in CBF and cilia-driven flow. These findings suggest that CFAP54 is required for assembly of the mammalian central apparatus and proper ciliary motility. In contrast, the male infertility results from a defect in spermiogenesis that prevents formation of mature sperm flagella, indicating that CFAP54 is also required for spermatogenesis.
The ciliary defects in Cfap54gt/gt mice are consistent with loss of C1d complex members in C. reinhardtii. In the case of either artificial microRNA knockdown of FAP74 (DiPetrillo and Smith, 2010) or a FAP46 null mutant (Brown et al., 2012), absence of the C. reinhardtii C1d projection and reduced flagellar motility were reported. Assembly of the C1d complex was perturbed in each case, suggesting that complex assembly is critical for calcium-dependent regulation of axonemal dynein motor force (DiPetrillo and Smith, 2010, 2011; Brown et al., 2012). Based on the ciliary defect in Cfap54gt/gt mice (Figures 6 and 7), as well as expression of the genes encoding the complex members in motile ciliated mouse tissues (Figure 1) (Lee et al., 2008), it is likely that these proteins have a conserved role in assembly and function of the mammalian C1d projection. The apparent absence of an ultrastructural defect in cilia from mice lacking CFAP221 (Lee et al., 2008) suggests that CFAP54 is not functionally redundant with CFAP221 and that CFAP221 may be essential only for the function of the C1d projection rather than its assembly.
In contrast to the absence of the C1d projection in tracheal epithelial cilia, mature sperm flagella fail to form in Cfap54gt/gt mice, suggesting there are distinct differences in the formation and maintenance of these two organelles. Flagellar formation occurs during spermiogenesis, the complex final stage of spermatogenesis during which spermatids undergo substantial cellular rearrangement to form mature spermatozoa (Hermo et al., 2010a–c). Because there are only disorganized, abortive flagellar remnants present in the testis of Cfap54gt/gt mice, it is possible that elongating spermatids employ a quality-control mechanism that triggers abortion of spermiogenesis when the spermatids are defective. This hypothesis is consistent with several additional models, including mice lacking CFAP221, which also have male infertility due to a lack of mature sperm flagella despite the presence of cilia with only a reduction in beat frequency (Lee et al., 2008). Similarly, a reduced CBF and absence of mature sperm flagella were observed in mice lacking SPEF2 (Sironen et al., 2011) and SPAG6 (Sapiro et al., 2002; Teves et al., 2014), both of which are also predicted to localize to the central microtubule apparatus. The consistency of this phenotype extends beyond defects in the central apparatus. Dynein arm defects were observed in cilia from mice lacking DPCD/DNA polymerase lambda (Kobayashi et al., 2002; Zariwala et al., 2004) and Mns1 (Zhou et al., 2012), and both mutants display a loss of sperm flagella (Kobayashi et al., 2002; Zariwala et al., 2004; Zhou et al., 2012). In addition, mature flagella are absent from mice lacking adenylate kinase 7 (AK7), which possess cilia with a variety of ultrastructural abnormalities (Fernandez-Gonzalez et al., 2009). The differences between the ciliary and flagellar phenotypes in these models support the hypothesis that spermiogenesis is more stringently regulated than ciliogenesis. However, mature but abnormal sperm flagella were reported in two additional models of PCD. Mice lacking dynein heavy-chain MDNAH7 have ultrastructurally normal cilia with a reduced beat frequency and normal numbers of mature sperm with a reduced motility (Neesen et al., 2001). In addition, loss of Tektin-t results in both tracheal epithelial cilia and sperm flagella with inner dynein arm defects (Tanaka et al., 2004). Therefore, although less likely, it is alternatively possible that certain ciliary proteins, including CFAP54, could play a distinct role during spermatogenesis.
The strain-dependent severity of hydrocephalus and early mortality observed in Cfap54gt/gt mice is consistent with other PCD models, including those lacking CFAP221 and SPEF2, which we previously analyzed and reported for the B6, 129, and (B6×129)F1 backgrounds (Lee et al., 2008; Sironen et al., 2011; Finn et al., 2014). The severity of hydrocephalus on the B6 background compared with 129 or (B6×129)F1 mutants suggests that there are genetic modifiers of PCD-associated hydrocephalus segregating in the B6 strain, and several additional PCD and non-PCD models are consistent with this hypothesis (Lee, 2013). Further studies are required to identify these modifier genes and decipher their roles in conferring susceptibility to severe hydrocephalus in the context of ependymal ciliary dysfunction.
Despite the presence of PCD phenotypes in Cfap54gt/gt mice, there is no evidence of situs inversus or other laterality defects, which are typically associated with dysfunction of motile cilia on the embryonic node. This is likely due to the localization of CFAP54 to the central microtubule apparatus, which is absent in nodal cilia, and is consistent with mice lacking CFAP221. Biochemical and cell biological studies are required to fully uncover the role of these central apparatus proteins in regulation of mammalian ciliary assembly and function. To date, no human mutations have been identified in CFAP54 or the genes encoding the other members of this complex. However, given the extensive genetic heterogeneity of PCD and the complexity of the motile cilium, it is entirely plausible that CFAP54 mutations will be identified in the subset of PCD patients without situs inversus and ultimately contribute to the ever-growing spectrum of PCD genetics.
MATERIALS AND METHODS
Generation and maintenance of a Cfap54 gene-trapped mouse line
A B6 ES cell clone (IST10309B2) with a gene-trapping cassette inserted into the first intron of Cfap54, previously known as 4930485B16Rik, was obtained from the Texas A&M Institute of Genomic Medicine (College Station, TX; Figure 2A). The clone was injected into B6-Albino recipient blastocysts, which were implanted into CD-1 females. A male chimera with a high percentage of black coat color was identified and bred to WT C57BL/6Nhsd female mice to generate black F1 pups with germ-line transmission of the gene-trapped allele. Injections and chimera breeding were performed by Applied StemCell (Menlo Park, CA). Heterozygotes carrying the Cfap54<Gt(IST10309B2)Tigm>(MGI:3942561) allele were then backcrossed to B6 and 129 to congenicity. Mice homozygous for the gene-trapped allele are referred to herein as Cfap54gt/gt. Phenotypic or genetic analyses were performed on either B6 mice at 3–5 wk of age or (B6×129)F1 animals at 8 wk of age or later. All experiments involving animals were conducted with the approval of the Sanford Research Institutional Animal Care and Use Committee.
PCR
Cfap54gt/gt mice were genotyped by PCR. As depicted in Figure 2A, the WT allele was amplified using primers flanking the insertion site, while the gene-trapped allele was detected with primer pairs designed to the 5′ and 3′ ends of the cassette. Genomic DNA was purified using the DNeasy Blood and Tissue kit (Qiagen, Venlo, Netherlands) and amplified using all three primer pairs. PCR amplification products were visualized by agarose gel electrophoresis and imaged on a GelDoc-It Imaging System (UVP, Upland, CA).
RT PCR
For traditional RT-PCR, RNA was extracted from WT and Cfap54gt/gt testis using the Ambion RNAqueous Total RNA Isolation Kit (Life Technologies, Grand Island, NY). First-strand cDNA was synthesized from 1 μg RNA using either the SuperScript III First Strand kit (Life Technologies) or the GoScript Reverse Transcription System (Promega, Madison, WI). For sequencing the Cfap54 open reading frame, 21 overlapping segments spanning the full predicted cDNA sequence were amplified by PCR and sequenced through Eurofins Genomics (Huntsville, AL). The sequences were analyzed using the Sequencher software (Gene Codes, Ann Arbor, MI).
Quantitative RT-PCR was performed using the standard TaqMan approach. Total RNA was extracted from Cfap54gt/gt testis and WT brain, heart, kidney, liver, lung, and testis with TRIzol and purified using the PureLink RNA Mini Kit (Life Technologies) according to the manufacturer’s instructions. RNA integrity was assessed using a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA), and first-strand cDNA was synthesized from 1 μg RNA using the GoScript Reverse Transcription System (Promega). Quantitative PCR (qPCR) was performed in a Stratagene Mx3000P qPCR system (Agilent Technologies) using the ABsolute Blue QPCR Mix (Thermo Scientific, Waltham, MA) and a 1:20 dilution of cDNA following the manufacturer’s instructions. Gene expression levels were normalized to Hprt. TaqMan primers and probes were designed using Beacon Designer software (Premier Biosoft, Palo Alto, CA) for mouse Cfap74 (F: CCAGTATGGAAGAACCTAC; R: CTGGACTCTTTCGTCATC; probe: TGTCCACAAGGTCCTCATCAGAA), Cfap54 (F: CCTCATGTCATGCTACTG; R: CCACTTACTGATGTCCTT; probe: CTTGCTGCTGGAACATACCTGC), Cfap46 (F: CTCTCCATCTGCTGTACG; R: CTCTGTGTCTGTCTCCAG; probe: CTTAGTGCCTACCAAGAGTTCCTCC), Wdr93 (F: GACAGTCTTTATCACATCAA; R: GAGAGATTTCCATCTTCAG; Probe: CTGCTTGCTGCTATCATCCACTTC), and Hprt (F: GATCCATTCCTATGACTGTA; R: TCTCCACCAATAACTTTTATG; probe: TCATTACAGTAGCTCTTCAGTCTGAT). Relative gene expression data were analyzed using the delta-delta Ct method as previously described (Pfaffl, 2001). For tissue expression pattern analysis, n = 8 (Figure 1), and for mutation validation, n = 7 WT and 7 Cfap54gt/gt (Figure 2C).
Sequence analysis
First-strand cDNA spanning the entire predicted Cfap54 open reading frame was amplified in 21 overlapping segments and sequenced as described above. The overlapping sequences were assembled into a contig using the Sequencher software (Gene Codes) to determine the complete Cfap54 cDNA sequence. The contig was then aligned to the genomic sequence from the Ensembl database (Flicek et al., 2014) to identify the size of each exon and the location of each splice site. Domain analysis in the predicted gene product was accomplished using the NCBI Conserved Domain search tool (Marchler-Bauer and Bryant, 2004).
Histology
Heads from B6 and (B6×129)F1 WT and Cfap54gt/gt mice were immersion fixed in Bouin’s fixative until the bones were decalcified, after which coronal slices were cut through the lateral ventricles of the brain and the maxillary sinuses. Testes from (B6×129)F1 WT and Cfap54gt/gt mice were immersion fixed in 10% buffered Formalin. Fixed testes, brain slices, and sinus slices were embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E) as previously described (McKenzie et al., 2013; Finn et al., 2014). Stained sections were analyzed by light microscopy on an Olympus IX71 (Olympus, Tokyo, Japan) or a Nikon 90i (Nikon, Tokyo, Japan) upright microscope. For brains, n = 4 B6 WT, 7 B6 Cfap54gt/gt, 5 129 WT, 4 129 Cfap54gt/gt, 2 (B6×129)F1 WT, and 2 (B6×129)F1 Cfap54gt/gt; for sinuses, n = 4 B6 WT, 6 B6 Cfap54gt/gt, 6 (B6×129)F1 WT, and 8 (B6×129)F1 Cfap54gt/gt; for testes, n = 5 (B6×129)F1 WT and 11 (B6×129)F1 Cfap54gt/gt.
Spermatozoa preparations
Spermatozoa were collected from the epididymis of (B6×129)F1 WT and Cfap54gt/gt mice, diluted in phosphate-buffered saline (PBS), and spread onto slides. The slides were dried, fixed in methanol, stained with the Camco differential stain kit (Cambridge Diagnostic Products, Fort Lauderdale, FL), and analyzed by light microscopy on an Olympus IX71 upright microscope. Samples were n = 5 WT and 6 Cfap54gt/gt.
Immunohistochemistry
Tracheae from (B6×129)F1 WT and Cfap54gt/gt mice were immersion fixed in 10% buffered Formalin. Fixed tissue was embedded in paraffin, sectioned, and stained using the BenchMark XT automated-slide staining system (Ventana Medical Systems, Tucson, AZ) as previously described (McKenzie et al., 2013; Finn et al., 2014). The mouse acetylated tubulin antibody (Sigma-Aldrich, St. Louis, MO) was used at a 1:6000 dilution, and the Biotin SP-conjugated AffiniPure goat anti-mouse secondary antibody (Jackson ImmunoResearch, West Grove, PA) was used at 1:1000. The slides were visualized on a Nikon Ni-E upright microscope. Samples were n = 5 WT and 11 Cfap54gt/gt.
Transmission electron microscopy
Tracheae from (B6×129)F1 WT and Cfap54gt/gt mice were immersion fixed in 100 mM sodium cacodylate buffer (pH 7.2) with 2.5% glutaraldehyde and 2% paraformaldehyde at 4°C overnight. Testes from the same mice were perforated with a 30G needle and immersion fixed in the same fixative at 4°C for 1 h. The testis was then bisected and placed in fresh fixative overnight at 4°C. Fixed tissues were washed three times with 100 mM sodium cacodylate (pH 7.2), postfixed with 1% OsO4 in 75 mM sodium cacodylate for 1 h on ice, washed three times with cold water, and stained with 1% uranyl acetate at 4°C overnight. The samples were then dehydrated, embedded in epon resin, and thin-sectioned. Sections were analyzed by transmission electron microscopy using a Philips CM10 electron microscope (Philips Innovation Services, Eindhoven, Netherlands). For image averages, 10 WT or Cfap54gt/gt axonemal cross-sections were aligned and superimposed with 20% opacity using Adobe Photoshop software (Adobe Systems, San Jose, CA). Samples were n = 4 WT and 3 Cfap54gt/gt.
CBF analysis
Tracheae from (B6×129)F1 WT and Cfap54gt/gt mice were dissected into DMEM with 1% penicillin–streptomycin. CBF was analyzed using the Sisson-Ammons Video Analysis system as previously described (Sisson et al., 2003; Lee et al., 2008; Sironen et al., 2011). Statistical significance was determined by Student’s t test. Samples were n = 10 WT and 10 Cfap54gt/gt.
Ciliary clearance assay
Brains from (B6×129)F1 WT and Cfap54gt/gt mice were dissected in PBS, and flow of India ink over exposed ependymal cilia was analyzed as previously described (Finn et al., 2014). The ependymal flow rate was calculated from the distance traveled by the front of the cilia-driven ink stream over 2 s. Tracheae from (B6×129)F1 WT and Cfap54gt/gt mice were dissected in DMEM (HyClone, GE Healthcare Life Sciences, Pittsburgh, PA) and bisected longitudinally. Tracheae were then transferred to a Sylgard-coated dish (Dow Corning, Midland, MI) with PBS and immobilized. India ink was diluted as described (Finn et al., 2014) and deposited onto the exposed ciliated epithelia at the bottom of the trachea. Ink flow over the epithelial cilia was analyzed as described for the brain (Finn et al., 2014). The epithelial flow rate was calculated from the time it took the front of the ink stream to travel across a superimposed grid, with four separate measurements taken for each animal. Statistical analyses for both ependymal and epithelial ciliary clearance were performed using GraphPad Prism (GraphPad Software, La Jolla, CA), and statistical significance was determined by t test using the Holm-Sidak method (alpha = 5.000%). For brains, n = 7 WT and 9 Cfap54gt/gt; for tracheae, n = 9 WT and 5 Cfap54gt/gt.
Nucleotide sequence accession number
The mouse Cfap54 cDNA sequence was deposited in GenBank as accession number KM983399.
Supplementary Material
Acknowledgments
We thank Claire Evans, Sam Dooyema, and Taylor Maier for technical assistance. This work was supported by National Institutes of Health (NIH) grants P20GM103620 and R01AA008769 (J.H.S.) and R37GM030626 (G.B.W.), the Robert W. Booth Endowment at the University of Massachusetts Medical School (G.B.W.), and Sanford Research. T.V.K. was supported by the Sanford Research Science Educator Research Fellowship Program funded by NIH grant R25HD072596. The Sanford Research Molecular Genetics Core, the Sanford Research Molecular Pathology Core, the Sanford Research Imaging Core, and the University of Massachusetts Medical School Core Electron Microscopy Facility were all supported by the NIH (grants P20GM103620 for Molecular Genetics, P20GM103620 and P20GM103548 for Molecular Pathology and Imaging, and S10RR027897 for Electron Microscopy).
Abbreviations used:
- 129
129S6/SvEvTac
- AK7
adenylate kinase 7
- B6
C57BL/6J
- CBF
ciliary beat frequency
- CFAP221
ciliary and flagellar protein 221
- CFAP54
ciliary and flagellar protein 54
- CSF
cerebrospinal fluid
- H&E
hematoxylin and eosin
- PBS
phosphate-buffered saline
- PCD
primary ciliary dyskinesia
- PCDP1
primary ciliary dyskinesia protein 1
- qRT-PCR
quantitative reverse transcription PCR
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.3140) on July 29, 2015.
REFERENCES
- Berbari NF, O’Connor AK, Haycraft CJ, Yoder BK. The primary cilium as a complex signaling center. Curr Biol. 2009;19:R526–R535. doi: 10.1016/j.cub.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JM, Dipetrillo CG, Smith EF, Witman GB. A FAP46 mutant provides new insights into the function and assembly of the C1d complex of the ciliary central apparatus. J Cell Sci. 2012;125:3904–3913. doi: 10.1242/jcs.107151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPetrillo CG, Smith EF. Pcdp1 is a central apparatus protein that binds Ca2+-calmodulin and regulates ciliary motility. J Cell Biol. 2010;189:601–612. doi: 10.1083/jcb.200912009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPetrillo CG, Smith EF. The Pcdp1 complex coordinates the activity of dynein isoforms to produce wild-type ciliary motility. Mol Biol Cell. 2011;22:4527–4538. doi: 10.1091/mbc.E11-08-0739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Gonzalez A, Kourembanas S, Wyatt TA, Mitsialis SA. Mutation of murine adenylate kinase 7 underlies a primary ciliary dyskinesia phenotype. Am J Respiratory Cell Mol Biol. 2009;40:305–313. doi: 10.1165/rcmb.2008-0102OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R, Evans CC, Lee L. Strain-dependent brain defects in mouse models of primary ciliary dyskinesia with mutations in Pcdp1 and Spef2. Neuroscience. 2014;277:552–567. doi: 10.1016/j.neuroscience.2014.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, et al. Ensembl 2014. Nucleic Acids Res. 2014;42:D749–D755. doi: 10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermo L, Pelletier RM, Cyr DG, Smith CE. Surfing the wave, cycle, life history, and genes/proteins expressed by testicular germ cells. Part 1: background to spermatogenesis, spermatogonia, and spermatocytes. Microsc Res Tech. 2010a;73:241–278. doi: 10.1002/jemt.20783. [DOI] [PubMed] [Google Scholar]
- Hermo L, Pelletier RM, Cyr DG, Smith CE. Surfing the wave, cycle, life history, and genes/proteins expressed by testicular germ cells. Part 2: changes in spermatid organelles associated with development of spermatozoa. Microsc Res Tech. 2010b;73:279–319. doi: 10.1002/jemt.20787. [DOI] [PubMed] [Google Scholar]
- Hermo L, Pelletier RM, Cyr DG, Smith CE. Surfing the wave, cycle, life history, and genes/proteins expressed by testicular germ cells. Part 3: developmental changes in spermatid flagellum and cytoplasmic droplet and interaction of sperm with the zona pellucida and egg plasma membrane. Microsc Res Tech. 2010c;73:320–363. doi: 10.1002/jemt.20784. [DOI] [PubMed] [Google Scholar]
- Horani A, Brody SL, Ferkol TW. Picking up speed: advances in the genetics of primary ciliary dyskinesia. Pediatr Res. 2014;75:158–164. doi: 10.1038/pr.2013.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez-Tallon I, Heintz N, Omran H. To beat or not to beat: roles of cilia in development and disease. Hum Mol Genet. 2003;12(spec. no. 1):)R27–R35. doi: 10.1093/hmg/ddg061. [DOI] [PubMed] [Google Scholar]
- Knowles MR, Daniels LA, Davis SD, Zariwala MA, Leigh MW. Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease. Am J Respir Crit Care Med. 2013;188:913–922. doi: 10.1164/rccm.201301-0059CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi Y, Watanabe M, Okada Y, Sawa H, Takai H, Nakanishi M, Kawase Y, Suzuki H, Nagashima K, Ikeda K, Motoyama N. Hydrocephalus, situs inversus, chronic sinusitis, and male infertility in DNA polymerase lambda-deficient mice: possible implication for the pathogenesis of immotile cilia syndrome. Mol Cell Biol. 2002;22:2769–2776. doi: 10.1128/MCB.22.8.2769-2776.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechtreck KF, Delmotte P, Robinson ML, Sanderson MJ, Witman GB. Mutations in Hydin impair ciliary motility in mice. J Cell Biol. 2008;180:633–643. doi: 10.1083/jcb.200710162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee L. Mechanisms of mammalian ciliary motility: insights from primary ciliary dyskinesia genetics. Gene. 2011;473:57–66. doi: 10.1016/j.gene.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Lee L. Riding the wave of ependymal cilia: genetic susceptibility to hydrocephalus in primary ciliary dyskinesia. J Neurosci Res. 2013;91:1117–1132. doi: 10.1002/jnr.23238. [DOI] [PubMed] [Google Scholar]
- Lee L, Campagna DR, Pinkus JL, Mulhern H, Wyatt TA, Sisson JH, Pavlik JA, Pinkus GS, Fleming MD. Primary ciliary dyskinesia in mice lacking the novel ciliary protein Pcdp1. Mol Cell Biol. 2008;28:949–957. doi: 10.1128/MCB.00354-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Bryant SH. CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 2004;32:W327–W331. doi: 10.1093/nar/gkh454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie CW, Klonoski JM, Maier T, Trujillo G, Vitiello PF, Huber VC, Lee L. Enhanced response to pulmonary Streptococcus pneumoniae infection is associated with primary ciliary dyskinesia in mice lacking Pcdp1 and Spef2. Cilia. 2013;2:18. doi: 10.1186/2046-2530-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DR. Speculations on the evolution of 9+2 organelles and the role of central pair microtubules. Biol Cell. 2004;96:691–696. doi: 10.1016/j.biolcel.2004.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neesen J, Kirschner R, Ochs M, Schmiedl A, Habermann B, Mueller C, Holstein AF, Nuesslein T, Adham I, Engel W. Disruption of an inner arm dynein heavy chain gene results in asthenozoospermia and reduced ciliary beat frequency. Hum Mol Genet. 2001;10:1117–1128. doi: 10.1093/hmg/10.11.1117. [DOI] [PubMed] [Google Scholar]
- Ostrowski LE, Blackburn K, Radde KM, Moyer MB, Schlatzer DM, Moseley A, Boucher RC. A proteomic analysis of human cilia: identification of novel components. Mol Cell Proteomics. 2002;1:451–465. doi: 10.1074/mcp.m200037-mcp200. [DOI] [PubMed] [Google Scholar]
- Pazour GJ, Agrin N, Leszyk J, Witman GB. Proteomic analysis of a eukaryotic cilium. J Cell Biol. 2005;170:103–113. doi: 10.1083/jcb.200504008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapiro R, Kostetskii I, Olds-Clarke P, Gerton GL, Radice GL, Strauss IJ. Male infertility, impaired sperm motility, and hydrocephalus in mice deficient in sperm-associated antigen 6. Mol Cell Biol. 2002;22:6298–6305. doi: 10.1128/MCB.22.17.6298-6305.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satir P, Christensen ST. Overview of structure and function of mammalian cilia. Annu Rev Physiol. 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- Sironen A, Kotaja N, Mulhern H, Wyatt TA, Sisson JH, Pavlik JA, Miiluniemi M, Fleming MD, Lee L. Loss of SPEF2 function in mice results in spermatogenesis defects and primary ciliary dyskinesia. Biol Reprod. 2011;85:690–701. doi: 10.1095/biolreprod.111.091132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisson JH, Stoner JA, Ammons BA, Wyatt TA. All-digital image capture and whole-field analysis of ciliary beat frequency. J Microsc. 2003;211:103–111. doi: 10.1046/j.1365-2818.2003.01209.x. [DOI] [PubMed] [Google Scholar]
- Smith EF, Lefebvre PA. The role of central apparatus components in flagellar motility and microtubule assembly. Cell Motil Cytoskeleton. 1997;38:1–8. doi: 10.1002/(SICI)1097-0169(1997)38:1<1::AID-CM1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Iguchi N, Toyama Y, Kitamura K, Takahashi T, Kaseda K, Maekawa M, Nishimune Y. Mice deficient in the axonemal protein Tektin-t exhibit male infertility and immotile-cilium syndrome due to impaired inner arm dynein function. Mol Cell Biol. 2004;24:7958–7964. doi: 10.1128/MCB.24.18.7958-7964.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teves ME, Sears PR, Li W, Zhang Z, Tang W, van Reesema L, Costanzo RM, Davis CW, Knowles MR, Strauss JF, 3rd, Zhang Z. Sperm-associated antigen 6 (SPAG6) deficiency and defects in ciliogenesis and cilia function: polarity, density, and beat. PLoS One. 2014;9:e107271. doi: 10.1371/journal.pone.0107271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zariwala M, O’Neal WK, Noone PG, Leigh MW, Knowles MR, Ostrowski LE. Investigation of the possible role of a novel gene, DPCD, in primary ciliary dyskinesia. Am J Respiratory Cell Mol Biol. 2004;30:428–434. doi: 10.1165/rcmb.2003-0338RC. [DOI] [PubMed] [Google Scholar]
- Zhou J, Yang F, Leu NA, Wang PJ. MNS1 is essential for spermiogenesis and motile ciliary functions in mice. PLoS Genet. 2012;8:e1002516. doi: 10.1371/journal.pgen.1002516. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.