

MAPKs are activated by dual phosphorylation. Much of the MAPK Fus3 is monophosphorylated and acts to inhibit signaling in vivo. Computational models reveal how a kinase scaffold and phosphatase act together to dynamically regulate dual-phosphorylated and monophosphorylated MAPKs and the downstream signal.
Abstract
Protein kinases regulate a broad array of cellular processes and do so through the phosphorylation of one or more sites within a given substrate. Many protein kinases are themselves regulated through multisite phosphorylation, and the addition or removal of phosphates can occur in a sequential (processive) or a stepwise (distributive) manner. Here we measured the relative abundance of the monophosphorylated and dual-phosphorylated forms of Fus3, a member of the mitogen-activated protein kinase (MAPK) family in yeast. We found that upon activation with pheromone, a substantial proportion of Fus3 accumulates in the monophosphorylated state. Introduction of an additional copy of Fus3 lacking either phosphorylation site leads to dampened signaling. Conversely, cells lacking the dual-specificity phosphatase (msg5Δ) or that are deficient in docking to the MAPK-scaffold (Ste5ND) accumulate a greater proportion of dual-phosphorylated Fus3. The double mutant exhibits a synergistic, or “synthetic,” supersensitivity to pheromone. Finally, we present a predictive computational model that combines MAPK scaffold and phosphatase activities and is sufficient to account for the observed MAPK profiles. These results indicate that the monophosphorylated and dual-phosphorylated forms of the MAPK act in opposition to one another. Moreover, they reveal a new mechanism by which the MAPK scaffold acts dynamically to regulate signaling.
INTRODUCTION
All cells must detect, interpret, and respond to a broad array of environmental signals. Most signal transduction pathways depend on the phosphorylation of cellular proteins by protein kinases. Among the best known are the mitogen-activated protein kinases (MAPKs), which are phosphorylated and activated by a MAPK kinase (MAPKK), which are in turn phosphorylated and activated by a MAPKK kinase (MAPKKK). These cascades respond to a variety of stress conditions and secreted hormones and are conserved in organisms ranging from yeast to humans.
The budding yeast Saccharomyces cerevisiae uses a typical MAPK signaling pathway to initiate the mating response. Haploid cells of the opposite mating type (a- or α-cells) secrete peptide pheromones that bind to cell surface receptors. These receptors activate a G protein and a protein kinase cascade comprising Ste11 (MAPKKK), Ste7 (MAPKK), and Fus3 (MAPK). All three of these kinases interact with the scaffold protein Ste5, which recruits the constituent kinases to the activated G protein at the plasma membrane. In addition to Fus3, a second MAPK Kss1 is activated by Ste7 but does not interact directly with Ste5. Thus, whereas Ste5 is required for activation of Fus3 (Breitkreutz et al., 2001; Andersson et al., 2004; Kusari et al., 2004; Maleri et al., 2004; Flatauer et al., 2005), it binds poorly to Kss1 and is not required for Kss1-mediated processes (Choi et al., 1994; Printen and Sprague, 1994; Kusari et al., 2004). Once activated, Fus3 goes on to phosphorylate proteins necessary for new gene transcription, cell division arrest, polarized cell expansion (chemotropic growth and shmoo formation), and cell–cell fusion (Wang and Dohlman, 2004). In the absence of Fus3, Kss1 can mediate the pheromone-dependent gene induction program (Roberts et al., 2000; Breitkreutz et al., 2001), whereas other responses, such as chemotropic growth, are abrogated (Hao et al., 2008). Thus, although Fus3 and Kss1 are activated by the same upstream protein kinases, they have distinct cellular functions. Here our focus is on the function of Fus3 and its scaffold, Ste5.
Based on the specificity of binding, it was long assumed that Ste5 acts to direct signaling toward Fus3 and away from Kss1. Although clearly required for Fus3 activation, Ste5 does not prevent the activation of Kss1 by pheromone (Choi et al., 1994; Printen and Sprague, 1994; Breitkreutz et al., 2001; Andersson et al., 2004; Maleri et al., 2004; Flatauer et al., 2005). It was later proposed that Ste5 is an allosteric regulator of Fus3. In support of that model, a fragment of Ste5 was demonstrated to promote Fus3 autophosphorylation at Tyr-182 (Bhattacharyya et al., 2006), one of two sites normally phosphorylated upon full activation of the kinase (Gartner et al., 1992). The monophosphorylated form of Fus3 was partially (20–25%) activated and competent to phosphorylate a variety of substrates in vitro (Bhattacharyya et al., 2006).
If Ste5 were found to activate Fus3 in vivo, even partially, it would raise a host of new questions. What proportion of Fus3 is monophosphorylated in this manner? How does the pool of monophosphorylated kinase affect downstream signaling? Does autophosphorylation on one site facilitate or hinder subsequent phosphorylation at the second site? If autophosphorylation on one site were followed by a dissociation event and transphosphorylation on the second site, the scaffold might act to delay signaling. Alternatively, if the scaffold stabilizes the interaction of Ste7 and Fus3, phosphorylation might occur in a processive manner, without substrate dissociation and reassociation. This would have the effect of minimizing the monophosphorylated species and accelerating full activation of the MAPK (Ferrell, 2000; Levchenko et al., 2000). Thus scaffold proteins could either speed or slow signal transduction, depending on the mechanism used.
Apart from altering the kinetics of MAPK activation, a processive or distributive mechanism could influence the dose–response relationship for the pathway. Theoretical and biochemical studies indicate that processive phosphorylation should favor a graded output, one that aligns MAPK activity with the graded input stimulus. Accordingly, scaffold proteins might convert an inherently switch-like MAPK to one that is more graded (Ferrell, 2000; Levchenko et al., 2000). Conversely, distributive phosphorylation is believed to produce an all-or-none (switch-like) output (Huang and Ferrell, 1996). In support of this second model, a number of functional activity assays reveal that Ste5 is necessary for slow and ultrasensitive activation, as is observed for Fus3 but not Kss1 (Hao et al., 2008; Malleshaiah et al., 2010). Thus the distinction between processive and distributive phosphorylation has important implications for signal encoding (Piala et al., 2014).
Considering the importance of how MAPKs are turned on and off, relatively little is known about how this occurs in cells. For example, it is still not established whether Ste5, or scaffolds in general, dictate a processive versus distributive phosphorylation mechanism in vivo. To address this question, we employ a new method to quantify the abundance of monophosphorylated and dual-phosphorylated Fus3 in the cell over time and in the presence or absence of Fus3 regulators; these regulators include the scaffold Ste5, as well as the MAPK phosphatases Ptp2, Ptp3, and Msg5. We show that Ste5 and Msg5 in particular act to limit full activation of Fus3 and that nearly half of the Fus3 pool remains nonphosphorylated after pathway stimulation. Of the protein that is phosphorylated, nearly half is in the monophosphorylated state. Whereas phosphorylation appears to be processive, dephosphorylation appears to be distributive. Finally, we show that monophosphorylated Fus3 does not activate, but instead impedes, the pheromone response in vivo. Together these findings indicate that the monophosphorylated species acts in a dynamic and dominant manner to limit signal transduction.
RESULTS
The phosphorylation state of Fus3 determines mating pathway output
It has long been recognized that MAP kinases must be phosphorylated on two residues in order to achieve full catalytic activity. This was originally documented for ERK2, for which the protein is phosphorylated on Tyr-185 and subsequently on the neighboring Thr-183 (Haystead et al., 1992; Ferrell and Bhatt, 1997). This dual phosphorylation alters the conformation of the protein, thereby enabling ATP to bind to the catalytic site (Canagarajah et al., 1997). These “activation loop” residues are conserved in the yeast ERK2 homologue Fus3, and mass spectrometry analysis indicates that the same order of events occurs (Hur et al., 2008). Our goal here was to determine the relative abundance and potential function of the monophosphorylated MAPK species.
We began by constructing genomically integrated mutant forms of Fus3 that cannot be phosphorylated at either of the activation loop residues: Tyr-182, Thr-180, or both. We then monitored the transcription of a mating-specific gene reporter. As shown in Figure 1A, cells in which the endogenous FUS3 gene had been deleted produced a maximal response to pheromone resembling the response of wild-type cells. In the absence of Fus3, pathway induction is preserved due to the action of a homologous MAPK, Kss1, as reported previously (Bardwell, 2005). In contrast, cells expressing a variant of Fus3 that can only be monophosphorylated (Fus3T180A or Fus3Y182F) exhibited a substantially diminished response. This reduction in signal activity is particularly striking, given the near-normal response seen in the complete absence of Fus3 expression.
FIGURE 1:
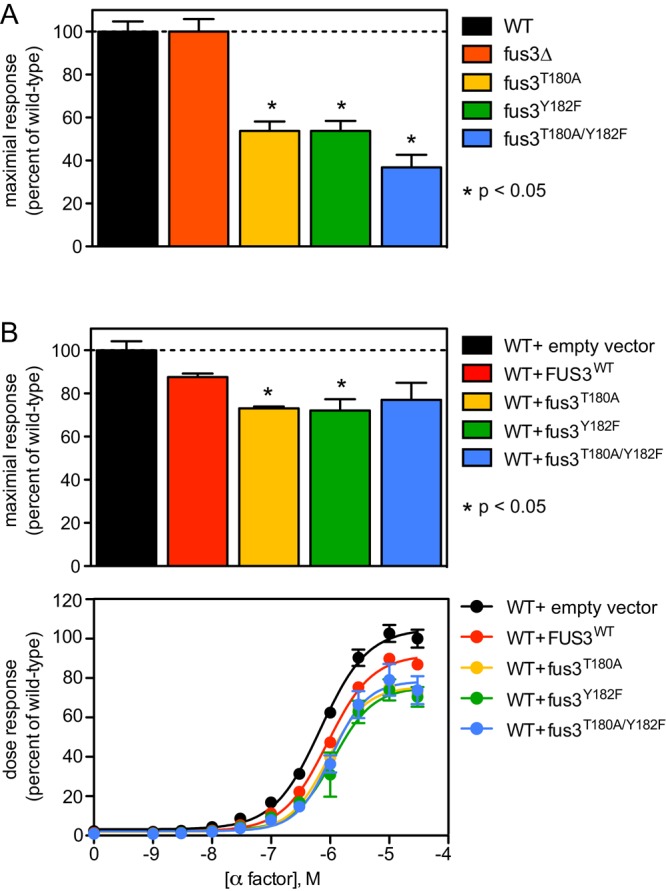
Signal inhibition by monophosphorylated Fus3. (A) Transcription reporter (FUS1-lacZ) activity in wild-type (WT), Fus3-deficient, or Fus3 activation loop mutant strains (fus3T180A, fus3Y182F, and combined fus3T180A/Y182F) stimulated with 10 μM α-factor. (B) Same analysis in wild-type cells bearing a single-copy plasmid with no insert (vector), wild-type FUS3, or mutations in the Fus3 activation loop. FUS1-lacZ data are presented as a percentage of maximum activity in wild-type cells (A and B, top) and as a full dose–response curve (B, bottom). Results report ±SEM (n = 3) for each data point, but this is not visible in every case. *Statistically significant Student’s t test of pairwise comparisons for wild type and the individual mutants.
To determine whether the Fus3 mutants limit signaling via the primary (Fus3-mediated) pathway, we repeated these experiments but this time in cells that coexpress endogenous wild-type Fus3 and either an additional copy of the MAPK or one of the phosphosite mutants. As shown in Figure 1B, signaling was diminished most substantially in cells in which the second copy of Fus3 was mutated (Fus3T180A or Fus3Y182F). Thus the monophosphorylated Fus3 dampens the mating response and does so in a genetically dominant manner. These findings suggest that the partially phosphorylated form of Fus3 acts to inhibit signaling.
The phosphorylation state of Fus3 is dynamically regulated
Having shown that monophosphorylated Fus3 inhibits the mating transcription response, we next sought to measure the relative abundance of the monophosphorylated protein in cells. To that end, we used the Phos-tag reagent. Phos-tag is a metal-coordinating small molecule with a high affinity for phosphorylated serine, threonine, and tyrosine (Kinoshita et al., 2006). Addition of Phos-tag to acrylamide gels slows the migration of polypeptides and does so in proportion to the number of phosphorylations on the molecule. As shown in Figure 2A, the Phos-tag method revealed three species of Fus3 (corresponding to the dual-phosphorylated, monophosphorylated, and nonphosphorylated forms of the protein) after stimulation with 10 μM pheromone, a dose that produces full pathway activation (Figure 1B). As expected, the dual-phosphorylated form of Fus3 was enriched after pheromone treatment, but monophosphorylated Fus3 was still readily detectable. As a control, we established that the individual phosphosite mutants yielded just two bands each, corresponding to the monophosphorylated and nonphosphorylated species (Figure 2B). Whereas the pool of monophosphorylated species was enriched in these mutants, the proportion of nonphosphorylated protein was unaffected (Figure 2B). The Fus3Y182F mutant migrated anomalously, equivalent to that of the activated wild-type form of the protein. In each case, only the slower-migrating phosphorylated species was evident using p44/p42 antibodies raised against a phosphorylated MAPK peptide. None of the phosphorylated species was detected in the absence of the upstream MAPK kinase Ste11 (Figure 2A). No bands were detected in a FUS3-deficient strain (Figure 2B).
FIGURE 2:

Analysis of differentially phosphorylated forms of Fus3. (A) Wild-type or ste11Δ (MAPKK) mutant cells untreated (0) or treated for 2 or 15 min with 10 μM α-factor were lysed and resolved by immunoblotting after SDS–PAGE and probed with Fus3 antibodies (top) or p44/p42 antibodies (top middle), with Phos-tag reagent and probed with Fus3 antibodies (bottom middle), or with G6PDH load control antibodies (bottom). Bands represent the dual-phosphorylated (ppKss1, ppFus3), monophosphorylated (pFus3), nonphosphorylated (npFus3), and total (Fus3) protein. Except for the topmost panel, this and all subsequent experiments were done using Phos-tag. (B) Wild-type, Fus3 activation loop (T180A, Y182F), and fus3Δ mutants were resolved by Phos-tag SDS–PAGE and immunoblotting with Fus3 antibodies (top), p44/p42 antibodies (middle), or G6PDH load control antibodies (bottom). Arrowheads indicate the bands of interest (i.e., those recognized by p44/42 antibodies). (C) Wild-type, fus3KR (catalytically inactive), and ste5ND (nondocking) mutants untreated or treated for 15 min with 10 μM α-factor were resolved by Phos-tag SDS–PAGE and immunoblotted with Fus3 antibodies. Representative data are shown below. Band intensity was quantified as a percentage of total Fus3 in each lane. Results are reported as ±SEM (n ≥ 3). (D) Wild-type, ste7Δ, and phosphatase-deficient ptp2Δ, ptp3Δ, and msg5Δ mutants, alone or in combination, were resolved by Phos-tag SDS–PAGE and immunoblotted with Fus3 antibodies (top) or G6PDH load control antibodies (bottom).
Ste5 is believed to act in part by stimulating the autophosphorylation of Fus3 on Tyr-182 (Bhattacharyya et al., 2006). Support for this model comes from in vitro studies using a peptide fragment of Ste5 (Bhattacharyya et al., 2006). To determine whether Fus3 is also autophosphorylated in vivo, we compared the mobility of wild-type Fus3 and a catalytically inactive form of the protein (Fus3K42R). As shown in Figure 2C, Fus3K54R is monophosphorylated despite the lack of catalytic activity; as with the wild-type protein, the monophosphorylated species was evident in unstimulated conditions and increased further upon pheromone stimulation. Thus autophosphorylation is not the only means by which Fus3 becomes monophosphorylated and activated, at least in vivo. To determine the contribution of the scaffold Ste5, we monitored Fus3 in the absence of Ste5–Fus3 interaction. Deletion of STE5 blocks signaling altogether, so as an alternative, we used a mutant form of Ste5 in which Fus3 docking is disrupted (‘‘nondocking’’ allele, Ste5ND). This mutant binds poorly to Fus3 (Bhattacharyya et al., 2006; Maeder et al., 2007) and yet produces a transcription-induction response that is actually enhanced (Bhattacharyya et al., 2006). Once again we were able to detect monophosphorylated Fus3 and at levels comparable to that of wild-type (STE5+) cells. However, the relative abundance of dual-phosphorylated Fus3 was increased (∼50%) in the Ste5ND strain compared with wild type (Figure 2C). This confirms previous observations that Ste5 does not activate but instead limits the full activation of Fus3 in vivo (Bhattacharyya et al., 2006; Hao et al., 2008). We infer that Ste5 acts in two opposing ways to regulate Fus3 activity. First, Ste5 binds to Fus3 indirectly via Ste7, thereby promoting activation. This is based on the observation that Fus3 binds poorly to Ste5ND but is nevertheless activated by Ste7. Second, Ste5 binds to Fus3 directly and inhibits activation. This is based on the enhanced signaling observed in Ste5ND cells.
Although Fus3 is phosphorylated by Ste7, Fus3 is also regulated by protein phosphatases. Accordingly, the cellular pool of monophosphorylated Fus3 is likely to be regulated through the combined action of kinases and phosphatases. To test this, we examined Fus3 in a panel of mutants lacking one or more of the known MAPK phosphatases: Ptp2 and Ptp3, which are Tyr-specific phosphatases, and Msg5, which is a dual-specificity (Tyr; Ser or Thr) phosphatase (Doi et al., 1994; Zhan et al., 1997; Remenyi et al., 2005). As shown in Figure 2D, cells lacking the dual-specificity phosphatase (msg5D) exhibited a decrease in monophosphorylated Fus3. Cells lacking the Tyr-specific phosphatases exhibited an increase in basal levels of dual-phosphorylated Fus3, but there was little or no effect of these mutants in pheromone-stimulated cells (Figure 2D). These data suggest that Fus3 is dephosphorylated by Msg5 to enrich the cellular pool of monophosphorylated Fus3.
Ste5 and Msg5 regulate the kinetics of Fus3 phosphorylation
To expand our analysis, we next examined how the relative abundance of the monophosphorylated and dual-phosphorylated forms of Fus3 change over time. As shown in Figure 3A, pheromone-activated cells exhibited a substantial (∼60%) drop in nonphosphorylated kinase within 5 min, accompanied by a corresponding increase in both the monophosphorylated and dual-phosphorylated forms of the protein. Whereas the pool of dual-phosphorylated Fus3 rose rapidly and showed an initial peak at ∼2–3 min, the pool of monophosphorylated Fus3 rose in a more gradual manner. Most strikingly, the relative abundance of monophosphorylated and dual-phosphorylated Fus3 was nearly equivalent after the initial peak of activation (from 5 to 60 min). Taken together, these data establish that a substantial proportion of Fus3 exists in the monophosphorylated state. Together with the data presented earlier (Figure 1), these findings suggest that stimulated cells produce a combination of Fus3 that is fully phosphorylated (activated), monophosphorylated, and nonphosphorylated (inhibitory).
FIGURE 3:

Dynamics of differentially phosphorylated forms of Fus3. (A) Left, wild-type cells were treated for the indicated times with 10 μM α-factor and resolved by Phos-tag SDS–PAGE and immunoblotted with Fus3 antibodies (top) or G6PDH load control antibodies (bottom). Right, dual-phosphorylated (ppFus3), monophosphorylated (pFus3), and nonphosphorylated (npFus3) quantified as a percentage of total Fus3 at 15 min. Results are reported as ±SEM (n ≥ 3). (B and C) Wild-type (WT), ste5FB (feedback-phosphorylation deficient), ste5ND (nondocking), ptp2Δ/ptp3Δ, and msg5Δ (phosphatase-deficient) mutants treated and resolved by Phos-tag SDS–PAGE, as described. Dual-phosphorylated Fus3 is quantified as a percentage of total Fus3 at 15 min. Results are reported as ±SEM (n ≥ 3).
To further establish the long-term consequences of differential phosphorylation, we monitored Fus3 in cells lacking the protein phosphatases or that express mutant forms of the scaffold Ste5. As noted earlier, Ste5ND cannot dock to Fus3 but nevertheless allows activation of Fus3 by the upstream kinase Ste7. Compared with wild type, the Ste5ND mutant cells exhibited higher overall accumulation of dual-phosphorylated Fus3 (Figure 3B). In addition, we tested another variant of the scaffold (Ste5FB), one that cannot undergo feedback phosphorylation and binds poorly to Fus3 (Malleshaiah et al., 2010). As with the Ste5ND mutant, the Ste5FB mutant exhibited higher accumulation of dual-phosphorylated Fus3 (Figure 3B), and this accumulation occurred more rapidly than in either the wild-type or Ste5ND strain. Thus binding to Ste5 or feedback phosphorylation of Ste5 limits the activation of Fus3 (Bhattacharyya et al., 2006; Malleshaiah et al., 2010). When binding or feedback is blocked, a greater proportion of Fus3 is fully activated. Given our goal of understanding the scaffolding function, we did all subsequent experiments with Ste5ND.
There are at least two ways by which Ste5 could impede Fus3 activation. One possibility is that Ste5 slows phosphorylation of Fus3 by the upstream MAPK kinase, Ste7. In support of this model, Ste5 was shown to impart some distinctive time- and dose-dependent behaviors to Fus3. Compared with Kss1, Fus3 is phosphorylated more slowly and in a more ultrasensitive manner (Hao et al., 2008; Malleshaiah et al., 2010). In the absence of docking to Ste5, Fus3 behaves like Kss1 (Hao et al., 2008). Another possibility is that Ste5 accelerates dephosphorylation, perhaps by recruiting one or more MAPK phosphatases. As compared with wild type, cells lacking the dual-specificity phosphatase (msg5Δ) exhibited more rapid accumulation of dual-phosphorylated Fus3, although the differences narrowed after 60 min (Figure 3C). In contrast, there was no effect of deleting PTP2 and PTP3. We infer that both Ste5 and Msg5 slow the accumulation of dual-phosphorylated Fus3.
Ste5 and Msg5 cooperate to maintain a pool of monophosphorylated Fus3
To further investigate the role of Ste5 in limiting Fus3 activity, we measured the accumulation of monophosphorylated versus dual-phosphorylated Fus3 in wild-type cells, in cells in which the interaction between Fus3 and Ste5 had been disrupted (ste5ND), and in cells lacking the phosphatase Msg5 (msg5Δ). For these experiments, we focused on early times and monitored the relative kinetics of Fus3 mono phosphorylation and dual phosphorylation. Compared with wild type, the peak of dual-phosphorylated Fus3 was more pronounced in the ste5ND mutant, but the monophosphorylated species remained roughly unchanged (Figure 4A, compare top left and right). In the msg5Δ strain, dual-phosphorylated Fus3 was again elevated, and in this case, the monophosphorylated pool of Fus3 was significantly diminished (Figures 4A, bottom left). These results are consistent with the view that both Ste5 and Msg5 limit the activity of Fus3. In contrast, a ptp2/3Δ double-deletion strain showed only small changes in the kinetics of monophosphorylated or dual-phosphorylated Fus3 (Figure 4A, bottom right).
FIGURE 4:

Dynamics and mathematical model of the differentially phosphorylated forms of Fus3. (A) Time series for wild-type (top left), ste5ND (nondocking) mutant (top right), msg5Δ (bottom left), and ptp2Δ/ptp3Δ (bottom right) cells treated with 10 μM α-factor and resolved by Phos-tag SDS–PAGE and immunoblotting with Fus3 antibodies. Dual-phosphorylated (ppFus3), monophosphorylated (pFus3), and nonphosphorylated (npFus3) quantified as a percentage of total Fus3. Results are reported as ±SEM (n ≥ 3). Circles are experimental results. Lines are simulation results of the mathematical model shown in B. (B) Left, diagram of the mathematical models. Black lines represent pathway components present in all models. The dashed line is included in the model in which feedback-phosphorylated Ste5 (pSte5) limits Fus3 phosphorylation (activation), and the red lines are included in the model in which feedback-phosphorylated Ste5 increases Fus3 dephosphorylation (deactivation). The combined model includes both effects. Top right, performance of all three models. This graph shows values for the sum of squared deviations (SSDs) for the 39,000 best parameter sets found by the Monte Carlo algorithm for parameter estimation. Bottom right, model predictions for the combined model and corresponding experimental results for the ste5ND msg5Δ double mutant.
In every case, the appearance of the dual-phosphorylated form of Fus3 preceded that of the monophosphorylated form. Although we can envision several mechanisms to account for this behavior, the simplest invokes processive phosphorylation and distributive dephosphorylation. To determine whether a distributive dephosphorylation mechanism can account for our experimental results and to quantify the relative roles of Ste5 and Msg5 in limiting MAPK activity, we developed three mathematical models to describe the kinetics of Fus3 activation and inactivation (Figure 4B). All of our models started with the pheromone-dependent recruitment of Ste5 to the plasma membrane. We did not explicitly model the activation of the kinases Ste11 and Ste7 but instead assumed a single rate-limiting step in the activation (dual phosphorylation) and recruitment of Fus3. Dephosphorylation of Fus3 was assumed to occur in a distributive manner. The abundances of Ste5, Ptp2, and Ptp3 were assumed to remain constant, whereas the production of Fus3 and Msg5 was assumed to increase as a function of Fus3 activity (feedback through gene induction).
Although our results and previous studies demonstrate that Ste5 dampens Fus3 activity, they do not establish a mechanism for this effect. Therefore we developed three models to test various scenarios for the mechanism by which feedback phosphorylation of Ste5 by Fus3 limits Fus3 activity. In the first scenario, feedback phosphorylation of Ste5 reduces the rate at which Fus3 is phosphorylated. In the second scenario, feedback phosphorylation of Ste5 increases the rate at which Fus3 is dephosphorylated (perhaps by recruiting a phosphatase). We tested each mechanism alone or in combination (third scenario).
The model equations (see Materials and Methods) were simulated using the NDSolve function in Wolfram Mathematica 9.0. To perform parameter estimations, we implemented a Markov chain Monte Carlo algorithm (Hao et al., 2012) to search parameter space for sets of model parameter values that minimize the “aggregated” sum of squared deviations (SSDs) between experimental time courses for nonphosphorylated (npFus3), monophosphorylated (pFus3), and dual-phosphorylated (ppFus3) protein, as measured in wild-type, ste5ND, msg5Δ, and ptp2/3Δ cells. Surprisingly, models that include increased Fus3 dephosphorylation generated the best fits to the experimental data (Figure 4B, top right) and accurately captured the individual contributions of both Ste5 and Msg5 to signal dampening (Figure 4A). Thus the models suggest that the primary mechanism by which Ste5 limits Fus3 activity is by increasing the rate of dephosphorylation, potentially by recruiting a phosphatase.
To further confirm our assumption that phosphorylation occurs through a processive mechanism and dephosphorylation follows distributive kinetics, we tested a model that allowed for the possibility of a distributive (sequential) mechanism for Fus3 phosphorylation. However, to generate a good fit to the data, this model required that the second phosphorylation event occur substantially faster (by three orders of magnitude) than the first phosphorylation event. Thus this model effectively reduced to a processive (single-step) mechanism, validating our use of a processive phosphorylation model.
Loss of a phosphatase and MAPK scaffolding confers synthetic supersensitivity to pheromone
To test the validity of the models, we used each model to predict Fus3 activity in a strain containing the nondocking Ste5 mutant and also lacking Msg5 (ste5ND msg5Δ). Of interest, the models in which Fus3 dephosphorylation is increased by feedback phosphorylation to Ste5 predicted that the double-mutant strain would accumulate substantially more dual-phosphorylated Fus3 than either of the single mutants alone (Figure 4B, bottom right). Experimental measurements made in the double mutant showed excellent agreement with these model predictions (Figures 4B, bottom right, and 5A). At the level of transcription, the effects of the double mutant were similarly magnified (Figure 5B). Whereas the individual mutants conferred approximately twofold decrease in the EC50 for pheromone, combining the two mutations resulted in greater than eightfold decrease in EC50. This increase in pheromone sensitivity is in marked contrast to the reduction in transcription activation presented in Figure 1, in which accumulation of monophosphorylated Fus3 dampened the transcription response by up to 60%, depending on the genetic background. These opposing effects on activity are qualitatively similar to those exhibited by the “benchmark” pathway regulator, the GTPase- activating protein Sst2. Whereas deletion of SST2 confers a decrease in the EC50, twofold overexpression of SST2 dampens the maximum response (Hao et al., 2003). Collectively these data support the predictions of the model and indicate that Ste5 and Msg5 cooperate to limit the accumulation of fully phosphorylated Fus3.
FIGURE 5:

Synergistic activation of Fus3. (A) Dual-phosphorylated Fus3 in wild type and ste5ND (nondocking) msg5Δ (phosphatase-deficient) mutants replotted from Figure 3 for comparison with the ste5ND msg5Δ mutant. Dual-phosphorylated Fus3 is quantified as a percentage of total Fus3 at 15 min. Results are reported as ±SEM (n ≥ 3). (B) Transcription reporter data in the same strains as in A, as a percentage of maximum activity in wild type. Inset, Hill slope and EC50 for each strain. Results are reported as ±SEM (n = 3). (C) Pheromone-induced growth arrest for cells treated with α-factor. Halo diameters are quantified for all strains at 5 μg (left). Results are reported as ±SEM (n = 4). Right, representative halo assays for the wild-type and ste5ND msg5Δ strains at 1.5, 5, and 15 μg. (D) Polarized growth in cells treated with 300 nM α-factor. Representative images for wild-type, msg5Δ, ste5ND, and ste5ND msg5Δ cells, each bearing the integrated polar cap marker Bem1-GFP (arrow), at the indicated times. The path of the polar cap was quantified for 10 cells 45–200 min after pheromone addition. Data are representative of two or more experiments. Scale bar, 10 μM.
Finally, we considered the role of Ste5 and Msg5 in the mating response. When exposed to high concentrations of mating pheromone, yeast cells will arrest in G1 and expand in a polarized manner. Cells lacking Sst2 arrest at concentrations much lower than that needed by wild-type cells (Chan and Otte, 1982a, b) and do not polarize properly (Segall, 1993). In addition, we found that cells bearing the msg5Δ and ste5ND mutations exhibited a greater sensitivity to pheromone in the growth arrest plate assay (Figure 5C). To monitor cell polarization, we used a custom-designed microfluidic device and tracked the distribution of Bem1–green fluorescent protein (GFP) as a marker of the polar cap (Dixit et al., 2014; Kelley et al., 2015). Whereas wild-type and msg5Δ cells polarized quickly and expanded in a single direction, ste5ND and msg5Δ ste5ND strains turned frequently and failed to orient in any one direction (Figure 5D). Thus the ste5ND msg5Δ strain exhibits transcription and polarization defects similar to those reported previously for sst2Δ. Whereas Sst2 limits activation of both Fus3 and Kss1, Ste5 and Msg5 act specifically to limit the accumulation of dual-phosphorylated Fus3.
DISCUSSION
It is well known that MAPKs are dually phosphorylated and activated in response to an extracellular signal. Here we demonstrate that Fus3 is both monophosphorylated and dually phosphorylated in response to pheromone stimulation. The dual-phosphorylated species accumulates rapidly and precedes the appearance of the monophosphorylated species, suggesting a processive mechanism of phosphorylation and a distributive mechanism of dephosphorylation. The scaffold Ste5 and the phosphatase Msg5 act together to maintain a substantial pool of monophosphorylated Fus3. Monophosphorylated Fus3 appears to be a functionally important protein species, one that inhibits signal transmission and does so in a functionally dominant manner.
Previous studies demonstrated that Ste5 and Ste7 compete for binding with Fus3, and, in such a case, there would have to be a dissociation event between steps of autophosphorylation (Ste5–Fus3) and transphosphorylation (Ste7–Fus3; Kusari et al., 2004). Thus, on the basis of the literature, we expected to observe distributive phosphorylation of Fus3. Moreover, a distributive mechanism had already been demonstrated for extracellular signal-regulated kinase (ERK) in vitro (Haystead et al., 1992; Ferrell and Bhatt, 1997). A more recent analysis, however, revealed that ERK is phosphorylated processively in cells (Aoki et al., 2011). As with Fus3, the monophosphorylated species of ERK does not precede but instead accompanies the appearance of the dual-phosphorylated species (Aoki et al., 2011). These differences suggest that there are additional proteins or processes in the cell that modulate MAPK phosphorylation in vivo. According to one model, the effects of molecular crowding, as exists in the cellular milieu, accelerate MAPK activation so as to appear processive in vivo. In support of that model, investigators showed that the addition of an osmolyte (to displace water and mimic the effects of molecular crowding) conferred a processive rate of activation in vitro (Aoki et al., 2011). Molecular crowding could just as easily be achieved by binding to scaffolds such as Ste5 or Pbs2 in yeast and kinase suppressor of Ras (KSR) or c-Jun N-terminal kinase inhibitory protein (JIP) in animals (Breitkreutz and Tyers, 2006). Pbs2 serves as both a scaffold and a MAPKK for the p38 orthologue Hog1, which, like Fus3 is phosphorylated in a processive manner in vivo (English et al., 2015). In this way, scaffolds might act to increase the thermodynamic activity, and thereby promote phosphorylation, of the kinase.
Another consideration is the effect of protein localization. Whereas activated MAPKs typically translocate into the nucleus, where they phosphorylate transcription regulators, other substrates are located at the plasma membrane or in the cytoplasm. For example, Fus3 directly phosphorylates the nuclear transcription factor Ste12 (Elion et al., 1993; Hung et al., 1997; Breitkreutz et al., 2001), as well as the cytoplasmic protein Sst2 (Garrison et al., 1999; Parnell et al., 2005). Moreover, subcellular localization is highly dynamic. Under conditions in which Fus3 translocates from the plasma membrane to the nucleus, Ste5 shuttles from the nucleus to the plasma membrane (Mahanty et al., 1999; van Drogen et al., 2001; Maeder et al., 2007). These movements are likely to affect local concentrations of enzyme, scaffold, and substrate and consequently affect signal output. For instance, when Ste5 is permanently tethered to the plasma membrane, the transcriptional output is more graded than it is in wild-type cells (Takahashi and Pryciak, 2008). Thus changes in scaffold binding, concentration, and localization can all have potentially important effects on signal output.
Given the difficulty of reconciling MAPK activities in vivo and in vitro, we took an alternative approach in silico. Specifically, we built a computational model in which Fus3 is phosphorylated processively and dephosphorylation depends on feedback interactions with the scaffold and phosphatases. Our model also posits that feedback phosphorylation of Ste5 slows the rate of downstream signaling and protects against signal saturation. We have shown experimentally that Ste5 cooperates with Msg5 to maintain the pool of monophosphorylated Fus3. Msg5 could be acting directly on Fus3 or indirectly through Ste5 or a Ste5-binding partner (Remenyi et al., 2005). Ste5ND on its own has little effect on the abundance of monophosphorylated Fus3. When interactions with both Ste5 and Msg5 are disrupted, however, nearly all of the Fus3 protein is dually phosphorylated. Fus3 and Ste5 also regulate the abundance of Msg5 through gene induction, thereby providing another mechanism to fine-tune MAPK activity over time. Independently, these mechanisms dynamically regulate Fus3 phosphorylation. Collectively these mechanisms generate a dynamic self-regulatory network. Whereas Fus3 is partially redundant with the MAPK Kss1, Kss1 does not bind to Ste5 and was not considered in this analysis.
Our findings add support to the proposal that monophosphorylated Fus3 down-regulates pathway activity and does so through phosphorylation of Ste5 (Bhattacharyya et al., 2006; Hao et al., 2008; Malleshaiah et al., 2010). Further, our data provide evidence that autophosphorylation is a minor contributor to the pool of monophosphorylated Fus3. Although our data point to the importance of Msg5 and dephosphorylation in this process, other (as-yet-unidentified) mechanisms may further contribute to the production of monophosphorylated Fus3 in vivo.
Collectively our findings reveal a substantial pool of nonphosphorylated and monophosphorylated Fus3 in pheromone-stimulated cells, and these have an inhibitory effect on signaling. This could account for the ability of a second copy of Fus3 to partially dampen the activity of the mating pathway. Although we do not know the mechanism of inhibition in this case, there is growing evidence that even catalytically inactive kinases can have important cellular functions. Approximately 10% of all human kinase genes lack one of three key residues necessary for activity and are therefore likely to be catalytically inert. Nevertheless, many of these “pseudokinase” proteins are expressed and have been demonstrated to perform functions that do not involve substrate phosphorylation (Leslie, 2013). For example, a pseudo-MAPK in yeast (Mlp1) was shown to regulate transcription (Kim et al., 2008) and to bind Msg5 through an unusual docking domain (Palacios et al., 2011). Here we have shown that an incompletely phosphorylated form of Fus3 exists in the absence of catalytic activity and that this monophosphorylated form of the protein acts in opposition to the fully phosphorylated kinase. Given the evolutionary conservation of structurally similar MAPKs, we anticipate finding functionally similar, differentially phosphorylated MAPKs in other systems. A deeper understanding of MAPK regulation should point to new mechanisms and new therapeutic strategies to influence stimulus–response behaviors in humans.
MATERIALS AND METHODS
Experimental procedures
Strains, plasmids, and growth conditions.
Standard methods were used throughout for growth, maintenance, and transformation of yeast and bacterial cultures as well as for the manipulation of DNA. Strains and plasmids used in this study are described in Tables 1 and 2. Mutagenesis was performed using the QuikChange site-directed mutagenesis kit (Life Technologies, Grand Island, NY). Yeast strains carrying integrated mutations were constructed using the Delitto Perfetto method (Storici et al., 2001). Oligonucleotides used for strain construction and DNA amplification of FUS3 T180 and Y182 mutants are listed in Table 3. Cells were grown in synthetic complete medium, absent specific nutrients to maintain plasmid selection, and containing 2% (wt/vol) dextrose.
TABLE 1:
Strains used in this study.
| Strain namea | Genotype | Source |
|---|---|---|
| BY4741 | MATa leu2Δ met15Δ his3-1 ura3Δ | Brachmann et al. (1998) |
| fus3Δ | MATa fus3::kanMX | Invitrogen |
| msg5Δ | MATa msg5::kanMX | Invitrogen |
| ptp2/3Δ | MATa ptp2::URA3 ptp3::kanMX | Hao et al. (2012) |
| ptp2/3Δ msg5Δ | MATa ptp2::URA3 ptp3::LEU2 msg5::kanMX | This study |
| ste5ND | MATa ste5ND | Hao et al. (2008) |
| ste5FB | MATa ste5FB | This study |
| ste5ND msg5Δ | MATa ste5ND msg5::kanMX | This study |
| ste11Δ | MATa ste11::kanMX | Invitrogen |
| ste7Δ | MATa ste7::kanMX | Invitrogen |
| fus3 T180A | MATa fus3 T180A | This study |
| fus3 Y182F | MATa fus3 Y182F | This study |
| fus3 T180A/Y182F | MATa fus3 T180A/Y182F | This study |
aAll mutant strains derived from BY4741.
TABLE 2:
Plasmids used in this study.
| Plasmid name | Description | Source |
|---|---|---|
| pRS316 | CEN URA3 vector | Sikorski and Hieter (1989) |
| pRS316-FUS3 | CEN URA3 FUS3 | Hao et al. (2012) |
| pRS316-FUS3-T180A | CEN URA3 FUS3T180A | This study |
| pRS316-FUS3-Y182F | CEN URA3 FUS3Y182F | This study |
| pRS316-FUS3-T180A/Y182F | CEN URA3 FUS3T180A/Y182F | This study |
| pRS425-FUS1-lacZ | 2 μ LEU2 PFUS1-lacZ | Hoffman et al. (2002) |
All plasmids used in Figure 1.
TABLE 3:
Oligonucleotides used in this study.
| Primer name | Sequence |
|---|---|
| P1 Fus3-CORE 60up | TTA GCA AGA ATC ATT GAC GAG TCA GCC GCG GAC AAT TCA GAG CCC ACA GGT CAG CAA AGC GGC GAG CTC GTT TTC GAC ACT GG |
| P2 Fus3-CORE 60dn | CAC GTC CAT GGC CCT TGA GTA TTT GGC AGA GGT TAA CAT CAC CTC TGG CGC CCT GTA CCA ACG TCC TTA CCA TTA AGT TGA TC |
| P1 Fus3 activation-loop IRO | CCA GAT GCT GAG TGA CGA |
| P2 Fus3 activation-loop IRO | GCA GGG TAC ATG GGA AGC |
Transcriptional reporter and halo assay.
Growth arrest (halo) and FUS1-LacZ levels were measured after treatment with mating pheromone, α-factor, as described previously (Hoffman et al., 2002). Cells grown to A600 nm ≈ 0.8 were stimulated for 90 min and then lysed, and β-galactosidase activity was measured spectrophotometrically after 60 min. Results are from two or three independent experiments of three or four colonies per strain read in quadruplicate.
Cell extracts and immunoblotting.
Protein extracts were produced by glass bead lysis in trichloroacetic acid as previously described (Hao et al., 2007). Protein concentration was determined by Dc protein assay (Bio-Rad Laboratories, Hercules, CA). Protein extracts were resolved by standard SDS–PAGE or 50 μM Mn2+-Phos-tag in 10% acrylamide SDS–PAGE according to manufacturer’s instructions (Wako Chemicals, Richmond, VA). Proteins were detected by immunoblotting with phospho-MAPK p44/42 antibodies (9101; Cell Signaling Technology, Beverly, MA) at 1:500, Fus3 antibodies (sc-6773, Santa Cruz Biotechnology, Dallas, TX) at 1:500, and glucose-6-phosphate dehydrogenase (G6PDH) antibodies (A9521; Sigma-Aldrich, St. Louis, MO) at 1:50,000. Immunoreactive species were visualized by fluorescence detection (Typhoon Trio+ Imager; GE Healthcare, Piscataway, NJ) of horseradish peroxidase–conjugated antibodies (sc-2006; Santa Cruz Biotechnology) at 1:10,000 using ECL-plus reagent (Life Technologies). Band intensity was quantified by scanning densitometry using ImageJ (National Institutes of Health, Bethesda, MD). Fus3 intensity values were first normalized to G6PDH loading control, and values for nonphosphorylated, monophosphorylated, and dual-phosphorylated bands were calculated as a percentage of the total for each lane. To capture changes in Fus3 protein induction over time and/or between different genetic backgrounds, samples were normalized to the 15-min time point. Briefly, the value of total Fus3 in the 15-min lane served as the denominator for calculating values of the three Fus3 forms at all time points. When comparing wild-type and mutant strains, all lanes were normalized to the 15-min time point of wild type.
Microscopy.
Cells expressing Bem1-GFP were examined in a microfluidic device as previously described (Kelley et al., 2015) in the presence of 300 nM pheromone. This was carried out on an Olympus IX83 with an Andor Revolution XD spinning disk unit. Images were taken every 5 min as a z-series of 1-μm step size, 5 μm around the focal plane. These images were aligned in FIJI (fiji.sc/Fiji) using the Descriptor-based series registration (2D/3D + t) plug-in. A single-pixel Gaussian blur was applied to the fluorescence images to remove camera noise, and a maximum intensity projection was created of the stacks. To plot the polar cap path, the Bem1-GFP images were thresholded to determine the centroid of the polar cap. The centroids were recorded for each time point for every cell from 0 to 200 min. Using MATLAB, the Cartesian coordinates of the polar cap centroids were converted to polar coordinates, and each path was rotated such that the final 50 min of time points had an average angle of 0. The first 45 min of experimental data was not included in the analysis to exclude data from mitotic events. The normalized paths were converted back into Cartesian coordinates and smoothed with the “smooth” function in MATLAB, using a window size of seven time points. Ten individual paths were plotted for each strain.
Mathematical model
We tested three different processive models for the time-dependent behavior of Fus3 activity. The models differ in the mechanism by which Ste5 negatively regulates Fus3 activity. The first model assumes that feedback-phosphorylated Ste5 (pSte5m) has no effect on the Fus3 phosphorylation rate. The second model assumes that pSte5m has no effect on the Fus3 dephosphorylation rate. The third model combines the effect of pSte5m on phosphorylation of npFus3 and dephosphorylation of ppFus3 and pFus3 and contains the other two models as limiting cases. We describe this model in detail and point out how the other two were obtained from it. We explicitly modeled the temporal evolution of three species of Ste5: 1) free and inactive (Ste5c), 2) plasma membrane bound and active (Ste5m), and 3) feedback-phosphorylated, plasma membrane bound, and less active (pSte5m). We assumed that the total population of Ste5 is conserved and that Ste5 is recruited to the plasma membrane upon stimulation with mating pheromone. The model assumed that Fus3 is dually phosphorylated in a processive manner. To test whether feedback phosphorylation of Ste5 affects the activation of Fus3, we assumed that the rate of Fus3 phosphorylation depends on the form of Ste5 (Ste5m or pSte5m) with which Fus3 is associated. We assumed that Fus3 is induced and degraded and that monophosphorylated (pFus3) or dual-phosphorylated (ppFus3) Fus3 feedback-phosphorylates Ste5m. We modeled Fus3 dephosphorylation as a distributive process. We assumed that the dephosphorylation rate depends on the current level of the induced dual-specificity phosphatase Msg5. Because the mechanism by which Ste5 limits Fus3 activity is not known, we also included the possibility that dephosphorylation rate depends on the phosphorylated Ste5 species (pSte5m).
The equations that describe the evolution of Ste5 species are as follows:
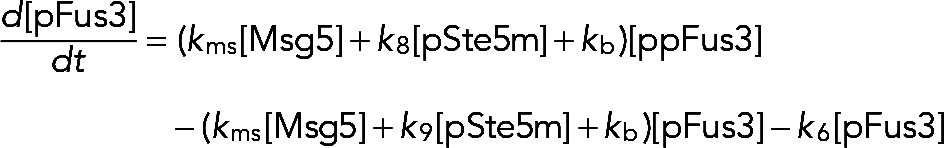 (1) (1)
|
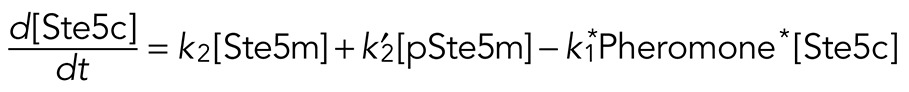 (2) (2)
|
 (3) (3)
|
where the first term of Eq. 1 describes the rate at which mating pheromone induces the recruitment of Ste5 to the plasma membrane. The second term defines the dephosphorylation rate for pSte5m. The third term defines the rate at which Ste5 transitions back to the cytosol. The fourth and fifth terms define rates of feedback phosphorylation of Ste5m by dual-phosphorylated Fus3 (ppFus3) and monophosphorylated Fus3 (pFus3), respectively. In addition, the second term in Eq. 2 defines the rate at which pSte5m returns to Ste5c.
The equation that describes the evolution of the nonphosphorylated Fus3 (npFus3) concentration is
 (4) (4)
|
The first term on the right-hand side of Eq. 4 accounts for basal induction of Fus3. The second and third terms correspond to the positive feedback resulting from induction of Fus3 via ppFus3 and pFus3, respectively. Positive-feedback follows Michaelis–Menten kinetics, where our parameter search suggests that the rate of Fus3 induction by ppFus3 is faster than that mediated by pFus3 (see Table 4 for all parameter values). The fourth term describes the rate at which monophosphorylated Fus3 (pFus3) is dephosphorylated. We assumed three mechanisms for dephosphorylation. The first depends on the phosphatase Msg5, whose expression is induced by active Fus3. The second is independent of Msg5 and is included to test the possibility that feedback-phosphorylated Ste5 (pSte5m) limits Fus3 activity by increasing the dephosphorylation rate (perhaps by recruiting another phosphatase). Finally, we include a basal dephosphorylation rate that is independent of Msg5 and Ste5. The fifth and sixth terms in Eq. (4) define rates of dual phosphorylation mediated by Ste5m and pSte5m, respectively. These terms allow us to test the possibility that feedback-phosphorylated Ste5 (pSte5m) limits Fus3 activity by decreasing the phosphorylation rate (i.e., k7′ < k7). The last term defines the degradation rate for Fus3.
TABLE 4:
Model rate parameters.
| Parameter | Complete model | Limiting case 1 | Limiting case 2 | Description |
|---|---|---|---|---|
| k1 | 1.15 × 101 | 8.51 | 7.71 | [Ste5m] activation |
| k2 | 1.25 × 10−4 | 1.12 × 10−4 | 2.43 × 10−2 | [Ste5m] deactivation |
| k2p | 1.66 × 10−2 | 1.1 × 10−4 | 2.43 × 10−2 | [pSte5m] deactivation |
| k3 | (2.73 × 10−4, 4.09 × 10−5) | (1.18 × 10−4, 1.77 × 10−5) | (1.5 × 10−4, 2.25 × 10−5) | [Ste5m] feedback phosphorylation |
| k3p | (4.81 × 10−2, 7.21 × 10−3) | (1.43 × 10−2, 2.15 × 10−3) | (4.78 × 10−2, 7.17 × 10−3) | [Ste5m] feedback phosphorylation |
| kb | 1.29 × 10−4 | 8.2 × 10−2 | 2.55 × 10−3 | [Fus3] dephosphorylation |
| k4 | 4.36 × 10−1 | 4.38 × 10−1 | 4.88 × 10−1 | [p-Ste5m] dephosphorylation |
| k5 | 1.15 | 1.48 | 1.32 | [Fus3] synthesis |
| k5p | 0.0 | 0.0 | 0.0 | |
| k6 | 2.64 × 10−3 | 2.49 × 10−3 | 3.26 × 10−3 | [Fus3] degradation |
| k7 | 1.43 × 10−1 | 1.46 × 10−1 | 1.94 × 10−1 | [pp-Fus3] phosphorylation |
| k7p | 9.13 × 10−2 | 1.46 × 10−1 | 7.73 × 10−1 | [pp-Fus3] feedback phosphorylation |
| k8 | 6.01 × 10−4 | 6.71 × 10−4 | 0.0 | [pp-Fus3] dephosphorylation |
| k9 | 1.16 × 10−3 | 1.64 × 10−3 | 0.0 | [p-Fus3] dephosphorylation |
| k10 | 5.19 × 10−4 | 1.06 × 10−3 | 1.14 × 10−3 | [Fus3] synthesis |
| k11 | 5.16 × 10−3 | 4.06 × 10−4 | 3.28 × 10−3 | [Msg5] synthesis |
| k12 | 1.09 × 10−4 | 5.39 × 10−4 | 1.11 × 10−4 | [Msg5] degradation |
| kms | (2.04 × 10−3, 0.) | (3.19 × 10−3, 0.) | (2.82 × 10−4, 0.) | [Msg5] dephosphorylation |
| kamsg5 | 1.06 × 10−4 | 2.16 × 10−2 | 1.09 × 10−4 | [Msg5] activation |
| K1 | 2.5 × 10−1 | 2.45 × 101 | 2.47 × 101 | Michaelis constant |
| K2 | 3.1 | 1.31 × 101 | 2.84 | Michaelis constant |
| K3 | 4.12 | 1.04 | 1.03 | Michaelis constant |
| K4 | 1.07 | 2.49 × 101 | 1.16 | Michaelis constant |
| Kmsg5 | 1.62 | 1.36 | 2.71 | Michaelis constant |
The name, numerical value, and function of each rate parameter in the model are given. The rate parameters k3 and k3p are shown for the wild-type (left), ste5ND (right), and msg5Δ plus ste5ND (right) models. The rate parameter kms is shown for the wild-type (left) and msg5Δ plus ste5ND (right) models. Parameters k3 and k3p are “knocked down” in the model and are assumed to take 5% of the original value for both the ste5ND and ste5ND plus msg5Δ mutant models. Parameter kms is set to 0 for both the msg5Δ and ste5ND plus msg5Δ mutant models. Parameters k5p and K2 have no appreciable affect on the model and are set to 0 in the model figures.
The rate equations that describe dual-phosphorylated and monophosphorylated Fus3 are
 (5) (5)
|
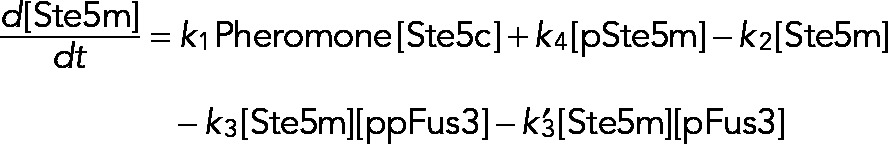 (6) (6)
|
where terms describing the rates of phosphorylation, dephosphorylation, and degradation have the same form as in Eq. 4.
The rate equation that describes evolution of the Msg5 concentration is
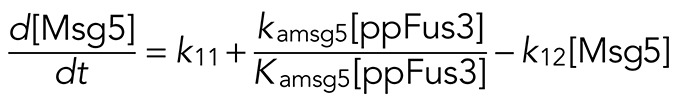 (7) (7)
|
where the first and second terms are the basal and ppFus3-dependent synthesis rates, respectively. The third term defines the rate of Msg5 degradation.
Equations 1– 7 define the full model that allows for the possibility that Ste5 affects both the phosphorylation and dephosphorylation rates of Fus3. The model in which pSte5m only decreases the phosphorylation rate is obtained by setting the parameters k8 and k9 equal to zero, and the model in which pSte5m only increases the dephosphorylation rate is achieved by setting k7′ = k7.
Acknowledgments
We thank John Houser for insightful discussions about mathematical modeling strategies. This work was supported, in whole or in part, by National Institutes of Health Grants R01-GM080739 (to H.G.D.), R01-GM079271 (to T.C.E.), T32-GM067553, and R25-GM05533612 (to P.C.M.).
Abbreviations used:
- BEM1
Bud EMergence 1
- FUS3
cell FUSion 3
- GFP
green fluorescent protein
- G6PDH
glucose-6-phosphate dehydrogenase
- KSS1
Kinase Suppressor of Sst2 mutations
- MAPK
mitogen-activated protein kinase
- MSG5
multicopy suppressor of GPA1
- PTP
protein tyrosine phosphatase
- SSD
sum of squared deviations
- SST2
SuperSensiTive 2
- STE
STErile
- Thr
threonine
- Tyr
tyrosine
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.3359) on July 15, 2015.
REFERENCES
- Andersson J, Simpson DM, Qi M, Wang Y, Elion EA. Differential input by Ste5 scaffold and Msg5 phosphatase route a MAPK cascade to multiple outcomes. EMBO J. 2004;23:2564–2576. doi: 10.1038/sj.emboj.7600250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki K, Yamada M, Kunida K, Yasuda S, Matsuda M. Processive phosphorylation of ERK MAP kinase in mammalian cells. Proc Natl Acad Sci USA. 2011;108:12675–12680. doi: 10.1073/pnas.1104030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell L. A walk-through of the yeast mating pheromone response pathway. Peptides. 2005;26:339–350. doi: 10.1016/j.peptides.2004.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya RP, Remenyi A, Good MC, Bashor CJ, Falick AM, Lim WA. The Ste5 scaffold allosterically modulates signaling output of the yeast mating pathway. Science. 2006;311:822–826. doi: 10.1126/science.1120941. [DOI] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Breitkreutz A, Boucher L, Tyers M. MAPK specificity in the yeast pheromone response independent of transcriptional activation. Curr Biol. 2001;11:1266–1271. doi: 10.1016/s0960-9822(01)00370-0. [DOI] [PubMed] [Google Scholar]
- Breitkreutz A, Tyers M. Cell signaling. A sophisticated scaffold wields a new trick. Science. 2006;311:789–790. doi: 10.1126/science.1124620. [DOI] [PubMed] [Google Scholar]
- Canagarajah BJ, Khokhlatchev A, Cobb MH, Goldsmith EJ. Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell. 1997;90:859–869. doi: 10.1016/s0092-8674(00)80351-7. [DOI] [PubMed] [Google Scholar]
- Chan RK, Otte CA. Isolation and genetic analysis of Saccharomyces cerevisiae mutants supersensitive to G1 arrest by a factor and a factor pheromones. Mol Cell Biol. 1982a;2:11–20. doi: 10.1128/mcb.2.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan RK, Otte CA. Physiological characterization of Saccharomyces cerevisiae mutants supersensitive to G1 arrest by a factor and a factor pheromones. Mol Cell Biol. 1982b;2:21–29. doi: 10.1128/mcb.2.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KY, Satterberg B, Lyons DM, Elion EA. Ste5 tethers multiple protein kinases in the MAP kinase cascade required for mating in S. cerevisiae. Cell. 1994;78:499–512. doi: 10.1016/0092-8674(94)90427-8. [DOI] [PubMed] [Google Scholar]
- Dixit G, Kelley JB, Houser JR, Elston TC, Dohlman HG. Cellular noise suppression by the regulator of G protein signaling Sst2. Mol Cell. 2014;55:85–96. doi: 10.1016/j.molcel.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi K, Gartner A, Ammerer G, Errede B, Shinkawa H, Sugimoto K, Matsumoto K. MSG5, a novel protein phosphatase promotes adaptation to pheromone response in S. cerevisiae. EMBO J. 1994;13:61–70. doi: 10.1002/j.1460-2075.1994.tb06235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elion EA, Satterberg B, Kranz JE. FUS3 phosphorylates multiple components of the mating signal transduction cascade: evidence for STE12 and FAR1. Mol Biol Cell. 1993;4:495–510. doi: 10.1091/mbc.4.5.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English JG, Shellhammer JP, Malahe M, McCarter PC, Elston TC, Dohlman HG. MAPK feedback encodes a switch and timer for tunable stress adaptation in yeast. Sci Signal. 2015;8:ra5. doi: 10.1126/scisignal.2005774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrell JE., Jr What do scaffold proteins really do. Sci STKE. 2000;2000:pe1. doi: 10.1126/stke.2000.52.pe1. [DOI] [PubMed] [Google Scholar]
- Ferrell JE, Jr, Bhatt RR. Mechanistic studies of the dual phosphorylation of mitogen-activated protein kinase. J Biol Chem. 1997;272:19008–19016. doi: 10.1074/jbc.272.30.19008. [DOI] [PubMed] [Google Scholar]
- Flatauer LJ, Zadeh SF, Bardwell L. Mitogen-activated protein kinases with distinct requirements for Ste5 scaffolding influence signaling specificity in Saccharomyces cerevisiae. Mol Cell Biol. 2005;25:1793–1803. doi: 10.1128/MCB.25.5.1793-1803.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison TR, Zhang Y, Pausch M, Apanovitch D, Aebersold R, Dohlman HG. Feedback phosphorylation of an RGS protein by MAP kinase in yeast. J Biol Chem. 1999;274:36387–36391. doi: 10.1074/jbc.274.51.36387. [DOI] [PubMed] [Google Scholar]
- Gartner A, Nasmyth K, Ammerer G. Signal transduction in Saccharomyces cerevisiae requires tyrosine and threonine phosphorylation of FUS3 and KSS1. Genes Dev. 1992;6:1280–1292. doi: 10.1101/gad.6.7.1280. [DOI] [PubMed] [Google Scholar]
- Hao N, Behar M, Parnell SC, Torres MP, Borchers CH, Elston TC, Dohlman HG. A systems-biology analysis of feedback inhibition in the sho1 osmotic-stress-response pathway. Curr Biol. 2007;17:659–667. doi: 10.1016/j.cub.2007.02.044. [DOI] [PubMed] [Google Scholar]
- Hao N, Nayak S, Behar M, Shanks RH, Nagiec MJ, Errede B, Hasty J, Elston TC, Dohlman HG. Regulation of cell signaling dynamics by the protein kinase-scaffold Ste5. Mol Cell. 2008;30:649–656. doi: 10.1016/j.molcel.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao N, Yildirim N, Nagiec MJ, Parnell SC, Errede B, Dohlman HG, Elston TC. Combined computational and experimental analysis reveals mitogen-activated protein kinase-mediated feedback phosphorylation as a mechanism for signaling specificity. Mol Biol Cell. 2012;23:3899–3910. doi: 10.1091/mbc.E12-04-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao N, Yildirim N, Wang Y, Elston TC, Dohlman HG. Regulators of G protein signaling and transient activation of signaling: experimental and computational analysis reveals negative and positive feedback controls on G protein activity. J Biol Chem. 2003;278:46506–46515. doi: 10.1074/jbc.M308432200. [DOI] [PubMed] [Google Scholar]
- Haystead TA, Dent P, Wu J, Haystead CM, Sturgill TW. Ordered phosphorylation of p42mapk by MAP kinase kinase. FEBS Lett. 1992;306:17–22. doi: 10.1016/0014-5793(92)80828-5. [DOI] [PubMed] [Google Scholar]
- Hoffman G, Garrison TR, Dohlman HG. Analysis of RGS proteins in Saccharomyces cerevisiae. Methods Enzymol. 2002;344:617–631. doi: 10.1016/s0076-6879(02)44744-1. [DOI] [PubMed] [Google Scholar]
- Huang CY, Ferrell JE., Jr Ultrasensitivity in the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1996;93:10078–10083. doi: 10.1073/pnas.93.19.10078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung W, Olson KA, Breitkreutz A, Sadowski I. Characterization of the basal and pheromone-stimulated phosphorylation states of Ste12p. Eur J Biochem. 1997;245:241–251. doi: 10.1111/j.1432-1033.1997.00241.x. [DOI] [PubMed] [Google Scholar]
- Hur JY, Kang GY, Choi MY, Jung JW, Kim KP, Park SH. Quantitative profiling of dual phosphorylation of Fus3 MAP kinase in Saccharomyces cerevisiae. Mol Cells. 2008;26:41–47. [PubMed] [Google Scholar]
- Kelley JB, Dixit G, Sheetz JB, Venkatapurapu SP, Elston TC, Dohlman HG. RGS proteins and septins cooperate to promote chemotropism by regulating polar cap mobility. Curr Biol. 2015;25:275–285. doi: 10.1016/j.cub.2014.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KY, Truman AW, Levin DE. Yeast Mpk1 mitogen-activated protein kinase activates transcription through Swi4/Swi6 by a noncatalytic mechanism that requires upstream signal. Mol Cell Biol. 2008;28:2579–2589. doi: 10.1128/MCB.01795-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics. 2006;5:749–757. doi: 10.1074/mcp.T500024-MCP200. [DOI] [PubMed] [Google Scholar]
- Kusari AB, Molina DM, Sabbagh W, Jr, Lau CS, Bardwell L. A conserved protein interaction network involving the yeast MAP kinases Fus3 and Kss1. J Cell Biol. 2004;164:267–277. doi: 10.1083/jcb.200310021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie M. Molecular biology. “Dead” enzymes show signs of life. Science. 2013;340:25–27. doi: 10.1126/science.340.6128.25. [DOI] [PubMed] [Google Scholar]
- Levchenko A, Bruck J, Sternberg PW. Scaffold proteins may biphasically affect the levels of mitogen-activated protein kinase signaling and reduce its threshold properties. Proc Natl Acad Sci USA. 2000;97:5818–5823. doi: 10.1073/pnas.97.11.5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder CI, Hink MA, Kinkhabwala A, Mayr R, Bastiaens PI, Knop M. Spatial regulation of Fus3 MAP kinase activity through a reaction-diffusion mechanism in yeast pheromone signalling. Nat Cell Biol. 2007;9:1319–1326. doi: 10.1038/ncb1652. [DOI] [PubMed] [Google Scholar]
- Mahanty SK, Wang Y, Farley FW, Elion EA. Nuclear shuttling of yeast scaffold Ste5 is required for its recruitment to the plasma membrane and activation of the mating MAPK cascade. Cell. 1999;98:501–512. doi: 10.1016/s0092-8674(00)81978-9. [DOI] [PubMed] [Google Scholar]
- Maleri S, Ge Q, Hackett EA, Wang Y, Dohlman HG, Errede B. Persistent activation by constitutive Ste7 promotes Kss1-mediated invasive growth but fails to support Fus3-dependent mating in yeast. Mol Cell Biol. 2004;24:9221–9238. doi: 10.1128/MCB.24.20.9221-9238.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malleshaiah MK, Shahrezaei V, Swain PS, Michnick SW. The scaffold protein Ste5 directly controls a switch-like mating decision in yeast. Nature. 2010;465:101–105. doi: 10.1038/nature08946. [DOI] [PubMed] [Google Scholar]
- Palacios L, Dickinson RJ, Sacristan-Reviriego A, Didmon MP, Marin MJ, Martin H, Keyse SM, Molina M. Distinct docking mechanisms mediate interactions between the Msg5 phosphatase and mating or cell integrity mitogen-activated protein kinases (MAPKs) in Saccharomyces cerevisiae. J Biol Chem. 2011;286:42037–42050. doi: 10.1074/jbc.M111.286948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnell SC, Marotti LA, Jr, Kiang L, Torres MP, Borchers CH, Dohlman HG. Phosphorylation of the RGS protein Sst2 by the MAP kinase Fus3 and use of Sst2 as a model to analyze determinants of substrate sequence specificity. Biochemistry. 2005;44:8159–8166. doi: 10.1021/bi0503091. [DOI] [PubMed] [Google Scholar]
- Piala AT, Humphreys JM, Goldsmith EJ. MAP kinase modules: the excursion model and the steps that count. Biophys J. 2014;107:2006–2015. doi: 10.1016/j.bpj.2014.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Printen JA, Sprague GF., Jr Protein-protein interactions in the yeast pheromone response pathway: Ste5p interacts with all members of the MAP kinase cascade. Genetics. 1994;138:609–619. doi: 10.1093/genetics/138.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remenyi A, Good MC, Bhattacharyya RP, Lim WA. The role of docking interactions in mediating signaling input, output, and discrimination in the yeast MAPK network. Mol Cell. 2005;20:951–962. doi: 10.1016/j.molcel.2005.10.030. [DOI] [PubMed] [Google Scholar]
- Roberts CJ, Nelson B, Marton MJ, Stoughton R, Meyer MR, Bennett HA, He YD, Dai H, Walker WL, Hughes TR, et al. Signaling and circuitry of multiple MAPK pathways revealed by a matrix of global gene expression profiles. Science. 2000;287:873–880. doi: 10.1126/science.287.5454.873. [DOI] [PubMed] [Google Scholar]
- Segall JE. Polarization of yeast cells in spatial gradients of alpha mating factor. Proc Natl Acad Sci USA. 1993;90:8332–8336. doi: 10.1073/pnas.90.18.8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storici F, Lewis LK, Resnick MA. In vivo site-directed mutagenesis using oligonucleotides. Nat Biotechnol. 2001;19:773–776. doi: 10.1038/90837. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Pryciak PM. Membrane localization of scaffold proteins promotes graded signaling in the yeast MAP kinase cascade. Curr Biol. 2008;18:1184–1191. doi: 10.1016/j.cub.2008.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Drogen F, Stucke VM, Jorritsma G, Peter M. MAP kinase dynamics in response to pheromones in budding yeast. Nat Cell Biol. 2001;3:1051–1059. doi: 10.1038/ncb1201-1051. [DOI] [PubMed] [Google Scholar]
- Wang Y, Dohlman HG. Pheromone signaling mechanisms in yeast: a prototypical sex machine. Science. 2004;306:1508–1509. doi: 10.1126/science.1104568. [DOI] [PubMed] [Google Scholar]
- Zhan XL, Deschenes RJ, Guan KL. Differential regulation of FUS3 MAP kinase by tyrosine-specific phosphatases PTP2/PTP3 and dual-specificity phosphatase MSG5 in Saccharomyces cerevisiae. Genes Dev. 1997;11:1690–1702. doi: 10.1101/gad.11.13.1690. [DOI] [PubMed] [Google Scholar]