

Abstract
To investigate the metabolic fate of HT-2 toxin (HT2) and T-2 toxin (T2) in wheat (Triticum aestivum L.), an untargeted metabolomics study utilizing stable isotopic labeling and liquid chromatography–high resolution mass spectrometry was performed. In total, 11 HT2 and 12 T2 derived in planta biotransformation products were annotated putatively. In addition to previously reported mono- and diglucosylated forms of HT2, evidence for the formation of HT2-malonyl-glucoside and feruloyl-T2, as well as acetylation and deacetylation products in wheat was obtained for the first time. To monitor the kinetics of metabolite formation, a time course experiment was conducted involving the Fusarium head blight susceptible variety Remus and the resistant cultivar CM-82036. Biotransformation reactions were observed already at the earliest tested time point (6 h after treatment), and formed metabolites showed different kinetic profiles. After ripening, less than 15% of the toxins added to the plants were determined to be unmetabolized.
Keywords: trichothecenes, masked mycotoxins, cereals, liquid chromatography−high resolution mass spectrometry, metabolomics, stable isotopes
Introduction
Mycotoxins are low molecular weight fungal metabolites that frequently play a crucial role in plant pathogenesis and spreading of fungal infection. Fusarium species employ an arsenal of compounds as virulence factors, including certain trichothecene mycotoxins, to infect cereals such as wheat and barley.1 These two crop plants account for approximately 80% of the European small-grain production and may be severely contaminated with trichothecenes.2 Plants, however, are equipped with mechanisms to counteract the phytotoxicity of xenobiotics, including fungal toxins, leading to the formation of conjugated metabolites that are deposited in the apoplast or vacuole.3
T-2 toxin (T2) and its deacetylated form HT-2 toxin (HT2, lacking the acetyl-group at C-4) are members of the large family of trichothecenes, sharing a common tetracyclic ring system. Fusarium langsethiae, F. poae, and F. sporotrichioides are the predominant species that invade cereal crops and produce T2 and HT2 under cool and moist conditions on the field.4 Similar to most trichothecenes, T2 and HT2 not only inhibit protein synthesis and cell proliferation in plants, but also cause acute or chronic intoxication of humans and animals.5 Toxic effects include growth retardation, myelotoxicity, hematotoxicity, and necrotic lesions on contact sites. Because of their toxic potential, the European Food Safety Authority (EFSA) established a tolerable daily intake (TDI) value of 100 ng/kg body weight per day for the sum of T2 and HT2.6 Furthermore, the European Commission Recommendation 2013/165/EU provides indicative levels for the sum of these two toxins in cereals and cereal products ranging from 15 μg/kg for cereal-based foods for infants and young children up to 2000 μg/kg for oat milling products.7
Exposure of animals and humans to HT2 and T2 triggers metabolic transformations, which are generally well-characterized for a number of species, namely, rodents, pigs, chickens, and cattle.8 The major metabolic pathway of T2, regardless of the affected animal species, is its rapid deacetylation at C-4 that results in the formation of HT2. Other reactions commonly involved during metabolism of this mycotoxin in mammals are hydrolysis (e.g., neosolaniol, T2-triol, and T2-tetraol), oxidation (e.g., 3′-hydroxy-HT2 and 3′-hydroxy-T2), de-epoxidation (e.g., de-epoxy-3′-hydroxy-HT2, de-epoxy-T2-triol, and de-epoxy-HT2), and glucuronide conjugation of biotransformation products such as HT2 and neosolaniol.9 De-epoxidation is an important detoxification mechanism and together with metabolic changes (e.g., conjugation) of the hydroxyl group at C-3 has the greatest impact on reducing the toxicity of trichothecenes.10
In plants, conjugation of xenobiotics with glucose, sulfate, glutathione, or amino acids constitute well-known and effective detoxification strategies.3 The resulting metabolites are chemically distinct compounds produced by enzyme-catalyzed reactions, belonging to the so-called “masked mycotoxins”.11 Formation of glucosylated trichothecenes in artificially and naturally contaminated cereals was first shown for the type-B trichothecenes deoxynivalenol (DON)12 and nivalenol,13 followed by HT2 and T2.14 Nonetheless, metabolic products similar to those reported in animals have been detected in T2-treated Baccharis spp., namely, HT2, 3′-hydroxy-HT2, and T2-tetraol.15 To date, very limited information exists with regard to the metabolic fate of HT2 and T2 in cereal grains and natural occurrence of their plant-derived metabolites. In a recent survey study, HT-2 toxin-3-O-β-glucoside (HT2-3-Glc) was quantified in naturally contaminated barley, wheat, and oats, reaching concentrations of up to 300 μg/kg.16
To study the metabolism of xenobiotics in planta comprehensively, unbiased methods offer the chance to probe the entire metabolic space of the biological system under investigation. While liquid chromatography–high resolution mass spectrometry (LC-HRMS) methods are principally suited to measure hundreds of metabolites simultaneously, they come with the drawback of primarily detecting unspecific signals of nonbiological origin.17 Stable isotope labeling (SIL)-assisted untargeted metabolomics may resolve several of these limitations. SIL workflows have been recently applied in conjunction with HRMS to investigate the metabolism of DON in wheat and identification of novel plant-derived DON-metabolites.18 This approach has also been useful for the characterization of the fungal secondary metabolomes of Aspergillus fumigatus(19) and F. graminearum.20
The aim of this study was to investigate the in planta metabolic fate of T2 and HT2 in wheat. Putative identification of novel metabolites was performed with untargeted SIL-assisted metabolic profiling using LC-HRMS. Data were processed with MetExtract,21 and annotation was based on accurate mass measurements as well as LC-HRMS/MS spectra.22 Time course kinetics of the identified metabolites were also studied in two wheat lines ranging from the flowering stage until full ripening.
Materials and Methods
Chemicals and Reagents
HPLC grade acetonitrile and methanol were obtained from VWR (Vienna, Austria), and formic acid (LC-MS grade) and TWEEN 20 were purchased from Sigma-Aldrich (Vienna, Austria). Ammonium formate solution (5 M) was provided by Agilent Technologies (Waldbronn, Germany). Water was purified successively by reverse osmosis using an ELGA Purelab Ultra-AN-MK2 system (Veolia Water, Vienna, Austria). Crystalline nonlabeled T2 (purity 85%) and HT2 (purity 92%), as well as uniformly labeled U-[13C24] T2 (purity 98%; degree of enrichment 99.6 atom % 13C) and uniformly labeled U-[13C22] HT2 (purity 86%; degree of enrichment 99.3 atom % 13C) standards were purchased from Romer Labs GmbH (Tulln, Austria). Solutions from lyophilized labeled and nonlabeled T2 (10.0 g/L) and HT2 (10.0 g/L) were prepared separately by dissolving the compounds in acetonitrile. Certified standard solutions of T2 and HT2 (100.4 mg/L and 100.6 mg/L, respectively, in acetonitrile) were also obtained from Romer Labs GmbH. The analytical standard of HT2-3-Glc was enzymatically produced from HT2 (Michlmayr et al., manuscript in preparation). 3-Acetyl-T2 was synthesized from T2 using acetic anhydride in dichloromethane/pyridine with 4-dimethylaminopyridine as catalyst. Stock solutions in acetonitrile were prepared and were stored in sealed vials at −20 °C until use.
Plant Materials
The plant experiments are described according to the Metabolomics Standard Initiative (MSI) for the Proposed Minimum Information Set for reporting plant biological experimental designs. For the metabolomics profiling experiment, the spring wheat (Triticum aestivum L.) variety “Remus” (“Sappo”/“Mex”/“Famos”) was selected. The awn-free variety was developed at the Bavarian State Institute for Agronomy in Freising, Germany. It possesses well-adapted agronomic characteristics for cultivation in Central Europe but is highly susceptible to Fusarium head blight (FHB) and DON.23,24 The time-course experiment was conducted on the wheat variety Remus and the FHB-resistant variety “CM-82036-1TP-10Y-OST-10Y-OM-OFC” (abbreviated to “CM-82036”) which originated from the cross of the varieties “Sumai-3” and “Thornbird” and possess awns.24 Seeds of Remus and CM-82036 were first germinated. No vernalization was required, but the wheat seedlings were routinely submitted to a cold treatment at 5 °C for 1 week to improve tillering. Pots (diameter 23 cm) were filled with 7 L of a substrate (consisting of 500 L heat-sterilized compost, 250 L peat, 10 kg sand, and 250 g rock flour). Five seedlings of the same wheat line were planted in each pot.
Experiments were conducted in a greenhouse with computer-controlled settings for light, temperature, and relative air humidity. Light intensity (outside dark) was 360 μmol/sm2 at 1 m above the soil throughout the complete experiment. Relative air humidity was set between 60 and 70% during plant growth. Temperature (day/night) and duration of illumination (hours) varied according to the development stage of the plants: from planting until the end of tillering (stage 3 on the Feekes scale25), 12 °C/10 °C/12 h; end tillering to midstem extension when the ear starts to swell (stage 8), 14 °C/10 °C/14 h; mid stem extension to start heading (stage 10.1), 16 °C/14 °C/14 h; from the start of heading to the start of flowering (stage 10.51), 18 °C/14 °C/14 h; from the start of flowering (stage 10.51) until the end of the experiment, 20 °C/18 °C/16 h. In total, the seedlings needed 9–10 weeks to reach the flowering stage.
During the experiment, the pots were watered if required (typically three times/week). Water was applied until the substrate was completely wet and the water started to seep out through the holes in the bottom of the pot. The soil substrate contained sufficient minerals to support seedling growth. At the end of tillering (stage 5 on the Feekes scale), 2 g of a mineral fertilizer (COMPO Blaukorn ENTEC, Münster, Germany; N/P/K/Mg: 14/7/17/2) was applied per pot. To prevent mildew, the cabinet was treated twice during plant cultivation with sulfur overnight (sulfur evaporator, Nivola, Lisse, The Netherlands).
Treatment of Wheat Plants and Sampling
The experimental design was a completely randomized block with three biological replicates. Upon reaching the correct developmental stage (stage 10.51 on the Feekes scale), flowering ears were selected and individually labeled. Only one ear per plant was used in the experiment to prevent possible systemic effects of an earlier treated ear on other later flowering ears of the same plant. A woolen thread was used to label the lowest treated spikelet in the lower part of the ear (approx. 2/3 from the ear tip). In general, spikelets were treated by injecting 10 μL of the respective test solution into each of the outer floret (hence, 20 μL per spikelet) after placing the pipet tip in the spikelet using an electronic pipet Multipette Xstream (Eppendorf, Hamburg, Germany). After each treatment, the ear was covered for 24 h with a small transparent plastic bag internally wetted by spraying ca. 2 mL of bidistilled water with a handhold sprayer. This assured a high relative humidity promoting diffusion of the mycotoxins into the plant cells.
In the metabolic profiling experiment a 50:50 (v/v) mixture of 13C-labeled and nonlabeled solutions of HT2 or T2 (in acetonitrile:water (50:50, v/v) and 1% TWEEN) was used. As controls, ears were treated with a mock solution containing only acetonitrile:water (50:50, v/v) and 1% TWEEN. At time point zero, two neighboring spikelets were treated, and 48 h later the second treatment on the same ears was performed using the next pair of adjacent spikelets located above those treated previously. At 96, 120, and 144 h after the first application, treatments on the same ear continued as described, always selecting the next pair of spikelets in acropetal direction. In total, 200 μg of the 12C/13C toxin mixture was applied per ear. Sampling was performed 24 h after the last round of toxin application by removing the ear with a surgical scissor and dividing the wheat ear into three parts, but only the middle part was later used for LC-MS analysis: lower part (upper part of the stem and nontreated spikelets in basipetal direction of the treated ones), middle part (spikelets treated with toxins), and upper part (spikelets above the treated spikelets). All samples were weighed, shock-frozen in liquid nitrogen, and stored at −80 °C until further processing.
For the time course study, wheat ears were treated with nonlabeled T2 or HT2 or with the mock solution (methanol:water (50:50, v/v) and 1% TWEEN) similar to the process described above. The only difference was that 10 pairs of neighboring spikelets were treated with 10 μL each in one treatment resulting in a single dose of 200 μg per ear. Samples were collected at eight time points (0 h, 6 h, 12 h, 1 day, 2 days, 3 days, 1 week, and at full ripening) in triplicate. Wheat ears were removed from the plants with a surgical scissor, weighed as a whole, and immediately frozen with liquid nitrogen to prevent any metabolic activity until analysis. All collected samples were stored at −80 °C until further processing.
Sample Preparation
The frozen wheat ears were ground into a fine powder using a ball mill (MM 301 Retsch, Haan, Germany). The samples were placed inside 10 mL stainless steel vessels, precooled with liquid nitrogen, and were milled for 30 s at 30 Hz. After homogenization, (100 ± 2) mg of wheat material were weighed into 1.5 mL Eppendorf tubes and extracted with 500 μL of acetonitrile:water:formic acid (79:20.9:0.1, v/v/v) for 90 min with 200 rpm at room temperature on a GFL3017 rotary shaker (Burgwedel, Germany). Afterward, the extracts were centrifuged for 10 min at 22 570g using an Awel centrifuge MF 48-R (Blein, France). The supernatants (ca. 200 μL) were transferred to HPLC vials containing microinserts. The time course samples were partly further diluted 1:10 or 1:50 (v/v) in acetonitrile:water (50:50, v/v) to allow quantification of target analytes.
UHPLC-QTOF-MS Analysis
Measurements were performed with a 1290 series ultra high performance liquid chromatography (UHPLC) system coupled to a 6550 iFunnel quadrupole time-of-flight (QTOF)-MS system (both Agilent Technologies, Waldbronn, Germany) equipped with an electrospray ionization (ESI) interface. Chromatographic separation was achieved on a reversed-phase Zorbax SB-C18 column (2.1 × 150 mm, 1.8 μm particle size; Agilent Technologies) kept at 30 °C. In the isotope-assisted, untargeted metabolic profiling experiment, gradient elution was carried out with water (solvent A) and methanol (solvent B), both containing 0.1% formic acid, and applying the following gradient 1: 0–0.5 min 10% B, 0.5–20 min linear gradient from 10 to 100% B, 20–22 min flushing at 100% B and column re-equilibration with the initial chromatographic conditions (10% B) from 22 to 25 min. During the first 2 min and after the 22 min point mark, the eluent flow was directed to the waste. The flow rate of the mobile phase was 0.25 mL/min, and the injection volume was set to 3 μL. To avoid carry over, the needle was flushed with acetonitrile:water (50:50, v/v) for 5 s immediately prior to injection. For the time course experiment, 5 mM ammonium formate was added to both solvents to promote the formation of ammonium adducts. Furthermore, gradient 2 was shortened to 10 min (0–0.5 min, 20% B; 0.5–6.0 min, 20–100% B; 6.0–8.0 min, 100% B; 8.0–8.1 min, 100–20% B; 8.1–10.0 min, 20% B) for faster analysis.
Full scan spectra were acquired in the range of m/z 50–1500 in positive and negative ESI modes at an acquisition rate of 3 spectra per second. The source conditions were set as follows: gas temperature, 130 °C; drying gas flow, 14 L/min; nebulizer, 30 psig; sheath gas temperature, 300 °C and flow, 10 L/min; capillary voltage, 4000 V; and nozzle voltage, 500 V. Targeted LC-HRMS/MS experiments for identification and characterization of the formed metabolites were performed with gradient 1, but 5 mM ammonium formate was added in the solvents to obtain ammonium adducts for improved fragmentation. An isolation width of m/z 1.3 was applied for precursor selection; the acquisition speed in MS/MS was set to 3 spectra per second, and the applied collision energies (5, 10, and 20 eV) depended on the analytes. The QTOF was tuned and calibrated prior to use on the same day, and during the run two reference masses (positive mode, m/z 121.0508 and 922.0098; negative mode, m/z 112.9855 and 966.0007) were constantly infused via a second nebulizer to ensure mass accuracy.
Data were acquired with Mass Hunter Data Acquisition version B.05.01, and evaluation of data was performed using Mass Hunter Qualitative and Quantitative Analysis version B.06.00 (all Agilent Technologies), as well as Excel 2010 (Microsoft Co., Redmond, WA).
Data Processing for Detection of HT2 and T2 Wheat Metabolites
Acquired raw, full-scan LC-HRMS data were centroided and converted to the mzXML format with MSConvert from the ProteoWizard package (version 3.0.3959; http://proteowizard.sourceforge.net/). The 13C-enrichment (∼99.5%) and the applied nonlabeled to 13C-labeled tracer ratio (∼1) were determined for the highly abundant [M + Na]+ adducts of the T2 and HT2 tracers using TOPPView (version 1.10).26 All biotransformation products of the T2 and HT2 tracer were expected to show a very similar 13C-enrichment and nonlabeled-to-13C-labeled ratio in the recorded LC-HRMS data. Successively, MetExtract21 was utilized to screen for T2 and HT2 derived biotransformation products present as nonlabeled and partially 13C-labeled ions. The principal processing steps of the software are as follows: Mass peak pairings of nonlabeled (M) and 13C-labeled (M′) biotransformation product ions were searched for in each recorded mass spectrum (min intensity abundance, 500 counts; M:M′ ratio, 0.5–1.5). Each mass peak pairing had to have an m/z difference that corresponded to the number of labeled carbon atoms (max allowed relative m/z tolerance window, ± 4 ppm). The isotope patterns of both the nonlabeled and the partially 13C-labeled ions were then tested against the determined number of labeled carbon atoms (one isotopolog tested; max allowed abundance deviation, ± 15%). Detected mass peak pairings were clustered (max m/z deviation of ions of a single cluster, ± 9 ppm; min number of detected mass peaks, 3), and for each cluster, both the extracted ion chromatogram (EIC) of the native and the EIC of the labeled biotransformation product ions were extracted (EIC window, ± 10 ppm). Chromatographic peaks were detected in both EICs using the wavelet algorithm of Du et al.27 Only chromatographic peak pairs present in both EICs that eluted at the same retention time (max allowed deviation, ± 10 scans) and had a highly similar peak shape (min Pearson correlation coefficient, 0.85) were considered to be biotransformation product ions of both nonlabeled and 13C-labeled T2 and HT2 tracers. Other also detected chromatographic peaks in either one of the EICs were discarded as these did not represent nonlabeled and 13C-labeled biotransformation product ions of T2 and HT2 but other metabolites of wheat or contaminants. Finally, detected biotransformation product ions were convoluted into feature groups (max allowed retention time deviation, ± 10 scans; min Pearson correlation coefficient, 0.85), each representing one biotransformation product of T2 and HT2 in wheat. Ions assigned to a single group were investigated with respect to the presence of known mass increments between the detected ion species with the aim to annotate ion species. Commonly observed species during ESI positive mode were [M + H]+, [M + NH4]+, [M + Na]+, and [M+K]+ ions, while in negative mode [M-H]−, [M + Cl]−, [M + HCOO]− ions were regularly detected.
Validation of the Method for Quantification Purposes
For those analytes for which analytical standards were available (T2, HT2, HT2-3-Glc, and 3-acetyl-T2), an in-house validation was performed. Mock samples of the variety Remus were weighed (100 ± 2 mg) using the biological triplicates and were spiked with a solution containing all target analytes in acetonitrile before extraction on one level (1500 μg/kg, corresponding to 300 μg/L in the “undiluted” sample extract). The samples were stored overnight at room temperature to ensure solvent evaporation and to achieve equilibrium between the analytes and the matrix. The next day, the samples were extracted according to the procedure described above. Calculating ratios of the EIC (target m/z ± 30 ppm) peak area achieved in these samples with those in a neat solvent standard (300 μg/L, mean value from triplicate analysis) multiplied with 100 resulted in the apparent recovery (RA). For the evaluation of matrix effects (expressed as signal suppression or enhancement, SSE), mock samples of the same type (again in biological replicates) were extracted, and the raw extracts of the undiluted samples and 1:10 and 1:50 (v/v) diluted samples were spiked at a concentration of 300 μg/L. A comparison of EIC abundance values with the neat solvent standard (300 μg/L) provided the SSE values of the respective dilution. The extraction recovery was calculated by dividing RA with the SSE of the undiluted sample and multiplying with 100. For the calculation of the corresponding relative standard deviation, the propagation of uncertainty was taken into account.
For the quantification of HT2, HT2-3-Glc, T2, and 3-acetyl-T2, neat solvent standards ranging from 3 to 300 μg/L for 3-acetyl-T2 and from 10 to 1000 μg/L for the other three compounds were prepared in acetonitrile–water (50:50, v/v). They were measured along with the different dilutions of the sample extracts, and calibration was performed using 1/x weighted calibration curves.
Results and Discussion
HT2 and T2 Metabolite Annotation and Characterization
Raw UHPLC-QTOF-MS data derived from HT2- and T2-treated samples were processed by MetExtract, and the automatically generated features and feature groups were further manually inspected for false positives. Erroneously picked features, which by chance fulfilled MetExtract screening criteria, were easily recognized by certain characteristics such as imperfect coelution of assumed 12C and 13C EIC traces, a 12C/13C mass shift higher than the number of C atoms contained in the intact tracer, or implausible isotopolog intensity ratios. Finally, a total of 15 (HT2) and 16 (T2) feature groups (i.e., metabolites) including the native, nonmodified toxins were obtained. Of those, 12 (HT2) and 13 (T2) feature groups were of sufficient intensity, enabling their further structural elucidation by MS/MS, verifying their tentative chemical structure (shown in Tables 1 and 2). Each of these feature groups consisted of up to 25 12C/13C ion pairs originating from adducts and/or in-source fragments that shared the same retention time and chromatographic peak shape. It is worth mentioning that in mock samples, no feature pairs corresponding to T2 or HT2 metabolic products were detected by automated data processing, demonstrating the high selectivity of the used isotope-assisted untargeted workflow. Annotation of biotransformation products was based on accurate m/z values, assumed ion species, number of toxin-derived 13C atoms per metabolite ion, and LC-HRMS/MS spectral interpretations. The detailed structural characterization procedure is described in Meng-Reiterer, Varga, and co-workers28 who performed a similar study on the metabolism of T2 and HT2 in barley. Briefly, characteristic fragments obtained from standard measurements (T2, HT2, HT2-3-Glc, and 3-acetyl-T2) or described in the literature were compared with those experimentally obtained here for the MetExtract derived metabolites. LC-HRMS/MS measurements of pairs of corresponding nonlabeled and labeled precursor provided further information. The LC-HRMS/MS spectra of the observed metabolites of HT2 and T2 in barley28 were compared with those obtained in wheat and were shown to be identical. Figure 1 shows an overlay of EICs of all detected biotransformation products, and Tables 1 and 2 provide a summary of the annotated metabolites in HT2- and T2-treated wheat ears of the variety Remus.
Table 1. Overview of Putative HT-2 Toxin (HT2) Metabolites Detected in Wheat Ears of the Variety Remus (Susceptible to Fusarium Head Blight)a.
| peak no. | RT (min)b | chemical formulac | ion speciesd | m/zd | mass accuracy (ppm) | 13C count | putative metabolite |
|---|---|---|---|---|---|---|---|
| 1 | 5.2 | C23H34O12 | [M + Na]+ | 525.1940 | –0.41 | 17 | 15-acetyl-T2-tetraol-glucoside*** |
| 2 | 10.2 | C28H42O14 | [M + Na]+ | 625.2461 | –0.92 | 22 | hydroxy-HT2-glucoside** |
| 3 | 10.9 | C31H44O17 | [M + Na]+ | 711.2447 | –3.33 | 22 | hydroxy-HT2-malonyl-glucoside** |
| 4 | 13.0 | C26H40O12 | [M+HCOO]− | 589.2498 | –0.64 | 20 | T2-triol-glucoside** |
| 5 | 13.2 | C28H40O13 | [M+HCOO]− | 629.2446 | –0.78 | 22 | dehydro-HT2-glucoside** |
| 6, 7 | 13.3, 14.0 | C34H52O18 | [M+HCOO]− | 793.3135, 793.3135 | –0.08, –0.08 | 22 | HT2-diglucoside**,e |
| 8 | 14.1 | C28H42O13 | [M + Na]+ | 609.2518 | +0.00 | 22 | HT2-3-glucoside* |
| 9 | 14.3 | C31H44O16 | [M + Na]+ | 695.2505 | –2.39 | 22 | HT2-malonyl-glucoside** |
| HT2 | 14.9 | C22H32O8 | [M + Na]+ | 447.1992 | +0.59 | 22 | HT-2 toxin* |
| T2 | 16.0 | C24H34O9 | [M + Na]+ | 489.2098 | +0.68 | 22 | T-2 toxin (T2)* |
| 10 | 16.2 | C24H34O9 | [M + Na]+ | 489.2083 | –2.46 | 22 | 3-acetyl-HT2** |
Confirmation with standard by comparison of retention time, accurate mass, and HRMS/MS spectra.
Annotation with accurate mass and HRMS/MS spectra.
Annotation with accurate mass and HRMS/MS spectra only in HT2-treated barley.28
Samples were collected after exposure to a mixture of 200 μg/ear 12C/13C HT2 (1:1, v/v) during days 1, 2, 3, 5, and 7 on the same ear, and annotation was performed with accurate mass measurements and HRMS/MS spectra.
Retention time.
Sum formula of neutral compound.
Accurate mass and ion species of the most abundant ion.
Structural isomers of HT2-diglucoside with presumably different conjugations of the glucose moiety.
Table 2. Overview of Putative T-2 Toxin (T2) Metabolites Detected in Wheat Ears of the Variety Remus (Susceptible to Fusarium Head Blight)a.
| peak no. | RT (min)b | chemical formulac | ion speciesd | m/zd | mass accuracy (ppm) | 13C count | putative metabolite |
|---|---|---|---|---|---|---|---|
| 2 | 10.2 | C28H42O14 | [M + Na]+ | 625.2473 | +1.00 | 22 | hydroxy-HT2-glucoside** |
| 3 | 10.9 | C31H44O17 | [M + Na]+ | 711.2459 | –1.64 | 22 | hydroxy-HT2-malonyl-glucoside** |
| 4 | 13.0 | C26H40O12 | [M+HCOO]− | 589.2497 | –0.81 | 20 | T2-triol-glucoside** |
| 5 | 13.2 | C28H40O13 | [M+HCOO]− | 629.2446 | –0.79 | 22 | dehydro-HT2-glucoside*** |
| 6, 7 | 13.3, 14.0 | C34H52O18 | [M+HCOO]− | 793.3131, 793.3131 | –0.63, –0.63 | 22 | HT2-diglucoside**,e |
| 8 | 14.2 | C28H42O13 | [M + Na]+ | 609.2519 | +0.23 | 22 | HT2-3-glucoside* |
| 9 | 14.4 | C31H44O16 | [M + Na]+ | 695.2508 | –1.95 | 22 | HT2-malonyl-glucoside** |
| HT2 | 14.9 | C22H32O8 | [M + Na]+ | 447.1993 | +0.81 | 22 | HT-2 toxin (HT2)* |
| T2 | 16.0 | C24H34O9 | [M + Na]+ | 489.2096 | +0.20 | 24 | T-2 toxin* |
| 11 | 17.3 | C26H36O10 | [M + Na]+ | 531.2187 | –2.57 | 24 | 3-acetyl-T2* |
| 12, 13 | 18.1, 18.4 | C34H42O12 | [M + Na]+ | 665.2561, 665.2561 | –1.12, −1.12 | 24 | feruloyl-T2**,f |
Confirmation with standard by comparison of retention time, accurate mass, and HRMS/MS-spectra.
Annotation with accurate mass and HRMS/MS-spectra.
Annotation with accurate mass and HRMS/MS-spectra only in HT2-treated wheat samples.
Samples were collected after exposure to a mixture of 200 μg/ear 12C/13C T2 (1:1, v/v) during days 1, 2, 3, 5, and 7 on the same ear, and annotation was performed with accurate mass measurements and HRMS/MS spectra.
Retention time.
Sum formula of neutral compound.
Accurate mass and ion species of the most abundant ion.
Structural isomers of HT2-diglucoside with different conjugations of the glucose moiety.
Probably trans-feruloyl-T2 and trans-isoferuloyl-T2.
Figure 1.
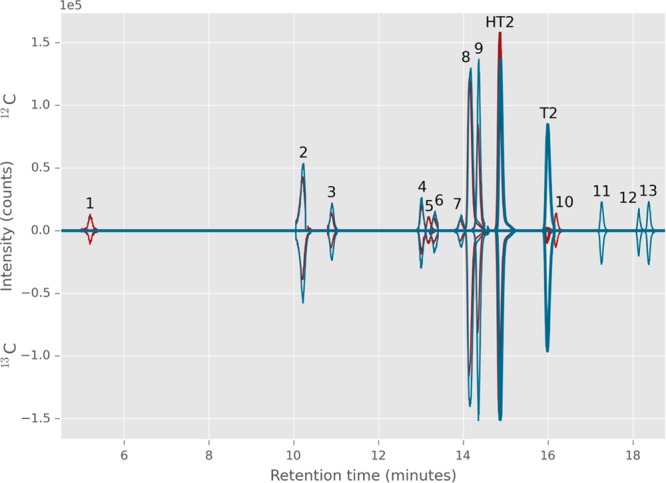
Overlaid extracted ion chromatograms (EICs) based on MetExtract data processing output showing the biotransformation products of a sample treated with a mixture of 12C/13C-HT-2 toxin (red trace) and one treated with a mixture of 12C/13C T-2 toxin (blue trace). EICs of nonlabeled metabolites are displayed with positive intensity values; those of the corresponding labeled metabolites are displayed as negative intensities. Perfect coelution, similar peak shape, and 12C/13C-intensities of EIC peaks verify the detected ions are truly tracer-derived metabolites.
In the samples which had been treated with HT2 (m/z 447.1989, [M + Na]+), a mass difference between the 12C and 13C ion pairs of 22.0738 Da (corresponding to the mass difference between 22 12C and 13C atoms) signified that the conjugated metabolites contained the intact molecule of HT2. Formation of HT2-3-Glc (Table 1, peak 8), an already known monoglucosylated HT2 metabolite, was verified by measurements of an authentic standard and was also detected in T2-treated samples (Table 2, peak 8). Only one peak for the m/z ratio corresponding to HT2-3-Glc was found in the treated wheat samples. This peak was perfectly matching in retention time and HR MS/MS fragmentation with the authentic standard of HT-2 toxin-3-O-β-glucoside. Additionally, two tentative diglucosylated HT2 isomers (Table 1, peaks 6 and 7) with slightly different retention times were detected in the wheat samples. We postulate that because monoglucosylation of HT2 was found to occur only at C-3, the additional sugar moiety is most likely transferred by a different enzyme, presumably leading to β-1,4 or β-1,6 diglucosides. Likewise, HT2-malonyl-glucoside is formed by conjugation of malonic acid to the glucose moiety of HT2-3-Glc. In addition to the metabolites identified in barley,28 also dehydro-HT2-glucoside was tentatively identified. Its HRMS/MS spectrum is provided as Figure S1 in Supporting Information. The absence of m/z 101.0608 and the occurrence of a fragment ion with m/z 99.0456 indicate the loss of two hydrogen atoms from the isovaleryl moiety, most likely after cleavage of water from hydroxy-HT2-glucoside. It should be noted that full structural elucidation of all detected metabolites would require purification and subsequent nuclear magnetic resonance (NMR) measurements, which was beyond the scope of this study.
The majority of the other metabolites were derived from HT2-3-Glc by additional conjugations and/or changes in the HT2 skeleton. Evaluation of mass shifts between corresponding 12C/13C feature pairs revealed that two metabolites contained less than 22 13C carbon atoms in their HT2 backbone (Table 1, peaks 1 and 4). These ions correspond to the putative metabolites 15-acetyl-T2-tetraol-glucoside (loss of the isovaleryl moiety at C-8) and T2-triol-glucoside (loss of the acetyl group at C-15). The fact that no 15-acetyl-T2-tetraol, T2-triol, or hydroxy-HT2 were detected even at the earlier time points indicates that the respective phase I type reactions hydrolysis, deacetylation, and oxidation, generally assumed to occur prior to conjugation of xenobiotics, took place after the conjugation of glucose to the HT2 backbone. Hence, direct conjugation of unaltered mycotoxins with plant constituents can result in metabolites susceptible to phase I reactions, under appropriate conditions, as has already been reported in the literature for pesticides.29
As previously described for HT2, metabolites containing the intact T2 carbon backbone (m/z 489.2095, [M + Na]+) should reveal a mass difference of 24.0805 between the 12C and 13C ion pairs. Nevertheless, only three such biotransformation products were found. Putative metabolites including an intact T2 skeleton were 3-acetyl-T2 and two tentatively isomeric peaks of feruloyl-T2 (Table 2, peaks 11–13). As ferulic acid (4-hydroxy-3-methoxycinnamic acid) naturally exists majorly as trans-ferulic acid,30 the second isomer is more likely isoferuloyl-T2 rather than a diastereomer of feruloyl-T2. It is worth mentioning that T2-glucoside could not be detected in this study as a biotransformation product of T2, although it has previously been documented in T2-contaminated wheat.14,31 The majority of metabolites in T2-treated wheat were formed after deacetylation of the toxin to HT2 and further metabolism according to the same metabolic pathways.
In Planta Acetylation of HT2
Many organisms, including plants, are capable of deacetylating T2 to HT2. However, with the 12C/13C experimental setup, we were able to show that wheat is also capable of acetylating HT2. The feature pair m/z 489.2083 (12C)/511.2823 (13C) recognized by MetExtract indicates the presence of 22 tracer-derived carbon atoms (as for the originally applied HT2), and the m/z value fits to the sum formula of the 13C-HT2 backbone to which a nonlabeled acetyl group has been conjugated (13C22C2H34O9; calculated m/z for Na-adduct 511.2833). HT2 has two hydroxyl groups at positions C-3 and C-4, which might be acetylated, and T2 differs from HT2 only by the acetyl-group at position C-4. Because the obtained retention time of 16.2 min (Figure 1, peak 10) was close to, but did not match with those of a T2 standard (16.0 min), it was postulated that the acetyl moiety of this HT2 metabolite results from acetylation at the position C-3 instead of C-4. With MS/MS measurements (see Figure S2 in Supporting Information) it was not possible to determine differences in fragmentation between native T2 and the putative 3-acetyl-HT2. Quantification of the presumed 3-acetyl-HT2 using T2 as standard resulted in about 40 μg/kg that accounts for less than 0.4% of the applied HT2. In a similar study with barley,28 no acetylated forms of HT2 could be confirmed. This acetylation reaction at C-3 can be regarded as a detoxification process because it has also been described to be a strategy employed by the trichothecene-producing fungus during toxin biosynthesis for self-protection.32 Finally, it should be mentioned that the small EIC-peak (m/z 511.2832) at 16.0 min could be T2. However, the second isotopolog of 13C-T2 (present as impurity of the 13C-HT2 used for spiking) will also lead to this signal, rendering this assumption inconclusive. To sum up, the presence of an acetylated form of HT2 other than T2, (presumably) 3-acetyl-HT2, was confirmed in wheat samples. Whether also T2 might be formed from HT2 remains unclear on the basis of our data. In any case, the deacetylation of T2 in wheat is seemingly far faster, compared to the potential reverse acetylation of HT2, so that the potential occurrence of 3-acetyl HT2 or T2 after metabolization from HT2 in wheat is only marginal.
Method Validation
A small validation study was performed with wheat samples of the variety Remus to ensure absolute quantification for HT2, HT2-3-Glc, T2, and 3-acetyl-T2. Biological triplicates (1 day and full ripening) from mock samples were spiked on one level before extraction to obtain RA. In the fully ripened samples, lower recoveries (56–124%) and higher relative standard deviations (up to 31%) were observed than in the samples harvested 1 day after treatment (74–132% and up to 3.5%, respectively; see Table S1 in Supporting Information). Additionally, different dilutions (undiluted, 1:10, and 1:50 (v/v)) of blank extracts of these biological triplicates were spiked on one level to assess matrix effects. For HT2 and HT2-3-Glc, some matrix enhancement was observed. The determined extraction recoveries (RE) were approximately 100%; therefore, obtained quantitative values in the kinetic experiment were only corrected for SSE in the respective dilution. The SSE values obtained for the samples harvested after full ripening were used for the correction of both varieties. For all other time points the SSE values determined for the day 1 samples were used.
Time Course Kinetics and Metabolic Pathways
The kinetic profiles of T2 and HT2 metabolites were studied with a time course experiment conducted in wheat lines susceptible (Remus) and resistant (CM-82036) to FHB. Treated wheat ears did not show premature bleaching after a single exposure to 200 μg of HT2 or T2, and in both wheat lines the same metabolites were determined. The kinetics of the major metabolites T2, HT2, and HT2-3-Glc in Remus and CM-82036 are illustrated in Figure 2 (HT2-treated sample) and Figure 3 (T2-treated sample). The absolute values were quantified with analytical standards, corrected for the observed SSE at the respective dilution, corrected for the individual ear weight, and calculated in nanomoles per ear. In T2-treated samples deacetylation of T2 to HT2 progresses rapidly, and already after 12 h only ca. 30% of the initially added toxin could be determined. This is in line with the study of Lattanzio et al.,33 reporting the enzymatic hydrolysis of T2 to HT2 after 120 min of incubation in wheat (conversion rate 89%), as well as other cereals. At the same time, rapid glucosylation of HT2 to form HT2-3-Glc occurred. Biotransformation of HT2 was also activated at a very early stage, regardless of whether it originated from direct application or deacetylation of T2. HT2-3-Glc was the major metabolite 6 h after exposure and accounted for approximately 33% of the added toxin after a treatment time of 1 week. Another interesting observation is that HT2-3-Glc reached a maximum concentration between 3 days and 1 week, after which its concentration decreased until harvest because of further metabolization.
Figure 2.

Results from the time course kinetics experiment. After exposure to a single dose of 200 μg/ear HT-2 toxin (HT2), the whole ears (n = 3) were harvested immediately after treatment (0 h), after 6 h, 12 h, 1 day, 2 days, 3 days, and 1 week, as well as at full ripening (ca. 8 weeks after treatment). For the parent toxin (HT2) and the major metabolite HT-2 toxin-3-glucoside, the absolute amounts (in nanomoles per ear) are provided together with the standard deviation of the biological triplicate. These values are based on quantitative measurements using the respective analytical standards and are corrected for matrix effects occurring during liquid chromatography–mass spectrometric analysis; the individual ear weights were taken into consideration. For all other metabolites no analytical standard was available; therefore, the relative abundances (in percent) are provided together with the standard deviation of the biological triplicate. Those values are based on the peak areas of the ammonium adduct (for 3-acetyl-HT-2 toxin the sodium adduct was used) multiplied with the individual ear weight. All values were set into correlation with the highest observed normalized area (usually the full ripening samples of Remus or CM-82036).
Figure 3.

Results from the time course kinetics experiment. After exposure to a single dose of 200 μg/ear T-2 toxin (T2), the whole ears (n = 3) were harvested immediately after treatment (0 h), after 6 h, 12 h, 1 day, 2 days, 3 days, and 1 week, as well as at full ripening (ca. 8 weeks after treatment). For the parent toxin (T2), the two major metabolites HT-2 toxin and HT-2 toxin-3-glucoside, as well as 3-acetyl-T-2 toxin, the absolute amounts (in nanomoles per ear) are provided together with the standard deviation of the biological triplicate. These values are based on quantitative measurements using the respective analytical standards and are corrected for matrix effects occurring during liquid chromatography–mass spectrometric analysis; the individual ear weights were taken into consideration. For all other metabolites, no analytical standard was available; therefore, the relative abundances (in percent) are provided together with the standard deviation of the biological triplicate. Those values are based on the peak areas of the ammonium adduct multiplied with the individual ear weight. All values were set into correlation with the highest observed normalized area (usually the full ripening samples of Remus or CM-82036).
For all other metabolites, analytical standards were not available; thus, the data obtained from EIC integrated peak areas were corrected only for the individual ear weight. The values were set into correlation with the highest observed area (usually the full ripening samples of Remus or CM-82036) (HT2-treated samples in Figure 2, T2-treated samples in Figure 3). According to these findings, C3-acetylation of both toxins and (iso)ferulic acid conjugation of T2 are the immediate responses of plant detoxification mechanisms, apart from glucosylation. However, these compounds were further metabolized or covalently bound to plant matrix, as their concentration decreased over time. To accurately assess their abundance under field conditions at harvest, natural occurrence studies in cereals for these compounds are necessary. Conjugations with an additional glucose moiety or malonic acid to existing HT2 monoglucosides occurred later. Hydrolysis of the acetyl group of HT2-3-Glc in C-15, leading to the formation of T2-triol glucoside, is presumably a consecutive later reaction as the compound was detectable only in ripened samples. The proposed metabolic pathway of HT2 and T2 metabolization is summarized in Figure 4.
Figure 4.

Proposed metabolic pathways of HT-2 toxin (HT2) and T-2 toxin (T2) in wheat.
The average weight of wheat ears of these two lines were significantly different. While CM-82036 ears on average weighed (0.92 ± 0.20) g, Remus ears weights were (1.61 ± 0.33) g on average. In addition, the ear weight (and the relative moisture content) changed over the investigated time frame. Therefore, data can be shown either as metabolite amounts per ear or as concentration (nanomoles of metabolite per gram of ear). Figures 2 and 3 represent data normalized to the weight of whole wheat ears. No significant differences in the amounts of HT2 and T2 metabolites in the two different wheat lines were determined. Another way of representing the same data would show that CM-82036 wheat plants more efficiently metabolize the toxins, as less biomass (of the treated ear) was able to produce approximately the same amount of metabolites. To compare the two wheat varieties in a truly quantitative manner it would be necessary to normalize the amount of added toxin relative to the different (dry) ears weights, which was beyond the scope of this study. Also, it should be mentioned that the applied toxins might not have had sufficient time to diffuse into the entire ear in the early time points.
Biological Role of Formed Metabolites and Toxicological Implications
According to these findings, the C12–C13 epoxy group of HT2 and T2 that is crucial for toxicity was not affected by wheat biotransformation. The conversion of T2 to HT2 is considered a detoxification step in mammals8 and also shown for plants in an Arabidopsis thaliana detached leaf assay.34 Formation of HT2-3-Glc was found to be the dominating biotransformation reaction of both HT2 and T2. Glucosylation of trichothecenes at the position C-3, with the anomeric carbon of glucose linked via a β-d-glucosidic bond, has been reported to greatly reduce phytotoxicity.35
Inhibition of the growth and spreading of Fusarium species due to antimicrobial properties of principal phenolic acids in grains and their role in cell wall thickening have been reported in the literature.36,37 Our results concerning conjugation of T2 with ferulic acid might be another beneficial property of ferulic acid, assuming a largely decreased toxicity of the conjugate.
In contrast to DON metabolism in wheat,18 no glutathione or other sulfur-containing conjugates of HT2 and T2 were detected in this study. This does obviously reflect the lack of an α,β-unsaturated carbonyl functionality at C-8 as T2 and HT2, a functionality that is presumed to be the primary conjugation target of glutathione,38 in type B trichothecenes such as DON.
Although glucosylation of trichothecenes is considered a detoxification process, the resulting conjugates may be reactivated within the mammalian gastrointestinal tract, a fact that is currently regarded as the main health risk associated with masked mycotoxins.39−41 In this context, limited and contradicting information is available concerning the toxic potential of type A trichothecene hydroxylation products, which could possibly emerge by microbial reactivation in the gut. Existing toxicological studies agree that 3′-hydroxy-HT2 is less toxic compared to its parent form,42 whereas 3′-hydroxy-T2 has been reported as being more acutely toxic in vivo than T2.43 These novel “activated” metabolites may constitute toxicologically relevant mycotoxin derivatives; thus, further investigations are required. Intestinal hydrolysis and uptake can vary for different conjugated forms. For instance, it has been shown that malonyl isoflavone glucosides are more effectively absorbed than nonmalonylated isoflavone glucosides in rats.44 Therefore, thorough toxicokinetic and overall toxicological assessment of novel and known masked mycotoxins should be conducted.
In summary, the metabolism of HT2 and T2 mycotoxins in wheat was presented according to data obtained by untargeted metabolomics profiling and time course kinetic studies. Exposure of wheat to either HT2 or T2 primarily activates metabolic reactions involving hydroxylation, (de)acetylation, and various conjugations. HT2 is the major metabolite resulting from exposure to T2 which follows the same pathway to that of directly inoculated HT2. Metabolites such as HT2-malonyl-glucoside, hydroxy-HT2-glucoside, dehydro-HT2-glucoside, T2-triol-glucoside, as well as (iso)feruloyl-T2 were documented for the first time. The interested reader is referred to Meng-Reiterer and Varga et al.28 for a detailed structural elucidation. Kinetic data revealed that detoxification progresses rapidly, resulting in the almost complete degradation of the toxins, within 1 week, after a single exposure in flowering wheat ears. No different metabolites were observed between the FHB-resistant wheat line CM-82036 and the FHB-susceptible wheat line Remus. Further investigations to compare the role of detoxification efficiency of wheat varieties in FHB resistance are required. Additional studies to assess the toxicological relevance and potential food safety concerns of the in planta metabolites emerging from T2 and HT2 mycotoxins are warranted.
Acknowledgments
The authors thank Imer Maloku for his support with the plant experiments.
Glossary
Abbreviations Used
- DON
deoxynivalenol
- EFSA
European Food Safety Authority
- EIC
extraction ion chromatogram
- ESI
electrospray ionization
- FHB
Fusarium head blight
- HRMS
high resolution mass spectrometry
- HT2
HT-2 toxin
- HT2-3-Glc
HT-2 toxin-3-O-β-glucoside
- LC
liquid chromatography
- MS/MS
tandem mass spectrometry
- MSI
Metabolomics Standard Initiative
- NMR
nuclear magnetic resonance
- QTOF
quadrupole time-of-flight
- RA
apparent recovery
- RE
extraction recovery
- SIL
stable isotope labeling
- SSE
signal suppression or enhancement
- T2
T-2 toxin
- TDI
tolerable daily intake
- UHPLC
ultra high performance liquid chromatography
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jafc.5b02697.
HRMS/MS spectra of the putative in planta HT2 metabolites dehydro-HT2-glucoside and 3-acetyl-HT-2 and results from the method validation (PDF)
Author Contributions
∥ A.V.N. and E.V. contributed equally to the work.
The financial support by the Austrian Federal Ministry of Science, Research and Economy, the National Foundation for Research, Technology and Development of Austria, BIOMIN Holding GmbH, as well as the Austrian Science Fund (FWF, grants P26213-B22 and SFB F3706) is gratefully acknowledged. Furthermore, this work has been supported by the Finnish Funding Agency for Technology and Innovation (TEKES; Myco-DETECT Project 401/31/2011) and the Walter Ehrström Foundation. The production of the HT2-3-Glc was performed within a Vienna Science and Technology Fund project (WWTF LS12–012) and the synthesis of 3-acetyl-T2 was performed within the FWF project SFB F3701.
The authors declare no competing financial interest.
Supplementary Material
References
- Desjardins A. E.; Hohn T. M. Mycotoxins in Plant Pathogenesis. Mol. Plant-Microbe Interact. 1997, 10, 147–152 10.1094/MPMI.1997.10.2.147. [DOI] [Google Scholar]
- Bottalico A.; Perrone G. Toxigenic Fusarium species and mycotoxins associated with head blight in small-grain cereals in Europe. Eur. J. Plant Pathol. 2002, 108, 611–624 10.1023/A:1020635214971. [DOI] [Google Scholar]
- Coleman J. O. D.; Blake-Kalff M. M. A.; Davies T. G. E. Detoxification of xenobiotics by plants: chemical modification and vacuolar compartmentation. Trends Plant Sci. 1997, 2, 144–151 10.1016/S1360-1385(97)01019-4. [DOI] [Google Scholar]
- Krska R.; Malachova A.; Berthiller F.; van Egmond H. P. Determination of T-2 and HT-2 toxins in food and feed: an update. World Mycotoxin J. 2014, 7, 131–142 10.3920/WMJ2013.1605. [DOI] [Google Scholar]
- Rocha O.; Ansari K.; Doohan F. M. Effects of trichothecene mycotoxins on eukaryotic cells: a review. Food Addit. Contam. 2005, 22, 369–378 10.1080/02652030500058403. [DOI] [PubMed] [Google Scholar]
- European Food Safety Authority (EFSA) Panel on Contaminants in the Food Chain (CONTAM) . Scientific Opinion on the risks for animal and public health related to the presence of T-2 and HT-2 toxin in food and feed. EFSA Journal 2011, 9, 12, 2481. [Google Scholar]
- Commission Recommendation 2013/165/EU of 27 March 2013 on the presence of T-2 and HT-2 toxin in cereals and cereal products Off. J. Eur. Commun. 2013, L 91, 12–15. [Google Scholar]
- Wu Q.; Dohnal V.; Kuca K.; Yuan Z. Trichothecenes: Structure-Toxic Activity Relationships. Curr. Drug Metab. 2013, 14, 641–660 10.2174/1389200211314060002. [DOI] [PubMed] [Google Scholar]
- Dohnal V.; Jezkova A.; Jun D.; Kuca K. Metabolic Pathways of T-2 Toxin. Curr. Drug Metab. 2008, 9, 77–82 10.2174/138920008783331176. [DOI] [PubMed] [Google Scholar]
- He J.; Zhou T.; Young J. C.; Boland G. J.; Scott P. M. Chemical and biological transformations for detoxification of trichothecene mycotoxins in human and animal food chains: a review. Trends Food Sci. Technol. 2010, 21, 67–76 10.1016/j.tifs.2009.08.002. [DOI] [Google Scholar]
- Rychlik M.; Humpf H.-U.; Marko D.; Dänicke S.; Mally A.; Berthiller F.; Klaffke H.; Lorenz N. Proposal of a comprehensive definition of modified and other forms of mycotoxins including “masked” mycotoxins. Mycotoxin Res. 2014, 30, 197–205 10.1007/s12550-014-0203-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthiller F.; Krska R.; Dall’Asta C.; Lemmens M.; Adam G.; Schuhmacher R. Determination of DON-3-Glucoside in artificially and naturally contaminated wheat with LC-MS/MS. Mycotoxin Res. 2005, 21, 205–208 10.1007/BF02959264. [DOI] [PubMed] [Google Scholar]
- Nakagawa H.; Ohmichi K.; Sakamoto S.; Sago Y.; Kushiro M.; Nagashima H.; Yoshida M.; Nakajima T. Detection of a new Fusarium masked mycotoxin in wheat grain by high-resolution LC–Orbitrap MS. Food Addit. Contam., Part A 2011, 28, 1447–1456 10.1080/19440049.2011.597434. [DOI] [PubMed] [Google Scholar]
- Busman M.; Poling S. M.; Maragos C. M. Observation of T-2 Toxin and HT-2 Toxin Glucosides from Fusarium sporotrichioides by Liquid Chromatography Coupled to Tandem Mass Spectrometry (LC-MS/MS). Toxins 2011, 3, 1554–1568 10.3390/toxins3121554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirocha C. J.; Abbas H. K.; Treeful L.; Bean G. T-2 Toxin and Diacetoxyscirpenol Metabolism by Baccharis spp. Appl. Environ. Microbiol. 1988, 54, 2277–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanail A. V.; Syvähuoko J.; Malachová A.; Jestoi M.; Varga E.; Michlmayr H.; Adam G.; Sieviläinen E.; Berthiller F.; Peltonen K. Simultaneous determination of major type A and B trichothecenes, zearalenone and certain modified metabolites in Finnish cereal grains with a novel liquid chromatography-tandem mass spectrometric method. Anal. Bioanal. Chem. 2015, 407, 4745–4755 10.1007/s00216-015-8676-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoridis G.; Gika H. G.; Wilson I. D. LC-MS-based methodology for global metabolite profiling in metabonomics/metabolomics. TrAC, Trends Anal. Chem. 2008, 27, 251–260 10.1016/j.trac.2008.01.008. [DOI] [Google Scholar]
- Kluger B.; Bueschl C.; Lemmens M.; Michlmayr H.; Malachova A.; Koutnik A.; Maloku I.; Berthiller F.; Adam G.; Krska R.; Schuhmacher R. Biotransformation of the Mycotoxin Deoxynivalenol in Fusarium Resistant and Susceptible Near Isogenic Wheat Lines. PLoS One 2015, 10, e0119656. 10.1371/journal.pone.0119656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano P. M.; Jamin E. L.; Tadrist S.; Bourdaud’hui P.; Pean M.; Debrauwer L.; Oswald I. P.; Delaforge M.; Puel O. New Untargeted Metabolic Profiling Combining Mass Spectrometry and Isotopic Labeling: Application on Aspergillus fumigatus Grown on Wheat. Anal. Chem. 2013, 85, 8412–8420 10.1021/ac401872f. [DOI] [PubMed] [Google Scholar]
- Bueschl C.; Kluger B.; Lemmens M.; Adam G.; Wiesenberger G.; Maschietto V.; Marocco A.; Strauss J.; Bödi S.; Thallinger G. G.; Krska R.; Schuhmacher R. A novel stable isotope labelling assisted workflow for improved untargeted LC–HRMS based metabolomics research. Metabolomics 2014, 10, 754–769 10.1007/s11306-013-0611-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueschl C.; Kluger B.; Berthiller F.; Lirk G.; Winkler S.; Krska R.; Schuhmacher R. MetExtract: a new software tool for the automated comprehensive extraction of metabolite-derived LC/MS signals in metabolomics research. Bioinformatics 2012, 28, 736–738 10.1093/bioinformatics/bts012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluger B.; Bueschl C.; Neumann N.; Stückler R.; Doppler M.; Chassy A. W.; Waterhouse A. L.; Rechthaler J.; Kampleitner N.; Thallinger G. G.; Adam G.; Krska R.; Schuhmacher R. Untargeted Profiling of Tracer-Derived Metabolites Using Stable Isotopic Labeling and Fast Polarity-Switching LC–ESI-HRMS. Anal. Chem. 2014, 86, 11533–11537 10.1021/ac503290j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buerstmayr H.; Lemmens M.; Grausgruber H.; Ruckenbauer P. Scab Resistance of International Wheat Germplasm. Cereal Res. Commun. 1996, 24, 195–202. [Google Scholar]
- Lemmens M.; Scholz U.; Berthiller F.; Dall’Asta C.; Koutnik A.; Schuhmacher R.; Adam G.; Buerstmayr H.; Mesterházy A.; Krska R.; Ruckenbauer P. The Ability to Detoxify the Mycotoxin Deoxynivalenol Colocalizes With a Major Quantitative Trait Locus for Fusarium Head Blight Resistance in Wheat. Mol. Plant-Microbe Interact. 2005, 18, 1318–1324 10.1094/MPMI-18-1318. [DOI] [PubMed] [Google Scholar]
- Large E. C. Growth stages in cereals illustration of the Feekes scale. Plant Pathol. 1954, 3, 128–129 10.1111/j.1365-3059.1954.tb00716.x. [DOI] [Google Scholar]
- Sturm M.; Kohlbacher O. TOPPView: An Open-Source Viewer for Mass Spectrometry Data. J. Proteome Res. 2009, 8, 3760–3763 10.1021/pr900171m. [DOI] [PubMed] [Google Scholar]
- Du P.; Kibbe W. A.; Lin S. M. Improved peak detection in mass spectrum by incorporating continuous wavelet transform-based pattern matching. Bioinformatics 2006, 22, 2059–2065 10.1093/bioinformatics/btl355. [DOI] [PubMed] [Google Scholar]
- Meng-Reiterer J.; Varga E.; Nathanail A. V.; Bueschl C.; Rechthaler J.; McCormick S. P.; Michlmayr H.; Malachová A.; Fruhmann P.; Adam G.; Berthiller F.; Lemmens M.; Schuhmacher R.. Tracing the metabolism of HT-2 toxin and T-2 toxin in barley by isotope assisted untargeted screening and quantitative LC-HRMS analysis. Anal. Bioanal Chem., in press, DOI: 10.1007/s00216-015-8975-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole D. J. Detoxification and Activation of Agrochemicals in Plants. Pestic. Sci. 1994, 42, 209–222 10.1002/ps.2780420309. [DOI] [Google Scholar]
- Sosulski F.; Krygier K.; Hogge L. Free, Esterified, and Insoluble-Bound Phenolic Acids. 3. Composition of Phenolic Acids in Cereal and Potato Flours. J. Agric. Food Chem. 1982, 30, 337–340 10.1021/jf00110a030. [DOI] [Google Scholar]
- McCormick S. P.; Kato T.; Maragos C. M.; Busman M.; Lattanzio V. M. T.; Galaverna G.; Dall-Asta C.; Crich D.; Price N. P. J.; Kurtzman C. P. Anomericity of T-2 Toxin-glucoside: Masked Mycotoxin in Cereal Crops. J. Agric. Food Chem. 2015, 63, 731–738 10.1021/jf504737f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander N. The TRI101 story: engineering wheat and barley to resist Fusarium head blight. World Mycotoxin J. 2008, 1, 31–37 10.3920/WMJ2008.x004. [DOI] [Google Scholar]
- Lattanzio V. M.; Solfrizzo M.; Visconti A. Enzymatic hydrolysis of T-2 toxin for the quantitative determination of total T-2 and HT-2 toxins in cereals. Anal. Bioanal. Chem. 2009, 395, 1325–1334 10.1007/s00216-009-2822-9. [DOI] [PubMed] [Google Scholar]
- Desjardins A. E.; McCormick S. P.; Appell M. Structure-Activity Relationships of Trichothecene Toxins in an Arabidopsis thaliana Leaf Assay. J. Agric. Food Chem. 2007, 55, 6487–6492 10.1021/jf0709193. [DOI] [PubMed] [Google Scholar]
- Poppenberger B.; Berthiller F.; Lucyshyn D.; Sieberer T.; Schuhmacher R.; Krska R.; Kuchler K.; Glössl J.; Luschnig C.; Adam G. Detoxification of the Fusarium Mycotoxin Deoxynivalenol by a UDP-glucosyltransferase from Arabidopsis thaliana. J. Biol. Chem. 2003, 278, 47905–47914 10.1074/jbc.M307552200. [DOI] [PubMed] [Google Scholar]
- McKeehen J. D.; Busch R. H.; Fulcher R. G. Evaluation of Wheat (Triticum aestivum L.) Phenolic Acids during Grain Development and Their Contribution to Fusarium Resistance. J. Agric. Food Chem. 1999, 47, 1476–1482 10.1021/jf980896f. [DOI] [PubMed] [Google Scholar]
- Gunnaiah R.; Kushalappa A. C.; Duggavathi R.; Fox S.; Somers D. J. Integrated Metabolo-Proteomic Approach to Decipher the Mechanisms by Which Wheat QTL (Fhb1) Contributes to Resistance against Fusarium graminearum. PLoS One 2012, 7, e40695. 10.1371/journal.pone.0040695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner S. A.; Boddu J.; Berthiller F.; Hametner C.; Stupar R. M.; Adam G.; Muehlbauer G. J. Transcriptome Analysis of the Barley-Deoxynivalenol Interaction: Evidence for a Role of Glutathione in Deoxynivalenol Detoxification. Mol. Plant-Microbe Interact. 2010, 23, 962–976 10.1094/MPMI-23-7-0962. [DOI] [PubMed] [Google Scholar]
- Dall’Erta A.; Cirlini M.; Dall’Asta M.; Del Rio D.; Galaverna G.; Dall’Asta C. Masked Mycotoxins Are Efficiently Hydrolyzed by Human Colonic Microbiota Releasing Their Aglycones. Chem. Res. Toxicol. 2013, 26, 305–312 10.1021/tx300438c. [DOI] [PubMed] [Google Scholar]
- De Angelis E.; Monaci L.; Mackie A.; Salt L.; Visconti A. Bioaccessibility of T-2 and HT-2 toxins in mycotoxin contaminated bread models submitted to in vitro human digestion. Innovative Food Sci. Emerging Technol. 2014, 22, 248–256 10.1016/j.ifset.2013.11.011. [DOI] [Google Scholar]
- Nagl V.; Woechtl B.; Schwartz-Zimmermann H. E.; Hennig-Pauka I.; Moll W.-D.; Adam G.; Berthiller F. Metabolism of the masked mycotoxin deoxynivalenol-3-glucoside in pigs. Toxicol. Lett. 2014, 229, 190–197 10.1016/j.toxlet.2014.06.032. [DOI] [PubMed] [Google Scholar]
- Islam Z.; Nagase M.; Ota A.; Ueda S.; Yoshizawa T.; Sakato N. Structure-Function Relationship of T-2 Toxin and Its Metabolites in Inducing Thymic Apoptosis in vivo in Mice. Biosci., Biotechnol., Biochem. 1998, 62, 1492–1497 10.1271/bbb.62.1492. [DOI] [PubMed] [Google Scholar]
- Yoshizawa T.; Sakamoto T.; Ayano Y.; Mirocha C. J. 3′-Hydroxy T-2 and 3′-Hydroxy HT-2 Toxins: New Metabolites of T-2 Toxin, a Trichothecene Mycotoxin, in Animals. Agric. Biol. Chem. 1982, 46, 2613–2615 10.1271/bbb1961.46.2613. [DOI] [Google Scholar]
- Yonemoto-Yano H.; Maebuchi M.; Fukui K.; Tsuzaki S.; Takamatsu K.; Uehara M. Malonyl Isoflavone Glucosides Are Chiefly Hydrolyzed and Absorbed in the Colon. J. Agric. Food Chem. 2014, 62, 2264–2270 10.1021/jf404378r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.