

Abstract
Tissue development and homeostasis are regulated by opposing pro-survival and pro-death signals. An interesting feature of the Tumor Necrosis Factor (TNF) family of ligands is that they simultaneously activate opposing signals within a single cell via the same ligand-receptor complex. The magnitude of pro-death events such as caspase activation and pro-survival events such as NF-κB activation vary not only from one cell type to the next but also among individual cells of the same type due to intrinsic and extrinsic noise. The molecules involved in these pro-survival/pro-death pathways, and the different phenotypes that result from their activities, have been recently reviewed. Here we focus on the impact of cell-to-cell variability in the strength of these opposing signals on shaping cell fate decisions.
Keywords: Death ligand, apoptosis, survival, life-death decision, variability, cell fate
Life-Death Decisions
During embryogenesis, development, and tissue turnover, some cells die by apoptosis while other cells avoid death and assume various cellular fates. What makes some cells die and others survive is not completely understood. In some cases, only specific cells receive the death signal, while in other cases, the signal is interpreted differently due to cell- or context-specific cues. Such cell-to-cell variability, which has various origins, has recently been shown to play an important role in cell fate decisions 1–3.
Similarly, stress-response signaling often has a dual role, activating survival pathways to buffer and repair damage, and death pathways to kill cells when the damage is beyond repair. Examples include pathways regulating heat shock proteins, p53, autophagy, and inflammation, and here, too, individual cells often respond with variable outputs 4–6. Thus, protective stress pathways and death signaling are tightly linked, and many cellular proteins have evolved to exert both functions, often in parallel 7,8.
Proteins that regulate cell death are also essential for normal cellular processes, including metabolism, proliferation, and differentiation 9,10. In some cases, these proteins “deviate” from their physiological function only when external cues point to cell death. In other cases, proteins exhibit both functions simultaneously (for example through interactions within different protein complexes), or play a survival role only when death is inhibited. Some of these proteins (e.g. caspases) belong to multiple signaling pathways (Box 1), while others, such as the TNF family of death ligands, activate parallel but opposing pathways through recruitment of different sets of signaling molecules. Thus, many proteins are essential for both life and death of cells, and the particular outcome may depend on cell type, exposure to external stimuli, or other context-dependent choices.
Box 1. Non-death roles for death proteins.
Many proteins exhibit both pro- and anti-apoptotic activities. For example, cytochrome c is important for both mitochondrial homeostasis and execution of cell death 125. The first caspase to be discovered, Interleukin-1-beta-Converting-Enzyme (ICE/Caspase-1), is responsible for cytokine processing and represents a subgroup of inflammatory caspases with functions in immune signaling 126. TNF was discovered for its role in tumor necrosis, but also acts as an inflammatory cytokine 127. Subsequently, non-apoptotic roles have been uncovered for most proteins associated with apoptosis.
Both initiator and effector caspases exhibit non-apoptotic functions 9. Caspase-8 promotes cell migration 128–130, T cell proliferation 131, wound healing 132, stem cell reprogramming 133 and macrophage differentiation 134. Caspase-3 plays a role in shaping cell morphology 135 and in differentiation of red blood cells, lens epithelial cells, and skeletal muscle cells, processes that involve degradation of intracellular organelles or substrates (“incomplete apoptosis”) 136–140. Caspases are required for spermatid differentiation, oogenesis, and wing development in Drosophila 141–143, and also play a role in neuronal sculpting, synaptic plasticity, and neural development 117,144–146.
Bcl-2 family members also have functions unrelated to apoptosis, such as regulation of mitochondrial homeostasis and glucose metabolism 147,148. Mcl-1, an anti-apoptotic Bcl-2 family member, was first discovered as a differentiation marker for myeloid cells, and is required for embryonic development and immune system function 149. In addition, Bcl-2 members allow rapid switching between states that favor life versus death. For example, alternatively-spliced isoforms and cleavage products of these proteins can promote either survival or death, and the fast degradation rate of Mcl-1 in particular allows cells to rapidly undergo cell death under conditions of stress 108.
Death-Inducing Signaling Complex (DISC) proteins such as c-FLIP, FADD and RIP also promote death or survival, and combinatorial regulation of these proteins may determine cell fate 150,151. c-FLIP can be pro- or anti-apoptotic, depending on levels and the particular isoforms expressed 27,152,153, and cleaved FLIP (p43) regulates activation of survival pathways via NF-κB 154. FLIP and FADD are both required for embryonic development and T cell proliferation 155, and FADD plays a role in cell cycle progression, differentiation, and innate immunity 151. Moreover, FADD, Caspase-8, and FLIP appear to promote cell survival during development through inhibition of necroptosis, inducing apoptosis only in response to certain stimuli 35. RIP1 can activate survival, apoptosis, or necroptosis, depending on its post-translational modifications 150. Finally, kinases associated with the DISC can have pro- or anti-apoptotic activity: p38, JNK, PKC and ERK either promote or inhibit apoptosis induced by death ligands, depending on context 59.
In this review, we examine how a “death” signal can lead to a non-death output, with a particular focus on the TNF family of death ligands. We also describe some non-apoptotic functions of “death” proteins and discuss potential advantages of this convergence. Finally, we review how the interplay between death and survival signaling has been studied at the level of single cells, how variability in these signals contributes to variability in cell fate, and the implications of these studies for understanding the roles of life-death signaling in development and disease.
Integrating Life-Death Signals
Cell-to-cell variability has been shown to play an important role in cell fate decisions 11. This variability can result from differences in cellular state (genetic, epigenetic, phenotypic, or due to stochastic fluctuations) as well as from cell cycle differences or effects of the cellular microenvironment 1–3. Cues external to the cell, such as death or survival stimuli, can be viewed as variable inputs acting on already variable cellular states. Together, these different sources of variability lead to downstream heterogeneity in phenotype.
The following simplified scenarios illustrate several ways in which competing pro-death/pro-survival signals can lead to variable cell fates. On the one hand, the relative strength of distinct and opposing stimuli may tip the balance in favor of survival or death, as in the case of a growth factor protecting cells from a death-inducing agent (Figure 1A). On the other hand, a single stimulus may induce both death and survival signals within a single cell; the internal state of the cell then determines which pathway is dominant at a given time (Figure 1B). For example, a block in apoptosis may unmask pro-survival signals triggered by a death ligand, or vice-versa; this may be true at the cell population level, or may vary among individual cells (Figure 1Ci). Alternatively, pro-death and pro-survival signaling may actively compete to determine whether a cell lives or dies, leading to cell-to-cell variability in response (Figure 1Cii). Finally, in cells exposed to a death-inducing agent, counter-balancing adaptive pathways may become activated to varying degrees in individual cells in a population, protecting against a future death stimulus (Figure 1D). Thus, a cell’s choice between life and death can be a function of both external context (e.g. signals from other ligands or cells) and its own internal state.
Figure 1. Scenarios illustrating how parallel or competing death and survival signals may influence cell fate decisions in individual cells.
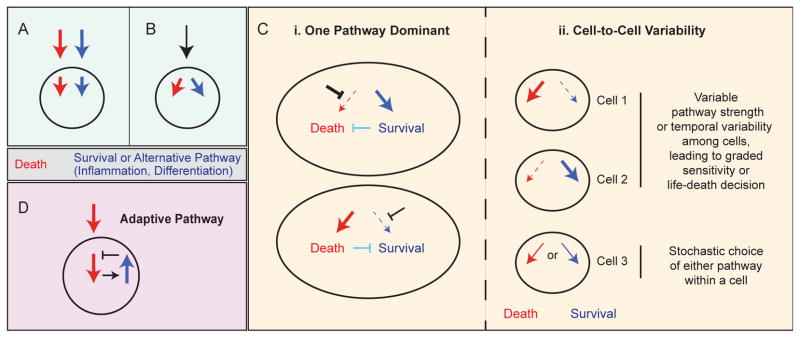
A, Separate death and survival signals (e.g. an inhibitor drug and growth factor ligand) activate parallel pathways within a cell. B, A single stimulus, such as a death ligand, activates both pro-death and pro-survival pathways. C, In the case of a single stimulus activating two pathways, one pathway might be dominant within a cell population, for example if the other pathway is not functioning (as in very high expression of an anti-apoptotic protein; bold black inhibition arrow), or if the first pathway negatively regulates the second pathway (aqua inhibition arrows)—leading to activation of only one of the pathways within the cell population. In this case, the strength of a single pathway would presumably vary among individual cells, leading to variability in cell fate (i). Alternatively, both pathways may be activated within cells, and cell-to-cell variability affects the relative strength or duration of each pathway, further leading to differences in cell fate (ii). In cells 1 and 2, variable pathway strength, or temporal variability among cells, leads to graded sensitivity within the cell population and variable life-death fate outcomes. In cell 3, one pathway or another is activated stochastically within a cell, also leading to variable life-death fate outcomes among cells. D, A drug treatment that can induce cell death leads to activation of a compensatory adaptive pathway that mediates resistance to the death signal in cells that escape death initially. The difference between B–C and D is that in B–C the death signal directly activates an alternative pathway alongside the death pathway (as in the case of death ligands), whereas in D, the adaptive pathway is indirectly activated in response to the death signal via a separate pathway. It should be noted that these two outcomes are not necessarily mutually exclusive.
Death Ligands and Death Receptors
Evasion of apoptosis is a hallmark of cancer cells and contributes to both cancer progression and resistance to chemotherapeutic drugs. Traditional chemotherapy targets the “intrinsic” pathway of cell death, activating apoptosis from within cells through induction of DNA damage or other cellular stresses. In contrast, the “extrinsic” apoptosis pathway is mediated by death ligands that bind to death receptors expressed at the surface of target cells 12. Apoptosis triggered by death ligands is thought to represent a form of innate immunity.
The three most-studied members of the death ligand family are TNF, Fas-Ligand (FasL), and TRAIL (TNF-Related Apoptosis-Inducing Ligand), in part because of their potential as cancer therapeutics. However, death ligands also activate non-apoptotic pathways, including inflammation and metastasis 13. The extent of activation of each of these pathways depends in part on the ligand: TNF is a strong inducer of inflammation, whereas FasL primarily induces apoptosis but exhibits hepatotoxicity, precluding its use as a cancer drug 12. The promise of TRAIL and agonist TRAIL receptor antibodies as cancer therapeutics reflects their specificity in targeting cancer cells for death. Nevertheless, many cancers exhibit resistance to these agents, and TRAIL can also activate “non-death” pathways under certain conditions 14,15. Thus, even the most potent apoptosis-inducing death ligand may successfully kill some cancer cell types but lead to unwanted effects in others.
Cell Death by Death Ligands
Death ligands bind to their cognate receptors and induce cell death via recruitment of a Death-Inducing Signaling Complex (DISC) and consequent activation of a caspase cascade that leads to permeabilization of mitochondria, further activation of caspases, and degradation of the proteome and genome (Box 2).
Box 2. Cell death by death ligands.
In the extrinsic apoptosis pathway, cell death is initiated by binding of ligand to receptors (DR4/5 for TRAIL and FasR for FasL) and subsequent recruitment of proteins to intracellular receptor tails. Ligand-receptor engagement leads to receptor aggregation, followed by a conformational change in receptor tails that promotes recruitment of the adaptor protein FADD and initiator caspases, and formation of the DISC 156,157. At the DISC, initiator caspases-8 and -10 (denoted as C8 in the figure) become activated via dimerization and autocatalytic cleavage, a process that can be either blocked or promoted by the presence of FLIP 26,158,159. In some cell types (Type I cells), initiator caspases directly cleave and activate effector caspases-3/7 (denoted as C3), leading to cell death. In contrast, Type II cells require mitochondrial outer membrane permeabilization (MOMP) for activation of effector caspases and cell death 160. In these cells, effector caspases are held in check by inhibitor of apoptosis protein XIAP 161,162, and this inhibition is relieved in a pathway involving MOMP 163,164. Initiator caspases cleave the pro-apoptotic BH3-only protein Bid into truncated Bid (tBid), which translocates to the mitochondria and activates pro-apoptotic Bcl-2 family members Bax and Bak. When the amount of activated Bax/Bak (denoted as Bax*) exceeds a threshold such that it overcomes the inhibitory effect of anti-apoptotic Bcl-2 family proteins, Bax/Bak forms pores in the mitochondrial membrane, leading to MOMP and release of Smac and cytochrome c into the cytosol 165. Cytochrome c promotes assembly of the caspase-9 (C9)-containing apoptosome, while Smac displaces XIAP from its inhibitory effect on caspase-3, leading to rapid cleavage of caspase-3 substrates and cell death 166 (see figure I, part A, shown here as a simplified representation of TRAIL-induced apoptosis). Each of these parts of the pathway (receptors, DISC, mitochondria, and IAP/C3) exhibits multiple levels of regulation that affect the strength of apoptotic signaling (see figure I, part B, and main text).
Figure I.

Cell death by death ligands.
Receptor-mediated apoptosis can be regulated and fine-tuned at many levels. Interactions among receptors (including anti-apoptotic decoy receptors for TRAIL), receptor post-translational modifications and localization to different membrane compartments determine the strength of pro-apoptotic signaling, for example by promoting or preventing the formation of higher-order receptor aggregates 16–22. The DISC is a complex structure containing multiple components that together determine whether apoptosis becomes activated in response to different stimuli in different cell types 23–25. c-FLIP, a DISC protein that has homology to caspase-8 but lacks proteolytic activity, is an anti-apoptotic regulator of initiator caspases, although it can exert pro-apoptotic activity in some contexts 26,27. The Bcl-2 family, which comprises more than 20 members, also undergoes complex regulation 28,29. Finally, extrinsic cell death is regulated by inhibitor of apoptosis proteins (IAPs), in particular XIAP. When XIAP levels are sufficiently high, cell death may be blocked; conversely, a low XIAP:caspase-3 ratio promotes cell death, diverting cells from Type II, mitochondrial outer membrane permeabilization (MOMP)-dependent death, to a Type I pathway in which caspase-3 is directly activated by caspase-8, leading to cell death even in the absence of MOMP (see Box 2) 30–32. It is likely that the relative levels of multiple proteins in the apoptosis pathway (and the interactions among them), rather than variation in a single protein, determines whether apoptosis occurs in response to death ligand. Thus, cells treated with death ligand may die or survive depending on their location in a multidimensional phase space of protein expression levels 31,33,34.
Alternative Pathways Induced by Death Ligands
The extrinsic cell death pathway has been described as a “node of coordination in immune signaling networks” because of its multifaceted role in the immune system 15. Thus, it is not surprising that death receptors can induce both apoptosis and cell survival, processes that are equally important in the regulation of inflammation and immunity (Figure 2A). TNF is a strong inducer of the pro-inflammatory transcription factor NF-κB and leads to upregulation of both inflammatory and pro-survival genes that inhibit caspase activation; TNF is thus generally pro-apoptotic only under conditions in which the NF-κB pathway is blocked 35 (Figure 2B). In contrast, TRAIL and FasL are strongly pro-apoptotic, and are thought to activate NF-κB relatively weakly, such that alternative signaling emerges mainly under conditions in which apoptosis is attenuated 23,36–39 (Figure 2C). Moreover, NF-κB may mediate inflammatory signaling in response to TRAIL and FasL more strongly than survival signaling per se 40–42.
Figure 2. Activation of opposing pathways by death ligands.

A, Death ligands activate opposing signals leading to apoptosis via a caspase pathway, or to alternative pathways via kinases and NF-κB. Alternative pathways include both survival and phenotypes such as inflammation, cell migration, and differentiation, and are mediated at least in part via transcription of target genes. B–C, Different ligands lead to sequential recruitment of distinct signaling complexes to receptor tails, with the primary complex promoting cell survival/inflammation in the case of TNF but apoptosis in the case of TRAIL/FasL. B, TNF leads to the formation of a primary signaling complex (complex I; bold blue arrow) that activates the NF-κB pathway, and induces cell death via the delayed formation of a secondary complex (complex II; dashed red arrow) that becomes active only when NF-κB signaling is inhibited. Apoptosis, in turn, negatively regulates the NF-κB pathway through cleavage of complex I proteins such as RIP1. When apoptosis is also inhibited, necroptosis may ensue through formation of an alternative secondary complex (IIb; not shown). C, TRAIL and Fas agonists primarily activate apoptotic DISCs (bold red arrow), but also activate NF-κB and kinase signaling through formation of secondary or alternative DISC structures (dashed blue arrow). In some cases NF-κB signaling in response to TRAIL/FasL is weak relative to apoptotic signaling, or becomes dominant only in cases when apoptotic signaling is dampened or blocked. Necroptosis may ensue when both apoptosis and survival signaling are inhibited in certain settings (not shown). D, Summary of factors affecting apoptotic vs. non-apoptotic DISC formation and signaling in response to TRAIL and FasL. Receptor clustering, which is controlled by ligand concentration, membrane vs. soluble ligand presentation, receptor localization in lipid rafts, and receptor post-translational modifications, reaches a threshold of activation that promotes pro-apoptotic DISC assembly, caspase-8 (C8) activation, and cell death (left). In contrast, sub-threshold receptor clustering leads to reduced formation of pro-apoptotic DISC structures and/or formation of anti-apoptotic DISCs (right). Stimulation strength and extent of receptor clustering can influence subsequent DISC structure and components, which are also regulated by the availability of different DISC factors. Some or all of these factors may impact signaling choices in different contexts and may vary with regards to TRAIL vs. FasL signaling. In some cases, cells may fail to die due to insufficient apoptotic signaling even in the absence of additional survival pathway activation.
In spite of being strongly pro-apoptotic, TRAIL and FasL have been shown to exhibit non-apoptotic activities in many settings 13,43. FasL promotes branching of cultured neurons, as well as liver regeneration following hepatic injury 44,45. TRAIL is involved in non-apoptotic forms of cellular differentiation, with reported roles in hematopoiesis and intestinal and skeletal muscle differentiation 46–48. TRAIL promotes survival, migration, and proliferation of human vascular endothelial cells and smooth muscle cells via a pathway involving ERK and AKT, suggesting roles in vascular physiology and promotion of angiogenesis 49–51. Both FasL and TRAIL also exhibit tumorigenic activities in cancers that have acquired resistance to apoptosis. FasL induces migration and invasion of glioblastoma and other apoptosis-resistant tumor cells, through activation of PI3K, ERK, NF-κB, and caspases, and also leads to secretion of inflammatory cytokines 39,52. Tumor growth in a mouse xenograft model has been shown to depend on constitutive FasL secretion by tumor cells, in a pathway involving JNK 53. TRAIL induces cell proliferation, migration, invasion, and secretion of pro-inflammatory cytokines in apoptosis-resistant cell lines, and promotes metastasis in a mouse xenograft model of pancreatic adenocarcinoma 42,54–58. Thus, both TRAIL and FasL appear to play physiological roles other than activation of apoptosis, and these pathways may be coopted by cancer cells that have become TRAIL-resistant 59.
What determines whether a particular cell type or tumor undergoes apoptosis or activation of non-apoptotic pathways in response to treatment with a death ligand? The relative apoptotic sensitivity of a particular cell type following treatment is tuned at many levels (see Box 2). In addition, many of these same components determine the extent to which alternative/pro-survival pathways become activated. For example, stimulation with TNF, FasL, or TRAIL initiates recruitment of different protein complexes to receptor tails, leading to different cell fates 20,36. These fates include not only apoptosis and survival/inflammation phenotypes, but also alternative forms of cell death such as necroptosis 60,61. TNF-receptor binding initially promotes recruitment of a “survival/inflammation” complex (TNF complex I) that contains the proteins TRADD and RIP1 and leads to activation of IKK, NF-κB, and transcription of pro-survival and pro-inflammatory genes; this is followed by recruitment of pro-apoptotic proteins FADD and caspase-8 in a secondary complex (TNF complex II) that is inhibited by anti-apoptotic NF-κB targets, thereby promoting cell survival. When NF-κB is inhibited, however, activity is switched toward complex II signaling and apoptosis; the apoptosis and survival pathways activated by TNF thus negatively regulate each other 23,62. When apoptosis is also blocked, an alternative complex (IIb) leads to cell death by necroptosis, a process that is dependent on RIP1 and RIP3 60.
In contrast, Fas agonists and TRAIL primarily lead to formation of pro-apoptotic caspase-8-containing DISCs, but subtle changes in DISC structure and composition can shift cells to a survival pathway that also involves activation of inflammatory signals, or to necroptosis 61,63. Which proteins are recruited to the DISC appears to depend on ligand presentation and strength as well as on the extent of receptor clustering and internalization, receptor post-translational modifications, and the availability of intracellular DISC proteins 16–20,64,65. For example, receptor sub-membrane localization and subsequent recruitment of distinct DISC complexes can determine TRAIL-mediated death vs. survival, with cell death favored when receptors localize to lipid rafts, and c-FLIP- and RIP1-dependent survival signaling when receptors are sequestered in non-raft fractions 66. Low FasL doses or receptor expression levels are sufficient to promote FasL-mediated survival but not cell death, presumably through a sub-threshold-dependent recruitment of pro-survival DISC components 67,68. Ligand presentation is also important since in vivo, membrane-bound FasL induces apoptosis whereas soluble FasL leads to NF-κB signaling and inflammation 69. In some cell types, E-cadherin-mediated cytoskeletal coupling is necessary for optimal TRAIL receptor clustering and pro-apoptotic DISC formation, and cells that have lost this coupling are resistant to cell death 21. Notably, expression levels of TRAIL receptors are generally not as good at predicting apoptotic sensitivity as is their ability to form high molecular weight complexes upon stimulation 19.
Within the DISC, multiple factors can affect the activation of caspase-8 and whether cells live or die. For example, small changes in the levels of different c-FLIP isoforms can shift cells toward apoptosis or survival 24,70; a variety of kinases (e.g. p38, JNK) that either promote or inhibit apoptosis may also be recruited and activated 52,71,72. In the case of TRAIL, increased levels of anti-apoptotic DISC components may shift cells toward activation of a secondary pro-inflammatory DISC complex, promoting activation of NF-κB, p38, and JNK 37. Ubiquitination of caspase-8 is required both for its activation and for subsequent dampening of its activity through degradation 73,74, and the extent of death effector domain (DED)-mediated caspase-8 chain formation at the DISC can influence the degree of apoptosis induction and potentially affect the activation of alternative pathways 75,76. Finally, it has been proposed that caspase-8 dimerization at the DISC, in the absence of proteolytic cleavage, can lead to survival signaling by processing a limited repertoire of substrates in the absence of cell death; intra- vs. inter-dimeric caspase-8 cleavage may also regulate this process 77,78. Thus, the decision between life and death is mediated by fine-tuning the extent of both cell-death pathway activation and survival pathway induction. Both the type of ligand-receptor interaction and the balance of pro- and anti-apoptotic proteins that come together as a result can determine cell fate by favoring one pathway over another (Figure 2D).
Cell Fate Decisions in Individual Cells
The fine-tuning of apoptotic and non-apoptotic pathway activation in response to treatment with death ligands impacts not only the overall behavior of cell populations, but also cell fate decisions of individual cells within a population. For example, in response to treatment with TRAIL or other death-inducing agents, many cell types exhibit a fractional response in which only a subset of cells dies even at saturating ligand doses 40,79. Such cell-to-cell variability has been observed both in tumors in vivo and in cultured cells and is a contributor to the challenges of using these agents as anti-cancer drugs 20,80,81. The origins of this variability have been extensively reviewed and contribute not only to fractional killing, but also to variable activation of non-apoptotic pathways leading to alternative cell fate outcomes (Box 3).
Box 3. Cell-to-cell variability and cell fate.
Populations of cells are inherently heterogeneous, and cell fate differences among cells are observed in diverse cellular processes including cell proliferation, differentiation, and cell death 167–169. Sources of variation can be genetic or non-genetic; non-genetic cell-to-cell variability includes both epigenetic differences and stochastic fluctuations in the levels or activities of factors within cells 1,2,170–172. Stochastic fluctuations result from either extrinsic noise, loosely defined as fluctuations in gene expression external to a particular pathway, or intrinsic noise, defined as fluctuations in levels of proteins in the pathway itself 173. Stochastic differences among cells typically have a “remixing time,” defined as the time it takes for a cell to switch from a given state back to the population average, which usually occurs on the order of hours or days 107. This is in contrast to epigenetic variation, with typically longer switching times between states (on the order of weeks or months), although these switches can also be caused by stochastic factors 81,174. In between these two types of variation are transiently induced or adaptive responses affecting changes in gene and protein expression or protein phosphorylation; these responses typically last longer than stochastic fluctuations but are shorter in duration than most epigenetic cell states (although this distinction is often not clearly defined, and both transiently-induced and epigenetic differences may be described as examples of phenotypic variation).
Each of these sources of variability also leads to temporal variation in the activity or localization of signaling proteins that ultimately affects cell fate decisions. This has been demonstrated not only for p53 and caspase activation dynamics during cell death 30,175,176, but also for proteins involved in transcription and cell survival. NF-κB exhibits cell-to-cell variability through an inhibitor of kappa B protein (IκB)-mediated negative feedback loop that drives oscillations in NF-κB nuclear translocation 177. The set of genes activated in an individual cell is determined by dose of the stimulus, fold-changes in the levels of nuclear NF-κB, frequency of pulsatile stimulations, and temporal patterns of the oscillations 178–181. Variability in expression and in phosphorylation and localization dynamics of ERK, PI3K and AKT also affect cell fate, influencing cell cycle and differentiation decisions as well as tuning the response to inducers of cell growth and death 182–185. Thus, a multitude of factors can affect, at any given time, a cell’s decision to live or die in response to a death stimulus, and if it survives, whether it takes on a new phenotype.
In the case of TRAIL, transient variation in the levels of proteins or other factors within cells has been shown to determine both the timing of cell death as well as whether cells live or die 79,82,83. This variation is non-genetic in origin, with pre-existing differences among cells (prior to treatment) determining, in large part, the timing of cell death as well as the fate of individual cells 79,84. Sister cells that have recently divided are highly correlated in their death times, but de-correlate within hours after cell division 79. Importantly, experiments involving the use of cycloheximide as well as modeling studies have demonstrated that such cell-to-cell variability and cell fate bifurcations can occur even in the absence of induced pro-survival signaling, resulting simply from differences in protein expression and degradation among cells, leading to differences in the strength of apoptotic signaling 30,85.
Other studies show, however, that cell-to-cell variability affects not only the extent of death pathway activation, but also the magnitude of survival pathway induction, and that both can contribute to fractional killing within a cell population. For example, cells that survive an initial TRAIL treatment exhibit a period of transient resistance one day later in which 90% of cells are completely resistant to a subsequent TRAIL treatment40. Cells enter this transiently resistant state even when cell death is inhibited during initial TRAIL exposure using caspase inhibitors, demonstrating that resistance is at least partially induced by TRAIL itself, rather than being a consequence of selection for a relatively resistant cell subpopulation. Resistance can be sustained by periodic TRAIL treatments, even in the absence of cell killing, but cells reset to a sensitive state following outgrowth in the absence of TRAIL 40. Such induced survival signaling may also play a role in responses to FasL, which has been shown to variably activate life-death pathways depending on the relative expression of different DISC proteins within individual cells 24,70. In these studies and others 86,87, death and survival signals (e.g. caspases and NF-κB/kinases) are activated simultaneously in individual cells treated with Fas agonists or TRAIL, and presumably the relative strength of pro- and anti-apoptotic factors determines whether a cell lives or dies.
Several other studies have also suggested that the balance between survival and death pathway signaling following treatment of cells with death-inducing agents may contribute to differences in cell fate among cells in a population. For example, one group measured the dynamic regulation of multiple fluorescently-labeled endogenous proteins in cancer cells treated with a topoisomerase inhibitor and showed that individual cells responded heterogeneously, leading to different life-death outcomes 88. In these cells, upregulation of an RNA helicase correlated with cell survival. Another group showed that PI3K/mTOR inhibition in matrix-attached mammary epithelial cells led to death of inner matrix-deprived cells but to activation of an adaptive survival program in outer cells 89. In yet another study, compensatory survival signaling protected a subset of an oncogene-addicted cell population from drug-induced death 90. Thus, cell fate in response to death-inducing agents likely depends, at least in some cases, on variable activation of both death and survival pathways. Moreover, variably-induced survival or adaptive pathways may re-enforce cell fate decisions in cells that initially survive a treatment due to prior variation in factors affecting sensitivity to death (Figure 3A–C).
Figure 3. Pre-existing variability in cell state and subsequent variability in induced pathways mediate cell fate decisions in cells treated with death ligands.

In this schematic, yellow-orange shadings depict non-genetic heterogeneity in protein levels or other factors in a naïve cell population. A, Following treatment, individual cells activate apoptosis signaling (red arrows) to varying degrees (depending on their initial cell state), in the absence of induced survival/alternative signaling (blue arrow, not activated in this cell population). The sensitive fraction of cells dies by apoptosis (unfilled cells) and the less sensitive fraction survives (dark yellow-orange cells). When the treatment is removed, protein levels re-distribute within several days, such that the new cell population is equivalent to the starting control population (dashed black arrow; reset). B, When apoptosis is blocked, survival/alternative signals (blue arrows) may become variably activated within the cell population, either maintaining cell survival, or leading to transient induction (green arrow) of alternative cell phenotypes or adaptive states (red cells with filled nuclei, indicating activation of a transcriptional state). Re-treating the cell population with death ligand may sustain these induced states (curved purple arrow), whereas removing treatment allows the induced signals to decay as cells divide, returning cells to their pre-treatment population distribution (dashed black arrow). C, Treatment with a death ligand activates both death and survival/alternative pathways to varying degrees in individual cells (red and blue arrows). If the signals are of equal strength, other factors within cells may influence cell fate (left, unfilled vs. yellow cell); if the signals are of unequal strength, the stronger signal may prevail to determine life or death of the cell. Similar to the scenario described in B, cells that survive may take on alternative phenotypes or adaptive states that are transiently resistant to death by a future stimulus (green arrow and red cells with filled nuclei), either through further activation of the initial survival signal or through compensatory survival signaling from another pathway. Again, the survival/adaptive state can either be sustained by continued treatment during the resistance stage or lost when treatment is removed, and this may occur across different timescales (taken with permission and adapted from Flusberg et al., MBOC, 2013). The schematic illustrates cell responses to treatment with TRAIL but is also relevant to signaling by other death ligands as well as more generally to any death-inducing agent that activates parallel or compensatory survival signals. D, Individual cells inhabit different areas in a multidimensional phase space, here shown for illustrative purposes as a projection in two dimensions. A cell’s location in this space is determined by multiple factors, including transcriptional states and protein expression levels or activity. The phase space can be divided into regions (separated by solid diagonal line) in which cells treated with TRAIL (filled arrows) either undergo apoptosis or survive. Some cells lying near the division line may rely on chance to determine whether their fate is death or survival (denoted by cells bounded by dashed diagonal lines). However, once “pushed” into the survival region following TRAIL treatment, surviving cells respond to repeated TRAIL treatments with further upregulation of survival pathways and remain in this region until TRAIL is removed and survival signals have decayed. The decay period (dashed arrow) may take hours, days, or weeks-months, depending on the type of survival signal initiated (e.g. kinase cascade: hours, transcriptional program: days, epigenetic change: weeks-months; see also Box 3). Thus, cell fate results from both random variation in initial cell state and subsequent response to a stimulus.
A combination of pre-existing cell-to-cell variability (leading to differences in apoptotic sensitivity) and induced survival or adaptive pathway signaling in cells that survive is also likely to play a role in reversible (dynamic) drug resistance of cancer cell subpopulations. For example, cells have been shown to switch between drug-sensitive and drug-resistant states, or to develop resistance following a drug treatment and then become sensitive again when given a “drug holiday” 91–95. Dynamic TRAIL-resistance has similarly been observed in several studies, and has been collectively attributed to changes at the level of receptors, DISC, or Bcl-2 family proteins 40,56,96–98. In some of these cases, cells in a population may pre-exist in a dynamic equilibrium of sensitive and resistant states and switch back and forth over time, whereas in other cases, reversible resistance in a fraction of cells results from a drug-induced adaptive state 90–95,99–101. Notably, this distinction is not always clear unless specific experiments are designed to probe the origins and durability of resistance. In the absence of live-cell reporters, it is technically challenging to observe both the initial state of a cell and its ultimate fate, and to determine whether markers that characterize survivors pre-date drug treatment or arise in response to it 88,102.
When changes in cell state are induced by a drug treatment, they may not only affect survival but also lead to development of new phenotypes. For example, cells surviving fractional killing by TRAIL take on an inflammatory phenotype in which cells migrate and differentiate as though responding to an injury 40. Such plasticity in cell subpopulations can be rationalized with reference to a protein expression “landscape,” in which individual cells lie in different parts of the landscape and therefore exhibit differential responses to stimuli. Phenotype switching of an individual cell would thus depend on its “initial conditions” (position in the landscape) and how susceptible it is to a stimulus-induced adaptive response. Memory of a previous stimulus could determine the response of cells to a subsequent stimulus by shifting a cell to a different area of protein expression space 103. Once pushed into a pro-survival region of the landscape (e.g. after surviving an initial TRAIL treatment), continued stimulation might keep cells in this region via activation of positive feedback loops or other regulatory networks, thereby preventing cells from relaxing back to the apoptosis-sensitive position they occupied prior to stimulation (Figure 3D). It will be interesting to ascertain the identities of these feedback loops using live-cell markers, ideally tagged at the endogenous locus, or by comparing time courses of transcript or protein profiles in populations of cells following treatment 104–107.
Advantages of Simultaneous Induction of Opposing Life-Death Signals
It has been suggested that in order for cell death to have evolved, proteins necessary for initiating death must originally have had non-apoptotic roles, making cells dependent on them for survival; hence, the close relationship between death and survival mechanisms. Alternatively, death and survival pathways may be coupled such that a chronic pro-death signal is inhibited until a survival signal is turned off, as occurs during the rapid degradation of anti-apoptotic Bcl-2 family proteins in stressed or infected cells 108,109. Indeed, evidence for both of these evolutionary strategies appears to exist 7,110.
The activation of opposing pathways by a single stimulus may be advantageous on several levels. First, induction of inflammation by death ligands may act as a “second line of defense” to eliminate damaged or infected cells (by recruiting engulfing phagocytes) when the primary outcome (death) has failed to transpire 35. Second, cells faced with a sub-lethal stress or injury may induce protective mechanisms against a future exposure to that stress, a concept known as preconditioning or disease tolerance 111,112. At the level of individual cells, a memory of the first stress (activated only in cells that respond with induction of a stress response rather than death) leads to preferential protection of some cells over others when a future stressor arises. Moreover, cells that survive an injury may activate not only signals that protect themselves against death by a future insult but also inflammation and differentiation programs that promote repair of damage, clearance of apoptotic cells, and healing of wounds caused by death of neighboring cells 37,113–115. Thus, depending on need, a secondary signal may function either to heal damage caused by cell death or to carry cell death to completion by an alternative mechanism.
In the case of tissue sculpting, it may be highly efficient for a single stimulus to kill some cells and induce differentiation in others. This may occur, for example, during lumen formation, mammary gland involution, neuronal differentiation, and immune responses 8,9,116,117. TNF family members are involved in many of these processes, and non-apoptotic physiological responses to death ligands may have been co-opted by cancer cells to protect themselves against death by these agents 35,59. Thus, alternative fates of cancer cells following TRAIL or FasL treatment may be a carry-over from higher order differentiation systems in vivo, as well as a form of “bet-hedging” by cells to survive in a dynamic environment. Such “bet-hedging” through non-genetic heterogeneity has been demonstrated in bacteria118 and has also been proposed to occur in cancer cells evading drug treatment 94,119. It seems likely that cancer cells exposed to death ligands may use a combination of bet-hedging and environmental sensing as a strategy to evade the immune system, by generating a fraction of cells that not only resist death, but also co-opt death signals for promotion of their own survival (via activation of death ligand-induced survival pathways and protection against future death stimuli). At any given time, a subset of cells may be predisposed to survive treatment due to natural fluctuation in protein levels; these cells then adapt (following death ligand exposure) and induce transient phenotypic changes associated with survival, inflammation, and tumorigenesis. The presence of such cells results in fractional killing by death ligands and therapeutic drugs and promotes residual disease.
Concluding remarks
Many proteins and pathways in cells have evolved to exhibit both death and non-death functions in different contexts. Non-death roles include signaling in inflammation, differentiation, proliferation, metabolism, survival, or any pathway integral to the life of a cell. These same proteins also regulate cell death, in the context of maintaining balance in cell populations.
Cell death-inducing agents have shown promise as cancer therapeutics because of their ability to eliminate damaged, “primed”, or rapidly proliferating cancer cells. However, these agents also paradoxically activate a variety of non-death signals that are likely to reduce the effectiveness of treatment. Alternative signals can become activated in apoptosis-resistant cell populations, or in subpopulations of cells exhibiting cell-to-cell variability. Cells that survive treatment may undergo an adaptive program that ensures their survival when faced with subsequent treatments, leading to a “memory” of their resistant state. This memory can occur on various timescales, depending on the type of adaptation.
The idea that a cell “remembers” previous exposure to a treatment implies that with long-term exposure, cell states can be sustained for extended periods. The question then arises as to when “acquired resistance” is actually a manifestation of transiently induced resistance that is reversible upon drug removal, versus when it results from a stable genetic mutation or from a more slowly reversible epigenetic adaptation. This and other interesting questions will need to be addressed in future studies, to better understand the contribution of transiently-induced factors to the development of drug-resistant states. Predicting the emergence of these states through measurements of heterogeneity within cell populations should help improve strategies for identifying biomarkers of drug resistance, as well as suggest how drug holidays and drug combinations can reduce resistance-promoting mechanisms 94,100,120,121. In addition, it will be important to understand how cell-to-cell variability impacts treatment schedules that aim to avoid the emergence of undesirable adaptive states 40,93,122.
Life-death decisions are influenced by multiple factors converging on a single cell, where each factor can itself support various outcomes. Understanding signaling systems as interacting components can thus be utilized to exploit therapeutic selectivity to the system rather than to single components. For example, targeting a protein complex or pathway rather than an individual protein may give better results if that protein takes on different functions under different conditions 123,124. A deeper understanding of these protein interactions and pathways will aid us in designing better therapeutics that take into account this complex regulation.
Highlights.
TNF family death ligands activate both death and non-death signaling pathways.
Cells surviving death stimuli may enter adaptive, death-resistant states.
Cell-to-cell variability in life-death signaling impacts cell fate decisions.
The evolutionary advantages of variability in life-death signaling are discussed.
Acknowledgments
We thank M. Hafner, C. Mills, L. Kleiman, V. Becker, S. Bandara, M. Fallahi-Sichani, and members of the Sorger laboratory for discussions. Work in the Sorger laboratory was supported by NIH grant P01-CA139980 to P.K.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brock A, Chang H, Huang S. Non-genetic heterogeneity--a mutation-independent driving force for the somatic evolution of tumours. Nat Rev Genet. 2009;10:336–342. doi: 10.1038/nrg2556. [DOI] [PubMed] [Google Scholar]
- 2.Eldar A, Elowitz MB. Functional roles for noise in genetic circuits. Nature. 2010;467:167–173. doi: 10.1038/nature09326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raj A, van Oudenaarden A. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell. 2008;135:216–226. doi: 10.1016/j.cell.2008.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beere HM. Death versus survival: functional interaction between the apoptotic and stress-inducible heat shock protein pathways. J Clin Invest. 2005;115:2633–2639. doi: 10.1172/JCI26471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carvajal LA, Manfredi JJ. Another fork in the road--life or death decisions by the tumour suppressor p53. EMBO reports. 2013;14:414–421. doi: 10.1038/embor.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nature reviews. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan J. Divergence from a dedicated cellular suicide mechanism: exploring the evolution of cell death. Mol Cell. 2006;23:1–12. doi: 10.1016/j.molcel.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 8.Munoz-Pinedo C. Signaling pathways that regulate life and cell death: evolution of apoptosis in the context of self-defense. Adv Exp Med Biol. 2012;738:124–143. doi: 10.1007/978-1-4614-1680-7_8. [DOI] [PubMed] [Google Scholar]
- 9.Kuranaga E. Caspase signaling in animal development. Development, growth & differentiation. 2011;53:137–148. doi: 10.1111/j.1440-169X.2010.01237.x. [DOI] [PubMed] [Google Scholar]
- 10.Sola S, Aranha MM, Rodrigues CM. Driving apoptosis-relevant proteins toward neural differentiation. Molecular neurobiology. 2012;46:316–331. doi: 10.1007/s12035-012-8289-2. [DOI] [PubMed] [Google Scholar]
- 11.Losick R, Desplan C. Stochasticity and cell fate. Science. 2008;320:65–68. doi: 10.1126/science.1147888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ashkenazi A. Targeting the extrinsic apoptosis pathway in cancer. Cytokine Growth Factor Rev. 2008;19:325–331. doi: 10.1016/j.cytogfr.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 13.Guicciardi ME, Gores GJ. Life and death by death receptors. FASEB J. 2009;23:1625–1637. doi: 10.1096/fj.08-111005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malhi H, Gores GJ. TRAIL resistance results in cancer progression: a TRAIL to perdition? Oncogene. 2006;25:7333–7335. doi: 10.1038/sj.onc.1209765. [DOI] [PubMed] [Google Scholar]
- 15.Wilson NS, Dixit V, Ashkenazi A. Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat Immunol. 2009;10:348–355. doi: 10.1038/ni.1714. [DOI] [PubMed] [Google Scholar]
- 16.Feig C, Tchikov V, Schutze S, Peter ME. Palmitoylation of CD95 facilitates formation of SDS-stable receptor aggregates that initiate apoptosis signaling. EMBO J. 2007;26:221–231. doi: 10.1038/sj.emboj.7601460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mazurek N, et al. Cell-surface galectin-3 confers resistance to TRAIL by impeding trafficking of death receptors in metastatic colon adenocarcinoma cells. Cell death and differentiation. 2011;19:523–533. doi: 10.1038/cdd.2011.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rossin A, Derouet M, Abdel-Sater F, Hueber AO. Palmitoylation of the TRAIL receptor DR4 confers an efficient TRAIL-induced cell death signalling. The Biochemical journal. 2009;419:185–192. 182. doi: 10.1042/BJ20081212. p following 192. [DOI] [PubMed] [Google Scholar]
- 19.Wagner KW, et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nature medicine. 2007;13:1070–1077. doi: 10.1038/nm1627. [DOI] [PubMed] [Google Scholar]
- 20.Gonzalvez F, Ashkenazi A. New insights into apoptosis signaling by Apo2L/TRAIL. Oncogene. 2010;29:4752–4765. doi: 10.1038/onc.2010.221. [DOI] [PubMed] [Google Scholar]
- 21.Lu M, et al. E-cadherin couples death receptors to the cytoskeleton to regulate apoptosis. Mol Cell. 2014;54:987–998. doi: 10.1016/j.molcel.2014.04.029. [DOI] [PubMed] [Google Scholar]
- 22.Merino D, et al. Differential inhibition of TRAIL-mediated DR5-DISC formation by decoy receptors 1 and 2. Mol Cell Biol. 2006;26:7046–7055. doi: 10.1128/MCB.00520-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muppidi JR, Tschopp J, Siegel RM. Life and death decisions: secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity. 2004;21:461–465. doi: 10.1016/j.immuni.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Neumann L, et al. Dynamics within the CD95 death-inducing signaling complex decide life and death of cells. Mol Syst Biol. 2010;6:352. doi: 10.1038/msb.2010.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peter ME, Krammer PH. The CD95(APO-1/Fas) DISC and beyond. Cell death and differentiation. 2003;10:26–35. doi: 10.1038/sj.cdd.4401186. [DOI] [PubMed] [Google Scholar]
- 26.Micheau O, et al. The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. The Journal of biological chemistry. 2002;277:45162–45171. doi: 10.1074/jbc.M206882200. [DOI] [PubMed] [Google Scholar]
- 27.Chang DW, et al. c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 2002;21:3704–3714. doi: 10.1093/emboj/cdf356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18:157–164. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Letai A, et al. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 30.Albeck JG, et al. Quantitative analysis of pathways controlling extrinsic apoptosis in single cells. Mol Cell. 2008;30:11–25. doi: 10.1016/j.molcel.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aldridge BB, Gaudet S, Lauffenburger DA, Sorger PK. Lyapunov exponents and phase diagrams reveal multi-factorial control over TRAIL-induced apoptosis. Mol Syst Biol. 2011;7:553. doi: 10.1038/msb.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jost PJ, et al. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature. 2009;460:1035–1039. doi: 10.1038/nature08229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gaudet S, Spencer SL, Chen WW, Sorger PK. Exploring the contextual sensitivity of factors that determine cell-to-cell variability in receptor-mediated apoptosis. PLoS Comput Biol. 2012;8:e1002482. doi: 10.1371/journal.pcbi.1002482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Passante E, Wurstle ML, Hellwig CT, Leverkus M, Rehm M. Systems analysis of apoptosis protein expression allows the case-specific prediction of cell death responsiveness of melanoma cells. Cell death and differentiation. 2013 doi: 10.1038/cdd.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walczak H. Death receptor-ligand systems in cancer, cell death, and inflammation. Cold Spring Harbor perspectives in biology. 2013;5:a008698. doi: 10.1101/cshperspect.a008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin Z, El-Deiry WS. Distinct signaling pathways in TRAIL- versus tumor necrosis factor-induced apoptosis. Mol Cell Biol. 2006;26:8136–8148. doi: 10.1128/MCB.00257-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Varfolomeev E, et al. Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. The Journal of biological chemistry. 2005;280:40599–40608. doi: 10.1074/jbc.M509560200. [DOI] [PubMed] [Google Scholar]
- 38.Wajant H, Pfizenmaier K, Scheurich P. Non-apoptotic Fas signaling. Cytokine Growth Factor Rev. 2003;14:53–66. doi: 10.1016/s1359-6101(02)00072-2. [DOI] [PubMed] [Google Scholar]
- 39.Barnhart BC, et al. CD95 ligand induces motility and invasiveness of apoptosis-resistant tumor cells. Embo J. 2004;23:3175–3185. doi: 10.1038/sj.emboj.7600325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flusberg DA, Roux J, Spencer SL, Sorger PK. Cells surviving fractional killing by TRAIL exhibit transient but sustainable resistance and inflammatory phenotypes. Mol Biol Cell. 2013 doi: 10.1091/mbc.E12-10-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu WH, Johnson H, Shu HB. Tumor necrosis factor-related apoptosis-inducing ligand receptors signal NF-kappaB and JNK activation and apoptosis through distinct pathways. The Journal of biological chemistry. 1999;274:30603–30610. doi: 10.1074/jbc.274.43.30603. [DOI] [PubMed] [Google Scholar]
- 42.Ishimura N, Isomoto H, Bronk SF, Gores GJ. Trail induces cell migration and invasion in apoptosis-resistant cholangiocarcinoma cells. Am J Physiol Gastrointest Liver Physiol. 2006;290:G129–136. doi: 10.1152/ajpgi.00242.2005. [DOI] [PubMed] [Google Scholar]
- 43.Yurchenko M, Shlapatska LM, Sidorenko SP. The multilevel regulation of CD95 signaling outcome. Experimental oncology. 2012;34:153–159. [PubMed] [Google Scholar]
- 44.Desbarats J, Newell MK. Fas engagement accelerates liver regeneration after partial hepatectomy. Nature medicine. 2000;6:920–923. doi: 10.1038/78688. [DOI] [PubMed] [Google Scholar]
- 45.Zuliani C, et al. Control of neuronal branching by the death receptor CD95 (Fas/Apo-1) Cell death and differentiation. 2006;13:31–40. doi: 10.1038/sj.cdd.4401720. [DOI] [PubMed] [Google Scholar]
- 46.Freer-Prokop M, O’Flaherty J, Ross JA, Weyman CM. Non-canonical role for the TRAIL receptor DR5/FADD/caspase pathway in the regulation of MyoD expression and skeletal myoblast differentiation. Differentiation. 2009;78:205–212. doi: 10.1016/j.diff.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rimondi E, et al. Involvement of TRAIL/TRAIL-receptors in human intestinal cell differentiation. Journal of cellular physiology. 2006;206:647–654. doi: 10.1002/jcp.20512. [DOI] [PubMed] [Google Scholar]
- 48.Secchiero P, Zauli G. Tumor-necrosis-factor-related apoptosis-inducing ligand and the regulation of hematopoiesis. Curr Opin Hematol. 2008;15:42–48. doi: 10.1097/MOH.0b013e3282f15fa6. [DOI] [PubMed] [Google Scholar]
- 49.Secchiero P, et al. TRAIL promotes the survival and proliferation of primary human vascular endothelial cells by activating the Akt and ERK pathways. Circulation. 2003;107:2250–2256. doi: 10.1161/01.CIR.0000062702.60708.C4. [DOI] [PubMed] [Google Scholar]
- 50.Secchiero P, et al. TRAIL promotes the survival, migration and proliferation of vascular smooth muscle cells. Cell Mol Life Sci. 2004;61:1965–1974. doi: 10.1007/s00018-004-4197-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cantarella G, Di Benedetto G, Ribatti D, Saccani-Jotti G, Bernardini R. Involvement of caspase 8 and c-FLIPL in the proangiogenic effects of the tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) FEBS J. 2014;281:1505–1513. doi: 10.1111/febs.12720. [DOI] [PubMed] [Google Scholar]
- 52.Kleber S, et al. Yes and PI3K bind CD95 to signal invasion of glioblastoma. Cancer Cell. 2008;13:235–248. doi: 10.1016/j.ccr.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 53.Chen L, et al. CD95 promotes tumour growth. Nature. 2010;465:492–496. doi: 10.1038/nature09075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ehrhardt H, et al. TRAIL induced survival and proliferation in cancer cells resistant towards TRAIL-induced apoptosis mediated by NF-kappaB. Oncogene. 2003;22:3842–3852. doi: 10.1038/sj.onc.1206520. [DOI] [PubMed] [Google Scholar]
- 55.Kim JH, Choi C, Benveniste EN, Kwon D. TRAIL induces MMP-9 expression via ERK activation in human astrocytoma cells. Biochem Biophys Res Commun. 2008;377:195–199. doi: 10.1016/j.bbrc.2008.09.095. [DOI] [PubMed] [Google Scholar]
- 56.Pavet V, et al. Plasminogen activator urokinase expression reveals TRAIL responsiveness and supports fractional survival of cancer cells. Cell Death Dis. 2014;5:e1043. doi: 10.1038/cddis.2014.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trauzold A, et al. TRAIL promotes metastasis of human pancreatic ductal adenocarcinoma. Oncogene. 2006;25:7434–7439. doi: 10.1038/sj.onc.1209719. [DOI] [PubMed] [Google Scholar]
- 58.Zhou DH, et al. The potential molecular mechanism of overexpression of uPA, IL-8, MMP-7 and MMP-9 induced by TRAIL in pancreatic cancer cell. Hepatobiliary Pancreat Dis Int. 2008;7:201–209. [PubMed] [Google Scholar]
- 59.Azijli K, Weyhenmeyer B, Peters GJ, de Jong S, Kruyt FA. Non-canonical kinase signaling by the death ligand TRAIL in cancer cells: discord in the death receptor family. Cell death and differentiation. 2013;20:858–868. doi: 10.1038/cdd.2013.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Christofferson DE, Li Y, Yuan J. Control of life-or-death decisions by RIP1 kinase. Annual review of physiology. 2014;76:129–150. doi: 10.1146/annurev-physiol-021113-170259. [DOI] [PubMed] [Google Scholar]
- 61.Dickens LS, Powley IR, Hughes MA, MacFarlane M. The ‘complexities’ of life and death: death receptor signalling platforms. Exp Cell Res. 2012;318:1269–1277. doi: 10.1016/j.yexcr.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 62.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 63.Holler N, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 64.Austin CD, et al. Death-receptor activation halts clathrin-dependent endocytosis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:10283–10288. doi: 10.1073/pnas.0604044103. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 65.Kohlhaas SL, Craxton A, Sun XM, Pinkoski MJ, Cohen GM. Receptor-mediated endocytosis is not required for tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis. The Journal of biological chemistry. 2007;282:12831–12841. doi: 10.1074/jbc.M700438200. [DOI] [PubMed] [Google Scholar]
- 66.Song JH, et al. Lipid rafts and nonrafts mediate tumor necrosis factor related apoptosis-inducing ligand induced apoptotic and nonapoptotic signals in non small cell lung carcinoma cells. Cancer research. 2007;67:6946–6955. doi: 10.1158/0008-5472.CAN-06-3896. [DOI] [PubMed] [Google Scholar]
- 67.Lavrik IN, et al. Analysis of CD95 threshold signaling: triggering of CD95 (FAS/APO-1) at low concentrations primarily results in survival signaling. The Journal of biological chemistry. 2007;282:13664–13671. doi: 10.1074/jbc.M700434200. [DOI] [PubMed] [Google Scholar]
- 68.Legembre P, et al. Induction of apoptosis and activation of NF-kappaB by CD95 require different signalling thresholds. EMBO reports. 2004;5:1084–1089. doi: 10.1038/sj.embor.7400280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.LAOR, et al. Membrane-bound Fas ligand only is essential for Fas-induced apoptosis. Nature. 2009;461:659–663. doi: 10.1038/nature08402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fricker N, et al. Model-based dissection of CD95 signaling dynamics reveals both a pro- and antiapoptotic role of c-FLIPL. J Cell Biol. 2010;190:377–389. doi: 10.1083/jcb.201002060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Azijli K, et al. MAPK p38 and JNK have opposing activities on TRAIL-induced apoptosis activation in NSCLC H460 cells that involves RIP1 and caspase-8 and is mediated by Mcl-1. Apoptosis. 2013;18:851–860. doi: 10.1007/s10495-013-0829-3. [DOI] [PubMed] [Google Scholar]
- 72.Son JK, Varadarajan S, Bratton SB. TRAIL-activated stress kinases suppress apoptosis through transcriptional upregulation of MCL-1. Cell death and differentiation. 2010;17:1288–1301. doi: 10.1038/cdd.2010.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gonzalvez F, et al. TRAF2 Sets a threshold for extrinsic apoptosis by tagging caspase-8 with a ubiquitin shutoff timer. Mol Cell. 2012;48:888–899. doi: 10.1016/j.molcel.2012.09.031. [DOI] [PubMed] [Google Scholar]
- 74.Jin Z, et al. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell. 2009;137:721–735. doi: 10.1016/j.cell.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 75.Dickens LS, et al. A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol Cell. 2012;47:291–305. doi: 10.1016/j.molcel.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schleich K, et al. Stoichiometry of the CD95 death-inducing signaling complex: experimental and modeling evidence for a death effector domain chain model. Mol Cell. 2012;47:306–319. doi: 10.1016/j.molcel.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 77.Hughes MA, et al. Reconstitution of the death-inducing signaling complex reveals a substrate switch that determines CD95-mediated death or survival. Mol Cell. 2009;35:265–279. doi: 10.1016/j.molcel.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 78.Kallenberger SM, et al. Intra- and interdimeric caspase-8 self-cleavage controls strength and timing of CD95-induced apoptosis. Science signaling. 2014;7:ra23. doi: 10.1126/scisignal.2004738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature. 2009;459:428–432. doi: 10.1038/nature08012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Falschlehner C, Emmerich CH, Gerlach B, Walczak H. TRAIL signalling: decisions between life and death. Int J Biochem Cell Biol. 2007;39:1462–1475. doi: 10.1016/j.biocel.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 81.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12:323–334. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 82.Bhola PD, Simon SM. Determinism and divergence of apoptosis susceptibility in mammalian cells. J Cell Sci. 2009;122:4296–4302. doi: 10.1242/jcs.055590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rehm M, et al. Dynamics of outer mitochondrial membrane permeabilization during apoptosis. Cell death and differentiation. 2009;16:613–623. doi: 10.1038/cdd.2008.187. [DOI] [PubMed] [Google Scholar]
- 84.Flusberg DA, Sorger PK. Modulating cell-to-cell variability and sensitivity to death ligands by co-drugging. Physical biology. 2013;10:035002. doi: 10.1088/1478-3975/10/3/035002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bertaux F, Stoma S, Drasdo D, Batt G. Modeling Dynamics of Cell-to-Cell Variability in TRAIL-Induced Apoptosis Explains Fractional Killing and Predicts Reversible Resistance. PLoS Comput Biol. 2014;10:e1003893. doi: 10.1371/journal.pcbi.1003893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tang W, et al. Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced chemokine release in both TRAIL-resistant and TRAIL-sensitive cells via nuclear factor kappa B. FEBS J. 2009;276:581–593. doi: 10.1111/j.1742-4658.2008.06809.x. [DOI] [PubMed] [Google Scholar]
- 87.Zauli G, et al. PI-3K/Akt and NF-kappaB/IkappaBalpha pathways are activated in Jurkat T cells in response to TRAIL treatment. Journal of cellular physiology. 2005;202:900–911. doi: 10.1002/jcp.20202. [DOI] [PubMed] [Google Scholar]
- 88.Cohen AA, et al. Dynamic proteomics of individual cancer cells in response to a drug. Science. 2008;322:1511–1516. doi: 10.1126/science.1160165. [DOI] [PubMed] [Google Scholar]
- 89.Muranen T, et al. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell. 2012;21:227–239. doi: 10.1016/j.ccr.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee HJ, et al. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell. 2014;26:207–221. doi: 10.1016/j.ccr.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 91.Raha D, et al. The cancer stem cell marker aldehyde dehydrogenase is required to maintain a drug-tolerant tumor cell subpopulation. Cancer research. 2014 doi: 10.1158/0008-5472.CAN-13-3456. [DOI] [PubMed] [Google Scholar]
- 92.Roesch A, et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141:583–594. doi: 10.1016/j.cell.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sabnis GJ, Macedo LF, Goloubeva O, Schayowitz A, Brodie AM. Stopping treatment can reverse acquired resistance to letrozole. Cancer research. 2008;68:4518–4524. doi: 10.1158/0008-5472.CAN-07-5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sharma SV, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sun C, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118–122. doi: 10.1038/nature13121. [DOI] [PubMed] [Google Scholar]
- 96.Kurbanov BM, Fecker LF, Geilen CC, Sterry W, Eberle J. Resistance of melanoma cells to TRAIL does not result from upregulation of antiapoptotic proteins by NF-kappaB but is related to downregulation of initiator caspases and DR4. Oncogene. 2007;26:3364–3377. doi: 10.1038/sj.onc.1210134. [DOI] [PubMed] [Google Scholar]
- 97.Song JJ, An JY, Kwon YT, Lee YJ. Evidence for two modes of development of acquired tumor necrosis factor-related apoptosis-inducing ligand resistance. Involvement of Bcl-xL. The Journal of biological chemistry. 2007;282:319–328. doi: 10.1074/jbc.M608065200. [DOI] [PubMed] [Google Scholar]
- 98.Song JJ, et al. c-Cbl acts as a mediator of Src-induced activation of the PI3K-Akt signal transduction pathway during TRAIL treatment. Cell Signal. 2009;22:377–385. doi: 10.1016/j.cellsig.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chakrabarti L, Abou-Antoun T, Vukmanovic S, Sandler AD. Reversible adaptive plasticity: a mechanism for neuroblastoma cell heterogeneity and chemo-resistance. Frontiers in oncology. 2012;2:82. doi: 10.3389/fonc.2012.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fallahi-Sichani M, Honarnejad S, Heiser LM, Gray JW, Sorger PK. Metrics other than potency reveal systematic variation in responses to cancer drugs. Nat Chem Biol. 2013;9:708–714. doi: 10.1038/nchembio.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wilson TR, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–509. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xia X, Owen MS, Lee RE, Gaudet S. Cell-to-cell variability in cell death: can systems biology help us make sense of it all? Cell Death Dis. 2014;5:e1261. doi: 10.1038/cddis.2014.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Acar M, Becskei A, van Oudenaarden A. Enhancement of cellular memory by reducing stochastic transitions. Nature. 2005;435:228–232. doi: 10.1038/nature03524. [DOI] [PubMed] [Google Scholar]
- 104.Bodenmiller B, et al. Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators. Nature biotechnology. 2012;30:858–867. doi: 10.1038/nbt.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Farkash-Amar S, et al. Noise genetics: inferring protein function by correlating phenotype with protein levels and localization in individual human cells. PLoS genetics. 2014;10:e1004176. doi: 10.1371/journal.pgen.1004176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Janes KA, Wang CC, Holmberg KJ, Cabral K, Brugge JS. Identifying single-cell molecular programs by stochastic profiling. Nat Methods. 2010;7:311–317. doi: 10.1038/nmeth.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sigal A, et al. Variability and memory of protein levels in human cells. Nature. 2006;444:643–646. doi: 10.1038/nature05316. [DOI] [PubMed] [Google Scholar]
- 108.Marriott HM, et al. Dynamic changes in Mcl-1 expression regulate macrophage viability or commitment to apoptosis during bacterial clearance. J Clin Invest. 2005;115:359–368. doi: 10.1172/JCI21766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cuconati A, Mukherjee C, Perez D, White E. DNA damage response and MCL-1 destruction initiate apoptosis in adenovirus-infected cells. Genes Dev. 2003;17:2922–2932. doi: 10.1101/gad.1156903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fernando P, Megeney LA. Is caspase-dependent apoptosis only cell differentiation taken to the extreme? FASEB J. 2007;21:8–17. doi: 10.1096/fj.06-5912hyp. [DOI] [PubMed] [Google Scholar]
- 111.Jang JH, Moritz W, Graf R, Clavien PA. Preconditioning with death ligands FasL and TNF-alpha protects the cirrhotic mouse liver against ischaemic injury. Gut. 2008;57:492–499. doi: 10.1136/gut.2007.137703. [DOI] [PubMed] [Google Scholar]
- 112.Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science. 2012;335:936–941. doi: 10.1126/science.1214935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Banno T, Gazel A, Blumenberg M. Effects of tumor necrosis factor-alpha (TNF alpha) in epidermal keratinocytes revealed using global transcriptional profiling. The Journal of biological chemistry. 2004;279:32633–32642. doi: 10.1074/jbc.M400642200. [DOI] [PubMed] [Google Scholar]
- 114.Wu NL, Lee TA, Tsai TL, Lin WW. TRAIL-induced keratinocyte differentiation requires caspase activation and p63 expression. J Invest Dermatol. 2011;131:874–883. doi: 10.1038/jid.2010.402. [DOI] [PubMed] [Google Scholar]
- 115.Li F, et al. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Science signaling. 2010;3:ra13. doi: 10.1126/scisignal.2000634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Clarkson RW, Wayland MT, Lee J, Freeman T, Watson CJ. Gene expression profiling of mammary gland development reveals putative roles for death receptors and immune mediators in post-lactational regression. Breast Cancer Res. 2004;6:R92–109. doi: 10.1186/bcr754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Miura M. Apoptotic and non-apoptotic caspase functions in neural development. Neurochemical research. 2011;36:1253–1260. doi: 10.1007/s11064-010-0341-x. [DOI] [PubMed] [Google Scholar]
- 118.Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. Bacterial persistence as a phenotypic switch. Science. 2004;305:1622–1625. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- 119.Kreso A, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science. 2013;339:543–548. doi: 10.1126/science.1227670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Singh DK, et al. Patterns of basal signaling heterogeneity can distinguish cellular populations with different drug sensitivities. Mol Syst Biol. 2010;6:369. doi: 10.1038/msb.2010.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Almendro V, Marusyk A, Polyak K. Cellular heterogeneity and molecular evolution in cancer. Annual review of pathology. 2013;8:277–302. doi: 10.1146/annurev-pathol-020712-163923. [DOI] [PubMed] [Google Scholar]
- 122.Foo J, Michor F. Evolution of acquired resistance to anti-cancer therapy. J Theor Biol. 2014;355:10–20. doi: 10.1016/j.jtbi.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jin L, Wang W, Fang G. Targeting protein-protein interaction by small molecules. Annual review of pharmacology and toxicology. 2014;54:435–456. doi: 10.1146/annurev-pharmtox-011613-140028. [DOI] [PubMed] [Google Scholar]
- 124.Pinto JP, Machado RS, Xavier JM, Futschik ME. Targeting molecular networks for drug research. Frontiers in genetics. 2014;5:160. doi: 10.3389/fgene.2014.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ow YP, Green DR, Hao Z, Mak TW. Cytochrome c: functions beyond respiration. Nature reviews. 2008;9:532–542. doi: 10.1038/nrm2434. [DOI] [PubMed] [Google Scholar]
- 126.Martinon F, Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell death and differentiation. 2007;14:10–22. doi: 10.1038/sj.cdd.4402038. [DOI] [PubMed] [Google Scholar]
- 127.Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell death and differentiation. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 128.Barbero S, et al. Identification of a critical tyrosine residue in caspase 8 that promotes cell migration. The Journal of biological chemistry. 2008;283:13031–13034. doi: 10.1074/jbc.M800549200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ehrenschwender M, et al. Mutant PIK3CA licenses TRAIL and CD95L to induce non-apoptotic caspase-8-mediated ROCK activation. Cell death and differentiation. 2010;17:1435–1447. doi: 10.1038/cdd.2010.36. [DOI] [PubMed] [Google Scholar]
- 130.Helfer B, et al. Caspase-8 promotes cell motility and calpain activity under nonapoptotic conditions. Cancer research. 2006;66:4273–4278. doi: 10.1158/0008-5472.CAN-05-4183. [DOI] [PubMed] [Google Scholar]
- 131.Kennedy NJ, Kataoka T, Tschopp J, Budd RC. Caspase activation is required for T cell proliferation. The Journal of experimental medicine. 1999;190:1891–1896. doi: 10.1084/jem.190.12.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lee P, et al. Dynamic expression of epidermal caspase 8 simulates a wound healing response. Nature. 2009;458:519–523. doi: 10.1038/nature07687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Li F, et al. Apoptotic caspases regulate induction of iPSCs from human fibroblasts. Cell Stem Cell. 2010;7:508–520. doi: 10.1016/j.stem.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cathelin S, et al. Identification of proteins cleaved downstream of caspase activation in monocytes undergoing macrophage differentiation. The Journal of biological chemistry. 2006;281:17779–17788. doi: 10.1074/jbc.M600537200. [DOI] [PubMed] [Google Scholar]
- 135.Brentnall M, Weir DB, Rongvaux A, Marcus AI, Boise LH. Procaspase-3 regulates fibronectin secretion and influences adhesion, migration and survival independent of catalytic function. J Cell Sci. 2014 doi: 10.1242/jcs.135137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Basu S, et al. alpha6 integrin transactivates insulin-like growth factor receptor-1 (IGF-1R) to regulate caspase-3-mediated lens epithelial cell differentiation initiation. The Journal of biological chemistry. 2014;289:3842–3855. doi: 10.1074/jbc.M113.515254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Droin N, et al. Various functions of caspases in hematopoiesis. Front Biosci (Landmark Ed) 2009;14:2358–2371. doi: 10.2741/3383. [DOI] [PubMed] [Google Scholar]
- 138.Fernando P, Kelly JF, Balazsi K, Slack RS, Megeney LA. Caspase 3 activity is required for skeletal muscle differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:11025–11030. doi: 10.1073/pnas.162172899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ishizaki Y, Jacobson MD, Raff MC. A role for caspases in lens fiber differentiation. J Cell Biol. 1998;140:153–158. doi: 10.1083/jcb.140.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Larsen BD, et al. Caspase 3/caspase-activated DNase promote cell differentiation by inducing DNA strand breaks. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:4230–4235. doi: 10.1073/pnas.0913089107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Arama E, Agapite J, Steller H. Caspase activity and a specific cytochrome C are required for sperm differentiation in Drosophila. Dev Cell. 2003;4:687–697. doi: 10.1016/s1534-5807(03)00120-5. [DOI] [PubMed] [Google Scholar]
- 142.Kuranaga E, et al. Drosophila IKK-related kinase regulates nonapoptotic function of caspases via degradation of IAPs. Cell. 2006;126:583–596. doi: 10.1016/j.cell.2006.05.048. [DOI] [PubMed] [Google Scholar]
- 143.McCall K, Steller H. Requirement for DCP-1 caspase during Drosophila oogenesis. Science. 1998;279:230–234. doi: 10.1126/science.279.5348.230. [DOI] [PubMed] [Google Scholar]
- 144.Hyman BT, Yuan J. Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nature reviews Neuroscience. 2012;13:395–406. doi: 10.1038/nrn3228. [DOI] [PubMed] [Google Scholar]
- 145.Li Z, Sheng M. Caspases in synaptic plasticity. Molecular brain. 2012;5:15. doi: 10.1186/1756-6606-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Williams DW, Kondo S, Krzyzanowska A, Hiromi Y, Truman JW. Local caspase activity directs engulfment of dendrites during pruning. Nature neuroscience. 2006;9:1234–1236. doi: 10.1038/nn1774. [DOI] [PubMed] [Google Scholar]
- 147.Danial NN, Gimenez-Cassina A, Tondera D. Homeostatic functions of BCL-2 proteins beyond apoptosis. Adv Exp Med Biol. 2010;687:1–32. doi: 10.1007/978-1-4419-6706-0_1. [DOI] [PubMed] [Google Scholar]
- 148.Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443:658–662. doi: 10.1038/nature05111. [DOI] [PubMed] [Google Scholar]
- 149.Opferman JT, et al. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426:671–676. doi: 10.1038/nature02067. [DOI] [PubMed] [Google Scholar]
- 150.Festjens N, Vanden Berghe T, Cornelis S, Vandenabeele P. RIP1, a kinase on the crossroads of a cell’s decision to live or die. Cell death and differentiation. 2007;14:400–410. doi: 10.1038/sj.cdd.4402085. [DOI] [PubMed] [Google Scholar]
- 151.Tourneur L, Chiocchia G. FADD: a regulator of life and death. Trends Immunol. 2010;31:260–269. doi: 10.1016/j.it.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 152.Shirley S, Micheau O. Targeting c-FLIP in cancer. Cancer Lett. 2010 doi: 10.1016/j.canlet.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 153.Ozturk S, Schleich K, Lavrik IN. Cellular FLICE-like inhibitory proteins (c-FLIPs): fine-tuners of life and death decisions. Exp Cell Res. 2012;318:1324–1331. doi: 10.1016/j.yexcr.2012.01.019. [DOI] [PubMed] [Google Scholar]
- 154.Matsuda I, et al. The C-terminal domain of the long form of cellular FLICE-inhibitory protein (c-FLIPL) inhibits the interaction of the caspase 8 prodomain with the receptor-interacting protein 1 (RIP1) death domain and regulates caspase 8-dependent nuclear factor kappaB (NF-kappaB) activation. The Journal of biological chemistry. 2014;289:3876–3887. doi: 10.1074/jbc.M113.506485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Dillon CP, et al. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell reports. 2012;1:401–407. doi: 10.1016/j.celrep.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Martin DA, Siegel RM, Zheng L, Lenardo MJ. Membrane oligomerization and cleavage activates the caspase-8 (FLICE/MACHalpha1) death signal. The Journal of biological chemistry. 1998;273:4345–4349. doi: 10.1074/jbc.273.8.4345. [DOI] [PubMed] [Google Scholar]
- 157.Kischkel FC, et al. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995;14:5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Boatright KM, et al. A unified model for apical caspase activation. Mol Cell. 2003;11:529–541. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 159.Donepudi M, Mac Sweeney A, Briand C, Grutter MG. Insights into the regulatory mechanism for caspase-8 activation. Mol Cell. 2003;11:543–549. doi: 10.1016/s1097-2765(03)00059-5. [DOI] [PubMed] [Google Scholar]
- 160.Barnhart BC, Alappat EC, Peter ME. The CD95 type I/type II model. Seminars in immunology. 2003;15:185–193. doi: 10.1016/s1044-5323(03)00031-9. [DOI] [PubMed] [Google Scholar]
- 161.Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 162.Suzuki Y, Nakabayashi Y, Takahashi R. Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:8662–8667. doi: 10.1073/pnas.161506698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Deng Y, Lin Y, Wu X. TRAIL-induced apoptosis requires Bax-dependent mitochondrial release of Smac/DIABLO. Genes Dev. 2002;16:33–45. doi: 10.1101/gad.949602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Li S, et al. Relief of extrinsic pathway inhibition by the Bid-dependent mitochondrial release of Smac in Fas-mediated hepatocyte apoptosis. The Journal of biological chemistry. 2002;277:26912–26920. doi: 10.1074/jbc.M200726200. [DOI] [PubMed] [Google Scholar]
- 165.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 166.Han Z, Hendrickson EA, Bremner TA, Wyche JH. A sequential two-step mechanism for the production of the mature p17:p12 form of caspase-3 in vitro. The Journal of biological chemistry. 1997;272:13432–13436. doi: 10.1074/jbc.272.20.13432. [DOI] [PubMed] [Google Scholar]
- 167.Chang HH, Hemberg M, Barahona M, Ingber DE, Huang S. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature. 2008;453:544–547. doi: 10.1038/nature06965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Feinerman O, Veiga J, Dorfman JR, Germain RN, Altan-Bonnet G. Variability and robustness in T cell activation from regulated heterogeneity in protein levels. Science. 2008;321:1081–1084. doi: 10.1126/science.1158013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Spencer SL, et al. The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell. 2013;155:369–383. doi: 10.1016/j.cell.2013.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Munsky B, Neuert G, van Oudenaarden A. Using gene expression noise to understand gene regulation. Science. 2012;336:183–187. doi: 10.1126/science.1216379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Snijder B, Pelkmans L. Origins of regulated cell-to-cell variability. Nature reviews. 2011;12:119–125. doi: 10.1038/nrm3044. [DOI] [PubMed] [Google Scholar]
- 172.Spencer SL, Sorger PK. Measuring and modeling apoptosis in single cells. Cell. 2011;144:926–939. doi: 10.1016/j.cell.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Pedraza JM, van Oudenaarden A. Noise propagation in gene networks. Science. 2005;307:1965–1969. doi: 10.1126/science.1109090. [DOI] [PubMed] [Google Scholar]
- 174.Rando OJ, Verstrepen KJ. Timescales of genetic and epigenetic inheritance. Cell. 2007;128:655–668. doi: 10.1016/j.cell.2007.01.023. [DOI] [PubMed] [Google Scholar]
- 175.Hellwig CT, et al. Real time analysis of tumor necrosis factor-related apoptosis-inducing ligand/cycloheximide-induced caspase activities during apoptosis initiation. The Journal of biological chemistry. 2008;283:21676–21685. doi: 10.1074/jbc.M802889200. [DOI] [PubMed] [Google Scholar]
- 176.Purvis JE, et al. p53 dynamics control cell fate. Science. 2012;336:1440–1444. doi: 10.1126/science.1218351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Nelson DE, et al. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science. 2004;306:704–708. doi: 10.1126/science.1099962. [DOI] [PubMed] [Google Scholar]
- 178.Ashall L, et al. Pulsatile stimulation determines timing and specificity of NF-kappaB-dependent transcription. Science. 2009;324:242–246. doi: 10.1126/science.1164860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Paszek P, et al. Population robustness arising from cellular heterogeneity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:11644–11649. doi: 10.1073/pnas.0913798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lee RE, Walker SR, Savery K, Frank DA, Gaudet S. Fold change of nuclear NF-kappaB determines TNF-induced transcription in single cells. Mol Cell. 2014;53:867–879. doi: 10.1016/j.molcel.2014.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Tay S, et al. Single-cell NF-kappaB dynamics reveal digital activation and analogue information processing. Nature. 2010;466:267–271. doi: 10.1038/nature09145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Albeck JG, Mills GB, Brugge JS. Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol Cell. 2013;49:249–261. doi: 10.1016/j.molcel.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Chen JY, Lin JR, Cimprich KA, Meyer T. A two-dimensional ERK-AKT signaling code for an NGF-triggered cell-fate decision. Mol Cell. 2012;45:196–209. doi: 10.1016/j.molcel.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Shankaran H, et al. Rapid and sustained nuclear-cytoplasmic ERK oscillations induced by epidermal growth factor. Mol Syst Biol. 2009;5:332. doi: 10.1038/msb.2009.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Yuan TL, Wulf G, Burga L, Cantley LC. Cell-to-cell variability in PI3K protein level regulates PI3K-AKT pathway activity in cell populations. Curr Biol. 2011;21:173–183. doi: 10.1016/j.cub.2010.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]