

Abstract
Background
Genetic renal diseases (GRD) are a heterogeneous and incompletely understood group of disorders accounting for approximately 10 % of those diagnosed with kidney disease. The advent of Next Generation sequencing and new approaches to disease modelling may allow the identification and validation of novel genetic variants in patients with previously incompletely explained or understood GRD.
Methods/Design
This study will recruit participants in families/trios from a multidisciplinary sub-specialty Renal Genetics Clinic where known genetic causes of GRD have been excluded or where genetic testing is not available. After informed patient consent, whole exome and/or genome sequencing will be performed with bioinformatics analysis undertaken using a customised variant assessment tool. A rigorous process for participant data management will be undertaken. Novel genetic findings will be validated using patient-derived induced pluripotent stem cells via differentiation to renal and relevant extra-renal tissue phenotypes in vitro. A process for managing the risk of incidental findings and the return of study results to participants has been developed.
Discussion
This investigator-initiated approach brings together experts in nephrology, clinical and molecular genetics, pathology and developmental biology to discover and validate novel genetic causes for patients in Australia affected by GRD without a known genetic aetiology or pathobiology.
Keywords: Chronic kidney disease, Nephrology, Nephrogenetics, Genetic sequencing, Induced pluripotent stem cell
Background
Renal disease is common with reported prevalence ranging from 10 to 16 % of Australian adults [1–3]. The impact of renal disease on the Australian community in terms of morbidity, mortality and health service requirements is significant [4, 5]. Renal disease is the most common cause of hospital admission, with more than 1,000,000 admissions per year, and the cumulative cost of treating current and new cases of end stage kidney disease 2009–2020 is estimated at AUD$11.3–12.3 billion [6]. Genetic renal disease (GRD) accounts for approximately 10 % of patients with renal disease [7, 8], with an additional proportion not as yet identified as being GRD or even as having yet developed renal disease. Up to half of presently identified GRD cases remain genetically unexplained.
The impact of GRD is well demonstrated by the most common and best understood form: Autosomal Dominant Polycystic Kidney Disease (ADPKD). ADPKD has a population prevalence of 1/400-800 and is the most common potentially lethal genetic disease in humans [9]. In Australia and New Zealand, ADPKD is the primary renal diagnosis in 6 % of the 12,968 prevalent dialysis patients and the fourth most common cause for dialysis commencement [10, 11]. Effective medical therapies for this condition are on the cusp of clinical translation after several decades of research, which initially elucidated the genetic aetiology of ADPKD, then explained its pathogenesis, and finally enabled translational applications via clinical therapeutic trials [12]. ADPKD, however, represents less than half of the estimated cases of GRD with many other renal diseases being unidentified and incompletely understood. Studies aiming at the detailed phenotypic description and explanation of these other and often very rare forms of GRD, are needed.
Enormous progress has been achieved in the field of molecular genetics and genomics since the discovery of deoxyribose nucleic acid (DNA) in 1953 [13]. The emergence of massively parallel sequencing (MPS) from approximately 2005 [14] has arguably been the next greatest advance since the development of Sanger sequencing in the late 1970s [15, 16]. Next generation sequencing (NGS) collectively describes these recently-developed genetic technologies that enable rapid and cost effective sequencing of large amounts of DNA. While sequencing of the first human genome took over 10 years and required an international collaboration at the approximate cost of USD $3 billion [17], NGS now allows a human genome to be sequenced in days at the cost of only $1–2,000 [18]. Whole exome sequencing (WES) is a variation on whole genome sequencing (WGS) that allows for targeted sequencing of only the approximately 1.5 % of the human genome that contains protein-coding genes, reducing costs even further [19].
NGS facilitates rapid identification of disease-causing genetic variants in patients with a suspected Mendelian disorder. This utility was first proven by the discovery of the gene involved in causing Freeman-Sheldon syndrome [20]. With reduced cost and time, WES has become the preferred initial method for disease gene identification in small pedigrees, replacing previous methods, such as linkage analysis and association studies. By 2012, more than 100 causative genes had been identified by means of WES [14]. WES is now able to elucidate the causal mutations in nearly 50 % of patients thought to have a genetic condition [21–23]. This diagnostic yield is further optimised when family-based studies such as the trio/family-based approach are employed [24]. In nephrology, NGS has identified disease-causing mutations responsible for atypical haemolytic uremic syndrome [25, 26], steroid-resistant nephrotic syndrome [27–29] and nephronophthisis [30–39]. However, the resolution within which any base change can be identified using this technique results in the identification of large numbers of variations, not all of which are pathogenic. Hence, it is now more critical than ever to validate the pathogenicity of any novel genetic variant. This may include appropriate Mendelian segregation and genotype/phenotype correlation via some form of biological validation.
With the advent of reprogramming of adult cells to a pluripotent state, termed induced pluripotent stem cells (iPSC), clinicians now have an opportunity to differentiate cells from the patient in question into relevant tissue types and in vitro [40]. Patient-specific iPSC differentiation allows for more effective modelling of the patient’s own disease [41–44]. For renal disease, this is a challenge as the kidney is an architecturally and functionally complex organ at both a microscopic and macroscopic level. However, recent studies have reported the directed differentiation of iPSC to podocytes [45], tubular structures [46, 47] and self-organising renal organoids [48], providing a potential in vitro model system with which to validate novel genetic findings in GRD patients. This also provides the potential to better understand underlying pathology with the aim of developing new treatments.
Recent advances in genetic sequencing technology have resulted in remarkable improvements in the speed, throughput and cost of sequencing all, or part, of an individual’s genome. While it would appear feasible to apply these new technologies to kidney disease, a clinical protocol is required to guide patient identification and recruitment, ethical acquisition of material, and appropriate counselling whilst coupling this with NGS for the identification of novel mutations and iPSC validation to discover the genetic basis for rare genetic diseases. The need for patients to be informed of outcomes must also be addressed. We hypothesize that emerging high-throughput sequencing technologies will lead to the rapid identification of novel causative genes in GRD and propose a study to begin realising this potential within Australia.
Methods
Study aim
This study aims to discover the genetic basis for disease in a cohort of patients thought clinically to have a genetic disorder causing renal dysfunction or disease. The cohort will be selected based upon family history and phenotype strongly suggesting a genetic aetiology and in whom routine genetic testing is not available, not feasible or has not identified a mutation/s in currently known genes. Our hypothesis is that we will be able to identify the disease-causing mutations in a proportion of these participants.
Study design
This is a translational study based at Royal Brisbane and Women’s Hospital (RBWH) and the University of Queensland (UQ) to discover, validate and explain new genetic causes for inherited kidney disease where investigation for known genes has been unsuccessful or is unavailable. Participants will be recruited from patients attending the RBWH Conjoint Renal Genetics and Inherited Kidney Disease Clinics, as part of standard clinical care (Fig. 1).
Fig. 1.
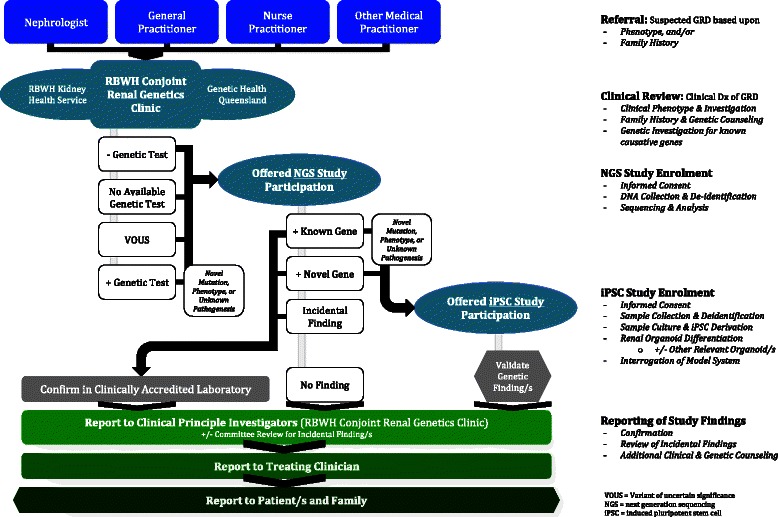
Study flow diagram
Ethical considerations
The RBWH Human Research and Ethics Committee (approval HREC/14/QRBW/34), the UQ Medical Research Ethics Committee (approval 2014000453) and the Children’s Health Queensland Hospital and Health Service Human Research and Ethics Committee (approval HREC/15/QRCH/126), reviewed and approved the study.
Target population
This study involves two groups of participants:
- An affected individual whose family history and/or phenotype strongly suggests a genetic aetiology and in whom routine genetic testing:
- Is not clinically available
- Is not feasible given the suspected disorder has high genetic heterogeneity, or
- Has already failed to arrive at a diagnosis
First degree and appropriate relatives of participants of group 1.
Patient identification and screening
Patients referred to the RBWH Kidney Health Service, RBWH Conjoint Renal Genetics Clinic and/or Genetic Health Queensland (RBWH) will be identified as potential research participants by their treating nephrologist and/or clinical geneticist.
Once a suitable individual from the target population is identified and consent obtained by the treating clinician, their first-degree relatives will be invited to participate in the study by consented individuals. The research team will then be available to discuss and provide full information about the study to the first-degree relatives and offer them the opportunity to participate, also by informed consent.
Patient/participant inclusion
In most cases inclusion in this study will require the participation of the patient and both parents. However, in some cases a combination of factors including the structure of the family pedigree, the suspected mode of inheritance, and information available regarding the specific disease may provide the required level of genetic information or require additional or alternate family members to be included in the study, and therefore warrant inclusion in the study. This will be determined on a case-by-case basis by the principal investigators.
Patient/participant exclusion
The criteria for participant exclusion include unwillingness to participate in the study and an insufficient number of direct relatives willing/able to participate in the study.
Patient/participant withdrawal
Participants may be withdrawn from the study in the following circumstances:
Withdrawal at participant request. All participants have the right to withdraw from the study at any point in time. If a participant withdraws from the study the analysis of their DNA and genetic information will cease at that point. DNA samples and any sequenced genomic data generated will be destroyed.
Withdrawal due to diagnosis from standard care. If a specific genetic diagnosis becomes available during the course of the study, the patient and the family will be withdrawn from the study. If the pathobiology of this diagnosis is not known or unclear, the participant may be offered the opportunity to continue with iPSC validation of the specific genetic diagnosis without participation in NGS.
Participant identifiers
Upon recruitment, all participants will be assigned a unique identifier for the purposes of this study. A confidential database, maintained on a Queensland Health (QH) server, will link participant identifying information and their study identifier code. Study team members from The University of Queensland will receive all samples and case details anonymously, with the exception of the unique identifier. Only the clinical service will know the identity of the patient samples.
DNA sample isolation
Peripheral blood is the preferred source of DNA for this study due to the relative high quality, reliability and yield of DNA compared to extractions from other tissues. Blood samples will be collected by a trained phlebotomist and DNA will be extracted by Pathology Queensland.
In some cases DNA samples will be required from a tissue other than blood in specific instances, for example, when validating the presence of mosaicism in a given individual. In these cases a buccal (cheek) cell swab or saliva sample will be taken by the treating nephrologist or clinical geneticist. Buccal samples will be taken using Qiagen Gentra Puregene Buccal Cell Kits (or equivalent) and saliva samples will be taken using Oragene saliva DNA collection kits.
Summary of clinical data, family structure and family history
The treating clinician of each affected index participant will prepare a summary of the clinical features of the participant, the family structure and any relevant family history. This information is required to make informed evaluations of the genetic variant data derived from each family. This summary will not include any identifying information other than participant study identifier codes. This clinical summary will be provided to the research team when DNA samples are provided.
DNA sequencing
Participant DNA samples will be sequenced using one or more next generation sequencing technologies. Participant samples may undergo whole genome sequencing, whole exome sequencing and/or targeted re-sequencing of restricted gene panels. The sequencing method will be customised depending on the clinical presentation of the condition and identity of any suspected candidate genes. It is expected that exome sequencing will be the predominant technique used to assess participant DNA due to the favourable equipoise between coverage of the genome and cost. Whole genomes will be sequenced to a minimum depth of 30× reads average coverage depth while exomes will be sequenced to a minimum depth such that >80 % of targeted regions are sequenced to a depth greater than 20× reads.
Sequence analysis
DNA sequence data will be analysed from all participating members of a family in parallel. Comprehensive quality control metrics are derived for each sample to ensure the quality of the sequence data. All sequence reads are compared to the human reference sequence and used to identify variant positions where the participant DNA is different from the reference genome. Each variant position within a family is then evaluated based on its likelihood to contribute to the disease of the affected individual/s. This assessment will be augmented and assisted by the Variant Assessment Tool [49–51], an internal software tool that integrates family genetic variant segregation details with external referenced information sources such as the frequency of the allele in published control populations, the predicted impact of the variant on protein coding sequences and genes known to be associated with genetic disorders.
Reporting of findings
At the completion of the analysis, the findings will be reported to the RBWH Kidney Health Service and RBWH Conjoint Renal Genetics Clinic principal investigators. These findings will then be presented to the treating clinician after patient re-identification using their unique study identifier. If candidate variants are identified that are believed to be involved in the patient’s renal disease, they will be validated using Sanger sequencing in an accredited clinical laboratory, if available. This information will then be provided to the treating clinician for discussion with their patient. If participants opted during the informed consent process not to receive their results, including any incidental findings discussed below, they will be asked to confirm this decision when the results of the research are available. Consent will be sought from the participant should relatives need to be approached regarding the research findings.
Validation using participant-derived iPSC lines
Identification of a predicted disease causing mutation via any NGS approach requires validation, particularly if the mutation is novel for this phenotype. This may include identification of a similar mutation in another family, validation of disease segregation within the affected family or in vitro cell based analyses. This is critical in the evaluation of any NGS-predicted mutation. In a limited number of cases, and with patient consent, participant-derived primary fibroblasts have been isolated from affected individuals and clearly unaffected relatives for the purpose of derivation of induced pluripotent stem cells. Such iPSC can be used to generate specific kidney cell types [46–48]. Comparisons will be made between lines derived from the patient and the unaffected relative to partially control for variations due to the underlying genetic background. This requires the collection of fibroblasts (skin cells), which can be cultured as a primary cell line, from the proband and an unaffected relative. These cell lines will only be used for direct functional assessment of the participants’ genetic variants or where the specific inherited kidney disease’s pathobiology is unknown or incompletely understood. The cell lines will not be used for commercial purposes and will not be shared with third parties not directly involved in this study.
After obtaining specific participant consent, medical staff will obtain skin samples using a punch or shave biopsy, or during a surgical procedure should a participant be having surgery for an unrelated reason. Fibroblast culture will be undertaken according to established protocol [52]. Blood [53], urine [54] or buccal swab samples may also or alternatively be used to obtain a participant sample. Samples will be labelled with participant study unique identifier codes and transferred directly to the research laboratories for fibroblast and iPSC culture.
iPSC will be created from cultured patient cells with non-integrative reprogramming using Sendai virus transfection [55]. Subsequent differentiation to self-organising renal progenitors, structures and organoids will be undertaken [48]. A minimum of three clones from each participant (both affected patient and unaffected relative) will be assessed for disease phenotypes, to account for interclonal differences. Additional differentiation to other tissue types of relevance to the presentation of the participant will be performed as appropriate depending on the variant/s discovered and the participant phenotype revealed in the summary of clinical information accompanying the participant study unique identifier code. This is anticipated to include but not be limited to neural, retinal, hepatic, pulmonary and osseous tissues.
Participants may decline to provide skin, blood, urine or buccal swab samples; this will not affect their ability to participate in the genetic sequencing component of this study.
Disclosure of results
Results will be returned to the treating clinician who will be able to re-identify the participant. The evaluation of the research finding and disclosure to the patient and their family will be the responsibility of the treating clinician. The research findings may include a result of uncertain significance or an uninformative result. Patients will be counselled on the potential clinical implications of the results, as is standard clinical care. Further testing in a clinically-accredited laboratory may need to take place should the result be required for clinical management.
The study may reveal results that are significant to family members. The research results will be provided to the patients in the clinic. They will be counseled, if relevant, on the need to inform their relatives of their results. These patients will be provided with a family letter they can send to their relatives. Relatives considered at risk can then obtain a referral to the RBWH Kidney Health Service, RBWH Conjoint Renal Genetics Clinic or Genetic Health Queensland (or their local nephrology or clinical genetics service) for counseling. Subsequent clinical genetic testing (cascade testing) may be arranged for these relatives.
Due to the nature of this analysis, any non-paternity/maternity present within a participating family will be detected during our analysis. In accordance with the Australian National Health and Medical Research Council guidelines (Medical Genetic Testing Information for Health Professionals 2010 [56]) non-paternity or non-maternity would be disclosed in only the most exceptional circumstances, as this may cause serious harm to individuals and families.
Results could potentially preclude participants from insurance such as life, income protection, and mortgage protection. It is an applicant’s responsibility to declare any known health information about themselves and their genetic relatives in insurance applications. This topic is covered in detail during the initial consent process.
Incidental findings
One or more disease-associated genetic variants that are not related to the genetic condition being investigated may be identified in a participant. These are termed “incidental findings”. An example of this could be a gene mutation associated with an increased risk of a serious health problem, such as cancer. There is growing recognition of particular genes for which there is consensus that mutations in those genes may represent clinically significant incidental findings [57, 58]. This study has a very small chance of discovering an individual/family has a heritable disposition to an unrelated disorder (eg, cancer). The frequency of such actionable unintended findings is estimated to be less than 3 % [59, 60]. Participants will undergo a rigorous prospective consent process informing them of this, with the option of opting out from receiving such information in their study results.
If a genetic variant is discovered that the research team believes may be medically relevant for one or more of the participants, and the participant has consented to receive such incidental findings, it will be referred to an expert panel for review. The panel will consist of one clinical nephrology principal investigator (AM, HH or delegate), one person expert in next generation sequencing bioinformatics (RT, CS or delegate), one coopted clinical geneticist (CP, MG, JM or delegate), and one coopted medical specialist in the field to which the potential disease pertains (eg oncologist, gastroenterologist). The committee will consider the incidental finding and determine if (a) the finding confers a high risk of disease in the future; and (b) interventions such as surveillance are available to decrease the risk of morbidity/mortality. Should both criteria be fulfilled, the result will be presented to the participant if they had consented to learn of such information.
All participants who have previously declined to be informed of incidental findings will be asked again if this is still their wish, regardless of whether an incidental finding has been uncovered or not. All participants are informed of this at the time of initial consent.
Discussion
Advances in NGS bring within reach the promise of identifying genetic changes associated with the majority of heritable diseases. The discovery of novel disease-causing genes and mutations for a specific condition results in substantial potential benefits to both the patient/family as well as other patients thought to have that condition (Table 1).
Table 1.
Potential benefits of identifying novel disease-causing mutations in kidney disease
| • Achieving a diagnosis |
| • Precise identification of the molecular defect for genotype/phenotype correlations |
| • Accurate genetic counselling and, where appropriate, subsequent prenatal testing |
| • Accurate cascade testing to identify risks to other relatives |
| • Enhanced gene lists for future diagnostic testing |
| • Elucidation of new genetic conditions/syndromes, improving diagnosis in further individuals |
| • Improved understanding of the molecular pathology, leading to the development of more effective therapies |
Recently Simons et al. have used a combination of WES and WGS to study the genetic basis of a cohort of patients with leukodystrophy; a group of rare disorders characterized by dysfunction of the white matter of the brain. This approach has described several new diseases, including “hypomyelination with brain stem and spinal cord involvement and leg spasticity”, due to autosomal recessive mutations in the aspartyl-tRNA synthetase gene DARS [51], and “leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum” due to de novo mutation in the gene TUBB4A [50]. This group has also recently identified the previously unknown genetic cause of Temple-Baraitser syndrome to be due to mutations in KCNH1 [49]. Our study utilises and builds upon this clear skillset and expertise to discover and describe new genetic causes of GRD.
There are examples however where disease-causing genotypes are recalcitrant to or are unexplored by the application of NGS, such as in Autosomal Dominant Tubulointerstitial Disease (formerly known as Medullary Cystic Kidney Disease type 1 [61, 62] and type 2 [63–66]). Furthermore, whole exome or genome approaches, as used in gene discovery experimentation, are inaccurate in several relatively common and well-described diseases such as ADPKD [67], which instead require a tailored MPS approach [67, 68]. On the other hand, the power of diagnostic NGS approaches have been exemplified in Alport Syndrome [69], particularly with respect to identifying mosaicism [70] and unclear clinicopathological associations [71–73].
Whilst utilising such rapidly evolving genomic approaches in clinical practice offers significant potential benefits [74], it also creates potential complications. The perceived benefits of this technology have become a source of concern, particularly with regard to unintended or incidental findings [75–82]. This is further exacerbated by the variety of legal and regulatory paradigms within a globalised society [83–89]. The frequency of such actionable unintended findings in comparable studies to ours has been less than 3 % [59, 60]. If the criteria for “actionability” is liberalised this may be higher [90, 91], though still of potential personal and cost benefit [92].
One cannot presently pretend to have durable or all-encompassing answers to the conundrums posed by incidental genetic findings. Prudence and increased emphasis on informed consent and appropriate reporting may however provide a way forward [93–95]. A less definable answer is that local multidisciplinary teams provide broad opportunities to consider such challenges from a plurality of perspectives thus improving clinical translation, maximising benefits, minimising risk and providing durable clinical supports. Accordingly these have been actively integrated into the study we propose.
Functional validation of novel genetic discoveries in rare diseases poses significant challenges. iPSC technologies, as first described in 2006 [40], provide opportunities and unique strategies to address this. Patient-derived iPSC approaches to create, identify, describe and differentiate renal progenitors and self-organising organoids [48] provide the opportunity to undertake such validation via patient-derived iPSC “disease in a dish” studies. Further experience with other tissue phenotypes, as may be encountered in inherited syndromic multisystem disease, will be utilised. Specifically, Wolvetang et al. have successfully modelled human genetic disease (Ataxia-Telangiectasia and Down syndrome) using patient derived iPSC in studies that have further described the cellular and tissue pathobiology underpinning how genetic aetiology translates into complex clinical phenotypes [96, 97]. The application of CRISPR-Cas9 technology [98–100] to introduce or correct mutations in such cellular or organoid disease models [101–103] may also be required and employed. We propose that this coordinated approach to in vitro patient-derived iPSC study will enable a greater ability to characterize rare GRD at a cellular level and presents a logical pathway and opportunity for functional validation of genetic variants discovered.
In summary, we describe a collaborative research approach to the elucidation of the underlying cause of previously refractory forms of GRD. The protocol requires integration between clinicians and scientists and the application of state-of-the-art genetic and stem cell technologies within diagnostic proximity of the patient in order to reduce the number of cases of unknown genetic causation. There remain significant challenges to delivering new knowledge to the clinician and patient, including the bioinformatics challenges of identifying a causative mutation out of the >60,000 variations likely to be detected in any individual, and the technical obstacles to the functional analysis of iPSC-derived organoids. If adopted more broadly, this approach can potentially translate into optimised diagnostic, therapeutic and clinical outcomes for affected patients.
Acknowledgments
This study is being funded by research grants from the RBWH Foundation; Pathology Queensland Study, Education and Research Trust Fund; Kidney Health Australia; Alport Foundation (Australia); and the RBWH Conjoint Kidney Research Laboratory. ML is a Senior Principal Research Fellow of the National Health and Medical Research Council, Australia.
The study investigators thank the patients who have and continue to participate in this study and the nephrologists who have referred patients to the RBWH Conjoint Renal Genetics and Inherited Kidney Disease Clinics from which participants have been recruited.
Abbreviations
- GRD
Genetic renal disease
- NGS
Next generation sequencing
- MPS
Massively parallel sequencing
- WES
Whole exome sequencing
- WGS
Whole genome sequencing
- iPSC
Induced pluripotent stem cell
- RBWH
Royal Brisbane and Women’s Hospital
- UQ
The University of Queensland
- VOUS
Variant of uncertain significance
Footnotes
Competing interests
Dr Andrew Mallett has received a travel grant and speaker’s honorarium from Amgen. Professor Melissa Little is an NHMRC Senior Principal Research Fellow and has received research funding for this work from NHMRC (APP1041277) and Organovo Inc.
Authors’ contributions
AM proposed the study, drafted and wrote the protocol, led all ethics submissions, is principal applicant on funding applications, drafted the first draft of the manuscript, and is the Lead and Coordinating Principal Investigator. RT, CS, ML, EW, WH, HH and VH participated in protocol editing and conduct of the study as Principal Investigators. CP, JM and MG participated in protocol drafting as Associate Investigators. BM, MT, PT, SA, GR, MT, GC, AC, GJ, CQ and HM approved of the protocol and contributed to the conduct of the study as Associate Investigators. All authors have approved the final manuscript and certify that this manuscript represents valid work and has not been previously published.
References
- 1.Chadban SJ, Briganti EM, Kerr PG, Dunstan DW, Welborn TA, Zimmet PZ, Atkins RC. Prevalence of kidney damage in Australian adults: The AusDiab kidney study. J Am Soc Nephrol. 2003;14(7 Suppl 2):S131–138. doi: 10.1097/01.ASN.0000070152.11927.4A. [DOI] [PubMed] [Google Scholar]
- 2.White SL, Polkinghorne KR, Atkins RC, Chadban SJ. Comparison of the prevalence and mortality risk of CKD in Australia using the CKD Epidemiology Collaboration (CKD-EPI) and Modification of Diet in Renal Disease (MDRD) Study GFR estimating equations: the AusDiab (Australian Diabetes, Obesity and Lifestyle) Study. Am J Kidney Dis. 2010;55(4):660–670. doi: 10.1053/j.ajkd.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 3.ABS. Australian Health Survey: Biomedical Results for Chronic Diseases, 2011–12. In. Canberra: Australian Bureau of Statistics; 2013.
- 4.AIHW . Chronic kidney disease - Regional variation in Australia. Canberra: Australian Institute of Health and Welfare; 2013. [Google Scholar]
- 5.AIHW . End-stage kidney disease in Australia: total incidence 2003–2007. Canberra: Australian Institute of Health and Welfare; 2011. [Google Scholar]
- 6.Cass A, Chadban S, Gallagher M, Howard K, Jones A, McDonald S, Snelling P, White S. The economic impact of end-stage kidney disease in Australia: projections to 2020. Melbourne: Kidney Health Australia; 2010. [Google Scholar]
- 7.Mallett A, Patel C, Salisbury A, Wang Z, Healy H, Hoy W. The prevalence and epidemiology of genetic renal disease amongst adults with chronic kidney disease in Australia. Orphanet J Rare Dis. 2014;9:98. doi: 10.1186/1750-1172-9-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fletcher J, McDonald S, Alexander SI, Australian, New Zealand Pediatric Nephrology A Prevalence of genetic renal disease in children. Pediatr Nephrol. 2013;28(2):251–256. doi: 10.1007/s00467-012-2306-6. [DOI] [PubMed] [Google Scholar]
- 9.Levy M, Feingold J. Estimating prevalence in single-gene kidney diseases progressing to renal failure. Kidney Int. 2000;58(3):925–943. doi: 10.1046/j.1523-1755.2000.00250.x. [DOI] [PubMed] [Google Scholar]
- 10.ANZDATA . The 34th annual report: ANZDATA registry report 2011. Adelaide: Australia and New Zealand Dialysis and Transplant Registry; 2011. [Google Scholar]
- 11.ANZDATA . The 35th annual report: ANZDATA registry report 2012. Adelaide: Australia and New Zealand Dialysis and Transplant Registry; 2012. [Google Scholar]
- 12.Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, Perrone RD, Krasa HB, Ouyang J, Czerwiec FS. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367(25):2407–2418. doi: 10.1056/NEJMoa1205511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171(4356):737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 14.Rabbani B, Mahdieh N, Hosomichi K, Nakaoka H, Inoue I. Next-generation sequencing: impact of exome sequencing in characterizing Mendelian disorders. J Hum Genet. 2012;57(10):621–632. doi: 10.1038/jhg.2012.91. [DOI] [PubMed] [Google Scholar]
- 15.Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol. 1975;94(3):441–448. doi: 10.1016/0022-2836(75)90213-2. [DOI] [PubMed] [Google Scholar]
- 16.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74(12):5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmutz J, Wheeler J, Grimwood J, Dickson M, Yang J, Caoile C, Bajorek E, Black S, Chan YM, Denys M, et al. Quality assessment of the human genome sequence. Nature. 2004;429(6990):365–368. doi: 10.1038/nature02390. [DOI] [PubMed] [Google Scholar]
- 18.Hayden EC. Technology: The $1,000 genome. Nature. 2014;507(7492):294–295. doi: 10.1038/507294a. [DOI] [PubMed] [Google Scholar]
- 19.Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, Bakkaloglu A, Ozen S, Sanjad S, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009;106(45):19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, Shaffer T, Wong M, Bhattacharjee A, Eichler EE, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461(7261):272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Need AC, Shashi V, Hitomi Y, Schoch K, Shianna KV, McDonald MT, Meisler MH, Goldstein DB. Clinical application of exome sequencing in undiagnosed genetic conditions. J Med Genet. 2012;49(6):353–361. doi: 10.1136/jmedgenet-2012-100819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369(16):1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, Ward P, Braxton A, Wang M, Buhay C, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312(18):1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, Das K, Toy T, Harry B, Yourshaw M, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014;312(18):1880–1887. doi: 10.1001/jama.2014.14604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bu F, Maga T, Meyer NC, Wang K, Thomas CP, Nester CM, Smith RJ. Comprehensive genetic analysis of complement and coagulation genes in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2014;25(1):55–64. doi: 10.1681/ASN.2013050453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lemaire M, Fremeaux-Bacchi V, Schaefer F, Choi M, Tang WH, Le Quintrec M, Fakhouri F, Taque S, Nobili F, Martinez F, et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet. 2013;45(5):531–536. doi: 10.1038/ng.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCarthy HJ, Bierzynska A, Wherlock M, Ognjanovic M, Kerecuk L, Hegde S, Feather S, Gilbert RD, Krischock L, Jones C, et al. Simultaneous sequencing of 24 genes associated with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2013;8(4):637–648. doi: 10.2215/CJN.07200712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hinkes BG, Mucha B, Vlangos CN, Gbadegesin R, Liu J, Hasselbacher K, Hangan D, Ozaltin F, Zenker M, Hildebrandt F, et al. Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2) Pediatrics. 2007;119(4):e907–919. doi: 10.1542/peds.2006-2164. [DOI] [PubMed] [Google Scholar]
- 29.Ashraf S, Gee HY, Woerner S, Xie LX, Vega-Warner V, Lovric S, Fang H, Song X, Cattran DC, Avila-Casado C, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest. 2013;123(12):5179–5189. doi: 10.1172/JCI69000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chaki M, Airik R, Ghosh AK, Giles RH, Chen R, Slaats GG, Wang H, Hurd TW, Zhou W, Cluckey A, et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell. 2012;150(3):533–548. doi: 10.1016/j.cell.2012.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taskiran EZ, Korkmaz E, Gucer S, Kosukcu C, Kaymaz F, Koyunlar C, Bryda EC, Chaki M, Lu D, Vadnagara K, et al. Mutations in ANKS6 cause a nephronophthisis-like phenotype with ESRD. J Am Soc Nephrol. 2014;25(8):1653–61. doi: 10.1681/ASN.2013060646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoff S, Halbritter J, Epting D, Frank V, Nguyen TM, van Reeuwijk J, Boehlke C, Schell C, Yasunaga T, Helmstadter M, et al. ANKS6 is a central component of a nephronophthisis module linking NEK8 to INVS and NPHP3. Nat Genet. 2013;45(8):951–956. doi: 10.1038/ng.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Halbritter J, Bizet AA, Schmidts M, Porath JD, Braun DA, Gee HY, McInerney-Leo AM, Krug P, Filhol E, Davis EE, et al. Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am J Hum Genet. 2013;93(5):915–925. doi: 10.1016/j.ajhg.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Otto EA, Hurd TW, Airik R, Chaki M, Zhou W, Stoetzel C, Patil SB, Levy S, Ghosh AK, Murga-Zamalloa CA, et al. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat Genet. 2010;42(10):840–850. doi: 10.1038/ng.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chaki M, Hoefele J, Allen SJ, Ramaswami G, Janssen S, Bergmann C, Heckenlively JR, Otto EA, Hildebrandt F. Genotype-phenotype correlation in 440 patients with NPHP-related ciliopathies. Kidney Int. 2011;80(11):1239–1245. doi: 10.1038/ki.2011.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otto EA, Ramaswami G, Janssen S, Chaki M, Allen SJ, Zhou W, Airik R, Hurd TW, Ghosh AK, Wolf MT, et al. Mutation analysis of 18 nephronophthisis associated ciliopathy disease genes using a DNA pooling and next generation sequencing strategy. J Med Genet. 2011;48(2):105–116. doi: 10.1136/jmg.2010.082552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Halbritter J, Porath JD, Diaz KA, Braun DA, Kohl S, Chaki M, Allen SJ, Soliman NA, Hildebrandt F, Otto EA, et al. Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum Genet. 2013;132(8):865–884. doi: 10.1007/s00439-013-1297-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Halbritter J, Diaz K, Chaki M, Porath JD, Tarrier B, Fu C, Innis JL, Allen SJ, Lyons RH, Stefanidis CJ, et al. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J Med Genet. 2012;49(12):756–767. doi: 10.1136/jmedgenet-2012-100973. [DOI] [PubMed] [Google Scholar]
- 39.Otto EA, Tory K, Attanasio M, Zhou W, Chaki M, Paruchuri Y, Wise EL, Wolf MT, Utsch B, Becker C, et al. Hypomorphic mutations in meckelin (MKS3/TMEM67) cause nephronophthisis with liver fibrosis (NPHP11) J Med Genet. 2009;46(10):663–670. doi: 10.1136/jmg.2009.066613. [DOI] [PubMed] [Google Scholar]
- 40.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 41.Sallam K, Kodo K, Wu JC. Modeling inherited cardiac disorders. Circ J. 2014;78(4):784–794. doi: 10.1253/circj.CJ-14-0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suzuki T, Mayhew C, Sallese A, Chalk C, Carey BC, Malik P, Wood RE, Trapnell BC. Use of induced pluripotent stem cells to recapitulate pulmonary alveolar proteinosis pathogenesis. Am J Respir Crit Care Med. 2014;189(2):183–193. doi: 10.1164/rccm.201306-1039OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leung A, Nah SK, Reid W, Ebata A, Koch CM, Monti S, Genereux JC, Wiseman RL, Wolozin B, Connors LH, et al. Induced pluripotent stem cell modeling of multisystemic, hereditary transthyretin amyloidosis. Stem Cell Reports. 2013;1(5):451–463. doi: 10.1016/j.stemcr.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chamberlain SJ, Li XJ, Lalande M. Induced pluripotent stem (iPS) cells as in vitro models of human neurogenetic disorders. Neurogenetics. 2008;9(4):227–235. doi: 10.1007/s10048-008-0147-z. [DOI] [PubMed] [Google Scholar]
- 45.Song B, Smink AM, Jones CV, Callaghan JM, Firth SD, Bernard CA, Laslett AL, Kerr PG, Ricardo SD. The directed differentiation of human iPS cells into kidney podocytes. PLoS One. 2012;7(9):e46453. doi: 10.1371/journal.pone.0046453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lam AQ, Freedman BS, Morizane R, Lerou PH, Valerius MT, Bonventre JV. Rapid and efficient differentiation of human pluripotent stem cells into intermediate mesoderm that forms tubules expressing kidney proximal tubular markers. J Am Soc Nephrol. 2014;25(6):1211–1225. doi: 10.1681/ASN.2013080831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xia Y, Nivet E, Sancho-Martinez I, Gallegos T, Suzuki K, Okamura D, Wu MZ, Dubova I, Esteban CR, Montserrat N, et al. Directed differentiation of human pluripotent cells to ureteric bud kidney progenitor-like cells. Nat Cell Biol. 2013;15(12):1507–1515. doi: 10.1038/ncb2872. [DOI] [PubMed] [Google Scholar]
- 48.Takasato M, Er PX, Becroft M, Vanslambrouck JM, Stanley EG, Elefanty AG, Little MH. Directing human embryonic stem cell differentiation towards a renal lineage generates a self-organizing kidney. Nat Cell Biol. 2014;16(1):118–126. doi: 10.1038/ncb2894. [DOI] [PubMed] [Google Scholar]
- 49.Simons C, Rash LD, Crawford J, Ma L, Cristofori-Armstrong B, Miller D, Ru K, Baillie GJ, Alanay Y, Jacquinet A, et al. Mutations in the voltage-gated potassium channel gene KCNH1 cause Temple-Baraitser syndrome and epilepsy. Nat Genet. 2015;47(1):73–77. doi: 10.1038/ng.3153. [DOI] [PubMed] [Google Scholar]
- 50.Simons C, Wolf NI, McNeil N, Caldovic L, Devaney JM, Takanohashi A, Crawford J, Ru K, Grimmond SM, Miller D, et al. A de novo mutation in the beta-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am J Hum Genet. 2013;92(5):767–773. doi: 10.1016/j.ajhg.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taft RJ, Vanderver A, Leventer RJ, Damiani SA, Simons C, Grimmond SM, Miller D, Schmidt J, Lockhart PJ, Pope K, et al. Mutations in DARS cause hypomyelination with brain stem and spinal cord involvement and leg spasticity. Am J Hum Genet. 2013;92(5):774–780. doi: 10.1016/j.ajhg.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raya A, Rodriguez-Piza I, Navarro S, Richaud-Patin Y, Guenechea G, Sanchez-Danes A, Consiglio A, Bueren J, Izpisua Belmonte JC. A protocol describing the genetic correction of somatic human cells and subsequent generation of iPS cells. Nat Protoc. 2010;5(4):647–660. doi: 10.1038/nprot.2010.9. [DOI] [PubMed] [Google Scholar]
- 53.Ye L, Muench MO, Fusaki N, Beyer AI, Wang J, Qi Z, Yu J, Kan YW. Blood cell-derived induced pluripotent stem cells free of reprogramming factors generated by Sendai viral vectors. Stem Cells Transl Med. 2013;2(8):558–566. doi: 10.5966/sctm.2013-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou T, Benda C, Dunzinger S, Huang Y, Ho JC, Yang J, Wang Y, Zhang Y, Zhuang Q, Li Y, et al. Generation of human induced pluripotent stem cells from urine samples. Nat Protoc. 2012;7(12):2080–2089. doi: 10.1038/nprot.2012.115. [DOI] [PubMed] [Google Scholar]
- 55.Fusaki N, Ban H, Nishiyama A, Saeki K, Hasegawa M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85(8):348–362. doi: 10.2183/pjab.85.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trent R, Otlowski M, Ralston M, Lonsdale L, Young M, Suthers G, Griffiths P, Delatycki M, Christodoulou J, Barlow-Stewart K. Medical genetic testing information for health professionals. Canberra: NHMRC Publications; 2010. [Google Scholar]
- 57.Smith LA, Douglas J, Braxton AA, Kramer K. Reporting Incidental Findings in Clinical Whole Exome Sequencing: Incorporation of the 2013 ACMG Recommendations into Current Practices of Genetic Counseling. J Genet Couns. 2014. [DOI] [PubMed]
- 58.Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, McGuire AL, Nussbaum RL, O’Daniel JM, Ormond KE, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15(7):565–574. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dorschner MO, Amendola LM, Turner EH, Robertson PD, Shirts BH, Gallego CJ, Bennett RL, Jones KL, Tokita MJ, Bennett JT, et al. Actionable, pathogenic incidental findings in 1,000 participants’ exomes. Am J Hum Genet. 2013;93(4):631–640. doi: 10.1016/j.ajhg.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johnston JJ, Rubinstein WS, Facio FM, Ng D, Singh LN, Teer JK, Mullikin JC, Biesecker LG. Secondary variants in individuals undergoing exome sequencing: screening of 572 individuals identifies high-penetrance mutations in cancer-susceptibility genes. Am J Hum Genet. 2012;91(1):97–108. doi: 10.1016/j.ajhg.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kirby A, Gnirke A, Jaffe DB, Baresova V, Pochet N, Blumenstiel B, Ye C, Aird D, Stevens C, Robinson JT, et al. Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat Genet. 2013;45(3):299–303. doi: 10.1038/ng.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bleyer AJ, Kmoch S, Antignac C, Robins V, Kidd K, Kelsoe JR, Hladik G, Klemmer P, Knohl SJ, Scheinman SJ, et al. Variable clinical presentation of an MUC1 mutation causing medullary cystic kidney disease type 1. Clin J Am Soc Nephrol. 2014;9(3):527–535. doi: 10.2215/CJN.06380613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zivna M, Hulkova H, Matignon M, Hodanova K, Vylet’al P, Kalbacova M, Baresova V, Sikora J, Blazkova H, Zivny J, et al. Dominant renin gene mutations associated with early-onset hyperuricemia, anemia, and chronic kidney failure. Am J Hum Genet. 2009;85(2):204–213. doi: 10.1016/j.ajhg.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hart TC, Gorry MC, Hart PS, Woodard AS, Shihabi Z, Sandhu J, Shirts B, Xu L, Zhu H, Barmada MM, et al. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J Med Genet. 2002;39(12):882–892. doi: 10.1136/jmg.39.12.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ekici AB, Hackenbeck T, Moriniere V, Pannes A, Buettner M, Uebe S, Janka R, Wiesener A, Hermann I, Grupp S, et al. Renal fibrosis is the common feature of Autosomal Dominant Tubulointerstitial Kidney Diseases caused by mutations in mucin 1 or uromodulin. Kidney Int. 2014;86(3)):589–99. doi: 10.1038/ki.2014.72. [DOI] [PubMed] [Google Scholar]
- 66.Bollee G, Dahan K, Flamant M, Moriniere V, Pawtowski A, Heidet L, Lacombe D, Devuyst O, Pirson Y, Antignac C, et al. Phenotype and outcome in hereditary tubulointerstitial nephritis secondary to UMOD mutations. Clin J Am Soc Nephrol. 2011;6(10):2429–2438. doi: 10.2215/CJN.01220211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qi XP, Du ZF, Ma JM, Chen XL, Zhang Q, Fei J, Wei XM, Chen D, Ke HP, Liu XZ, et al. Genetic diagnosis of autosomal dominant polycystic kidney disease by targeted capture and next-generation sequencing: utility and limitations. Gene. 2013;516(1):93–100. doi: 10.1016/j.gene.2012.12.060. [DOI] [PubMed] [Google Scholar]
- 68.Paul BM, Consugar MB, Ryan Lee M, Sundsbak JL, Heyer CM, Rossetti S, Kubly VJ, Hopp K, Torres VE, Coto E, et al. Evidence of a third ADPKD locus is not supported by re-analysis of designated PKD3 families. Kidney Int. 2014;85(2):383–392. doi: 10.1038/ki.2013.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Artuso R, Fallerini C, Dosa L, Scionti F, Clementi M, Garosi G, Massella L, Epistolato MC, Mancini R, Mari F, et al. Advances in Alport syndrome diagnosis using next-generation sequencing. Eur J Hum Genet. 2012;20(1):50–57. doi: 10.1038/ejhg.2011.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beicht S, Strobl-Wildemann G, Rath S, Wachter O, Alberer M, Kaminsky E, Weber LT, Hinrichsen T, Klein HG, Hoefele J. Next generation sequencing as a useful tool in the diagnostics of mosaicism in Alport syndrome. Gene. 2013;526(2):474–477. doi: 10.1016/j.gene.2013.05.045. [DOI] [PubMed] [Google Scholar]
- 71.Gibson J, Gilbert RD, Bunyan DJ, Angus EM, Fowler DJ, Ennis S. Exome analysis resolves differential diagnosis of familial kidney disease and uncovers a potential confounding variant. Genet Res. 2013;95(6):165–173. doi: 10.1017/S0016672313000220. [DOI] [PubMed] [Google Scholar]
- 72.Fallerini C, Dosa L, Tita R, Del Prete D, Feriozzi S, Gai G, et al. Unbiased next generation sequencing analysis confirms the existence of autosomal dominant Alport syndrome in a relevant fraction of cases. Clin Genet. 2014;86(3):252–7. [DOI] [PubMed]
- 73.Chatterjee R, Hoffman M, Cliften P, Seshan S, Liapis H, Jain S. Targeted exome sequencing integrated with clinicopathological information reveals novel and rare mutations in atypical, suspected and unknown cases of Alport syndrome or proteinuria. PLoS One. 2013;8(10):e76360. doi: 10.1371/journal.pone.0076360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wright C, Burton H, Hall A, Moorthie S, Pokorska-Bocci A, Sagoo G, Sanderson S, Skinne R. Next steps in the sequence - the implications of whole genome sequencing for health in the UK. Cambridge: Foundation for Genomics and Population Health; 2011. [Google Scholar]
- 75.Ross LF, Rothstein MA, Clayton EW. Mandatory extended searches in all genome sequencing: “incidental findings,” patient autonomy, and shared decision making. JAMA. 2013;310(4):367–368. doi: 10.1001/jama.2013.41700. [DOI] [PubMed] [Google Scholar]
- 76.Klitzman R, Appelbaum PS, Chung W. Return of secondary genomic findings vs patient autonomy: implications for medical care. JAMA. 2013;310(4):369–370. doi: 10.1001/jama.2013.41709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McGuire AL, Joffe S, Koenig BA, Biesecker BB, McCullough LB, Blumenthal-Barby JS, Caulfield T, Terry SF, Green RC. Point-counterpoint. Ethics and genomic incidental findings. Science. 2013;340(6136):1047–1048. doi: 10.1126/science.1240156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wolf SM, Annas GJ, Elias S. Point-counterpoint. Patient autonomy and incidental findings in clinical genomics. Science. 2013;340(6136):1049–1050. doi: 10.1126/science.1239119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Christenhusz GM, Devriendt K, Dierickx K. To tell or not to tell? A systematic review of ethical reflections on incidental findings arising in genetics contexts. Eur J Hum Genet. 2013;21(3):248–255. doi: 10.1038/ejhg.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Christenhusz GM, Devriendt K, Dierickx K. Disclosing incidental findings in genetics contexts: a review of the empirical ethical research. Eur J Med Genet. 2013;56(10):529–540. doi: 10.1016/j.ejmg.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 81.Abdul-Karim R, Berkman BE, Wendler D, Rid A, Khan J, Badgett T, Hull SC. Disclosure of incidental findings from next-generation sequencing in pediatric genomic research. Pediatrics. 2013;131(3):564–571. doi: 10.1542/peds.2012-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cho MK. Understanding incidental findings in the context of genetics and genomics. J Law Med Ethics. 2008;36(2):280–285. doi: 10.1111/j.1748-720X.2008.00270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wilkie T. Genetics and insurance in Britain: why more than just the Atlantic divides the English-speaking nations. Nat Genet. 1998;20(2):119–121. doi: 10.1038/2416. [DOI] [PubMed] [Google Scholar]
- 84.Malpas PJ. Is genetic information relevantly different from other kinds of non-genetic information in the life insurance context? J Med Ethics. 2008;34(7):548–551. doi: 10.1136/jme.2007.023101. [DOI] [PubMed] [Google Scholar]
- 85.Wilkinson R. Unjustified discrimination: is the moratorium on the use of genetic test results by insurers a contradiction in terms? Health Care Anal. 2010;18(3):279–293. doi: 10.1007/s10728-009-0137-9. [DOI] [PubMed] [Google Scholar]
- 86.Keogh LA, Otlowski MF. Life insurance and genetic test results: a mutation carrier’s fight to achieve full cover. Med J Aust. 2013;199(5):363–366. doi: 10.5694/mja13.10202. [DOI] [PubMed] [Google Scholar]
- 87.Wright GE, Koornhof PG, Adeyemo AA, Tiffin N. Ethical and legal implications of whole genome and whole exome sequencing in African populations. BMC Med Ethics. 2013;14:21. doi: 10.1186/1472-6939-14-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McDonald SA, Mardis ER, Ota D, Watson MA, Pfeifer JD, Green JM. Comprehensive genomic studies: emerging regulatory, strategic, and quality assurance challenges for biorepositories. Am J Clin Pathol. 2012;138(1):31–41. doi: 10.1309/AJCPXBA69LNSCVMH. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hook CC, DiMagno EP, Tefferi A. Primer on medical genomics. Part XIII: Ethical and regulatory issues. Mayo Clin Proc. 2004;79(5):645–650. doi: 10.4065/79.5.645. [DOI] [PubMed] [Google Scholar]
- 90.Ding LE, Burnett L, Chesher D. The impact of reporting incidental findings from exome and whole-genome sequencing: predicted frequencies based on modeling. Genet Med. 2015;17(3):197–204. [DOI] [PMC free article] [PubMed]
- 91.Tabor HK, Auer PL, Jamal SM, Chong JX, Yu JH, Gordon AS, Graubert TA, O’Donnell CJ, Rich SS, Nickerson DA, et al. Pathogenic variants for Mendelian and complex traits in exomes of 6,517 European and African Americans: implications for the return of incidental results. Am J Hum Genet. 2014;95(2):183–193. doi: 10.1016/j.ajhg.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bennette CS, Gallego CJ, Burke W, Jarvik GP, Veenstra DL. The cost-effectiveness of returning incidental findings from next-generation genomic sequencing. Genet Med. 2015;17(7):587–95. [DOI] [PMC free article] [PubMed]
- 93.Green RC, Lupski JR, Biesecker LG. Reporting genomic sequencing results to ordering clinicians: incidental, but not exceptional. JAMA. 2013;310(4):365–366. doi: 10.1001/jama.2013.41703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.van El CG, Dondorp WJ, de Wert GM, Cornel MC. Call for prudence in whole-genome testing. Science. 2013;341(6149):958–959. doi: 10.1126/science.341.6149.958-b. [DOI] [PubMed] [Google Scholar]
- 95.Platt J, Cox R, Enns GM. Points to Consider in the Clinical Use of NGS Panels for Mitochondrial Disease: An Analysis of Gene Inclusion and Consent Forms. J Genet Couns. 2014. [DOI] [PubMed]
- 96.Nayler S, Gatei M, Kozlov S, Gatti R, Mar JC, Wells CA, Lavin M, Wolvetang E. Induced pluripotent stem cells from ataxia-telangiectasia recapitulate the cellular phenotype. Stem Cells Transl Med. 2012;1(7):523–535. doi: 10.5966/sctm.2012-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Briggs JA, Sun J, Shepherd J, Ovchinnikov DA, Chung TL, Nayler SP, Kao LP, Morrow CA, Thakar NY, Soo SY, et al. Integration-free induced pluripotent stem cells model genetic and neural developmental features of down syndrome etiology. Stem Cells. 2013;31(3):467–478. doi: 10.1002/stem.1297. [DOI] [PubMed] [Google Scholar]
- 98.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013;31(3):230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- 100.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Roy A, Goodman JH, Begum G, Donnelly BF, Pittman G, Weinman EJ, Sun D, Subramanya AR. Generation of WNK1 knockout cell lines by CRISPR/Cas-mediated genome editing. Am J Physiol Renal Physiol. 2015;308(4):F366–376. doi: 10.1152/ajprenal.00612.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schwank G, Koo BK, Sasselli V, Dekkers JF, Heo I, Demircan T, Sasaki N, Boymans S, Cuppen E, van der Ent CK, et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 2013;13(6):653–658. doi: 10.1016/j.stem.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 103.Matano M, Date S, Shimokawa M, Takano A, Fujii M, Ohta Y, Watanabe T, Kanai T, Sato T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat Med. 2015;21(3):256–262. doi: 10.1038/nm.3802. [DOI] [PubMed] [Google Scholar]