

The nexin-dynein regulatory complex (N-DRC) is an axonemal structure that regulates ciliary motility. A Chlamydomonas mutant lacking the N-DRC subunit DRC3 has abnormal waveform and lacks portions of the N-DRC linker, including the L1 protrusion that connects to dynein g. DRC3 interacts with dynein g to control ciliary waveform.
Abstract
The nexin-dynein regulatory complex (N-DRC), which is a major hub for the control of flagellar motility, contains at least 11 different subunits. A major challenge is to determine the location and function of each of these subunits within the N-DRC. We characterized a Chlamydomonas mutant defective in the N-DRC subunit DRC3. Of the known N-DRC subunits, the drc3 mutant is missing only DRC3. Like other N-DRC mutants, the drc3 mutant has a defect in flagellar motility. However, in contrast to other mutations affecting the N-DRC, drc3 does not suppress flagellar paralysis caused by loss of radial spokes. Cryo–electron tomography revealed that the drc3 mutant lacks a portion of the N-DRC linker domain, including the L1 protrusion, part of the distal lobe, and the connection between these two structures, thus localizing DRC3 to this part of the N-DRC. This and additional considerations enable us to assign DRC3 to the L1 protrusion. Because the L1 protrusion is the only non-dynein structure in contact with the dynein g motor domain in wild-type axonemes and this is the only N-DRC–dynein connection missing in the drc3 mutant, we conclude that DRC3 interacts with dynein g to regulate flagellar waveform.
INTRODUCTION
The beating of cilia and flagella, terms here used interchangeably, generates a flow of fluid over the surface of cells and plays important roles wherever motile cilia occur. In mammals, ciliary beating moves mucus and trapped particles up and out of the airway as the first line of defense of the lungs. Ciliary activity also is important for embryo development, circulation of cerebrospinal fluid in the brain, and the sperm motility necessary for fertilization of the ovum. Impaired motility of cilia in humans gives rise to diverse disorders, including primary ciliary dyskinesia, situs inversus, male infertility, and hydrocephalus (Fliegauf et al., 2007; Brown and Witman, 2014).
The core structure of motile cilia is the axoneme, which contains the machinery that generates ciliary beating. In most motile cilia, the axoneme is a highly conserved “9 + 2” structure that consists of 9 outer doublet microtubules surrounding a pair of central microtubules; each doublet microtubule is composed of an A-tubule and a B-tubule. Rows of dynein motors are located on both outer and inner edges of the A-tubules (outer and inner dynein arms, respectively). These dyneins interact with the B-tubule of the adjacent doublet microtubule to generate an interdoublet sliding that forms the basis for ciliary movement. The axoneme also contains stalk-like structures called radial spokes that extend from the A-tubule toward the center of the axoneme, where they interact with projections from the central pair of microtubules. The doublet-associated machinery repeats at regular intervals along the doublet microtubules, giving rise to a fundamental 96-nm axial repeat unit that includes four outer dynein arms, seven different inner dynein arms, and a triplet of radial spokes. It is believed that mechanical and/or chemical signals from the central pair propagate through the radial spokes to the doublet microtubules to control dynein activity in order to generate a coordinated ciliary beat (Porter and Sale, 2000; Smith and Yang, 2004; Lindemann and Lesich, 2010; Oda et al., 2014).
Chlamydomonas reinhardtii mutants lacking the central pair or radial spokes have paralyzed flagella (Witman et al., 1978). The discovery of suppressor mutations (pf2, pf3, sup-pf-3, sup-pf-4, and sup-pf-5; Table 1) that restore flagellar activity to these paralyzed strains revealed the presence of an additional axonemal structure, termed the dynein regulatory complex (DRC), that inhibits dynein arm activity in the absence of signals from the radial spokes and central pair and is disrupted to various degrees in the suppressor strains (Huang et al., 1982; Gardner et al., 1994). Biochemical analysis of axonemes of the suppressor strains identified six polypeptides, termed DRC1–6, that are missing or reduced in those strains (Huang et al., 1982); subsequent studies identified at least five additional components of the DRC, termed DRC7–11 (Table 1; Piperno et al., 1994; Lin et al., 2011; Bower et al., 2013). All DRC proteins are highly conserved in most organisms with motile cilia or flagella, and the Chlamydomonas proteins have high BLAST E-values compared with their human homologues (Bower et al., 2013). Mutations in the human homologues of DRC1 (CCDC164) and DRC2 (CCDC65) are found in primary ciliary dyskinesia patients, indicating that the DRC is essential for normal development and health (Austin-Tse et al., 2013; Horani et al., 2013; Wirschell et al., 2013).
TABLE 1:
Affected proteins and structures in select N-DRC mutant strains.
| Strain | Affected N-DRC subunitsa | Affected N-DRC structural domainsb |
|---|---|---|
| pf2 | DRC3-7, (8–10), 11 | Entire proximal and distal lobe, L1 and L2 protrusions, OID linker, central linker, base plate protrusion |
| pf3 | DRC1, 2, (3, 4), 5, 6, (7–10), 11 | Entire proximal and distal lobe, (L1) and L2 protrusions, (OID linker), (central linker), entire base plate |
| sup-pf-3 | DRC(3–9), 11 | Entire proximal and distal lobe, (L1) and L2 protrusions, (OID linker), (central linker) |
| sup-pf-4 | DRC5, 6 | Part of distal lobe, L2 protrusion |
| sup-pf-5 | DRC1, 2, 5, 6 | Not determined |
| drc3 | DRC3 | Part of distal lobe, L1 protrusion |
Data for strains other than drc3 are from Table 3 of Piperno et al. (1994), Table 3 of Heuser et al. (2009), and Supplemental Table 4 of Bower et al. (2013).
aProteins in parentheses are reduced.
bStructures in parentheses are reduced.
Conventional electron microscopy and cryo–electron tomography (cryo-ET) of the DRC in axonemes of wild-type and the suppressor strains have revealed that the DRC is a large (∼1.5 MDa), architecturally complex structure located near the base of one of the radial spokes and with connections to multiple axonemal components (Gardner et al., 1994; Heuser et al., 2009). The DRC has two distinct parts—a “base plate” by which it is attached to the A-tubule, and a “linker” that projects away from the A-tubule and terminates in proximal and distal lobes that contact the B-tubule of the opposing doublet microtubule (Figure 1; Heuser et al., 2009). Other parts of the DRC connect to two and possibly three different inner dynein arms, to an outer dynein arm, and, through the calmodulin- and spoke-associated complex, to radial spoke RS2 and the shorter spoke RS3S (Heuser et al., 2009, 2012). The discovery that the DRC extends all the way to the opposing B-tubule led to the realization that the DRC linker corresponds to an axonemal structure previously termed the “nexin link” that connects the A-tubule of one doublet microtubule to the B-tubule of the adjacent doublet microtubule; as a result, the DRC was renamed the nexin-dynein regulatory complex (N-DRC; Heuser et al., 2009). Possible functions for the nexin link include resisting interdoublet sliding to convert this sliding into axonemal bending, monitoring sliding or doublet microtubule curvature to provide feedback to the dynein arms, and maintaining the arrangement of outer doublet microtubules (Warner, 1983; Heuser et al., 2009; Bower et al., 2013). Thus the N-DRC is ideally located to be a major hub for monitoring important parameters of axonemal bending and distributing regulatory signals to diverse parts of the axonemal motility machinery.
FIGURE 1:
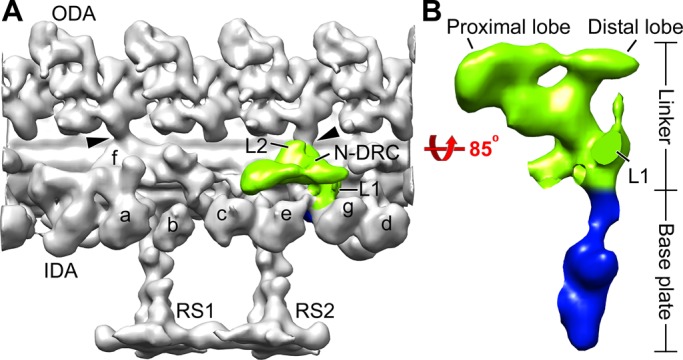
The architecture of the N-DRC in the Chlamydomonas wild-type axoneme. (A) Three-dimensional isosurface rendering of the averaged 96-nm repeat shows the nexin linker (green) of the N-DRC and other axonemal structures (gray) in a longitudinal front view (proximal is on the left). (B) Magnified surface rendering of the N-DRC shows the nexin linker (green) and the base plate (blue) in a longitudinal bottom view (from the central pair toward the doublet microtubule). For better visualization of the N-DRC, other axonemal structures are not shown in B. Arrowheads indicate OID linkers. a–g, inner arms a–g; IDA, inner dynein arm; L1, L1 protrusion; L2, L2 protrusion; ODA, outer dynein arm; RS1 and RS2, radial spokes 1 and 2.
Despite the importance of the N-DRC for ciliary function, its structural and biochemical complexity has presented a major challenge to understanding the specific functions of the N-DRC's subdomains and subunits. In this article, we characterize a newly discovered Chlamydomonas N-DRC mutant null for DRC3 to clarify its function.
RESULTS
Identification of a DRC3-null mutant
We previously reported a Chlamydomonas strain (B1179) in which a large insertion deleted several genes, including DRC3, NPHP4 (encoding the flagellar transition-zone protein nephrocystin-4 [NPHP4]), and four predicted genes that are of unknown function but apparently not associated with flagellar function (Awata et al., 2014). To ensure that the phenotype being analyzed in the studies that follow was not compromised by the absence of nephrocystin-4, we backcrossed B1179 twice to a wild-type strain and then transformed some of the mutant progeny with a DNA fragment containing the full-length NPHP4 gene. One of the resulting transformants, rescued for NPHP4, was used in this study as the drc3 strain.
The mutation in drc3 completely removes the first seven of 10 exons in the DRC3 gene (Figure 2A). To determine whether the remaining portion of DRC3 is expressed in the mutant, we made a polyclonal antibody to a fusion protein containing DRC3 amino acids 216–383 encoded by exons 5–9. In Western blots of isolated wild-type flagella, the antibody reacted with a single protein of the expected size (60 kDa); the protein was missing from whole-cell lysates and isolated flagella of the drc3 mutant and restored in flagella of a rescued strain (termed DRC3-R) generated by transforming drc3 with the full-length DRC3 gene (Figure 2B and Supplemental Figure S1). The drc3 mutant also was transformed with a construct expressing DRC3 with a C-terminal SNAP-tag. In Western blots of isolated flagella of the DRC3-SNAP strain, the antibody reacted with a single protein of the expected molecular mass for DRC3 plus the SNAP-tag (Figure 2B). Therefore the antibody is specific, and DRC3 is completely missing from drc3 flagella.
FIGURE 2:

Characterization of Chlamydomonas drc3 mutant. (A) Diagram of Chlamydomonas DRC3 gene. The black rectangles are exons. Double-headed arrows indicate the positions of PCR products used to delimit the deleted region; plus and minus marks indicate whether the PCR products were amplified in the wild type (WT) or the mutant (drc3[B1179]). (B) Western blot of isolated flagella probed with anti-DRC3. The antibody recognized a band of the predicted size for DRC3 (60 kDa) in WT; the band was absent from the drc3 flagella but was restored in the rescued strain (DRC3-R). In the DRC3-SNAP strain, the band recognized by anti-DRC3 shifted upward because of the addition of the SNAP-tag (20 kDa). The outer dynein arm intermediate chain IC2 served as a loading control. Note that the levels of DRC3 and DRC3-SNAP in flagella of the rescued strains are similar to that of DRC3 in wild-type flagella. (C) Means ± SDs of swimming speed determined from 50 cells each of WT, drc3, DRC3-R, and DRC3-SNAP strains. The swimming speed of drc3 cells was slower than that of WT and was rescued by transformation of the mutant with either the wild-type DRC3 gene or a gene expressing SNAP-tagged DRC3. Statistical significance was determined by the Tukey–Kramer method: NS, p ≥ 0.05; ***p < 0.001. (D) Swimming paths of WT, drc3, and DRC3-R cells. Positions of cells while swimming in the focal plane were plotted with Manual Tracking in ImageJ. Recording time (1.5 s) was identical in all samples; interval between dots is 33 ms. Even though the swimming speed of drc3 was only slightly less than that of WT, the tracks of drc3 cells are much shorter than those of WT cells because the former tended to swim out of the focal plane more rapidly.
Loss of DRC3 increases beat frequency but reduces swimming speed
As reported for other N-DRC mutants (Piperno et al., 1992), the drc3 cells have full-length flagella (Supplemental Figure S2). However, the swimming speed of drc3 cells (∼110 μm/s) is reduced compared with that of wild-type cells (∼140 μm/s; Figure 2C). Moreover, swimming drc3 cells did not remain in the microscope focal plane as long as wild-type cells (Figure 2D), suggesting that their swimming paths are more erratic. These abnormal swimming phenotypes were rescued in the DRC3-R and DRC3-SNAP strains (Figure 2, C and D), indicating that the phenotypes are caused specifically by lack of DRC3.
To understand why drc3 cells swim more slowly than wild-type cells, we analyzed the beat frequencies of wild-type, drc3, DRC3-R, and DRC3-SNAP cells by the fast Fourier transform (FFT) method. The average beat frequency of wild-type cells was ∼55 Hz (Figure 3A). Surprisingly, the average beat frequency of drc3 cells was ∼70 Hz (Figure 3A). Beat frequency was returned to <60 Hz in DRC3-R and DRC3-SNAP cells. In contrast to the drc3 cells, the beat frequencies of previously studied N-DRC mutants were either reduced or comparable to that of wild type (Figure 3B and Table 2).
FIGURE 3:

Flagellar beat frequency of drc3 cells is higher than that of WT and other N-DRC mutant cells. (A) Beat frequency of WT, drc3, DRC3-R, and DRC3-SNAP cells was analyzed by the FFT method. Loss of DRC3 caused increased beat frequency compared with WT cells. Beat frequency was restored to nearly normal by transformation of the drc3 strain with the WT gene or the construct expressing SNAP-tagged DRC3. (B) FFT spectra of other N-DRC mutants. Except for pf2, beat frequency is comparable to WT. The vertical solid and broken lines are aligned to the peaks of the spectra of the WT and the drc3 mutant, respectively.
TABLE 2:
Swimming speed, flagellar beat frequency, and waveform of N-DRC mutants.
| Strain | Swimming speed | Beat frequency | Waveform |
|---|---|---|---|
| drc3 | Slightly reduced | Increased | Reduced amplitude |
| pf2 | Reduceda | Reducedb | Reduced amplitudec |
| pf3 | Reduceda | Normal | Reduced amplitudec |
| sup-pf-3 | Reduceda | Nearly normal | Normald |
| sup-pf-4 | Slightly reduceda | Normal | Not determined |
aData from Gardner et al. (1994) and Kubo et al. (2012).
bReduced but variable in Brokaw and Kamiya (1987); normal in Kubo et al. (2012).
cData from Brokaw and Kamiya (1987)
dData from Brokaw et al. (1982) and Brokaw and Luck (1985).
Waveform of the drc3 mutant is abnormal
Because swimming speed is a function of flagellar length, beat frequency, and waveform, the combination of normal flagellar length and increased beat frequency suggested that the reduced swimming speed of drc3 cells is due to an altered waveform that causes them to swim less efficiently than wild-type cells. To assess this directly, we observed and recorded forward-swimming wild-type, drc3, and DRC3-R cells (Supplemental Movies S1–S3). Wild-type cells swim with an asymmetric flagellar bending pattern, as previously reported (Figure 4A; Brokaw et al., 1982). During the effective stoke, the flagella sweep down until they are nearly parallel to the sides of the cell; during the recovery stroke, the principal bend exceeds 180°. The drc3 mutant also swims with an asymmetric flagellar bending pattern, but the effective stroke typically ends when the flagella are perpendicular to the swimming direction rather than continuing down alongside the cell body. Moreover, during the recovery stroke, the principal bend is more open than in wild type, usually not exceeding 180° (Figure 4A). Thus the flagella of the drc3 cells have a smaller beat envelope (Figure 4B). A normal flagellar waveform is nearly restored in DRC3-R cells (Figure 4A). The abnormal flagellar waveform of drc3 cells would generate less force, possibly leading to the relatively erratic swimming paths of these cells.
FIGURE 4:
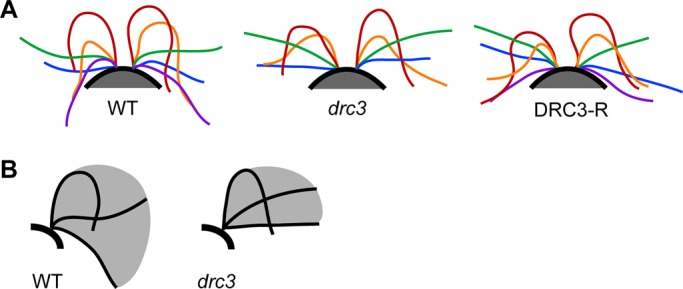
Loss of DRC3 affects flagellar waveform. (A) Representative flagellar waveforms of WT, drc3, and DRC3-R cells. Waveforms of at least six free-swimming cells were analyzed for each strain; cells were observed over 2–10 beat cycles. For each cell type, cis- and trans-flagella from the same digital image have the same color; tracings of different colors are not necessarily from the same beat cycle. Flagellar length is not consistent through the beat cycle because the flagellar tip was sometimes out of focus. (B) Schematic diagrams illustrating typical flagellar beat envelopes for the WT and the drc3 mutant. The drc3 cells have a smaller beat envelope.
It is of interest to compare the swimming velocity, flagellar beat frequency, and waveform of drc3 cells with those of other N-DRC mutants (Table 2). Despite the elevated beat frequency of drc3 cells, their swimming speed is a little lower than that of wild-type cells because their effective stroke is truncated and their recovery stroke also is likely to be less efficient. Cells of the N-DRC mutants pf2 and pf3 also swim with a smaller principal bend and reduced beat envelope than wild type, but their beat frequencies are either less than or similar to that of wild type (Figure 3; see also Brokaw and Kamiya, 1987; Kubo et al., 2012). As a result, the swimming velocities of pf2 and pf3 cells are reduced. Cells of sup-pf-4 have a normal beat frequency and swim at nearly normal speed; their flagellar waveform has not yet been determined. Cells of sup-pf-3 have a normal waveform (Brokaw et al., 1982; Brokaw and Luck, 1985) and normal or only slightly reduced beat frequency (Kubo et al., 2012; Figure 3), yet they have greatly reduced swimming velocity (Gardner et al., 1994; Kubo et al., 2012). Therefore further study is necessary to understand the reason for the reduced swimming velocity of sup-pf-3.
The drc3 mutation does not suppress the paralyzed phenotype of pf14
The N-DRC mutants were first identified as suppressors that restored some flagellar activity to paralyzed mutants with defects in the radial spokes or central pair of microtubules (Huang et al., 1982), and all previously identified N-DRC mutants have this suppressor activity (Huang et al., 1982; Piperno et al., 1992). To determine whether the drc3 mutation can similarly suppress paralysis caused by such defects, we generated recombinant strains carrying drc3 and pf14, a well-characterized mutation that causes loss of radial spokes RS1 and RS2 and is suppressed by other N-DRC mutations. The drc3 pf14 double mutant cells were well flagellated but remained completely paralyzed (Figure 5). To confirm that the flagellar activity of drc3 pf14 cells differs from that of a previously studied strain with mutations in both the N-DRC and radial spokes (Huang et al., 1982), we compared the drc3 pf14 cells to sup-pf-3 pf14 cells. In contrast to the drc3 pf14 strains, the sup-pf-3 pf14 cells showed considerable flagellar activity; consistent with previous findings (Piperno et al., 1994), most sup-pf-3 pf14 cells vibrated rapidly, some spun in place, and a few swam (Figure 5). The drc3 pf14 cells occasionally twitched their flagella, but this movement could not be distinguished from that of the pf14 single mutant (Hayashibe et al., 1997). Therefore, in contrast to all other N-DRC mutants, the drc3 mutation cannot suppress paralysis caused by the absence of radial spokes.
FIGURE 5:

Loss of DRC3 does not suppress the paralyzed-flagella phenotype of pf14. Stacked bar graphs showing percentage of cells of the indicated strains that were paralyzed (red), vibrating (yellow), spinning (green), or swimming (blue). Two independently isolated strains of the drc3 pf14 double mutant were analyzed. For each strain, 118 vegetative (left) and 113 gametic (right) cells were scored.
All known DRC components except DRC3 are present in the drc3 axoneme
Comparison of isolated axonemes from wild-type and N-DRC mutants by two-dimensional (2D) gel electrophoresis identified seven N-DRC subunits (DRC1–7); more recently, immunoprecipitation of the N-DRC from axonemal extracts combined with mass spectrometric analysis of the immunoprecipitates identified four additional subunits (DRC8–11; Huang et al., 1982; Piperno et al., 1992; Lin et al., 2011; Bower et al., 2013). To identify proteins missing in drc3 axonemes, we purified axonemes from wild-type and drc3 cells and compared them by 2D gel electrophoresis (Figure 6A). Consistent with our Western blot results (Figure 2B), the spot corresponding to DRC3 was missing from the drc3 axonemes. In contrast, DRC1, 2, and 4–7 were present in normal amounts. Moreover, spots corresponding to candidate N-DRC or N-DRC–associated proteins (FAP206, FAP230, FAP252, FAP61/IDA7, and spot 11; Lin et al., 2011) were not obviously altered in the mutant axonemes.
FIGURE 6:

N-DRC proteins in the drc3 axoneme. (A) Silver-stained 2D gels of WT and drc3 axonemes. The drc3 axonemes lack DRC3 (right, white arrowhead) but contain normal amounts of DRC1, 2, and 4–7 and of FAP206, 230, and 252, previously identified as candidate N-DRC proteins (Lin et al., 2011; Bower et al., 2013). The drc3 axonemes also contain normal amounts of FAP61/IDA7, which are reduced in many other N-DRC mutants, and of “spot 11,” an unidentified protein also reduced in many other N-DRC mutants (Lin et al., 2011). (B) Western blots of isolated flagella from WT and drc3 cells probed with anti-DRC8 and anti-DRC11 antibodies. DRC8 and DRC11 are present at normal levels in the drc3 flagella. IC2 was used as a loading control.
DRC8–11 are not readily identified by 2D gel electrophoresis. To determine whether DRC8 and DRC11 are missing from drc3 axonemes, we probed Western blots of the mutant axonemes with antibodies specific for each of these proteins and found that both proteins are present at normal levels in the drc3 axonemes (Figure 6B).
Antibody probes are not yet available for DRC9 and DRC10. To determine whether DRC9 and DRC10 are present in drc3 axonemes, we extracted both wild-type and drc3 axonemes with 0.4 M KI, which solubilizes many N-DRC proteins (Bower et al., 2013), and separated the extracted proteins by SDS–PAGE (Supplemental Figure S3). We then excised slices from the region of the gel (40–45 kDa) where these proteins (FAP122 and FAP84, respectively) were found in the analysis of the Chlamydomonas flagellar proteome (Pazour et al., 2005; Supplemental Figure S3). Analysis of the slices by mass spectrometry (MS) indicated that both DRC9 and DRC10 are present in the drc3 axoneme at levels comparable to those of the wild-type axoneme (Table 3). Thus all of the known or candidate N-DRC subunits except DRC3 are present in drc3 axonemes. These results indicate that drc3 is biochemically the least affected of the N-DRC mutants.
TABLE 3:
Select proteins identified from gel slices at locations corresponding to DRC9 and DRC10 relative mobilities.
| Number of exclusive spectra | |||
| Protein | Mass (kDa) | Wild type | drc3 |
| DRC9/FAP122 | 46 | 55 | 48 |
| DRC10/FAP84 | 41 | 49 | 39 |
| α-Tubulin | 50 | 411 | 445 |
| β-Tubulin | 50 | 304 | 309 |
| PF16 (central pair protein) | 50 | 89 | 95 |
| RSP16 | 39 | 30 | 25 |
| DRC5/FAP155 | 43 | 12 | 10 |
Total number of spectra exclusive to each protein is shown. Five other representative axonemal proteins are included as standards. A total of 176 and 180 proteins were identified in the wild-type and drc3 samples, respectively.
Structure of the N-DRC in the drc3 mutant
To determine how loss of DRC3 affected the structure of the N-DRC, we analyzed drc3 axonemes by cryo-ET and subtomogram averaging (Figure 7 and Supplemental Figure S4). We found missing densities only within the N-DRC linker domain: the L1 protrusion (which links directly to the motor domain of dynein g; Heuser et al., 2009), a small part of the distal lobe, and the connection between these two structures appeared to be missing (Figure 7, D–F, and Supplemental Figure S4, C and D). However, the part of the distal lobe that forms the “1b” connection to the opposing B-tubule appeared to be intact. The total mass of the densities missing in the drc3 N-DRC is estimated to be ∼140 kDa. The missing structures were restored in the axonemes of the DRC3-SNAP strain (Figure 7, G–I), indicating that their absence was caused specifically by loss of DRC3. Other parts of the N-DRC, including the proximal lobe and the base plate, resembled wild-type morphology. Therefore DRC3 most likely is located within the volume corresponding to the L1 protrusion, the distal part of the distal lobe, and the connection between these two densities.
FIGURE 7:

Structure of the N-DRC in the drc3 axoneme. Three-dimensional isosurface renderings of cryo-ET averages of the 96-nm repeats from WT (A–C), drc3 (D–F), and DRC3-SNAP (G–I) axonemes. The N-DRC linker (red, green) and base plate (blue) are shown in cross-sectional (A, D, G) and longitudinal (B, E, H) views of the doublet microtubule. (C, F, I) Enlarged N-DRC images in a longitudinal bottom view (looking from the axoneme center outward); to show the entire N-DRC clearly, the inner dynein arms and radial spoke RS2 were removed from these three images. A portion of the distal lobe, the L1 protrusion, and the connection between these two structures (red) are missing in the drc3 axonemes but are specifically restored in the strain rescued with DRC3-SNAP. Outer dynein arms and inner arm dyneins a–g are labeled. Inner arm dynein g, to which the L1 protrusion connects, is indicated in pink.
DRC3 is present in a subcomplex with DRC4, DRC7, and DRC11
We also sought to determine whether DRC3 was tightly associated with a specific subset of N-DRC proteins. Significant amounts of the N-DRC can be released from the axoneme by treatment with >0.2 M NaI (Bower et al., 2013; Wirschell et al., 2013). Experiments in which 0.4 M KI extracts from wild-type and drc3 axonemes were immunoprecipitated with our anti-DRC3 antibody followed by analysis of the immunoprecipitate by SDS–PAGE, MS, and Western blotting indicated that DRC4, DRC7, and DRC11 were specifically coprecipitated with DRC3 (Supplemental Figure S5 and Supplemental Table S1). To confirm these results, we rescued the drc3 mutant with a construct expressing DRC3 with a C-terminal hemagglutinin (HA) tag. Western blotting showed that DRC3-HA was present at wild-type levels in isolated flagella of the DRC3-HA strain (Supplemental Figure S6). We then extracted isolated axonemes of wild-type and DRC3-HA strains with 0.3 M KI, carried out immunoprecipitations using an anti-HA antibody, and separated the immunoprecipitated proteins by SDS–PAGE. Polypeptides of 55, 65, 85, and 180 kDa were specifically pulled down from the DRC3-HA sample but not the wild-type control sample (Figure 8A, arrow and arrowheads). Western blotting of the immunoprecipitate confirmed that these bands corresponded to DRC4, DRC3-HA, DRC11, and DRC7, respectively (Figure 8B). Therefore DRC4, DRC7, and DRC11 are tightly associated with DRC3 in a subcomplex within the N-DRC.
FIGURE 8:
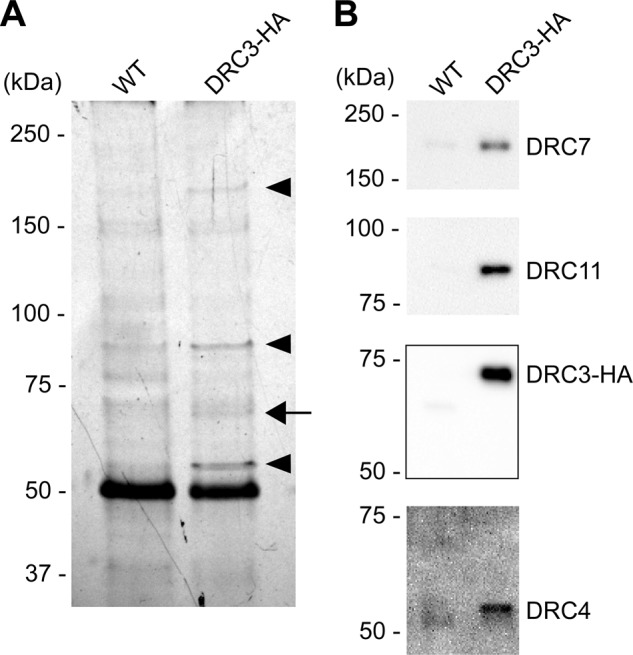
DRC3 is in a subcomplex with DRC4, DRC7, and DRC11. Isolated axonemes of WT and the DRC3-HA strain were extracted with KI and the extract subjected to immunoprecipitation using the anti-HA antibody. (A) Silver-stained SDS–polyacrylamide gel of the resulting immunoprecipitate. An arrow indicates a band likely to contain HA-tagged DRC3. Arrowheads indicate bands with polypeptides specifically coprecipitated with HA-tagged DRC3. (B) The same samples as in A were analyzed by Western blotting using anti-DRC3, 4, 7, and 11 antibodies.
DISCUSSION
Both structurally and biochemically, the drc3 mutant is the least affected of all known N-DRC mutants. As a result, unique aspects of the drc3 phenotype combined with the relatively limited nature of the drc3 lesions allow us to draw important conclusions about the location of DRC3 and closely associated N-DRC subunits, as well as about the role of specific N-DRC components and structures in flagellar waveform control.
Location of DRC3
Our cryo-ET analysis revealed that the N-DRC of drc3 axonemes is lacking the L1 protrusion, which is located in the central part of the linker, as well as the distal edge of the distal lobe and the connection between these two structures (Figure 7 and Supplemental Figure S4). The mass of DRC3 is predicted to be 60 kDa, whereas the mass of the densities missing in the drc3 N-DRC is estimated to be ∼140 kDa. Therefore the missing densities could accommodate either a dimer of DRC3 or one copy of DRC3 together with one or more yet-to-be-identified N-DRC proteins. A partial N-DRC isolated from wild type lacked the distal lobe but retained DRC3 in equimolar ratios with the other N-DRC subunits (Oda et al., 2015), so DRC3 most likely is located in the L1 protrusion.
DRC3 is required for normal flagellar waveform
The drc3 mutant has an abnormal waveform that is less effective than that of wild-type cells. Of the 11 known N-DRC subunits, only DRC3 is missing in the drc3 axoneme. Therefore DRC3 appears to be essential for generating a normal flagellar waveform. As noted in the preceding section, DRC3 probably is localized to the L1 protrusion. In the wild type, the L1 protrusion is the only non-dynein structure in contact with the linker of the dynein g motor domain, which is important for the head-swinging mechanism of the dynein powerstroke (Lin et al., 2014), and it is the only N-DRC–dynein connection that is missing in the drc3 mutant. Therefore the simplest interpretation is that loss of DRC3 and protrusion L1 eliminates control of dynein g by the N-DRC, with loss of coordination between dynein g and outer and/or other inner arm dyneins, resulting in aberrant flagellar waveform. It may be relevant that the smaller angle of the principal bend and smaller beat envelope of drc3 are similar to those of cells with defects in inner arm dynein f/I1 (Brokaw and Kamiya, 1987; Bayly et al., 2010; VanderWaal et al., 2011) or inner arms a and c–e (Kubo et al., 2012).
The N-DRC's controlled inhibition of dynein arm activity is retained in the drc3 mutant
One of the most striking features of the drc3 mutation is that it allows the flagellar paralysis caused by loss of radial spokes to be maintained. Because all previously studied N-DRC mutations restore some flagellar activity to these mutants, it has been proposed that the N-DRC keeps the dynein arms in an inactive state in the absence of signals from the radial spokes and that the previously studied N-DRC lesions prevent this inhibition (Brokaw et al., 1982). Because this inhibition is still present in the drc3 pf14 double mutant, the mutant must retain that part of the N-DRC responsible for this function. The subdomains of the N-DRC that are missing in all previously examined N-DRC mutants but remaining in drc3 are the L2 protrusion and the proximal portion of the distal lobe, likely including the part of the distal lobe that connects to the opposing B-tubule (Heuser et al., 2009). Therefore it is likely that one or both of these structures are critical for the failure to suppress the paralysis resulting from radial spoke mutations. The proximal portion of the distal lobe could be involved in monitoring changes in doublet microtubule curvature or spacing to provide feedback to the dynein arms during axonemal bending. The L2 protrusion does not connect to any structure outside the N-DRC; however, it is located between the proximal and distal lobes and the outer-inner dynein (OID) linker that connects to the outer dynein arm. The OID linker appears to control outer dynein arm activity so that the outer arms are not active on all doublets at once (Oda et al., 2013). Therefore the L2 protrusion may be important for communicating or integrating information between different parts of the N-DRC. In this context, it should be noted that paralysis caused by loss of the radial spokes or central pair also can be suppressed by mutations in the outer arm dynein heavy chains (Porter et al., 1994; Rupp et al., 1996) or IC2 (Oda et al., 2013), strongly suggesting that outer arm dynein is involved in maintaining the axoneme in a quiescent state in the absence of signals from the radial spokes. Thus it is possible that signals from the proximal lobe are relayed through L2 to the OID and thence to the outer arm to keep axonemes quiescent in the absence of radial spoke signals. In the wild-type axoneme, these signals would be integrated with signals from the radial spokes to cyclically activate and inactivate the outer dynein arms at appropriate times in the flagellar beat cycle.
DRC3, 4, 7, and 11 form a subcomplex in the N-DRC linker
We found that DRC3, 4, 7, and 11 occur in a subcomplex after extraction from wild-type axonemes. Previously, correlation of N-DRC proteins and cryo-ET densities present or absent in wild type and previously known N-DRC mutants placed DRC3, 4, and 7 together in the N-DRC linker region, with DRC4 near the base of the linker (Heuser et al., 2009). More recently, cryo-EM localization of DRC4 with structural tags on its N-terminal, middle, and C-terminal regions showed that it extends from the base plate all the way to the proximal lobe; similar localization of DRC5 placed its C-terminus near the center of the linker (Oda et al., 2015). Consistent with this, detailed biochemical analysis of the drc mutants suggested that DRC4 facilitates the assembly of DRC3, 5, 6, and 7 into the N-DRC (Lin et al., 2011). Finally, cross-linking of isolated axonemes from a strain expressing DRC4-HA showed that three polypeptides of ∼65, 70, and 85 kDa are closely associated with HA-tagged DRC4 (Rupp and Porter, 2003). Although the proteins cross-linked to HA-tagged DRC4 were not identified, the apparent mass of DRC3 as determined by SDS–PAGE is ∼60 kDa (Figure 2B; Piperno et al., 1994) and that of DRC11 is ∼85 kDa (Figure 6B). Taken together, the results indicate that DRC3, 4, 7, and 11 form a subcomplex within the N-DRC that likely extends from the base plate to the furthest part of the nexin linker.
DRC11 is likely located in the central part or proximal lobe of the nexin linker
DRC11 is missing in all previously examined N-DRC mutant axonemes except for those of sup-pf-4 (Bower et al., 2013). Therefore DRC11 should be located in a subdomain of the N-DRC that is missing in pf2, pf3, and sup-pf-3 but present in sup-pf-4 and drc3. By this criterion, possible locations for DRC11 are the proximal lobe and the central portion of the nexin linker that connects the proximal and distal lobes (Heuser et al., 2009). The distal lobe is almost completely missing from sup-pf-4 (with the exception of the most distal edge), and the distal part of the same lobe is missing from drc3, yet the amount of DRC11 appears to be normal in the axonemes of both mutants (Figure 6B; Bower et al., 2013), excluding the distal lobe as a possible location. Therefore DRC11 can be tentatively assigned to the proximal lobe or the central part of the nexin linker close to the L1 protrusion. This is consistent with our finding that DRC11 is in a subcomplex with DRC3 (which is located in the L1 protrusion) and DRC4 (which reaches to the proximal lobe).
Prospective
An essential step toward elucidating the roles of the different parts of the N-DRC will be to map the locations and functions of the individual subunits within this very large complex. An important advance toward this goal was recently made by cryo-EM localization of biotin carboxyl carrier protein tags on various parts of DRC1, DRC2, DRC4, and DRC5 (Oda et al., 2015). Here our demonstration that a null mutant for DRC3 can be structurally and functionally rescued with wild-type DRC3 fused to a SNAP-tag provides a foundation for similar high-resolution cryo-EM mapping using commercially available, readily detectable electron-dense gold nanoparticles that can be ligated to the SNAP-tag with high affinity and high efficiency (Song et al., 2015). Application of this approach to other N-DRC subunits should enable generation of a highly detailed architectural blueprint of the N-DRC, providing a basis for future work to decipher structure–function relationships within the N-DRC. The approach is similarly applicable to other axonemal proteins. More broadly, the versatile and well-developed SNAP technology has the potential to facilitate analysis of axonemal protein transport and turnover and protein–protein interactions (Keppler et al., 2004).
MATERIALS AND METHODS
Strains
Chlamydomonas reinhardtii wild-type strains 137c (nit1, nit2, mt+) and CC-124 (nit1, nit2, mt-) and mutant strains CC-613 (pf14, mt-) and CC-3000 (sup-pf-3/drc4, pf14, mt-) were obtained from the Chlamydomonas Resource Center (University of Minnesota, St. Paul, MN). The strain B1179 (nphp4-1, drc3-1, arg7, ARG7, mt+), in which the DRC3 gene is deleted, was described previously (Awata et al., 2014). The drc3 strain (drc3-1, nphp4-1, NPHP4, mt+), rescued for NPHP4, was produced by crossing B1179 sequentially to the wild-type strains CC124 and 137c and then transforming selected progeny with NPHP4. drc3 cells rescued with wild-type DRC3 (DRC3-R: drc3-1, DRC3, nphp4-1, NPHP4, mt+) or with constructs expressing SNAP-tagged DRC3 (DRC3-SNAP: drc3-1, DRC3-SNAP, nphp4-1, NPHP4, mt+) or HA-tagged DRC3 (DRC3-HA: drc3-1, DRC3-HA, nphp4-1, NPHP4, mt+) were produced as described under Transformation. The drc3 strains in the pf14 background (drc3-1, nphp4-1, NPHP4, pf14) were generated as described in the next section. Cells were grown in M medium I (Sager and Granick, 1953) modified with 2.2 mM KH2PO4 and 1.7 mM K2HPO4 (Witman, 1986) or TAP medium (Gorman and Levine, 1965).
Generation of drc3 pf14 double mutant
To produce drc3 pf14 double mutants, drc3 was crossed to pf14. Progeny were randomly picked and grown in 96-well plates. To screen for the drc3-1 allele, cultures were analyzed by PCR using the Phire Plant Direct PCR Kit (Life Technologies, Carlsbad, CA) with a set of primers specific to gene 531300, which is a close neighbor of DRC3 and was deleted by the same deletion that made drc3-1 (Awata et al., 2014). The point mutation in pf14 was screened with the Phire Plant Direct PCR Kit using a set of primers specific for RSP3, followed by digestion of the amplified DNA with MseI. To select strains expressing NPHP4, cultures were analyzed by indirect immunofluorescence microscopy with anti-NPHP4. Genotypes of drc3 pf14 strains were confirmed by real-time-PCR with sets of primer pairs specific to DRC3 and NPHP4 (DRC3Ex1F and DRC3Ex2R, NPHP4-F1 and -R1, and NPHP4-F2 and -R2; primers are listed in Supplemental Table S2).
Plasmid construction
pDRC3-BglII.
A plasmid containing full-length DRC3 (pDRC3; Awata et al., 2014) was modified by PCR-based mutagenesis (Ko and Ma, 2005) to eliminate the stop codon and create a BglII site after the altered codon as follows: ∼990 and 900 base pairs around the stop codon of DRC3 were amplified by PCR with primer sets DRC3PFL-DRC3stopSap and DRC3BglIISap-DRC3EcoNIDN, respectively, using Phusion High-Fidelity DNA polymerase (New England BioLabs, Ipswich, MA). These PCR products were digested with SapI and fused together with T4 ligase. The resultant 1890–base pair fragment was digested with BsiWI and cloned into pDRC3 at the BsiWI sites to create plasmid pDRC3-BglII.
pDRC3-SNAP.
A cassette of cDNA encoding SNAP (codon-optimized for C. reinhardtii; kind gift of Winfield Sale [Emory University, Atlanta, GA] and Maureen Wirschell [University of Mississippi Medical Center, Jackson, MI]) in pUC57 was excised at the BstYI sites and inserted into pDRC3-BglII at the BglII sites to create plasmid pDRC3-SNAP encoding a fusion protein in which the SNAP-tag is linked to the last amino acid of DRC3 by the sequence GDPN.
pDRC3-HAC.
To create plasmid pDRC3-HAC encoding DRC3 with a 3xHA tag on its C-terminus, the SmaI-NaeI fragment of p3xHA (Silflow et al., 2001) was cloned into pDRC3-BglII at the BglII site modified to have blunt ends.
Finally, the selectable marker gene aph7” (Berthold et al., 2002) was cloned into pDRC3, pDRC3-SNAP, and pDRC3-HA at the HindIII site. Sequences of all PCR products and regions around the restriction enzyme sites used for subcloning were confirmed. Primers are listed in Supplemental Table S2.
Transformation
The drc3 mutant was generated by transforming BC2 progeny of B1179 with NPHP4 and screened by immunofluorescence microscopy with anti-NPHP4, as previously described (Awata et al., 2014).
To generate DRC3-R, DRC3-SNAP, and DRC3-HA cells, the drc3 mutant was transformed (Pollock et al., 2004) as follows: About 2 × 108 drc3 cells were collected and resuspended in 10 ml of TAPS (TAP medium containing 40 mM sucrose). Cells were concentrated in 0.5 ml of the TAPS solution. An 85-μl aliquot of the cells was mixed with 1 μg of linearized pDRC3, pDRC3-SNAP, or pDRC3-HA fused to the marker gene aph7′′ and placed into a 1.0-mm-gap cuvette. After the cuvette was incubated on ice for 5 min, a pulse was applied with a BTX ECM 600 electroporator (Harvard Apparatus, Holliston, MA) at 200 V, 13 Ω, and 1000 μF. The cuvette was left on ice for 15 min, and the cells were then diluted with 10 ml of TAPS and incubated overnight under dim light. The next day, cells were plated on TAP plates containing hygromycin B. Colonies on the plates were randomly picked and screened by immunofluorescence microscopy with anti-DRC3, anti-SNAP, or anti–HA peptide.
Antibodies
Rabbit antibodies to C. reinhardtii DRC3 were raised against a part of the protein fused to maltose-binding protein (MBP). cDNA encoding DRC3 amino acids 206–383 was amplified by PCR with template cDNA (Pazour et al., 2005) and primers DRC3cDNA-5′ and -3′ linked to BamHI and HindIII sites, respectively. The amplified DNA fragment was cut with those enzymes and put into pMAL-cRI (New England BioLabs). The fusion proteins expressed in Escherichia coli BL21 were purified with amylose resin and injected into New Zealand White rabbits to produce polyclonal antibodies (Covance, Dedham, MA). The DRC3 antibodies were purified by affinity chromatography with the antigen attached to agarose resin. Antibodies bound to the affinity column were eluted and applied to an agarose column containing MBP to remove anti-MBP antibodies. The flowthrough fraction of the column was used in this study. Other antibodies used were anti-DRC4, anti-DRC8, and anti-DRC11 (Bower et al., 2013), anti-DRC7/FAP50 (Yang et al., 2011), anti-IC2 (King et al., 1985), anti–HA peptide (3F10; Roche, Basal, Switzerland), anti-NPHP4 (Awata et al., 2014), and anti–acetylated tubulin (Sigma-Aldrich, St. Louis, MO).
Measurement of flagella length
Wild-type and drc3 cells were grown in M medium to ∼105 cells/ml. Cells were fixed with an equal volume of M medium containing 2% glutaraldehyde and observed with an inverted microscope (ECLIPSE Ti U; Nikon, Tokyo, Japan) equipped with a differential interference contrast (DIC) polarizer (T-P2; Nikon), DIC analyzer (T-A2; Nikon), a numerical aperture (NA) 1.4 oil condenser, and a 60×/1.49 NA objective. DIC images through a 1.5× relay lens were captured with a Clara interline camera (Andor, Belfast, United Kingdom) and NIS-Elements BR software (Nikon). Flagella lengths were measured with the Segmented Line tool in ImageJ (National Institutes of Health, Bethesda, MD).
Motility analysis
To analyze swimming speeds and paths, aliquots of cell cultures were transferred to a plastic chamber (0.127 mm deep Fisherbrand UriSystem DeciSlide; Thermo Fisher Scientific, Waltham, MA). Movies were taken at 30 images/s with a digital charge-coupled device (CCD) camera (UP-610; UNIQ Vision, Santa Clara, CA) attached to an inverted microscope (Carl Zeiss) equipped with a 16×/0.35 NA Plan objective. Video Savant 3.0 software (IO industries, London, ON, Canada) was used to take the movies. Swimming speeds and paths were determined by ImageJ with Manual Tracking plug-in.
To record motility of drc3 pf14 cells and control strains, cells were transferred to DeciSlide chambers. Images with 1.5-s exposures were obtained at room temperature with an AxioCam MRm camera (Carl Zeiss, Jena, Germany) attached to an Axioskop2 plus microscope (Carl Zeiss) equipped with a 20×/0.50 NA Plan-Neofluar objective. AxioVision 3.1 software (Carl Zeiss) was used to collect images. Gamma adjustments were done with Photoshop CS2 (Adobe, San Jose, CA).
Flagellar beat frequency analysis
Cells in M medium were transferred to DeciSlide chambers and beat frequencies analyzed by the FFT method as described previously (Kamiya, 2000; Wakabayashi and King, 2006).
Flagellar waveform analysis
Wild-type, drc3, and DRC3-R cells were grown in M medium to 104–105 cells/ml. To make a thin, narrow chamber for observation of swimming cells, the surface of a 45 × 50 mm No. 1 coverslip was tightly covered with plastic wrap. A strip of 2-mm width was then excised from the center of the plastic wrap, and the remaining plastic wrap then firmly covered with a 22 × 22 mm No. 1 coverslip to complete the chamber. Cells were placed in the chamber and observed with DIC optics and a 60× oil immersion objective (PlanApo N 1.42 NA; Olympus, Tokyo, Japan) mounted on a motorized inverted microscope (Olympus IX81). To reduce behavioral responses to the illuminating beam, cells were observed and recorded with light passed through a red filter glass (#2408; Kopp Glass, Pittsburgh, PA). Cells were recorded at 200 images/s with a high-speed, progressive scan CCD camera (TM-6740CL; JAI, Yokohama, Japan) and Video Savant software (IO Industries) as previously described (Zhang and Sanderson, 2003; Delmotte and Sanderson, 2006). Serial images of swimming cells were extracted with ImageJ, and the flagellar configurations in individual images were duplicated with the pencil tool in Illustrator CS2 (Adobe).
Immunoprecipitations
Flagella were isolated with dibucaine as previously described (Witman, 1986). To prepare axonemes, flagella in HMDEK buffer (30 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.4, 5 mM MgSO4, 1 mM dithiothreitol [DTT], 1 mM ethylene glycol tetraacetic acid [EGTA], and 25 mM KCl) were demembranated with a final concentration of 1% NP-40 on ice for 10 min and centrifuged (26,890 × g for 10 min at 4°C). The pellet containing axonemes was resuspended in HMDEK buffer and the suspension centrifuged again. For immunoprecipitation with the anti–HA peptide antibody, the pellet was resuspended with buffer containing 30 mM HEPES, pH 7.4, 5 mM MgSO4, 1 mM DTT, 1 mM EGTA, and 0.3 M KI and incubated on ice for 15 min. The suspension was then centrifuged (26,890 × g for 10 min at 4°C) and the supernatant collected. Anti–HA Affinity Matrix (Roche) was added, and the mixture was incubated with rotation for 30 min at 4°C. For immunoprecipitation with anti-DRC3, the axonemal pellet was resuspended with buffer containing 30 mM HEPES, pH 7.4, 5 mM MgSO4, 1 mM DTT, 1 mM EGTA, and 0.4 M KI and incubated on ice for 25 min. The suspension was then centrifuged as described, and the supernatant was collected and diluted with an equal volume of HMDEK buffer lacking KI. The diluted solution was then mixed with anti-DRC3 bound to Dynabeads Protein G (Life Technologies) and incubated with rotation for 1 h at 4°C. The anti–HA Affinity Matrix or Dynabeads were next washed three times with buffer containing 30 mM HEPES, pH 7.4, 5 mM MgSO4, 1 mM DTT, 1 mM EGTA, and 0.3 or 0.2 M KI, respectively, and the proteins bound to anti-HA or anti-DRC3 were then eluted with 2.5× SDS-sample solution (0.15 M Tris-HCl, pH 6.8, 2.5 mM EDTA, 5% SDS, 12.5% sucrose, and 0.005% bromophenol blue). The samples were analyzed by SDS–PAGE and Western blotting. Protein bands in SDS–acrylamide gels were visualized with silver stain (Silver Stain Plus; Bio-Rad, Hercules, CA).
Western blotting
For Western blotting of isolated flagella, samples were prepared by mixing a suspension of isolated flagella with either an equal or one-fourth volume of 5× SDS-sample solution (0.3 M Tris-HCl, pH 6.8, 5 mM EDTA, 10% SDS, 25% sucrose, and 0.01% bromophenol blue), and DTT was added to a final concentration of 50 mM. The mixture was incubated at 75°C for 20 min and the proteins then separated by SDS–PAGE and transferred onto Immobilon-FL (EMD Millipore, Billerica, MA). The membrane was blocked with 5% skimmed milk in Tris-buffered saline containing 0.05% Tween (TBST) and then incubated with primary antibodies diluted with 0.3% bovine serum albumin (BSA) in TBST. After four TBST washes, the membrane was then incubated with secondary antibodies. The following secondary antibodies were used: for DRC3 and 11, anti-rabbit immunoglobulin G (IgG) conjugated with Dylight 649 (Life Technologies); for IC2, anti-mouse IgG conjugated with Dylight 549 (Life Technologies); for the HA peptide and DRC8, anti-rat and anti-rabbit IgG conjugated with horseradish peroxidase (HRP; Life Technologies), respectively. Both primary and secondary antibodies for DRC8 were diluted with HIKARI (Nacalai Tesque, Kyoto, Japan).
For Western blot analysis of immunoprecipitation experiments, proteins bound to anti-HA and anti-DRC3 were subjected to SDS–PAGE and transferred onto Immobilon-P and -FL (EMD Millipore), respectively. The Immobilon-P was blocked with 5% skimmed milk and 0.45% fish gelatin in TBST and was incubated with primary and secondary antibodies diluted with 0.3% BSA in TBST. The Immobilon-FL was treated as described for Western blotting of isolated flagella. Proteins on Immobilon-P and -FL were detected with anti-rabbit IgG conjugated with HRP and Dylight 649, respectively.
To prepare whole-cell samples, cells were collected by centrifugation, lysed with the 5× SDS-sample solution, and incubated at 75°C for 20 min in the presence of 50 mM DTT. Proteins were separated by SDS–PAGE and transferred onto Immobilon-FL; the membrane was then blocked with 5% skimmed milk and 0.45% fish gelatin in TBST. Both primary and secondary antibodies for DRC3 were diluted with HIKARI, and those for α-tubulin were in 0.3% BSA in TBST. Anti-rabbit IgG conjugated with HRP and anti-mouse IgG conjugated with Dylight 549 were used to detect DRC3 and α-tubulin, respectively.
After incubation with secondary antibodies, membranes were washed four times with TBST. SuperSignal West Dura Extended Duration Substrate (Life Technologies) was used as the substrate for HRP. The chemiluminescence or fluorophore signals from the secondary antibodies were captured with a FluorChem Q equipped with a CCD camera (ProteinSimple, San Jose, CA).
Mass spectrometry
To determine whether DRC9 and DRC10 are present in the drc3 axoneme, slices were excised from an SDS–polyacrylamide gel, cut into 1 × 1 mm pieces, and placed in 1.5-ml Eppendorf tubes with 1 ml of water for 30 min. The water was removed, and 50 μl of 250 mM ammonium bicarbonate was added. For reduction, 20 μl of a 45 mM solution of DTT was added, and the samples were incubated at 50°C for 30 min. The samples were cooled to room temperature, and then for alkylation 20 μl of a 100 mM iodoacetamide solution was added and allowed to react for 30 min. The gel slices were washed twice with 1-ml water aliquots. The water was removed, 1 ml of 50:50 (50 mM ammonium bicarbonate:acetonitrile) was placed in each tube, and samples were incubated at room temperature for 1 h. The solution was then removed, and 200 μl of acetonitrile was added to each tube, at which point the gels slices turned opaque white. The acetonitrile was removed, and gel slices were further dried in a SpeedVac (Savant, Thermo Scientific). Gel slices were rehydrated in 75 μl of 2 ng/μl trypsin (Sigma-Aldrich, St. Louis, MO) in 0.01% ProteaseMAX Surfactant (Promega, Madison, WI):50 mM ammonium bicarbonate. Additional bicarbonate buffer was added to ensure complete submersion of the gel slices. Samples were incubated at 37ºC for 21 h. The supernatant of each sample was then removed and placed in a separate 1.5-ml Eppendorf tube. Gel slices were further dehydrated with 100 μl of 80:20 (acetonitrile:1% formic acid). The extract was combined with the supernatants of each sample. The samples were then dried down in a SpeedVac. Samples were dissolved in 25 μl of 5% acetonitrile in 0.1% trifluoroacetic acid before injection for liquid chromatography/tandem MS.
A 4.0-μl aliquot was directly injected onto a custom-packed 2 cm × 100 μm C18 Magic 5-μm particle trap column. Peptides were then eluted and sprayed from a custom-packed emitter (75 μm × 25 cm C18 Magic 3-μm particle) with a linear gradient from 95% solvent A (0.1% formic acid in water) to 35% solvent B (0.1% formic acid in acetonitrile) in 60 min at a flow rate of 300 nl/min on a Waters Nano Acquity UPLC system. Data-dependent acquisitions were performed on a Q Exactive Mass Spectrometer (Thermo Scientific) according to an experiment in which full MS scans from 300 to 1750 m/z were acquired at a resolution of 70,000, followed by 10 MS/MS scans acquired under higher-energy collisional dissociation (HCD) fragmentation at a resolution of 17,500 with an isolation width of 1.6 Da. Raw data files were processed with Proteome Discoverer (version 1.4; Thermo Scientific) before searching with Mascot Server (version 2.5; Matrix Science, London, United Kingdom) against the C. reinhardtii index of the NCBInr database. Search parameters used were fully tryptic with two missed cleavages, parent mass tolerances of 10 ppm, and fragment mass tolerances of 0.05 Da. A fixed modification of carbamidomethyl cysteine and variable modifications of acetyl (protein N-terminus), pyroglutamic acid for N-terminal glutamine, and oxidation of methionine were considered. Search results were loaded into the Scaffold Viewer (Proteome Software, Portland, OR).
To identify proteins immunoprecipitated with anti-DRC3, protein bands of interest were excised from silver-stained gels and subjected to trypsin digestion as previously described (Spiess et al., 2011). MS to identify proteins in each band was performed as previously described (Awata et al., 2014).
2D gel electrophoresis
Axonemes were isolated from drc3 and wild-type cells by pH shock as previously described (Witman et al., 1972; Lin et al., 2011). The axoneme pellets were directly dissolved in 2D gel electrophoresis lysis buffer (7 M urea, 2 M thiourea, 4% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, 65 mM DTT, and 2% IPG buffer [pH 3–10 NL; GE Healthcare, Little Chalfont, United Kingdom]) and clarified by centrifugation at 46,000 × g for 1 h. We carried out 2D gel electrophoresis as previously described (Lin et al., 2011). Briefly, 70 μg of axoneme proteins was separated on a 13-cm immobilized pH 3–10 nonlinear gradient dry strip (GE Healthcare) for a total of 24 kV-h. Then the strip was subjected to two-step equilibration in equilibration buffer containing 6 M urea, 30% glycerol, 2% SDS, and 50 mM Tris-HCl (pH 8.8) with 1% DTT for the first step and 2.5% iodoacetamide for the second step. The second-dimension separation was performed on a 10% SDS–polyacrylamide gel (260 mm × 200 mm × 1 mm). After electrophoresis, gels were stained with silver nitrate (Yan et al., 2000). All samples were run at least in triplicate to confirm reproducibility.
Cryo-electron tomography
Cryosamples were prepared as previously described (Yamamoto et al., 2013). Briefly, axonemes were isolated using the same method as that used for 2D gel electrophoresis. Glow-discharged (30 s at −40 mA) copper holey carbon grids (200 mesh, 2-μm hole size; Quantifoil Micro Tools, Jena, Germany) were loaded in a home-made plunge freezer. A 3-μl amount of axoneme sample and 1 μl of 10-fold concentrated 10-nm colloidal gold solution (Sigma-Aldrich; used as fiducial markers during tomogram reconstruction) were added and mixed on the grid. After blotting for ∼2 s with a filter paper, the grid was immediately plunge-frozen in liquid ethane and then stored in liquid nitrogen until visualized by cryo-ET.
The cryosamples were visualized in a transmission electron microscope (Tecnai F30; FEI, Hillsboro, OR) operated at 300 keV. Tilt series of axonemes were collected in low-dose mode using SerialEM software (Mastronarde, 2005) as previously described (Yamamoto et al., 2013). Briefly, the sample was tilted stepwise from −65 to +65° in 1.5–2.5° angular increments and was imaged with −6 or −8 μm defocus using an energy filter (Gatan, Pleasanton, CA) operated in zero-loss mode with a slit width of 20 eV. The accumulative electron dose for each tilt series was restricted to ∼100 e/Å2. All images were recorded at a magnification of 13,500 with a 2k × 2k CCD camera (Gatan), resulting in a pixel size of ∼1 nm.
Image processing
The tilt series of axonemes were reconstructed into three-dimensional cryo–electron tomograms using the IMOD software package by fiducial marker alignment and weighted backprojection (Kremer et al., 1996). Only undistorted tomograms of each strain were used for further image processing. Subtomograms containing individual 96-nm repeats were extracted from the raw tomograms, and averaged using the software program PEET (Nicastro et al., 2006) to overcome the inherent low signal-to-noise ratio of cryo-tomograms. The UCSF Chimera software package (Pettersen et al., 2004) was used to display the averaged axonemal structures by isosurface rendering and to measure the volume size of the density defect in drc3 axonemes after normalizing the isosurface rendering threshold to the mass of the doublet microtubule (Heuser et al., 2009). The molecular mass of the defect in drc3 axonemes was estimated by assuming an average protein density of 1.43 g/cm3 (Quillin and Matthews, 2000).
Supplementary Material
Acknowledgments
Maureen Wirschell (University of Mississippi, Jackson, MS) and Winfield Sale (Emory University, Atlanta, GA) generously provided the pUC57 plasmid containing a Chlamydomonas codon-optimized sequence encoding SNAP. We thank Mary Porter (University of Minnesota, Minneapolis, MN) for providing antibodies to DRC4, DRC8, and DRC11 and Xiangyi Lu (Wayne State University, Detroit, MI) for providing antibodies to DRC7/FAP50. We thank Chen Xu for training and management of the electron microscopes in the Louise Mashal Gabbay Cellular Visualization Facility at Brandeis University (Waltham, MA). For MS, we are grateful to John Leszyk in the Proteomics and Mass Spectrometry Facility at the University of Massachusetts Medical School and to Bin Deng of the Vermont Genetics Network Proteomics Facility (Burlington, VT), which is supported by National Institutes of Health National Institute of General Medical Sciences Institutional Development Award (IDeA) P20 GM103449. This study also was supported by National Institutes of Health Grants R01 GM083122 (to D.N.), R01 GM051293 (to S.K.), HL103405 and HL119836 (to M.S.), and R37 GM030626 (to G.W.), by the March of Dimes Foundation (to D.N.), and by the Robert W. Booth Endowment at the University of Massachusetts Medical School (to G.W.).
Abbreviations used:
- cryo-ET
cryo–electron tomography
- DIC
differential interference contrast
- DRC
dynein regulatory complex
- FFT
fast Fourier transform
- MBP
maltose-binding protein
- MS
mass spectrometry
- N-DRC
nexin-dynein regulatory complex
- NPHP4
nephrocystin-4.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-01-0018) on June 10, 2015.
*These authors contributed equally to this work.
REFERENCES
- Austin-Tse C, Halbritter J, Zariwala MA, Gilberti RM, Gee HY, Hellman N, Pathak N, Liu Y, Panizzi JR, Patel-King RS, et al. Zebrafish ciliopathy screen plus human mutational analysis identifies C21orf59 and CCDC65 defects as causing primary ciliary dyskinesia. Am J Hum Genet. 2013;93:672–686. doi: 10.1016/j.ajhg.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awata J, Takada S, Standley C, Lechtreck KF, Bellvé KD, Pazour GJ, Fogarty KE, Witman GB. NPHP4 controls ciliary trafficking of membrane and large soluble proteins at the transition zone. J Cell Sci. 2014;127:4714–4727. doi: 10.1242/jcs.155275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayly PV, Lewis BL, Kemp PS, Pless RB, Dutcher SK. Efficient spatiotemporal analysis of the flagellar waveform of Chlamydomonas reinhardtii. Cytoskeleton (Hoboken) 2010;67:56–69. doi: 10.1002/cm.20424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthold P, Schmitt R, Mages W. An engineered Streptomyces hygroscopicus aph 7” gene mediates dominant resistance against hygromycin B in Chlamydomonas reinhardtii. Protist. 2002;153:401–412. doi: 10.1078/14344610260450136. [DOI] [PubMed] [Google Scholar]
- Bower R, Tritschler D, Vanderwaal K, Perrone CA, Mueller J, Fox L, Sale WS, Porter ME. The N-DRC forms a conserved biochemical complex that maintains outer doublet alignment and limits microtubule sliding in motile axonemes. Mol Biol Cell. 2013;24:1134–1152. doi: 10.1091/mbc.E12-11-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brokaw CJ, Kamiya R. Bending patterns of Chlamydomonas flagella: IV. mutants with defects in inner and outer dynein arms indicate differences in dynein arm function. Cell Motil Cytoskeleton. 1987;8:68–75. doi: 10.1002/cm.970080110. [DOI] [PubMed] [Google Scholar]
- Brokaw CJ, Luck DJ. Bending patterns of Chlamydomonas flagella: III. a radial spoke head deficient mutant and a central pair deficient mutant. Cell Motil. 1985;5:195–208. doi: 10.1002/cm.970050303. [DOI] [PubMed] [Google Scholar]
- Brokaw CJ, Luck DJ, Huang B. Analysis of the movement of Chlamydomonas flagella: the function of the radial-spoke system is revealed by comparison of wild-type and mutant flagella. J Cell Biol. 1982;92:722–732. doi: 10.1083/jcb.92.3.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JM, Witman GB. Cilia and diseases. Bioscience. 2014;64:1126–1137. [Google Scholar]
- Delmotte P, Sanderson MJ. Ciliary beat frequency is maintained at a maximal rate in the small airways of mouse lung slices. Am J Respir Cell Mol Biol. 2006;35:110–117. doi: 10.1165/rcmb.2005-0417OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- Gardner LC, O'Toole E, Perrone CA, Giddings T, Porter ME. Components of a “dynein regulatory complex” are located at the junction between the radial spokes and the dynein arms in Chlamydomonas flagella. J Cell Biol. 1994;127:1311–1325. doi: 10.1083/jcb.127.5.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman DS, Levine RP. Cytochrome f and plastocyanin: their sequence in the photosynthetic electron transport chain of Chlamydomonas reinhardi. Proc Natl Acad Sci USA. 1965;54:1665–1669. doi: 10.1073/pnas.54.6.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashibe K, Shingyoji C, Kamiya R. Induction of temporary beating in paralyzed flagella of Chlamydomonas mutants by application of external force. Cell Motil Cytoskeleton. 1997;37:232–239. doi: 10.1002/(SICI)1097-0169(1997)37:3<232::AID-CM5>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Heuser T, Dymek EE, Lin J, Smith EF, Nicastro D. The CSC connects three major axonemal complexes involved in dynein regulation. Mol Biol Cell. 2012;23:3143–3155. doi: 10.1091/mbc.E12-05-0357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser T, Raytchev M, Krell J, Porter ME, Nicastro D. The dynein regulatory complex is the nexin link and a major regulatory node in cilia and flagella. J Cell Biol. 2009;187:921–933. doi: 10.1083/jcb.200908067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horani A, Brody SL, Ferkol TW, Shoseyov D, Wasserman MG, Ta-shma A, Wilson KS, Bayly PV, Amirav I, Cohen-Cymberknoh M, et al. CCDC65 mutation causes primary ciliary dyskinesia with normal ultrastructure and hyperkinetic cilia. PLoS One. 2013;8:e72299. doi: 10.1371/journal.pone.0072299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Ramanis Z, Luck DJ. Suppressor mutations in Chlamydomonas reveal a regulatory mechanism for flagellar function. Cell. 1982;28:115–124. doi: 10.1016/0092-8674(82)90381-6. [DOI] [PubMed] [Google Scholar]
- Kamiya R. Analysis of cell vibration for assessing axonemal motility in Chlamydomonas. Methods. 2000;22:383–387. doi: 10.1006/meth.2000.1090. [DOI] [PubMed] [Google Scholar]
- Keppler A, Pick H, Arrivoli C, Vogel H, Johnsson K. Labeling of fusion proteins with synthetic fluorophores in live cells. Proc Natl Acad Sci USA. 2004;101:9955–9959. doi: 10.1073/pnas.0401923101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King SM, Otter T, Witman GB. Characterization of monoclonal antibodies against Chlamydomonas flagellar dyneins by high-resolution protein blotting. Proc Natl Acad Sci USA. 1985;82:4717–4721. doi: 10.1073/pnas.82.14.4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko JK, Ma J. A rapid and efficient PCR-based mutagenesis method applicable to cell physiology study. Am J Physiol Cell Physiol. 2005;288:C1273–C1278. doi: 10.1152/ajpcell.00517.2004. [DOI] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Kubo T, Yagi T, Kamiya R. Tubulin polyglutamylation regulates flagellar motility by controlling a specific inner-arm dynein that interacts with the dynein regulatory complex. Cytoskeleton (Hoboken) 2012;69:1059–1068. doi: 10.1002/cm.21075. [DOI] [PubMed] [Google Scholar]
- Lin J, Okada K, Raytchev M, Smith MC, Nicastro D. Structural mechanism of the dynein power stroke. Nat Cell Biol. 2014;16:479–485. doi: 10.1038/ncb2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Tritschler D, Song K, Barber CF, Cobb JS, Porter ME, Nicastro D. Building blocks of the nexin-dynein regulatory complex in Chlamydomonas flagella. J Biol Chem. 2011;286:29175–29191. doi: 10.1074/jbc.M111.241760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindemann CB, Lesich KA. Flagellar and ciliary beating: the proven and the possible. J Cell Sci. 2010;123:519–528. doi: 10.1242/jcs.051326. [DOI] [PubMed] [Google Scholar]
- Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Nicastro D, Schwartz C, Pierson J, Gaudette R, Porter ME, McIntosh JR. The molecular architecture of axonemes revealed by cryoelectron tomography. Science. 2006;313:944–948. doi: 10.1126/science.1128618. [DOI] [PubMed] [Google Scholar]
- Oda T, Yagi T, Yanagisawa H, Kikkawa M. Identification of the outer-inner dynein linker as a hub controller for axonemal dynein activities. Curr Biol. 2013;23:656–664. doi: 10.1016/j.cub.2013.03.028. [DOI] [PubMed] [Google Scholar]
- Oda T, Yanagisawa H, Kikkawa M. Detailed structural and biochemical characterization of the nexin-dynein regulatory complex. Mol Biol Cell. 2015;26:294–304. doi: 10.1091/mbc.E14-09-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda T, Yanagisawa H, Yagi T, Kikkawa M. Mechanosignaling between central apparatus and radial spokes controls axonemal dynein activity. J Cell Biol. 2014;204:807–819. doi: 10.1083/jcb.201312014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Agrin N, Leszyk J, Witman GB. Proteomic analysis of a eukaryotic cilium. J Cell Biol. 2005;170:103–113. doi: 10.1083/jcb.200504008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Piperno G, Mead K, LeDizet M, Moscatelli A. Mutations in the “dynein regulatory complex” alter the ATP-insensitive binding sites for inner arm dyneins in Chlamydomonas axonemes. J Cell Biol. 1994;125:1109–1117. doi: 10.1083/jcb.125.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piperno G, Mead K, Shestak W. The inner dynein arms I2 interact with a “dynein regulatory complex” in Chlamydomonas flagella. J Cell Biol. 1992;118:1455–1463. doi: 10.1083/jcb.118.6.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock SV, Prout DL, Godfrey AC, Lemaire SD, Moroney JV. The Chlamydomonas reinhardtii proteins Ccp1 and Ccp2 are required for long-term growth, but are not necessary for efficient photosynthesis, in a low-CO2 environment. Plant Mol Biol. 2004;56:125–132. doi: 10.1007/s11103-004-2650-4. [DOI] [PubMed] [Google Scholar]
- Porter ME, Knott JA, Gardner LC, Mitchell DR, Dutcher SK. Mutations in the SUP-PF-1 locus of Chlamydomonas reinhardtii identify a regulatory domain in the beta-dynein heavy chain. J Cell Biol. 1994;126:1495–1507. doi: 10.1083/jcb.126.6.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter ME, Sale WS. The 9 + 2 axoneme anchors multiple inner arm dyneins and a network of kinases and phosphatases that control motility. J Cell Biol. 2000;151:F37–F42. doi: 10.1083/jcb.151.5.f37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quillin ML, Matthews BW. Accurate calculation of the density of proteins. Acta Crystallogr D Biol Crystallogr. 2000;56:791–794. doi: 10.1107/s090744490000679x. [DOI] [PubMed] [Google Scholar]
- Rupp G, O'Toole E, Gardner LC, Mitchell BF, Porter ME. The sup-pf-2 mutations of Chlamydomonas alter the activity of the outer dynein arms by modification of the gamma-dynein heavy chain. J Cell Biol. 1996;135:1853–1865. doi: 10.1083/jcb.135.6.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp G, Porter ME. A subunit of the dynein regulatory complex in Chlamydomonas is a homologue of a growth arrest-specific gene product. J Cell Biol. 2003;162:47–57. doi: 10.1083/jcb.200303019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sager R, Granick S. Nutritional studies with Chlamydomonas reinhardi. Ann NY Acad Sci. 1953;56:831–838. doi: 10.1111/j.1749-6632.1953.tb30261.x. [DOI] [PubMed] [Google Scholar]
- Silflow CD, LaVoie M, Tam LW, Tousey S, Sanders M, Wu W, Borodovsky M, Lefebvre PA. The Vfl1 Protein in Chlamydomonas localizes in a rotationally asymmetric pattern at the distal ends of the basal bodies. J Cell Biol. 2001;153:63–74. doi: 10.1083/jcb.153.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EF, Yang P. The radial spokes and central apparatus: mechano-chemical transducers that regulate flagellar motility. Cell Motil Cytoskeleton. 2004;57:8–17. doi: 10.1002/cm.10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song K, Awata J, Tritschler D, Bower R, Witman GB, Porter ME, Nicastro D. In situ localization of N and C termini of subunits of the flagellar nexin-dynein regulatory complex (N-DRC) using SNAP tag and cryo-electron tomography. J Biol Chem. 2015;290:5341–5353. doi: 10.1074/jbc.M114.626556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiess PC, Deng B, Hondal RJ, Matthews DE, van der Vliet A. Proteomic profiling of acrolein adducts in human lung epithelial cells. J Proteomics. 2011;74:2380–2394. doi: 10.1016/j.jprot.2011.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderWaal KE, Yamamoto R, Wakabayashi K, Fox L, Kamiya R, Dutcher SK, Bayly PV, Sale WS, Porter ME. bop5 mutations reveal new roles for the IC138 phosphoprotein in the regulation of flagellar motility and asymmetric waveforms. Mol Biol Cell. 2011;22:2862–2874. doi: 10.1091/mbc.E11-03-0270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi K, King SM. Modulation of Chlamydomonas reinhardtii flagellar motility by redox poise. J Cell Biol. 2006;173:743–754. doi: 10.1083/jcb.200603019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner F D. Organization of interdoublet links in Tetrahymena cilia. Cell Motil. 1983;3:321–332. [Google Scholar]
- Wirschell M, Olbrich H, Werner C, Tritschler D, Bower R, Sale WS, Loges NT, Pennekamp P, Lindberg S, Stenram U, et al. The nexin-dynein regulatory complex subunit DRC1 is essential for motile cilia function in algae and humans. Nat Genet. 2013;45:262–268. doi: 10.1038/ng.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witman GB. Isolation of Chlamydomonas flagella and flagellar axonemes. Methods Enzymol. 1986;134:280–290. doi: 10.1016/0076-6879(86)34096-5. [DOI] [PubMed] [Google Scholar]
- Witman GB, Carlson K, Berliner J, Rosenbaum JL. Chlamydomonas flagella. I. Isolation and electrophoretic analysis of microtubules, matrix, membranes, and mastigonemes. J Cell Biol. 1972;54:507–539. doi: 10.1083/jcb.54.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witman GB, Plummer J, Sander G. Chlamydomonas flagellar mutants lacking radial spokes and central tubules. Structure, composition, and function of specific axonemal components. J Cell Biol. 1978;76:729–747. doi: 10.1083/jcb.76.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto R, Song K, Yanagisawa HA, Fox L, Yagi T, Wirschell M, Hirono M, Kamiya R, Nicastro D, Sale WS. The MIA complex is a conserved and novel dynein regulator essential for normal ciliary motility. J Cell Biol. 2013;201:263–278. doi: 10.1083/jcb.201211048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan JX, Wait R, Berkelman T, Harry RA, Westbrook JA, Wheeler CH, Dunn MJ. A modified silver staining protocol for visualization of proteins compatible with matrix-assisted laser desorption/ionization and electrospray ionization-mass spectrometry. Electrophoresis. 2000;21:3666–3672. doi: 10.1002/1522-2683(200011)21:17<3666::AID-ELPS3666>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Yang Y, Cochran DA, Gargano MD, King I, Samhat NK, Burger BP, Sabourin KR, Hou Y, Awata J, Parry DA, et al. Regulation of flagellar motility by the conserved flagellar protein CG34110/Ccdc135/FAP50. Mol Biol Cell. 2011;22:976–987. doi: 10.1091/mbc.E10-04-0331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Sanderson MJ. Oscillations in ciliary beat frequency and intracellular calcium concentration in rabbit tracheal epithelial cells induced by ATP. J Physiol. 2003;546:733–749. doi: 10.1113/jphysiol.2002.028704. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.