

Abstract
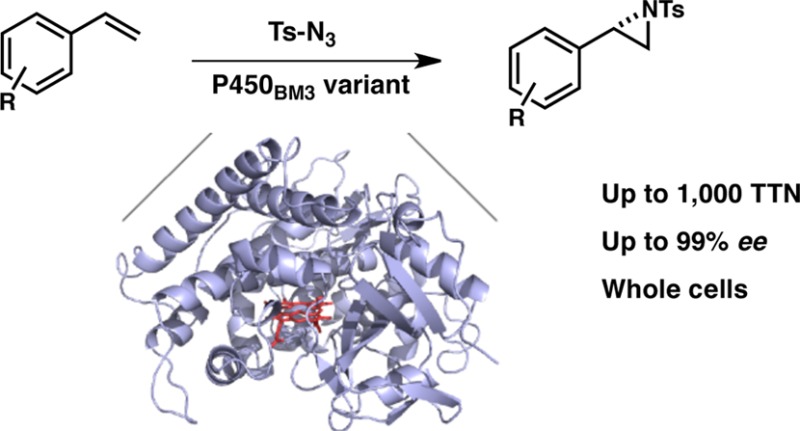
One of the greatest challenges in protein design is creating new enzymes, something evolution does all the time, starting from existing ones. Borrowing from nature’s evolutionary strategy, we have engineered a bacterial cytochrome P450 to catalyze highly enantioselective intermolecular aziridination, a synthetically useful reaction that has no natural biological counterpart. The new enzyme is fully genetically encoded, functions in vitro or in whole cells, and can be optimized rapidly to exhibit high enantioselectivity (up to 99% ee) and productivity (up to 1,000 catalytic turnovers) for intermolecular aziridination, demonstrated here with tosyl azide and substituted styrenes. This new aziridination activity highlights the remarkable ability of a natural enzyme to adapt and take on new functions. Once discovered in an evolvable enzyme, this non-natural activity was improved and its selectivity tuned through an evolutionary process of accumulating beneficial mutations.
Short abstract
Engineered cytochrome P450 variants take on challenging non-natural reaction, aziridination of olefins, demonstrating the power of evolution to expand biocatalytic reaction space.
Introduction
The impressive catalytic diversity of enzymes illustrates the breadth of reaction space explored throughout biological evolution. Chemists are taking advantage of this vast repository of biocatalysts for chemical synthesis; they are also mimicking nature’s engineering strategy of mutation and selection to optimize enzyme function for a myriad of applications.1 A remaining challenge is to create new enzymes for the many useful reactions that are not known in the biological world. We and others have argued that learning from nature’s mechanisms for catalytic innovation—exploiting enzyme catalytic promiscuity and retaining key elements of structure and mechanism while exploring new functions—can help us discover new biocatalysts starting from nature’s vast repertoire.2 The discovery and optimization of enzyme catalysts for non-natural reactions has provided alternative routes to important molecules that would otherwise be unattainable through biocatalysis.3
Our group4 and that of Fasan5 recently reported that heme proteins can form reactive metal–nitrenoid species and catalyze intramolecular C–H amination. We extended this novel biocatalytic activation mode to intermolecular sulfimidation and showed that the enzyme, derived from a cytochrome P450, could be tuned by mutation to catalyze nitrene transfer to various aryl sulfides with good activity and moderate enantioselectivity (Scheme 1A).6 Because heme proteins do not encounter nitrene precursors in their natural environments, they never had the chance to explore and evolve this capability. But the cytochrome P450’s versatile catalytic framework is clearly capable of catalytic activities well beyond what nature has explored.
Scheme 1. Enzyme-Catalyzed Intermolecular Nitrogen Atom Transfer.

The extensive literature on reactions catalyzed by metalloporphyrins provides inspiration for reactivities that should be investigated.7−10 Among the nitrene transfer reactions used in synthetic chemistry, aziridination with azide nitrene sources is particularly attractive (Scheme 1B), given its high atom economy and the utility of the aziridine functional group in chemical synthesis.8 Metalloporphyrin-catalyzed aziridination of olefins with iminoiodinanes or haloamines as the nitrene source dates to the 1980s or early 2000s, respectively.9 Advances in the past ten years have expanded metalloporphyrin-catalyzed aziridination to include systems that use azide-based nitrene sources.10
In contrast, enzymes that use electrophilic nitrogen species are extremely rare (e.g., proposed mechanism of TxtE-catalyzed nitration of tryptophan11). As a result, natural aziridination follows an entirely different strategy from the metalloporphyrin-catalyzed aziridination of olefins described above. Indeed, the biosynthesis of naturally occurring aziridine rings is believed to proceed by intramolecular nucleophilic displacement.12 Enzyme catalysts for the convergent synthesis of aziridines from olefins and azide nitrene precursors would be highly attractive and would fill a significant gap in nature’s biosynthetic repertoire.
During our previous work on sulfide imidation catalyzed by variants of cytochrome P450BM3, two factors appeared to affect reactivity. First, the reaction was promoted by electron-rich sulfides, and second, enzyme-catalyzed azide reduction to sulfonamide represented a significant side reaction, particularly with less reactive, electron-deficient sulfides (Scheme 1).6 Increasing the sulfide substrate loading improved the ratio of sulfimide to sulfonamide side product, which suggested to us that increasing the effective concentration of sulfide nitrene acceptor in the enzyme active site might improve sulfimidation productivity. Given the less reactive nature of olefins relative to sulfides along with operational limitations to increasing olefin effective concentration (i.e., their limited solubility in aqueous media), we surmised that intermolecular aziridination would be an even more challenging activation mode for the enzyme. We hypothesized, however, that protein engineering would allow us to circumvent these limitations if the enzyme could more effectively bind and orient the substrates in the enzyme active site for productive nitrene transfer or, alternatively, catalyze the competing azide reduction less efficiently. Here we demonstrate that active-site engineering can indeed enable a heme protein to catalyze efficient, highly enantioselective intermolecular nitrene transfer to olefins to make aziridines.
Results and Discussion
We started this investigation of enzyme-catalyzed aziridination with an engineered variant of cytochrome P450BM3, P411BM3-CIS-T438S, previously found to be effective for intramolecular C–H amination and sulfide imidation (see Table S1 for mutations from wild-type P450BM3).4,6 We call this enzyme a “P411” due to the change in the characteristic CO-bound Soret peak from 450 to 411 nm effected by mutation of the cysteine residue that coordinates the heme iron to serine (C400S).4,13 This axial cysteine is completely conserved in cytochrome P450s and is required for the native monooxygenase activity.14 Thus, the P411 enzyme is no longer a “cytochrome P450”, nor does it exhibit its native hydroxylation activity.13b,15 However, the C400S mutation increases the non-natural carbene and nitrene transfer activities of P450BM34,6,13 and other P450s.16 Two crystal structures of P411 variants of P450BM3 show that S400 coordinates the iron and causes no significant structural perturbation in the substrate binding pocket.13
Previous work on enzymatic sulfimidation suggested that electron-rich sulfides promote nitrene transfer.6 Reasoning that the electronic properties of the olefin substrate could influence aziridination as well, we tested the activity of P411BM3-CIS-T438S toward the electron-rich 4-methoxystyrene using tosyl azide (TsN3) as the nitrene precursor (Table 1). Tosyl azide was completely consumed in this reaction, the major product of which was the azide reduction product p-toluenesulfonamide (>300 total turnovers (TTN), not shown in Table 1). Amido-alcohol 2 appeared as a minor product. Control experiments showed that the desired aziridine product rapidly decomposes under aqueous reaction conditions to the corresponding amido-alcohol 2 (Figure S1). Degradation of this aziridine product has also been observed in studies with small-molecule catalysts.17 We therefore inferred that production of 2 is directly related to the nitrene transfer activity of the enzyme toward olefin 1.
Table 1. Total Turnovers (TTN) to Product for Aziridination Catalyzed by Purified Holoenzymes P411BM3-CIS-T438S (P) and P411BM3-CIS-T438S-I263F (P-I263F) with Selected Styrenyl Olefins 1, 3, and 5 and Tosyl Azidea.

| TTNb |
|||
|---|---|---|---|
| enzyme | 2 | 4 | 6 |
| P411BM3-CIS-T438S (P) | 15 | 8 | 5 |
| P-I263F | 150 | 160 | 190 |
Reactions were performed in 0.1 M KPi buffer pH = 8.0 using 0.2 mol % enzyme and NADPH as reductant, with 2.5 mM tosyl azide and 7.5 mM olefin. Detailed reaction conditions can be found in the Supporting Information.
TTN = total turnover number. TTNs were determined by HPLC analysis.
This low level of nitrene transfer activity to 4-methoxystyrene of the P411BM3-CIS-T438S enzyme prompted us to investigate other variants. We chose a small set of cytochrome P450BM3 variants and heme proteins prepared for other studies in order to assess how changes in the protein sequence affect nitrene transfer to olefin substrates (Tables S2 and S3). P450BM3 sequences lacking the C400S and/or T268A mutations were not active, nor did the Fe(II)-protoporphyrin IX (PPIX) cofactor catalyze aziridination under these conditions. Mutants differing from P411BM3-CIS-T438S by 2–5 alanine mutations in the active site showed some aziridination activity (4–8 TTN), but none was more productive than P411BM3-CIS-T438S. We also tested a set of enzymes containing different axial mutations, including the S400H, S400D, and S400 M mutants of P411BM3-CIS-T438S. These enzymes were also only weakly active, giving 2 at levels lower than P411BM3-CIS-T438S (3–4 TTN). Myoglobin (horse heart), cytochrome c (bovine heart), and cytochrome P450Rhf (from Rhodococcus sp. NCIMB 9784) were all inactive for this intermolecular aziridination (Table S3). An engineered variant of the thermostable cytochrome P450 from Sulfolobus acidocaldarius, CYP119, that contained an axial cysteine-to-serine mutation (C317S) did catalyze low levels of aziridination (∼7 TTN). This demonstrates that mutations previously described to activate non-natural nitrene-transfer activity in P450BM3 may confer measurable activity on other P450s as well. In turn, these enzymes should be suitable starting points for further engineering.16
Of all the enzymes tested, a variant of P411BM3-CIS-T438S having a single active-site substitution, I263F, was the most active toward 4-methoxystyrene by a wide margin, providing 150 total turnovers in the formation of amido-alcohol 2 from 4-methoxystyrene (Table 1). This variant, which was found to promote regioselective intramolecular C–H amination in a previous study,13a supported aziridination at 10-fold increased total turnovers compared to the P411BM3-CIS-T438S parent enzyme (henceforth referred to as the P enzyme). Comparison of these two enzymes’ activities with a set of styrenyl olefins showed that the P-I263F variant was also more productive with the less electron-rich 4-methylstyrene (3) and styrene (5) substrates. Furthermore, P-I263F is more productive with styrene (5) than with 4-methoxystyrene (1). This trend in substrate preference is inverted compared to that of the parent enzyme (Table 1) and the previously observed trend for intermolecular imidation of sulfides.6 Unlike the aziridine product that leads to amido-alcohol 2, aziridine products 4 and 6 were stable under the reaction conditions, although some amido-alcohol product was detected when standards and samples were analyzed by LC–MS (Figure S2).
P-I263F was even more productive when the reactions were carried out using whole Escherichia coli cells expressing this enzyme (Figure S3), consistent with our previous observations that enzyme-catalyzed metal–nitrenoid and metal–carbenoid transfer activities improved when the reactions were performed with whole cells.4,13b (No aziridine product was observed when cells not expressing the P411 catalyst were used, although tosyl azide was converted to sulfonamide 7 over the course of the reaction (Table 2), as reported previously for intramolecular C–H amination.4) The P-I263F enzyme provided enough aziridine product in whole-cell reactions to allow for screening variants in 96-well plate format. Thus, we sought further improvement by mutagenesis and screening for aziridination productivity. Site-saturation mutagenesis (SSM) libraries were created at several active site positions that were previously shown to influence productivity and enantioselectivity in other non-natural reactions (A78, L181, T438, A328).3a,13a Screening of these single SSM libraries for aziridination of 4-methylstyrene (3) identified P-I263F-A328V, with slightly improved yield and substantially improved % ee (96% ee; entry 4, Table 2). Another round of SSM performed on this variant at additional active site positions (F87, T268, L437) resulted in P-I263F-A328V-L437V with improved aziridine yield and a further increase in enantioselectivity (99% ee (S)). The P-I263F-L437V and P-I263F-A328V mutants were both less selective than P-I263F-A328V-L437V, demonstrating that both new mutations contribute to the very high enantioselectivity. Importantly, the yield of sulfonamide side product 7 diminished over the course of active site evolution, to the extent that aziridine 4 became the major product of the reaction catalyzed by P-I263F-A328V-L437V.
Table 2. Improvement in Yield and % ee for Aziridine Product 4 with Active-Site Evolution of P411BM3-CIS-T438S (P)a.

| % yield |
||||
|---|---|---|---|---|
| entry | enzyme | 4 | 7 | % ee of 4b |
| 1 | no enzymec | 0 | 95 | nd |
| 2 | P411BM3-CIS-T438S | 1.1 | 95 | 25 |
| 3 | P-I263F | 40 | 54 | 55 |
| 4 | P-I263F-A328V | 43 | 50 | 96 |
| 5 | P-I263F-L437V | 37 | 52 | 95 |
| 6 | P-I263F-A328V-L437V | 55 | 43 | 99 |
Reactions were carried out using whole E. coli cells resuspended in M9-N reaction buffer under anaerobic conditions, with 2.5 mM tosyl azide and 7.5 mM 4-methylstyrene. Yield is based on tosyl azide. See Supporting Information methods for detailed reaction setup and quantification procedures.
% ee determined by SFC analysis and calculated as (S – R)/(S + R).
“No enzyme” reactions were carried out using whole cells with no P411 enzyme expressed, as described in the Supporting Information methods.
Because the azide is fully consumed in these reactions, the improved aziridine yield could result from either an increase in the rate of aziridine formation or a decrease in the rate of competing azide reduction, or from a combination of both. To address this, we measured initial rates of reaction with the P-I263F, P-I263F-A328V, and P-I263F-A328V-L437V enzymes as purified holoenzymes (Table S4, Figure S4). Initial rates of aziridination for the purified enzymes reflected the yield improvements observed in whole cells: P-I263F and P-I263F-A328V have similar turnover frequencies (15–16 min–1), while P-I263F-A328V-L437V, having both new mutations, was improved (TOF ∼ 24 min–1). The initial turnover frequency of sulfonamide formation in vitro was similar for all the enzymes, and faster than aziridine formation (TOFs ∼ 26–29 min–1).
Having obtained a variant capable of high productivity and enantioselectivity for the aziridination of 4-methylstyrene (3), we next investigated whole-cell reactions with different substituted styrene substrates (Table 3). In contrast to enzyme-catalyzed imidation of sulfides,6 we saw no correlation between the electronics of the aryl substituent and productivity. In general, the evolved enzyme was more productive with styrenes substituted at the 4-position, though the highest productivity was observed with styrene itself. The evolved enzyme provided 600 catalytic turnovers for the formation of aziridine 6, corresponding to a 70% yield of 6 (entry 3 in Table 3). With higher styrene and tosyl azide loading, the enzyme catalyzed 1,000 turnovers for aziridination, while retaining high (S)-selectivity (99% ee) (Figure S5). Both 3-methylstyrene and 3-chlorostyrene were significantly less reactive than their 4-substituted counterparts, giving 85 and 21 turnovers, respectively, compared to 450 and 290 turnovers (entries 2, 4, 5, 6 in Table 3). The evolved enzyme is an exceptionally enantioselective aziridination catalyst with styrene entries 2–4 (Table 3), giving 99% ee in favor of the (S)-enantiomer with these three substrates. Both 4-methoxystyrene and α-methylstyrene (entries 1 and 8 in Table 3) gave exclusively racemic amido-alcohol product. Formation of the amido-alcohol product from these substrates may result from carbocation stabilization at the benzylic position due to the resonance and hyperconjugative stabilization provided by the respective p-OMe and α-Me groups, leading to decomposition of the aziridine product and subsequent carbocation quenching with water.
Table 3. Substrate Scope of Aziridination with P-I263F-A328V-L437V Showing Productivity and Selectivity for Each Producta.

Reactions were carried out with whole cells expressing P-I263F-A328V-L437V under anaerobic conditions, with 2.5 mM tosyl azide and 7.5 mM olefin.
% ee determined as (S – R)/(S + R). Absolute configurations were assigned based on analogy to 6. rac = racemic.
The highly enantioselective P-I263F-A328V-L437V variant has three mutations in its active site relative to the P enzyme used in initial reaction characterization (P411BM3-CIS-T438S). The crystal structure of the heme domain of P-I263F was recently solved and shows how the F263 side chain is oriented toward the heme cofactor within the active site (PDB ID: 4WG2, Figure S6).13a The effect of the I263F mutation on the active site is significant: the F263 side chain fills space above the heme cofactor whereas the I263 side chain is pointed up and away from the heme (Figure S6). Given the more conservative nature of the A328V and L437V mutations, these residues likely exert more subtle influences on active site structure, yet their impact on enantioselectivity is substantial (55% ee with P-I263F vs 99% ee with P-I263F-A328V-L437V, using 4-methylstyrene, Table 2).
Beyond I263F, several other mutations in the distal heme environment are necessary for high aziridination activity. In particular, reversion of either T268A or F87V in P-I263F markedly reduces aziridination productivity (Table S2) and demonstrates some nonadditivity among the activating mutations (i.e., I263F is activating only in the context of T268A). Moreover, although previous work has highlighted the importance of modulating heme electronic properties to access non-natural reactivity,4,13 here we observe that strong gains in aziridination activity are brought about by mutations on the distal heme side, suggesting that their effect may be the result of improving substrate binding and orientation, a hallmark of enzyme catalysis that is notable for a new-to-nature reaction such as P450-catalyzed nitrene transfer.
We propose that aziridine formation begins with reduction of the iron(III) heme from gaining an electron from NADPH via the P450BM3 reductase domain, analogous to the mechanism we proposed for enzyme-catalyzed sulfimidation.6 (Control experiments show that NADPH is not capable of reducing P-I263F in the absence of the reductase domain (Figure S7).) Reaction of tosyl azide with ferrous heme results in the formation of an iron nitrenoid species in the active site (formally in the +4 oxidation state), which can be either intercepted by olefin to produce aziridine or reduced by a second electron transfer from the reductase to form an unreactive iron(III) sulfonamide complex. In the event that sulfonamide is formed, the iron(III) heme must then consume another reducing equivalent to return to the reactive ferrous state. Given the key role of the reductase in catalyzing azide reduction, it should also be possible to improve aziridine yields by decreasing the rate of electron transfer, thereby selectively slowing the rate of azide reduction. Electron transfer in P450BM3 is well studied, and several amino acids have been identified to affect this process.18 We are currently exploring this additional route to enhancing the productivity of enzyme-catalyzed nitrene transfer reactions.
Conclusions
We report the first example of enzyme-catalyzed olefin aziridination. This challenging intermolecular reaction is catalyzed by a serine-ligated “P411” variant of cytochrome P450BM3. That the iron-heme cofactor has no measurable activity highlights the critical role of the protein in promoting this activity. Mutations in the enzyme active site resulted in a variant that exhibits significantly improved azide utilization compared to the parental enzyme and high (S)-selectivity (up to 99% ee). These results demonstrate the critical role of protein engineering in optimizing non-natural reactivity and suggest that the well-known plasticity of the P450 active site can be leveraged to target progressively more challenging non-natural reactions.
This new aziridination biocatalyst is likely just one of many new catalysts that will be discovered when researchers start systematically exploring the new functions that existing enzymes can take on.2 Exploiting the catalytic promiscuity of natural enzymes combined with evolutionary optimization will enable us to greatly expand the reaction space of genetically encoded biocatalysts.
Acknowledgments
We thank Dr. S. Virgil and the 3CS Catalysis Center at Caltech for assistance with HPLC, chiral HPLC, and LC–MS analysis. We thank Yufan Liang for assistance with chiral HPLC, and Hans Renata, Christopher Prier, and Sheel Dodani for helpful discussions. This work was supported by the Department of the Navy, Office of Naval Research (Grant N00014-11-0205), and the Jacobs Institute for Molecular Engineering for Medicine at Caltech. C.C.F and R.K.Z. are supported by NSF graduate research fellowships. T.K.H and J.A.M. are supported by Ruth L. Kirschstein National Research Service Awards (F32GM108143) (F32GM101792).
Supporting Information Available
The following file is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.5b00056.
Figures S1–S19, Tables S1–S4, and experimental procedures (PDF)
Author Contributions
† C.C.F. and R.K.Z.: equal contribution.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Bornscheuer U. T.; et al. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [DOI] [PubMed] [Google Scholar]; b Savile C. K.; et al. Biocatalytic asymmetric synthesis of chiral amines from ketones applied to Sitagliptin manufacture. Science 2010, 329, 305–309. [DOI] [PubMed] [Google Scholar]
- a Bornscheuer U. T.; Kazlauskas R. J. Catalytic promiscuity in biocatalysis: using old enzymes to form new bonds and follow new pathways. Angew. Chem., Int. Ed. 2004, 43, 6032–6040. [DOI] [PubMed] [Google Scholar]; b McIntosh J. A.; Farwell C. C.; Arnold F. H. Expanding P450 catalytic reaction space through evolution and engineering. Curr. Opin. Chem. Biol. 2014, 19, 126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Renata H.; Wang Z. J.; Arnold F. H. Expanding the enzyme universe: accessing non-natural reactions by mechanism-guided directed evolution. Angew. Chem., Int. Ed. 2015, 54, 3351–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Toscano M. D.; Woycechowsky K. J.; Hilvert D. Minimalist active-site redesign: teaching old enzymes new tricks. Angew. Chem., Int. Ed. 2007, 46, 3212–3236. [DOI] [PubMed] [Google Scholar]
- a Wang Z. J.; et al. Improved cyclopropanation activity of histidine-ligated cytochrome P450 enables the enantioselective formal synthesis of levomilnacipran. Angew. Chem., Int. Ed. 2014, 53, 6810–6813. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Seitz M.; et al. Synthesis of heterocyclic terpenoids by promiscuous squalene-hopene cyclases. ChemBioChem 2013, 14, 436–439. [DOI] [PubMed] [Google Scholar]; c Hammer S. C.; et al. Stereoselective friedel-crafts alkylation catalyzed by squalene hopene cyclases. Tetrahedron 2012, 68, 7624–7629. [Google Scholar]
- McIntosh J. A.; et al. Enantioselective intramolecular C–H amination catalyzed by engineered cytochrome P450 enzymes in vitro and in vivo. Angew. Chem., Int. Ed. 2013, 52, 9309–9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Singh R.; Bordeaux M.; Fasan R. P450-catalyzed intramolecular sp3 C–H amination with arylsulfonyl azide substrates. ACS Catal. 2014, 4, 546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Singh R.; Kolev J. N.; Sutera P. A.; Fasan R. Enzymatic C(sp3)-H amination: P450-catalyzed conversion of carbonazidates into oxazolidinones. ACS Catal. 2015, 5, 1685–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farwell C. C.; et al. Enantioselective imidation of sulfides via enzyme-catalyzed intermolecular nitrogen-atom transfer. J. Am. Chem. Soc. 2014, 136, 8766–8771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che C.-M.; Lo V. K.-Y.; Zhou C.-Y.; Huang J.-S. Selective functionalisation of saturated C–H bonds with metalloporphyrin catalysts. Chem. Soc. Rev. 2011, 40, 1950–1975. [DOI] [PubMed] [Google Scholar]
- a Degennaro L.; Trinchera P.; Luisi R. Recent advances in the stereoselective synthesis of aziridines. Chem. Rev. 2014, 114, 7881–7929. [DOI] [PubMed] [Google Scholar]; b Yudin A. K.Aziridines and Epoxides in Organic Synthesis; Wiley-VCH Verlag GmbH: 2006. [Google Scholar]
- a Mansuy D.; Mahy J. P.; Dureault A.; Bedi G.; Battioni P. Iron- and manganese-porphyrin catalysed aziridination of alkenes by tosyl- and acyl-iminoiodobenzene. J. Chem. Soc., Chem. Commun. 1984, 17, 1161–1163. [Google Scholar]; b Vyas R.; Goa G.-Y.; Harden J. D.; Zhang X. P. Iron(III) porphyrin catalyzed aziridination of alkenes with bromamine-T as nitrene source. Org. Lett. 2004, 6, 1907–1910. [DOI] [PubMed] [Google Scholar]
- a Gao G.-Y.; Jones J. E.; Vyas R.; Harden J. D.; Zhang X. P. Cobalt-catalyzed aziridination with diphenylphosphoryl azide (DPPA): direct synthesis of N-phosphorus-substituded aziridines from alkenes. J. Org. Chem. 2006, 71, 6655–6658. [DOI] [PubMed] [Google Scholar]; b Ruppel J. V.; Jones J. E.; Huff C. A.; Kamble R. M.; Chen Y.; Zhang X. P. A highly effective cobalt catalyst for olefin aziridination with azides: hydrogen bonding guided catalyst design. Org. Lett. 2008, 10, 1995–1998. [DOI] [PubMed] [Google Scholar]; c Jin L. M.; Xu X.; Lu H.; Cui X.; Wojtas L.; Zhang X. P. Effective synthesis of chiral N-fluoroaryl aziridines through enantioselective aziridnation of alkenes with fluoroaryl azides. Angew. Chem., Int. Ed. 2013, 52, 5309–5313. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lui Y.; Che C. M. [FeIII(F20-tpp)Cl] is an effective catalyst for nitrene transfer reactions and amination of saturated hydrocarbons with sulfonyl and aryl azides as nitrogen source under thermal and microwave-assisted conditions. Chem.—Eur. J. 2010, 16, 10494–10501. [DOI] [PubMed] [Google Scholar]
- a Dodani S. C.; et al. Structural, functional, and spectroscopic characterization of the substrate scope of the novel nitrating cytochrome P450 TxtE. ChemBioChem 2014, 15, 2259–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Barry S. M.; et al. Cytochrome P450-catalyzed L-tryptophan nitration in thaxtomin phytotoxin biosynthesis. Nat. Chem. Biol. 2012, 8, 814–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibodeaux C. J.; Chang W.-c.; Liu H.-w. Enzymatic chemistry of cyclopropane, epoxide, and aziridine biosynthesis. Chem. Rev. 2012, 112, 1681–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hyster T. K.; et al. Enzyme-controlled nitrogen-atom transfer enables regiodivergent C–H amination. J. Am. Chem. Soc. 2014, 136, 15505–15508. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Coelho P. S.; et al. A serine-substituted P450 catalyzes highly efficient carbene transfer to olefins in vivo. Nat. Chem. Biol. 2013, 9, 485–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yosca T. H.; et al. Iron(IV)hydroxide pKa and the role of thiolate ligation in C–H bond activation by cytochrome P450. Science 2013, 342, 825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sono M.; Andersson L. A.; Dawson J. H. Sulfur donor ligand binding to ferric cytochrome P-450-cam and myoglobin. Ultraviolet-visible absorption, magnetic circular dichroism, and electron paramagnetic resonance spectroscopic investigation of the complexes. J. Biol. Chem. 1982, 257, 8308–8320. [PubMed] [Google Scholar]
- Perera R.; et al. Molecular basis for the inability of an oxygen atom donor ligand to replace the natural sulfur donor heme axial ligand in cytochrome P450 catalysis. Spectroscopic characterization of the Cys436Ser CYP2B4 mutant. Arch. Biochem. Biophys. 2011, 507, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heel T.; et al. Non-natural olefin cyclopropanation catalyzed by diverse cytochrome P450s and other hemoproteins. ChemBioChem 2014, 15, 2556–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ando T.; et al. Iodine-catalyzed aziridination of alkenes using chloramine-T as a nitrogen source. Tetrahedron 1998, 54, 13485–13494. [Google Scholar]; b Kiyokawa K.; Kosaka T.; Minakata S. Metal-free aziridination of styrene derivatives with iminoiodinane catalyzed by a combination of iodine and ammonium iodide. Org. Lett. 2013, 15, 4858–4861. [DOI] [PubMed] [Google Scholar]
- a Sevrioukova I. F.; et al. Structure of cytochrome P450-redox partner electron-transfer complex. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 1863–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ost T. W.; et al. Phenylalanine 393 exerts thermodynamic control over the heme of flavocytochrome P450 BM3. Biochemistry 2001, 40, 13421–13429. [DOI] [PubMed] [Google Scholar]; c Roitel O.; Scrutton N. S.; Munro A. W. Electron transfer in flavocytochrome P450 BM3: kinetics of flavin reduction and oxidation, the role of cysteine 999, and relationships with mammalian cytochrome P450 reductase. Biochemistry 2003, 42, 10809–10821. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.