

Background: Pygo2 is a chromatin effector of the Wnt signaling pathway that regulates cell growth and differentiation.
Results: Pygo2 is degraded through the ubiquitin/proteasome pathway and is stabilized by Akt phosphorylation at its serine 48.
Conclusion: Stabilizing phosphorylation by Akt regulates Pygo2 protein expression.
Significance: Pygo2 is a common nodule downstream of Wnt and Akt pathways.
Keywords: E3 ubiquitin ligase, phosphorylation, ubiquitin ligase, ubiquitylation (ubiquitination), Wnt signaling, Akt, Cul4, DDB1, Pygo2
Abstract
Pygopus 2 (Pygo2/PYGO2) is an evolutionarily conserved coactivator and chromatin effector in the Wnt/β-catenin signaling pathway that regulates cell growth and differentiation in various normal and malignant tissues. Although PYGO2 is highly overexpressed in a number of human cancers, the molecular mechanism underlying its deregulation is largely unknown. Here we report that Pygo2 protein is degraded through the ubiquitin/proteasome pathway and is posttranslationally stabilized through phosphorylation by activated phosphatidylinositol 3-kinase/Akt signaling. Specifically, Pygo2 is stabilized upon inhibition of the proteasome, and its intracellular level is regulated by Cullin 4 (Cul4) and DNA damage-binding protein 1 (DDB1), components of the Cul4-DDB1 E3 ubiquitin ligase complex. Furthermore, Pygo2 is phosphorylated at multiple residues, and Akt-mediated phosphorylation at serine 48 leads to its decreased ubiquitylation and increased stability. Finally, we provide evidence that Akt and its upstream growth factors act in parallel with Wnt to stabilize Pygo2. Taken together, our findings highlight chromatin regulator Pygo2 as a common node downstream of oncogenic Wnt and Akt signaling pathways and underscore posttranslational modification, particularly phosphorylation and ubiquitylation, as a significant mode of regulation of Pygo2 protein expression.
Introduction
Wnt signaling is fundamentally important for both normal and malignant development (1). In the canonical pathway, binding of Wnt ligands to membrane receptor/co-receptor triggers intracellular events culminating in the inactivation of an axin-APC-GSK3 destruction complex, which normally directs β-catenin for phosphorylation and degradation (1, 2). β-Catenin is thus stabilized, translocates to the nucleus, binds to TCF/LEF transcription factors, and recruits chromatin-modifying/remodeling complexes to transcribe Wnt target genes (3). Among the myriad β-catenin/TCF-interacting nuclear factors are Pygopus proteins, the prototype of which was identified in Drosophila as a dedicated Wg/Wnt pathway coactivator (4–7). Subsequent studies have revealed that these proteins act as histone methylation readers by directly binding to lysine 4-methylated histone H3 (8–10) and participate in “writing” of the histone code by recruiting histone-modifying enzymes to the target chromatin (9, 11–14). Two mammalian Pygopus homologs exist, with the function of Pygopus 2 (Pygo2)4 being required for the development of multiple tissues in both Wnt-dependent and -independent manners (15–18). Furthermore, Pygo2 connects Wnt and Notch signaling to regulate the lineage differentiation of basal stem cells in the mammary gland (9, 19). Pygo2 is needed for efficient tumor initiation, and in its absence, MMTV-Wnt1 transgenic mice produce mammary tumors with a microacinar-like histopathology rather than the typical histopathology that resembles the aggressive, difficult to treat basal-like breast cancer subtype (20). Additionally, Pygo2 facilitates β-catenin-induced hair follicle stem cell activation and is required for skin overgrowth in K14-ΔN-β-catenin transgenic mice (21). Overexpression of PYGO2 has been reported for human breast, lung, colon, brain, cervical, and ovarian cancer cells (22–28), and PYGO2 resides in a chromosomal region that is frequently amplified in breast cancer (29, 30). These findings implicate the importance of controlling Pygo2/PYGO2 levels for proper Wnt signaling function in development, regeneration, and tumorigenesis.
Numerous studies suggest that Wnt signaling cross-talks with the phosphatidylinositol 3 kinase (PI3K)/Akt pathway, a major signaling pathway that is triggered by myriad growth factors, such as insulin and epidermal growth factor (EGF), and that modulates cellular growth, proliferation, metabolism, and survival (31–44). Dysregulation of the PI3K/Akt pathway occurs in many human cancers (45) as well as in metabolic diseases, such as type 2 diabetes (46). PI3K/Akt pathway activation is initiated at the plasma membrane, where phosphatidylinositol trisphosphate, generated by PI3K and degraded by phosphatase PTEN, recruits Akt to the membrane (47). Akt is activated through phosphorylation at threonine 308 and serine 473 by PDK1 and mTORC2, respectively (48–51), and proceeds to phosphorylate a wide variety of target proteins, including itself, to regulate diverse cellular processes (52, 53). Exemplifying the Wnt-Akt cross-talk, Akt phosphorylates GSK3β at serine 9 and inhibits its activity, leading to increased stabilization and nuclear translocation of β-catenin (44). Akt also directly phosphorylates β-catenin at serine 552, thereby increasing its cytoplasmic and nuclear accumulation (34).
In this work, we report findings showing that Akt phosphorylates Pygo2 at serine 48 to reduce its ubiquitylation and proteasome-dependent degradation. We identify Cul4-DDB1 E3 ligases as the candidate enzymatic complexes that are responsible for the ubiquitylation of Pygo2. Moreover, we provide evidence that the Akt regulation of Pygo2 occurs in the context of growth factor/PI3K signaling and show that the Pygo2-stabilizing effects of Wnt and Akt activation are additive. Taken together, our data uncover a new layer of mechanistic link between two key oncogenic signaling pathways, namely a convergence between Wnt signaling and the Akt pathway on chromatin regulator Pygo2.
Experimental Procedures
Cell Culture
HEK293 human kidney epithelial cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, 12100-046), supplemented with 10% fetal bovine serum (FBS) (Omega Scientific (Tarzana, CA), FB-02). HC11 mouse mammary epithelial cells were cultured in RPMI 1640 medium (Invitrogen, 31800-022) supplemented with 10% FBS. MCF10A-Er-Src cells were grown in DMEM/F-12 medium (Invitrogen, 11039), supplemented with 5% charcoal stripped horse serum (Invitrogen, 16050-122), 20 ng/ml EGF (Sigma, E5036), 10 μg/ml insulin (Sigma, I-1882), 0.5 μg/ml hydrocortisone (Sigma, H-0888), and 100 ng/ml cholera toxin (Sigma, C-8052). Src induction and cellular transformation were achieved by treatment of cells with 1 μm 4-OH-tamoxifen (TAM) (Sigma, H7904), typically for 36 h, as described (54). All cell lines were grown at 37 °C with 5% carbon dioxide. The Cul4A and Cul4B mutant mouse embryonic fibroblast cell lines were derived from the respective germ line knock-out mouse embryos and cultured in DMEM supplemented with 10% FBS (55, 56).
Antibodies, Recombinant Proteins, and Chemicals
Anti-Pygo2 rabbit polyclonal antibody was as described previously (9). Mouse monoclonal Pygo2 antibody was from Santa Cruz Biotechnology (Sc-390506). Mouse monoclonal anti-FLAG antibody (F1804) and anti-FLAG M2 affinity gel (A2220) were from Sigma. Antibodies against total Akt (catalog no. 4691), phospho-Akt (Ser-473) (catalog no. 4060), or phosphorylated Akt substrate (catalog no. 9614) were from Cell Signaling Technology (Boston, MA). Anti-DDB1 (GTX100130), anti-GFP (GTX113617), and anti-Myc (GTX10827) antibodies were from GeneTex (Irvine, CA). Glutathione S-transferase (GST)-tagged, truncated Pygo2 protein containing N-terminal amino acids 6–115 (GST-Pygo2(6–115)) was expressed in BL21(DE3)/pLysS strain and purified as described previously (9). GSK-3 fusion protein (catalog no. 9237) was from Cell Signaling Technology. λ-Protein phosphatase (P0753) was from New England Biolabs (Ipswich, MA). Insulin, EGF, cycloheximide (CHX), triciribine hydrate (TCN), and MG132 were from Sigma. Wnt3A-conditioned medium and control medium were generated from L1 cells following the ATCC (CRL-2647TM) instructions.
Plasmids and Primers
The HA-ubiquitin plasmid was purchased from Addgene (Cambridge, MA) (catalog no. 18712). pcDNA3.1-FLAG-Pygo2, pcDNA3.1-FLAG-Pygo2(6–115),and pGEX4.1-GST-Pygo2(6–115) constructs were generated as reported previously (9). GFP-DDB1 and Myc-Cul4A were from Dr. Zhen-Qiang Pan (Mount Sinai School of Medicine, New York). pUSE-Akt1(K179M) was from Millipore (Billerica, MA) (catalog no. 21-152). Constructs expressing FLAG-tagged Pygo2 mutant proteins were generated with site-directed PCR mutagenesis using DNA3.1-FLAG-Pygo2 as a template (except that the Pygo2 S40E construct was used as a PCR template for generating the Pygo2 triple E mutation) and the following primers: Pygo2 S40A, 5′-GCCGGTCTGCAAATGAAGGCGCCTGAAAAGAAGCGAAGA-3′ and 5′-TCTTCGCTTCTTTTCAGGCGCCTTCATTTGCAGACCGGC-3′; Pygo2 S40E, 5′-GGTCTGCAAATGAAGGAGCCTGAAAAGAAGCGA-3′ and 5′-TCGCTTCTTTTCAGGCTCCTTCATTTGCAGACC-3′; Pygo2 T50A, 5′-CGAAGAAAGTCCAATGCTCAGGGTCCTGCATAT-3′ and 5′-ATATGCAGGACCCTGAGCATTGGACTTTCTTCG-3′; Pygo2 S59E, 5′-GCATATTCACATCTGGAGGAGTTCGCCCCACCC-3′ and 5′-GGGTGGGGCGAACTCCTCCAGATGTGAATATGC-3′; Pygo2 S97A, 5′-CCTCCGTTCCTCGGCGCCCCGGTGCCCTTTGGA-3′ and 5′-TCCAAAGGGCACCGGGGCGCCGAGGAACGGAGG-3′; Pygo2 T120A, 5′-CCCCCAGGCTACGGCGCCGGAGGAGGAGGGGGT-3′ and 5′-ACCCCCTCCTCCTCCGGCGCCGTAGCCTGGGGG-3′; Pygo2 T301A, 5′-GGTCGAGGTGGGGGCGCCCCAGATGCCAACAGT-3′ and 5′-ACTGTTGGCATCTGGGGCGCCCCCACCTCGACC-3′; Pygo2 S48A/T50A, 5′-GAAGAGCCCTGAAAAGAAGCGAAGAAAGGCCAATGCGCAGGGTCCTGCATATTCACATCTG-3′ and 5′-CAGATGTGAATATGCAGGACCCTGCGCATTGGCCTTTCTTCGCTTCTTTTCAGGGCTCTTC-3′; Pygo2 S48E/T50E, 5′-GAAGAGCCCTGAAAAGAAGCGAAGAAAGGAGAATGAGCAGGGTCCTGCATATTCACATCTG-3′ and 5′-CAGATGTGAATATGCAGGACCCTGCTCATTCTCCTTTCTTCGCTTCTTTTCAGGGCTCTTC-3′; Pygo2 S48A, 5′-GAGCCCTGAAAAGAAGCGAAGAAAGGCCAATACTCAGGGTCCTGCATATTCAC-3′ and 5′-GTGAATATGCAGGACCCTGAGTATTGGCCTTTCTTCGCTTCTTTTCAGGGCTC-3′; Pygo2 S48E, 5′-GAGCCCTGAAAAGAAGCGAAGAAAGGAGAATACTCAGGGTCCTGCATATTCAC-3′ and 5′-GTGAATATGCAGGACCCTGAGTATTCTCCTTTCTTCGCTTCTTTTCAGGGCTC-3′.
Generation of a Stable HC11 Cell Line That Expresses FLAG-Pygo2 and Purification of FLAG-Pygo2
A lentiviral vector expressing FLAG-Pygo2 was co-transfected with packaging vectors pHR and pVSVG into 293T cells using Lipofectamine 2000 (Invitrogen, 11668019). Recombinant lentiviruses were harvested from the medium 48 h after transfection. HC11 cells at 50–70% confluence were infected with viral supernatants containing 10 μg/ml Polybrene for 24 h, after which fresh medium was added to the infected cells, which were subsequently selected with puromycin to establish the stable cell line expressing FLAG-Pygo2 protein.
FLAG-Pygo2 was affinity-purified from FLAG-Pygo2-expressing HC11 cells. Briefly, cells were grown to ∼90% confluence and lysed by radioimmune precipitation assay buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) with an anti-phosphatase mixture (50 mm NaF, 100 μm Na3VO4, 30 mm NaPPi) and a mixture of protease inhibitors (1 mm PMSF, 1 μg/ml aprotenin, 1 μg/ml leupeptin, 1 μg/ml pepstatin). The cleared lysate was incubated with anti-FLAG M2-agarose beads (Sigma, A2220) for 1 h at 4 °C. The beads were washed four times in cold lysis buffer and eluted with FLAG peptide (Sigma, F4799). The purified sample was then separated by a 10% SDS-polyacrylamide gel, and the protein bands were visualized by Coomassie Brilliant Blue staining to validate purity.
Protein Identification and Characterization by Liquid Chromatography and Tandem Mass Spectrometry (LC MS/MS)
Protein bands were cut for in-gel trypsin digestion as described previously (57). The resulting peptides were subjected to LC MS/MS analysis, which was carried out by nanoflow reverse phase HPLC (Eksigent, CA) coupled on-line to a Linear Ion Trap (LTQ)-Orbitrap XL mass spectrometer (Thermo Fisher Scientific) as described (58). The acquired MS/MS spectra were extracted and submitted to the development version (version 5.10.0) of Protein Prospector (University of California, San Francisco) for database searching. A concatenated database (22,268,936 sequence entries) composed of a normal UniProt database (2010.03.30) and a random form of the normal database was used for database searching. Mouse was selected as the restricted species. Trypsin was set as the enzyme with a maximum of two missed cleavage sites. The mass tolerance for parent ion was set at ±20 ppm, whereas ±0.8 Da tolerance was chosen for the fragment ions. Chemical modifications such as protein N-terminal acetylation, methionine oxidation, N-terminal pyroglutamine, and phosphorylation on serine and threonine were selected as variable modifications. The Search Compare program in Protein Prospector was used for summarization, validation, and comparison of results. The identified phosphorylated peptides have been further validated by manual inspection of their MS/MS spectra.
In Vitro Kinase Assay
In vitro assays for Akt kinase activity were performed in 50 μl of kinase buffer (25 mm Tris-HCl, 5 mm β-glycerophosphate, 2 mm DTT, 0.1 mm Na3VO4, 10 mm MgCl2) containing 10 mm ATP, 5 μg of protein substrates, and 500 ng of activated Akt1 (Millipore, 14-453). The reactions were incubated at 30 °C for 1 h, separated by 10% SDS-PAGE, and analyzed by immunoblotting using the phospho-Akt substrate antibody.
Pygo2 Ubiquitylation Assay
HEK293T cells were co-transfected with HA-ubiquitin and FLAG-Pygo2 constructs (or empty vector) with or without MG132 as indicated. Twenty-four hours after transfection, cells were incubated with MG132 for another 4 h before harvest. For FLAG-Pygo2 purification, cells were lysed under denaturing conditions (8 m urea, 200 mm NaCl, 100 mm Tris (pH 7.5), 0.2% SDS, 50 mm NaF, 100 μm Na3VO4, 30 mm NaPPi, and protease inhibitors (1 mm PMSF, 1 μg/ml aprotenin, 1 μg/ml leupeptin, 1 μg/ml pepstatin)), and lysates then diluted and incubated with the FLAG M2-agarose beads. Ubiquitylated Pygo2 was detected by Western blotting using the anti-HA antibody.
In Vitro GST Pull-down of Pygo2-interacting Proteins
Nuclear extract was prepared from 60 10-cm dishes of actively growing HEK293 cells as described previously (9). After the salt concentration was adjusted to 100 mm NaCl, the extract was divided into two equal aliquots and incubated with 10 μg of purified GST-Pygo2(6–115) or GST on beads. Following agitation at 4 °C for 2 h, the bound protein complex was extensively washed and eluted with 10 mm glutathione plus 0.1% Nonidet P-40. The samples were separated by SDS-PAGE and visualized by silver staining. Selected bands were excised, trypsinized, and subjected to LC MS/MS as described above.
Co-immunoprecipitation, Western Blotting, and Immunofluorescence
HEK293T cells were plated (5–6 × 106 cells/10-cm dish), and 20–24 h later, FLAG-Pygo2 plasmid was co-transfected with either GFP-DDB1, Myc-Cul4A, or pUSE-Akt1(K179M) using the calcium phosphate method (9). Cells were harvested 20–24 h posttransfection. The FLAG-Pygo2 protein was affinity-purified as described above, and bound proteins were analyzed by SDS-PAGE and immunoblotting.
For Western blotting, cells were lysed in Laemmli sample buffer (60 mm Tris-HCl, pH 8.0, 2% SDS, 4 mm EDTA, 5% β-mercaptoethanol, 10% glycerol), and lysates were analyzedas described (9). Immunofluorescence was performed as described previously (12).
Lentivirus-mediated shRNA Knockdown
Recombinant lentiviruses expressing DDB1 shRNAs were generated following the instructions provided by Addgene and as reported previously (9). DDB1 shRNAs were gifts from the laboratory of Dr. Anand Ganesan (University of California, Irvine). Clone identification numbers for these shRNAs are as follows: TRCN0000082853, TRCN0000082854, TRCN0000082855, TRCN0000082856, and TRCN0000082857.
Results
The Intracellular Protein Level of Pygo2/PYGO2 Is Regulated by Proteasome Activity and Cul4-DDB1 Ubiquitin E3 Ligase
In examining the expression of Pygo2 in mouse mammary tumors, we observed elevated Pygo2 protein levels in MMTV-Wnt1 mammary hyperplastic tissues, whereas alteration at the mRNA level was minimal (20). We also examined PYGO2 expression in an established human mammary epithelial cell (MEC) line, MCF10A-Er-Src, which carries ER-Src, a derivative of the Src kinase oncoprotein (v-Src) that is fused to the ligand-binding domain of the estrogen receptor (ER) (59). As expected, based on previous reports (59, 60), treatment of MCF10A-Er-Src cells with TAM induced transformation within 24–36 h, which was manifested by their phenotypic alterations and elevated Akt activity, as measured by phospho-Akt level (Fig. 1, A and B). Interestingly, PYGO2 protein level was increased 3.5-fold upon TAM treatment compared with that in untransformed cells (Fig. 1, B and C). In contrast, the levels of β-catenin protein and PYGO2 mRNA were unaffected (Fig. 1, B–D). These findings suggest that Pygo2/PYGO2 expression may be up-regulated at a posttranscriptional level in transformed MECs, and they point to a positive correlation between Akt activity and PYGO2 protein level. We will return to the latter notion later.
FIGURE 1.

Pygo2/PYGO2 protein is up-regulated upon transformation of MCF10A-Er-Src cells and is degraded in a proteasome-dependent manner. A, morphology of MCF10A-Er-Src cells with and without TAM induction. B, Western blot analysis of the indicated proteins in whole cell lysates. C and D, quantification from three independent experiments of PYGO2 and β-catenin protein levels (C) and PYGO2 mRNA level (D) in MCF10A-Er-Src cells with and without induction. In A–D, cells were treated with either TAM or ethanol control (−TAM) and analyzed 36 h later. β-actin protein and 18S rRNA levels were used for normalization in C and D, respectively. The normalized values from before induction were set as 1. Values are means ± S.E.; *, p < 0.01, Student's t test. E–G, effects of CHX and MG132 on exogenous Pygo2. HEK293T cells were transfected with FLAG-Pygo2 and pEGFP-C1; treated 24 h later with 100 μm CHX (E), 10 μm MG132 (F), or both (G); and harvested at the indicated time points for analysis. H and K, effects of CHX (H and J) or MG132 (I and K) on endogenous PYGO2 in MCF10A-Er-Src (H and I) or HEK293T (J and K) cells. MCF10A-Er-Src cells were treated with (or without) TAM for 36 h, followed by treatment with CHX (H) or MG132 (I) for the indicated periods of time. HEK293T cells were treated with CHX for the indicated periods of time (J) or with MG132 for 14 h (K). Pygo2/PYGO2 levels were determined by Western blotting using FLAG (E–G) or Pygo2 (H–K) antibody. The blot was reprobed with GFP or β-actin antibody as a loading control.
To explore how Pygo2 expression is modulated at the protein level, we analyzed the effects of CHX (an inhibitor of de novo protein synthesis) and MG132 (a proteasome inhibitor) on the level of FLAG-tagged Pygo2 protein (FLAG-Pygo2) in HEK293T cells. Blockage of new protein synthesis by CHX led to a gradual decrease in the level of FLAG-Pygo2 (estimated half-life is ∼4 h), whereas proteasome inhibition resulted in FLAG-Pygo2 accumulation (Fig. 1, E and F). In addition, co-treatment with CHX and MG132 canceled out their opposite effects, thus maintaining the cellular level of FLAG-Pygo2 (Fig. 1G). Similar results were obtained for endogenous PYGO2 protein in MCF10A-Er-Src and HEK293T cells (Fig. 1, H–K). Interestingly, the half-life of PYGO2 in MCF10A-Er-Src cells was increased from ∼4 h to ∼7 h upon TAM-induced transformation, and MG132-induced PYGO2 accumulation was more prominent in transformed cells compared with untransformed controls (Fig. 1, H and I). Together, these data suggest that intracellular Pygo2/PYGO2 is degraded in a proteasome-dependent manner.
Ubiquitylation earmarks proteins for proteasome-dependent degradation, and ubiquitin ligases catalyze such a tagging reaction (61). To identify the possible E3 ligase that mediates Pygo2 ubiquitylation, we performed an in vitro GST pull-down experiment coupled with mass spectrometric analysis to identify potential Pygo2-interacting proteins in HEK293T cells. GST-tagged N-terminal homology domain (GST-NHD) was used as a bait, and this analysis identified six unique peptides for DDB1 and two peptides for Vpr (HIV-1)-binding protein (VPRBP) (Fig. 2A), both of which are components of the Cul4-DDB1-VPRBP E3 ligase complex (62). Reciprocal co-immunoprecipitation experiments confirmed the physical association between FLAG-Pygo2 and DDB1 (Fig. 2, B and C). Interaction was also detected between endogenous Pygo2 and DDB1 proteins in HC11 cells, a mouse MEC line with stem/progenitor-like activities (68, 69) (Fig. 2D).
FIGURE 2.

Pygo2 interacts with DDB1. A, identification of DDB1 and VPRBP as candidate Pygo2-interacting proteins. A GST pull-down assay was performed as described under “Experimental Procedures.” Left, silver staining showing the banding patterns of GST versus GST-NHD complexes. Right, unique peptides identified for DDB1 and VPRBP. B–D, physical association between Pygo2 and DDB1 in HEK293T (B and C) or HC11 (D) cells. For B and C, HEK293T cells were transfected with FLAG-Pygo2 and GFP-DDB1 as indicated. Twenty-four hours later, whole cell lysates were immunoprecipitated (IP) using FLAG M2 beads (B) or anti-GFP antibody (C) and then immunoblotted using the indicated antibodies. D, whole cell lysates from HC11 cells were subjected to immunoprecipitation with anti-DDB1 antibody or IgG as indicated. The immunocomplexes were probed separately for Pygo2 and DDB1.
To further validate the interaction between Pygo2 and DDB1, we examined their co-localization with indirect immunofluorescence using HC11 cells stably expressing FLAG-Pygo2. Both FLAG-Pygo2 and DDB1 proteins showed diffuse nuclear staining excluding the DAPI-dense, heterochromatic territories, with staining signals significantly reduced when cells were pretreated with 0.5% Triton X-100 (Fig. 3A). Triton pre-extraction is expected to remove cell and nuclear membranes as well as most of the cytoplasm, leaving cytoskeleton and chromatin behind (63, 64). Interestingly, the addition of MG132 did not affect the Triton-soluble Pygo2 signals but elicited very prominent Triton-insoluble Pygo2 signals in discrete subnuclear clusters. DDB1 was also concentrated in such clusters in MG132/Triton-treated cells. Similarly, endogenous Pygo2 co-localizes with DDB1 either diffusely in the nuclei without treatment or in subnuclear clusters upon Triton extraction and proteasome inhibition (Fig. 3B).
FIGURE 3.
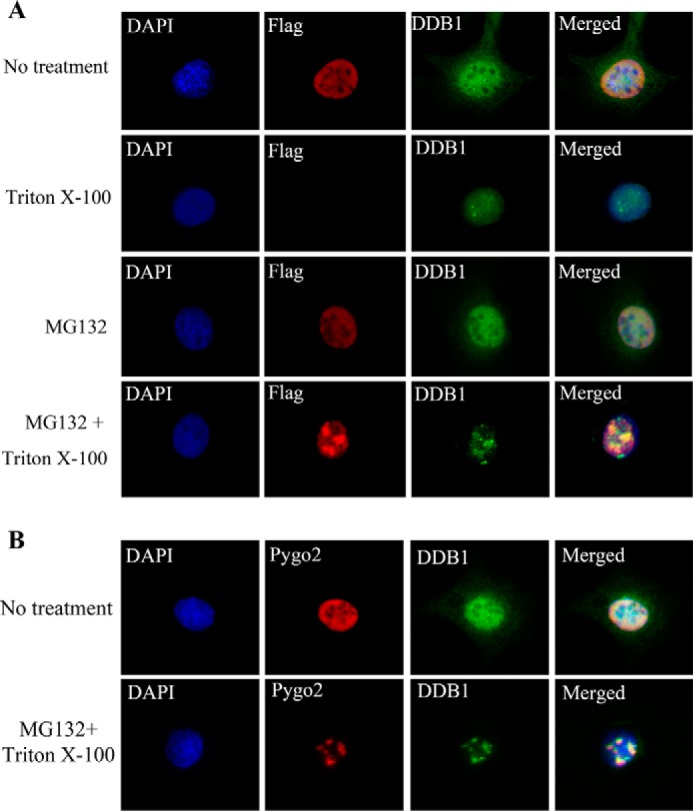
Pygo2 co-localizes with DDB1. Shown are results of indirect immunofluorescence of FLAG-Pygo2 (A) or endogenous Pygo2 (B) and DDB1 under different treatment conditions. A, HC11 cells that stably express FLAG-Pygo2 were incubated with or without MG132 for 4 h, pre-extracted with 0.5% Triton X-100, and immunostained using the indicated antibodies. B, parental HC11 cells were treated and immunostained as indicated. DAPI stains the nuclei.
To determine whether Pygo2 also associates with Cul4, we co-expressed FLAG-Pygo2 with Myc-Cul4A in HEK293T cells. Myc-Cul4A was found to co-purify with FLAG-Pygo2, and this interaction was weakened when DDB1 was depleted from the cells using a DDB1-specific shRNA (Fig. 4A). Therefore, Pygo2 interacts with Cul4 in a manner that requires DDB1, well known for its function as an adapter in Cul4-DDB1 E3 ligase complexes (65–67).
FIGURE 4.

Cul4-DDB1 E3 ligase complex mediates Pygo2/PYGO2 degradation. A, Pygo2 interacts with Cul4A in a DDB1-dependent manner. HEK293T cells were transfected with the indicated constructs with or without DDB1 shRNA, and immunoprecipitation was performed using anti-Myc antibody. B, effect of DDB1 knockdown on Pygo2 level. HEK293T cells that stably express FLAG-Pygo2 were transfected with the indicated shRNAs, and whole cell lysates were used for immunoblotting with the indicated antibodies. C, Cul4A overexpression decreases Pygo2 level. HEK293T cells were transfected with increasing amounts of a Myc-tagged Cul4A-expressing construct, and protein expression was examined 24 h later, as indicated. D, Pygo2 expression in Cul4A−/− or Cul4B−/Y mouse embryonic fibroblasts. Note that the Cul4B gene resides in the X chromosome. The upper bands of the doublets (indicated by brackets) represent neddylated forms. o in the Cul4B panels, nonspecific species. E, effects of Cul4A and Cul4B knockdown on PYGO2 level in BT474 cells. Cells were transfected with the indicated shRNAs, and whole cell lysates were used for immunoblotting with the indicated antibodies. F and G, stabilization of PYGO2 in BT474 cells upon silencing of Cul4B but not Cul4A. The half-lives of PYGO2 were determined in control or BT474 cell lines with stable Cul4A or Cul4B knockdown. Cells were treated with 100 μm CHX for the indicated time period. PYGO2 levels were determined by Western blotting (F), quantified by an Odyssey Infrared imaging system, and normalized to α-tubulin levels (G) at the indicated time points.
We next evaluated the impact of varying DDB1 and Cul4 levels on Pygo2 protein expression. Depletion of DDB1 using three different shRNAs resulted in increased levels of FLAG-Pygo2 in HEK293T cells, and the extent of this effect seemed to correlate with the degree of DDB1 knockdown (Fig. 4B). Two Cul4 paralogs, Cul4A and Cul4B, exist in mammals; they share 82% sequence identity, both interact with DDB1, and they have been shown to play redundant roles in some contexts (55, 56, 70, 71). Overexpression of Cul4A led to reduced FLAG-Pygo2 protein level in a dosage-dependent manner (Fig. 4C). Conversely, loss of Cul4A (compare Cul4a−/− with Cul4af/f) led to a minimal (1.2-fold), whereas loss of Cul4B (compare Cul4b−/Y with Cul4bf/Y) led to a significant, increase in the level of endogenous Pygo2 in mouse embryonic fibroblast cells (Fig. 4D). Moreover, siRNA depletion of Cul4B, but not Cul4A, in BT474 breast cancer cells led to a moderate accumulation of endogenous PYGO2 (Fig. 4E) and significantly extended PYGO2 half-life (Fig. 4, F and G). Taken together, our data suggest Cul4B-DDB1 as the predominant E3 ligase that directly or indirectly mediates Pygo2/PYGO2 degradation.
Pygo2 Is Phosphorylated at Multiple Residues, with Serine 48 Being Critical for Its Stability
Protein phosphorylation is often found to trigger ubiquitylation/proteasome-dependent degradation (72–74). Pygo2 has been identified as a phosphoprotein through a systematic phosphoproteome screen (75). On Western blots of tissue and cell lysates, Pygo2 often displayed as slowly migrating bands (13) (Fig. 5A), leading us to wonder whether such bands represent phosphorylated forms of Pygo2. To test this notion, we treated FLAG-Pygo2 protein purified from HC11 cells with λ-phosphatase, prior to SDS-PAGE and Western blot analyses. As a result of this treatment, the multiple Pygo2 bands collapsed to a single, faster migrating species, but the addition of phosphatase inhibitors restored the slowly migrating bands (Fig. 5A). These results demonstrate that Pygo2 is phosphorylated in MECs.
FIGURE 5.

Mapping phosphorylated residues in Pygo2 and mutational analysis of their importance in protein stability. A, Pygo2 is a phosphoprotein. FLAG-Pygo2 was purified by immunoprecipitation (IP) from HC11 cells that stably express it and then incubated with λ-phosphatase (PPase) or its inhibitors, as indicated. The bracket on the right marks the slowly migrating forms of Pygo2. B, top, schematic diagram of the domain structure of, and phosphorylated residues in, Pygo2. Bottom, the putative Akt phosphorylation consensus sequence in Pygo2 encompasses both Ser-48 and Thr-50 and is evolutionarily conserved. C and F, HEK293T cells were transfected with pEGFP-C1 and FLAG-Pygo2 or its mutant derivatives, and whole cell lysates were collected 24 h later for Western blot analysis. E, decreased interaction between Pygo2 triple E mutant and DDB1. HEK293T cells were transfected with the indicated constructs, immunoprecipitated using anti-DDB1 antibody, followed by immunoblotting with FLAG or DDB1 antibodies. Note that the Pygo2 triple A mutant displayed wild-type-level association with DDB1. D and G, quantitative analysis of three independent experiments as shown in C and F, respectively. The y axis indicates relative protein expression level after normalization by GFP, with the value for wild-type FLAG-Pygo2 expression being set as 1. Shown are mean ± S.E. (error bars). *, p < 0.01, Student's t test. H–J, CHX chase assays to measure the half-lives (J) of FLAG-Pygo2 and its mutant derivatives. HEK293T cells were transfected with pEGFP-C1 and FLAG-Pygo2 or the indicated mutants. Twenty-four hours after transfection, cells were treated with CHX, and samples were collected at the indicated time points for immunoblotting analysis. The level of remaining Pygo2 (normalized against GFP) at different time points was quantified as the percentage of the initial Pygo2 level (at time 0). Shown are mean ± S.E. from three independent experiments. *, p < 0.01, Student's t test.
To identify the phosphorylation site(s) on Pygo2, we performed LC MS/MS analysis on purified FLAG-Pygo2. Pygo2 was unambiguously identified with 46 unique peptides, resulting in 54% sequence coverage. Eight phosphorylated residues were found, five of which (Ser-40, Ser-48, Thr-50, Thr-59, and Ser-97) reside in the conserved NHD domain (Fig. 5B). In contrast, no phosphorylated residues were detected within the conserved plant homology domain (PHD) at the C terminus of Pygo2.
To assess whether phosphorylation affects Pygo2 protein stability, we performed site-directed mutagenesis to mutate the above Ser/Thr residues, singly or in combination, to alanine or glutamate. We first checked protein expression following transfection of the mutant constructs into HEK293T cells. Notably, simultaneous mutation of Ser-40, Ser-48, and Thr-50 residues to Glu (termed the triple E mutant) led to a slight but statistically significant increase in Pygo2 protein level compared with the wild type and other mutants (Fig. 5, C and D). Additionally, compared with wild-type Pygo2, the interaction between its triple E mutant and DDB1 was weakened (Fig. 5E). The S40E single mutant showed slightly decreased protein expression level compared with the wild type; however, this difference is not statistically significant (Fig. 5D). Moreover, S40A, T50A, and T50E did not show a consistent effect on Pygo2 protein level (Fig. 5, C, D, F, and G). In contrast, S48E resembled the triple E mutation in giving rise to higher, whereas S48A resulted in lower, Pygo2 protein level (Fig. 5, F and G).
An analysis of Pygo2 protein half-life revealed that both the triple E and S48E mutants displayed significantly enhanced stability (estimated half-lives: 6.2 and 6.1 h, respectively; p = 0.03 and 0.02, respectively, compared with wild type), whereas the S48A mutant displayed decreased stability (estimated half-life: ∼2.5 h; p = 0.02, compared with wild type) (Fig. 5, H–J). In contrast, the other mutations did not show any significant effect on Pygo2 half-life.
A previous study has reported that the N-terminal nuclear localization sequence (NLS) and the C-terminal PHD domain of Pygopus function redundantly to mediate its nuclear localization (76). Given that Ser-48 is within the NLS, we also tested whether its phosphorylation affects Pygo2 nuclear localization. First, we transfected several deletion constructs of Pygo2 (Fig. 6A) into cells and performed indirect immunofluorescence to determine the resulting protein's subcellular localization. Deletion of the C-terminal domain including PHD (ΔPHD) did not affect nuclear localization, whereas deletion of the N-terminal domain, including NHD (115-C), resulted in cytoplasmic localization of the protein (Fig. 6B). Conversely, the N-terminal domain alone (amino acids 1–115) was sufficient to confer nuclear localization. Thus, in our hands, it appears that only the N-terminal domain of Pygo2 contains essential nuclear localization determinants. Indeed, deletion of the NLS (ΔNLS) resulted in loss of nuclear localization. However, neither the triple A/E nor the S48A/E mutations affected Pygo2 nuclear localization (Fig. 6C), suggesting that Ser-48 phosphorylation does not interfere with the functionality of the Pygo2 NLS.
FIGURE 6.

Nuclear localization of Pygo2 deletion mutants. A, schematic diagram of the deletion mutants. B and C, indirect immunofluorescence showing the subcellular localization of FLAG-Pygo2 and its mutant derivatives in HEK293T cells. Cells were transfected with the indicated constructs, and staining was performed 12 h later using anti-FLAG antibody. DAPI stains the nuclei.
Akt Phosphorylates Pygo2 at Ser-48, and This Phosphorylation Inhibits Pygo2 Ubiquitylation
Sequence analysis revealed that both Ser-48 and Thr-50 residues are part of a highly conserved consensus substrate motif (RXRXX(S/T)) for Akt phosphorylation (Fig. 5B). Because Akt phosphorylates several key players in Wnt signaling, such as GSK3 and β-catenin, we sought to determine whether Akt can phosphorylate Pygo2 as well. An antibody that specifically recognizes the Akt consensus phosphorylation motif reacted with FLAG-Pygo2 purified from HC11 (Fig. 7A) or HEK293T (Fig. 7B) cells. The level of phosphorylated Pygo2 decreased after λ-phosphatase treatment, and this decrease was abolished when phosphatase inhibitors were present (Fig. 7C). The Akt phosphosubstrate antibody also reacted with a truncated Pygo2 protein containing only the N-terminal domain (amino acids 6–115) (Fig. 7D), indicating that Akt phosphorylation probably occurs within this region.
FIGURE 7.

Akt phosphorylates Pygo2. A and B, FLAG-Pygo2 is a phospho-Akt substrate. HC11 cells that stably express FLAG-Pygo2 (A) or HEK293T cells transiently transfected with the FLAG-Pygo2 construct (B) were immunoprecipitated (IP) by FLAG M2 beads, followed by immunoblotting with anti-phospho-Akt substrate antibody. C, λ-protein phosphatase (λ-PPase) treatment decreases the level of Akt-phosphorylated FLAG-Pygo2. Immunoprecipitated FLAG-Pygo2 from HC11 cells was treated with λ-protein phosphatase in the presence or absence of inhibitors and visualized by immunoblotting with anti-phospho-Akt substrate antibody. The blot was reprobed for Pygo2 to ensure an equal level of total Pygo2. D, the phospho-Akt substrate antibody recognizes the N-terminal domain of Pygo2. HEK293T cells were transfected with the FLAG-Pygo2(6–115) construct. Twenty-four hours later, whole cell lysates (WCL) were immunoprecipitated using FLAG M2 beads, followed by immunoblotting with the anti-phospho-Akt substrate antibody. E, Pygo2 interacts with Akt1(K179M) in HEK293T cells. F and G, Akt phosphorylates the N-terminal domain of Pygo2 in vitro (F) and in HEK293T cells (G). F, bacterially purified GST-Pygo2(6–115) was incubated with Akt1 as described under “Experimental Procedures” and probed with anti-phospho-Akt substrate antibody (top). The blot was reprobed for Pygo2 (note that the antibody was generated against a GST-Pygo2 fusion protein). G, HEK293T cells were transfected with the indicated plasmids in serum-free media for 24 h, followed by immunoprecipitation using FLAG M2 beads and an Akt kinase assay. Samples were immunoblotted with anti-phospho-Akt substrate (top) or anti-Pygo2 (bottom) antibody. H, mutation of Ser-48 but not Thr-50 abolishes Akt phosphorylation. HEK293T cells were transfected with the indicated constructs, and immunoprecipitation was performed 24 h later using FLAG M2 beads. Shown are results of immunoblotting of the immunoprecipitates (IP; top) or whole cell lysates (WCL; bottom) using the indicated antibodies.
To determine whether Pygo2 is a direct substrate of Akt, we first examined whether they interact with each other. Indeed, FLAG-Pygo2 co-immunoprecipitated with a kinase-dead form of Akt1 protein where the active site lysine 179 (Lys-179) was mutated to methionine (Fig. 7E), indicating a physical association between Pygo2 and Akt1 that does not require the enzymatic activity of Akt. Next, we performed in vitro kinase assay, where a bacterially expressed GST-Pygo2(6–115) fusion protein was purified and incubated with recombinant, active Akt1. Akt1 efficiently phosphorylated GST-Pygo2(6–115) and known substrate GSK3β but not the GST control (Fig. 7F). Further, FLAG-Pygo2(6–115) purified from serum-starved HEK293T cells was also phosphorylated by Akt1 in vitro (Fig. 7G).
We next sought to examine whether both Ser-48 and Thr-50 of Pygo2 are phosphorylated by Akt. To do this, we transfected into HEK293T cells constructs expressing FLAG-tagged, full-length Pygo2 or its Ser-48 and Thr-50 mutant derivatives. The FLAG-tagged proteins were purified and subjected to Western blot analysis using the Akt phosphosubstrate antibody. Mutating Ser-48 to either Ala or Glu, alone or in combination with mutating Thr-50 to Ala or Glu, abolished Akt phosphorylation of Pygo2, whereas Thr-50 mutation alone had no detectable effect (Fig. 7H). Taken together, these data demonstrate that Ser-48 is the predominant Akt phosphorylation site on Pygo2.
To understand how Ser-48 phosphorylation affects Pygo2 stability, we examined the impact of Ser-48 mutations on Pygo2 ubiquitylation. Specifically, we co-expressed FLAG-Pygo2 or its Ser-48 mutant derivatives with HA-tagged ubiquitin in HEK293T cells. The FLAG-Pygo2 proteins were purified under denaturing conditions for Western blot analysis using an anti-HA antibody. High molecular weight, ubiquitylated forms of FLAG-Pygo2 were detected when transfected cells were treated with MG132 (Fig. 8A). Compared with wild-type Pygo2, its S48A mutant showed slightly increased, whereas its S48E mutant showed dramatically decreased, ubiquitylation (Fig. 8B). Thus, a phosphomimetic substitution of Ser-48 inhibits Pygo2 ubiquitylation.
FIGURE 8.
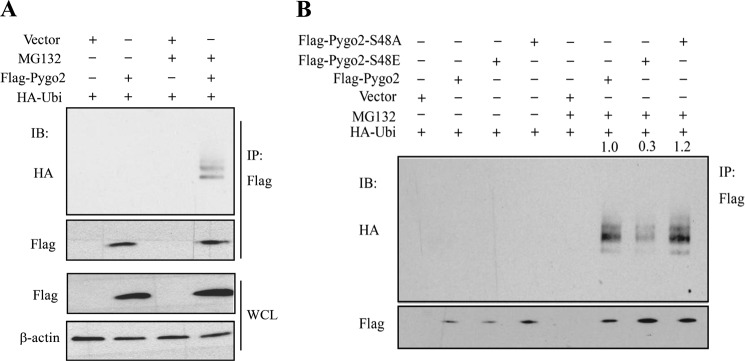
Pygo2 is ubiquitylated and the extent of its ubiquitylation is decreased by the S48E mutation. FLAG-Pygo2 (A) or its Ser-48 mutant derivatives (B) was co-transfected with HA-ubiquitin, and cells were treated 24 h later with MG132, followed by immunoprecipitation using FLAG M2 beads. FLAG vector was used as a negative control. See also the legend to Fig. 7, G and H. IP, immunoprecipitation; IB, immunoblot; WCL, whole cell lysate.
Akt and Its Upstream Growth Factors Act in Parallel with Wnt to Stabilize Pygo2
It has been shown that Wnt3A inhibits Pygopus degradation, and a Wnt signaling inhibitor that promotes CK1α activity counters this effect (77). We therefore wondered whether Wnt stabilizes Pygo2 in a manner that requires Akt phosphorylation. The addition of Wnt3A conditioned medium to HC11 cells stably expressing FLAG-Pygo2 resulted in an increase in the levels of not only β-catenin but also FLAG-Pygo2 (Fig. 9A), whereas the level of Pygo2 mRNA was not altered (Fig. 9B). A protein stability assay using CHX treatment confirmed that Wnt3A increased the half-life of FLAG-Pygo2 (Fig. 9C). Inhibition of Akt activation by TCN exerted a significant negative effect on Pygo2 protein stability regardless of whether Wnt3A was present or not (Fig. 9C). As expected (78, 79), Akt itself was not activated when HC11 cells were treated with GSK3β inhibitors LiCl and 6-bromoindirubin-3′-oxime (Bio) (Fig. 9D). Although the addition of serum-containing medium led to increased Ser-473 phosphorylation of Akt, the extent of increase was similar with the Wnt3A conditioned medium (Fig. 9D). Together, these data suggest that the stabilizing effects of Wnt3A and Akt activation on Pygo2 are additive and that they act in a parallel rather than linear fashion.
FIGURE 9.

Effects of growth factors on Pygo2 level and Akt phosphorylation. A and B, the addition of Wnt3A conditioned medium increases Pygo2 protein but not mRNA level. HC11 cells that stably express FLAG-Pygo2 were treated with control medium generated from L cells (0), supplemented with the indicated amounts of Wnt3A conditioned medium for 12 h, at which time cells were harvested for whole cell lysates for immunoblotting (A) or mRNAs for RT-PCR (B). C, effects of Wnt3A and Akt inhibition on the half-life of Pygo2. HC11 cells were switched to fresh L cell-derived medium (−), 50% Wnt3A conditioned medium (Wnt3A), 50% Wnt3A conditioned medium containing 2 μm TCN (Wnt3A + TCN), or L cell-derived medium containing 2 μm TCN (TCN) and treated with CHX 30 min later. Whole cell lysates were collected for immunoblotting at the indicated time points following CHX addition. The Pygo2 expression levels were normalized to β-actin, and values at time 0 for each treatment were set as 100%. The levels of Pygo2 at 2 and 4 h posttreatment were shown as a percentage of the initial levels for that particular treatment. Error bars, S.E. D, Wnt signaling does not activate Akt. HC11 cells were treated with GSK3β inhibitor LiCl or Bio or with Wnt3A conditioned medium, and whole cell lysates were collected for immunoblotting using Akt or phospho-Akt (Ser-473) antibodies. E, Pygo2 up-regulation and Akt activation upon insulin or EGF treatment. Serum-starved HC11 cells were stimulated with insulin or EGF for 15 min with or without TCN, and whole cell lysates were harvested for immunoblotting using the indicated antibodies. F, increased phospho-Pygo2 upon insulin treatment. HC11 cells were serum-starved for 16 h and stimulated with 50 nm insulin for 15 min in the absence or presence of 0.2 μm wortmannin (or DMSO as a control). Cell extracts were immunoprecipitated (IP) using FLAG M2 beads followed by immunoblotting. Bio, 6-bromoindirubin-3′-oxime. WCL, whole cell lysate.
We next tested whether the classical Akt-activating growth factors stabilize Pygo2. As expected (80), treatment of HC11 cells with insulin or EGF led to increased levels of the activated form of Akt (3.8-fold for EGF, 5.4-fold for 50 nm insulin, and 6.7-fold for 100 nm insulin, using total Akt as control) (Fig. 9E). Coinciding with Akt activation, the FLAG-Pygo2 protein level was elevated (Fig. 9E). The addition of TCN or wortmannin, a selective inhibitor of PI3K (81), abrogated the growth factor-induced Akt activation and Akt phosphorylation of Pygo2 (Fig. 9, E and F). These findings suggest that Akt acts downstream of insulin and EGF to phosphorylate and stabilize Pygo2.
Discussion
In this work, we provide convincing evidence that chromatin regulator Pygo2 is degraded through the ubiquitin/proteasome pathway and is posttranslationally stabilized through phosphorylation by activated PI3K/Akt signaling. In addition, our findings highlight Pygo2 as a common node downstream of oncogenic Wnt and Akt signaling pathways.
Posttranslational modifications are known to play a vital role in regulating protein stability (82, 83). Phosphorylation has been recognized as a prominent mechanism for modulating protein stability via enhancing substrate ubiquitylation and proteasome-dependent degradation. This process is often mediated through phosphorylation-dependent interactions between substrates and their E3 ubiquitin ligases (61, 84, 85). Here, we put forward a rare case where phosphorylation leads to reduced ubiquitylation and elevated protein stability. The reduced interaction between Pygo2 triple E mutant and DDB1 suggests a model in which Ser-48 phosphorylation compromises Pygo2 ubiquitylation by weakening its interaction with the E3 ubiquitin ligase complex. Ser-48 phosphorylation may also act via additional mechanisms, including those independent of protein-protein interactions (61, 85). Pygo2 is a medium sized protein (∼41 kDa) with only 13 lysine residues, 7 of which are located at the N terminus between positions 10 and 47, in close proximity to Ser-48. Therefore, it is also possible that Ser-48 phosphorylation introduces steric hindrance to prevent nearby lysine ubiquitylation, thus inhibiting Pygo2 degradation.
Our findings imply, although they do not directly prove, that the Cul4-DDB1 E3 ligase complex ubiquitylates Pygo2. Substrate recognition by the Cullin-RING E3 ligases often requires posttranslational modifications of the substrate (e.g. phosphorylation for Cul1 or hydroxylation for Cul2). However, these requirements are dependent on substrate receptors within specific Cullin-RING ligase complexes. Although the necessity of posttranslational modifications have not been well documented for Cul4-DDB1 substrates, methylation of orphan nuclear receptor RORα by enhancer of zeste homolog 2 (EZH2) methyltransferase has been shown to facilitate its recognition by VPRBP (or DCAF1) of the Cul4-DDB1-DCAF1 ubiquitin ligase for subsequent ubiquitylation and degradation (86). Therefore, Ser-48 phosphorylation may potentially interfere with methylation-mediated ubiquitylation. As such, our work suggests inhibitory phosphorylation of Pygo2 by Akt as a novel mechanism for regulation of substrate recognition by the Cul4-DDB1 ubiquitin ligases, an intriguing possibility worthy of future investigation.
Although both Cul4A and Cul4B may possess the biochemical capacity to ubiquitylate Pygo2 (indeed, Cul4A associates with Pygo2, and its overexpression decreases Pygo2 protein level), Cul4B probably takes the lion's share of this task under physiological conditions. This echoes previous work showing that depletion of Cul4B leads to elevated β-catenin level to an extent that is greater than depletion of Cul4A (87). We also note the reported finding that depletion of Cul4B but not Cul4A stabilizes, whereas overexpression of both enhances the ubiquitylation of, WDR5 (71), a core component of the histone H3 lysine 4 methyltransferase complex with which Pygo2 associates (9, 12). Multiple possibilities may account for the differential requirement under non-overexpressing, physiological conditions. The two Cul4 proteins may localize to different subcellular locations, thus having differential access to the substrates. Indeed, it has been reported that Cul4B is predominantly nuclear, whereas Cul4A is predominantly cytoplasmic (88). It is also possible that Pygo2 associates preferentially with the Cul4B complex due to higher concentration of the endogenous Cul4B protein and/or the existence of auxiliary factors. Future studies are needed to explore these possibilities.
By virtue of its ability to ubiquitylate a number of substrates with chromatin regulatory functions as well as histones, the Cul4-DDB1 complex has been long known to regulate DNA metabolism and chromatin structure (89). Of interest, Cul4-DDB1-mediated ubiquitylation is often initiated by chromatin-based signals and occurs on the chromatin. In light of this general theme, our finding of accumulation of Triton-insoluble forms of Pygo2 in discrete subnuclear domains upon proteasome inhibition is particularly intriguing. It is tempting to speculate that compared with “free” Pygo2, chromatin-bound Pygo2 proteins are preferred substrates of the Cul4-DDB1 complex. If so, then what is the functional significance of selectively turning over chromatin-bound Pygo2 in terms of Wnt signaling output and Wnt-regulated cellular events? What mechanisms distinguish “free” from chromatin-bound Pygo2? What is the physical and functional relationship between Cul4-DDB1-mediated ubiquitylation of Pygo2, β-catenin, and WDR5? Our findings here pave the way to address these important questions in the future.
Our work identifies Pygo2 as a direct target of Akt and Ser-48 as the predominant target site of Akt phosphorylation. Previously, Pygo2 has been implicated as a direct target of CK1 (77). We found that the Thr-59 residue of Pygo2, part of a CK1α consensus motif (90), is indeed phosphorylated. However, mutation of this residue fails to produce significant changes in Pygo2 protein level or stability, leaving open the possibility that CK1α stabilization of Pygo2 occurs via alternative phosphorylation sites.
This newly identified Akt regulation of Pygo2 adds yet another layer to the cross-talks between two of the most affected signaling pathways in cancer. The multitiered cross-talks between Wnt/β-catenin/Pygo2 signaling and PI3K/Akt signaling present an exquisite example of collaboration and synergy between distinct growth factor signals and are consistent with both pathways not only stimulating tumorigenesis but also promoting the self-renewal of embryonic and tissue stem cells (9, 19, 91–94). The notion that active Akt increases free and nuclear β-catenin levels, either indirectly via inhibitory phosphorylation of GSK3β or directly via stabilizing phosphorylation of β-catenin, has been proposed or accepted by many groups (31–40). However, it has also been argued that inside the cells, GSK3 kinases exist in different pools; thus, their actions in PI3K/Akt and Wnt/β-catenin pathways are spatially separate (95, 96). We observed a tight correlation between Akt activation and Pygo2 level but not β-catenin level in transformed MECs. It is possible that Akt acts on the different Wnt signaling components in a cell type-specific manner. Alternatively, Akt may regulate Pygo2 and GSK3β/β-catenin in a spatially and/or temporally sensitive fashion. Does Wnt signaling in turn regulate classical PI3K/Akt pathway components? Although some studies show this to be the case (32, 35), another study questions the notion that Wnt3A-induced β-catenin stabilization requires activation of Akt signaling (97). Consistently with the latter, we did not observe increased phosphorylation of Akt protein upon activating Wnt signaling. More importantly, our discovery of the control of Pygo2 protein stability by Akt phosphorylation, in addition to Akt phosphorylation of the more upstream components in Wnt signaling, implicates the critical importance of intricately regulating the molecular events that occur at the chromatin level.
Author Contributions
Q. L. and X. D. conceived, designed, and coordinated the study and wrote the manuscript. Q. L., Y. L., B. G., and L. F. performed the experiments and analyzed the data. P. Z., S. B., L. H., and X. D. contributed to experimental design and data analysis. L. H. edited the manuscript. All authors approved the final version of the manuscript.
Acknowledgments
We thank Yongsheng Shi for the MCF10A-Er-Src cells, Chris Hughes for Wnt3A-expressing L1 cells, Ping Wang for the Akt1(K179M) construct, Kazuhide Watanabe for suggestions, and Peter Kaiser and David Fruman for advice.
This work was supported, in whole or in part, by National Institutes of Health Grants R01-GM083089 (to X. D.) and R21CA161807 and R01GM074830 (to L. H.). This work was also supported by Susan G. Komen Grant KG110897 (to X. D.). The authors declare that they have no conflicts of interest with the contents of this article.
- Pygo2
- Pygopus 2
- CHX
- cycloheximide
- DDB1
- DNA damage-binding protein 1
- MEC
- mammary epithelial cell
- MG132
- carbobenzoxyl-Leu-Leu-leucinal
- MMTV
- murine mammary tumor virus
- NHD
- N-terminal homology domain
- NLS
- nuclear localization sequence
- PHD
- plant homology domain
- TAM
- tamoxifen
- TCN
- triciribine hydrate
- VPRBP
- Vpr (HIV-1)-binding protein.
References
- 1. Clevers H. (2006) Wnt/β-catenin signaling in development and disease. Cell 127, 469–480 [DOI] [PubMed] [Google Scholar]
- 2. Clevers H., Nusse R. (2012) Wnt/β-catenin signaling and disease. Cell 149, 1192–1205 [DOI] [PubMed] [Google Scholar]
- 3. Mosimann C., Hausmann G., Basler K. (2009) β-Catenin hits chromatin: regulation of Wnt target gene activation. Nat. Rev. Mol. Cell Biol. 10, 276–286 [DOI] [PubMed] [Google Scholar]
- 4. Belenkaya T. Y., Han C., Standley H. J., Lin X., Houston D. W., Heasman J., Lin X. (2002) pygopus encodes a nuclear protein essential for Wingless/Wnt signaling. Development 129, 4089–4101 [DOI] [PubMed] [Google Scholar]
- 5. Kramps T., Peter O., Brunner E., Nellen D., Froesch B., Chatterjee S., Murone M., Züllig S., Basler K. (2002) Wnt/wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear β-catenin-TCF complex. Cell 109, 47–60 [DOI] [PubMed] [Google Scholar]
- 6. Parker D. S., Jemison J., Cadigan K. M. (2002) Pygopus, a nuclear PHD-finger protein required for Wingless signaling in Drosophila. Development 129, 2565–2576 [DOI] [PubMed] [Google Scholar]
- 7. Thompson B., Townsley F., Rosin-Arbesfeld R., Musisi H., Bienz M. (2002) A new nuclear component of the Wnt signalling pathway. Nat. Cell Biol. 4, 367–373 [DOI] [PubMed] [Google Scholar]
- 8. Fiedler M., Sánchez-Barrena M. J., Nekrasov M., Mieszczanek J., Rybin V., Müller J., Evans P., Bienz M. (2008) Decoding of methylated histone H3 tail by the Pygo-BCL9 Wnt signaling complex. Mol. Cell 30, 507–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gu B., Sun P., Yuan Y., Moraes R. C., Li A., Teng A., Agrawal A., Rhéaume C., Bilanchone V., Veltmaat J. M., Takemaru K., Millar S., Lee E. Y., Lewis M. T., Li B., Dai X. (2009) Pygo2 expands mammary progenitor cells by facilitating histone H3 K4 methylation. J. Cell Biol. 185, 811–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kessler R., Hausmann G., Basler K. (2009) The PHD domain is required to link Drosophila Pygopus to Legless/β-catenin and not to histone H3. Mech. Dev. 126, 752–759 [DOI] [PubMed] [Google Scholar]
- 11. Andrews P. G., He Z., Popadiuk C., Kao K. R. (2009) The transcriptional activity of Pygopus is enhanced by its interaction with cAMP-response-element-binding protein (CREB)-binding protein. Biochem. J. 422, 493–501 [DOI] [PubMed] [Google Scholar]
- 12. Chen J., Luo Q., Yuan Y., Huang X., Cai W., Li C., Wei T., Zhang L., Yang M., Liu Q., Ye G., Dai X., Li B. (2010) Pygo2 associates with MLL2 histone methyltransferase and GCN5 histone acetyltransferase complexes to augment Wnt target gene expression and breast cancer stem-like cell expansion. Mol. Cell. Biol. 30, 5621–5635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gu B., Watanabe K., Dai X. (2012) Pygo2 regulates histone gene expression and H3 K56 acetylation in human mammary epithelial cells. Cell Cycle 11, 79–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nair M., Nagamori I., Sun P., Mishra D. P., Rhéaume C., Li B., Sassone-Corsi P., Dai X. (2008) Nuclear regulator Pygo2 controls spermiogenesis and histone H3 acetylation. Dev. Biol. 320, 446–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jessen S., Gu B., Dai X. (2008) Pygopus and the Wnt signaling pathway: a diverse set of connections. BioEssays 30, 448–456 [DOI] [PubMed] [Google Scholar]
- 16. Li B., Rhéaume C., Teng A., Bilanchone V., Munguia J. E., Hu M., Jessen S., Piccolo S., Waterman M. L., Dai X. (2007) Developmental phenotypes and reduced Wnt signaling in mice deficient for pygopus 2. Genesis 45, 318–325 [DOI] [PubMed] [Google Scholar]
- 17. Schwab K. R., Patterson L. T., Hartman H. A., Song N., Lang R. A., Lin X., Potter S. S. (2007) Pygo1 and Pygo2 roles in Wnt signaling in mammalian kidney development. BMC Biol. 5, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Song N., Schwab K. R., Patterson L. T., Yamaguchi T., Lin X., Potter S. S., Lang R. A. (2007) pygopus 2 has a crucial, Wnt pathway-independent function in lens induction. Development 134, 1873–1885 [DOI] [PubMed] [Google Scholar]
- 19. Gu B., Watanabe K., Sun P., Fallahi M., Dai X. (2013) Chromatin effector Pygo2 mediates Wnt-notch crosstalk to suppress luminal/alveolar potential of mammary stem and basal cells. Cell Stem Cell 13, 48–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Watanabe K., Fallahi M., Dai X. (2014) Chromatin effector Pygo2 regulates mammary tumor initiation and heterogeneity in MMTV-Wnt1 mice. Oncogene 33, 632–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sun P., Watanabe K., Fallahi M., Lee B., Afetian M. E., Rheaume C., Wu D., Horsley V., Dai X. (2014) Pygo2 regulates β-catenin-induced activation of hair follicle stem/progenitor cells and skin hyperplasia. Proc. Natl. Acad. Sci. U.S.A. 111, 10215–10220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Andrews P. G., Lake B. B., Popadiuk C., Kao K. R. (2007) Requirement of Pygopus 2 in breast cancer. Int. J. Oncol. 30, 357–363 [PubMed] [Google Scholar]
- 23. Brembeck F. H., Wiese M., Zatula N., Grigoryan T., Dai Y., Fritzmann J., Birchmeier W. (2011) BCL9–2 promotes early stages of intestinal tumor progression. Gastroenterology 141, 1359–1370 [DOI] [PubMed] [Google Scholar]
- 24. Chen Y. Y., Li B. A., Wang H. D., Liu X. Y., Tan G. W., Ma Y. H., Shen S. H., Zhu H. W., Wang Z. X. (2011) The role of Pygopus 2 in rat glioma cell growth. Med. Oncol. 28, 631–640 [DOI] [PubMed] [Google Scholar]
- 25. Liu Y., Dong Q. Z., Wang S., Fang C. Q., Miao Y., Wang L., Li M. Z., Wang E. H. (2013) Abnormal expression of Pygopus 2 correlates with a malignant phenotype in human lung cancer. BMC Cancer 13, 346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Popadiuk C. M., Xiong J., Wells M. G., Andrews P. G., Dankwa K., Hirasawa K., Lake B. B., Kao K. R. (2006) Antisense suppression of pygopus2 results in growth arrest of epithelial ovarian cancer. Clin. Cancer Res. 12, 2216–2223 [DOI] [PubMed] [Google Scholar]
- 27. Tzenov Y. R., Andrews P. G., Voisey K., Popadiuk P., Xiong J., Popadiuk C., Kao K. R. (2013) Human papilloma virus (HPV) E7-mediated attenuation of retinoblastoma (Rb) induces hPygopus2 expression via Elf-1 in cervical cancer. Mol. Cancer Res. 11, 19–30 [DOI] [PubMed] [Google Scholar]
- 28. Wang Z. X., Chen Y. Y., Li B. A., Tan G. W., Liu X. Y., Shen S. H., Zhu H. W., Wang H. D. (2010) Decreased pygopus 2 expression suppresses glioblastoma U251 cell growth. J. Neurooncol. 100, 31–41 [DOI] [PubMed] [Google Scholar]
- 29. Mesquita B., Lopes P., Rodrigues A., Pereira D., Afonso M., Leal C., Henrique R., Lind G. E., Jerónimo C., Lothe R. A., Teixeira M. R. (2013) Frequent copy number gains at 1q21 and 1q32 are associated with overexpression of the ETS transcription factors ETV3 and ELF3 in breast cancer irrespective of molecular subtypes. Breast Cancer Res. Treat. 138, 37–45 [DOI] [PubMed] [Google Scholar]
- 30. Shadeo A., Lam W. L. (2006) Comprehensive copy number profiles of breast cancer cell model genomes. Breast Cancer Res. 8, R9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Agarwal A., Das K., Lerner N., Sathe S., Cicek M., Casey G., Sizemore N. (2005) The AKT/IκB kinase pathway promotes angiogenic/metastatic gene expression in colorectal cancer by activating nuclear factor-κB and β-catenin. Oncogene 24, 1021–1031 [DOI] [PubMed] [Google Scholar]
- 32. Almeida M., Han L., Bellido T., Manolagas S. C., Kousteni S. (2005) Wnt proteins prevent apoptosis of both uncommitted osteoblast progenitors and differentiated osteoblasts by β-catenin-dependent and -independent signaling cascades involving Src/ERK and phosphatidylinositol 3-kinase/AKT. J. Biol. Chem. 280, 41342–41351 [DOI] [PubMed] [Google Scholar]
- 33. Desbois-Mouthon C., Cadoret A., Blivet-Van Eggelpoël M. J., Bertrand F., Cherqui G., Perret C., Capeau J. (2001) Insulin and IGF-1 stimulate the β-catenin pathway through two signalling cascades involving GSK-3β inhibition and Ras activation. Oncogene 20, 252–259 [DOI] [PubMed] [Google Scholar]
- 34. Fang D., Hawke D., Zheng Y., Xia Y., Meisenhelder J., Nika H., Mills G. B., Kobayashi R., Hunter T., Lu Z. (2007) Phosphorylation of β-catenin by AKT promotes β-catenin transcriptional activity. J. Biol. Chem. 282, 11221–11229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fukumoto S., Hsieh C. M., Maemura K., Layne M. D., Yet S. F., Lee K. H., Matsui T., Rosenzweig A., Taylor W. G., Rubin J. S., Perrella M. A., Lee M. E. (2001) Akt participation in the Wnt signaling pathway through Dishevelled. J. Biol. Chem. 276, 17479–17483 [DOI] [PubMed] [Google Scholar]
- 36. He X. C., Yin T., Grindley J. C., Tian Q., Sato T., Tao W. A., Dirisina R., Porter-Westpfahl K. S., Hembree M., Johnson T., Wiedemann L. M., Barrett T. A., Hood L., Wu H., Li L. (2007) PTEN-deficient intestinal stem cells initiate intestinal polyposis. Nat. Genet. 39, 189–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kobielak K., Stokes N., de la Cruz J., Polak L., Fuchs E. (2007) Loss of a quiescent niche but not follicle stem cells in the absence of bone morphogenetic protein signaling. Proc. Natl. Acad. Sci. U.S.A. 104, 10063–10068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bader A. G., Kang S., Zhao L., Vogt P. K. (2005) Oncogenic PI3K deregulates transcription and translation. Nat. Rev. Cancer 5, 921–929 [DOI] [PubMed] [Google Scholar]
- 39. Macdonald B. T., Semenov M. V., He X. (2007) SnapShot: Wnt/β-catenin signaling. Cell 131, 1204. [DOI] [PubMed] [Google Scholar]
- 40. Rochat A., Fernandez A., Vandromme M., Molès J. P., Bouschet T., Carnac G., Lamb N. J. (2004) Insulin and wnt1 pathways cooperate to induce reserve cell activation in differentiation and myotube hypertrophy. Mol. Biol. Cell 15, 4544–4555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sharma M., Chuang W. W., Sun Z. (2002) Phosphatidylinositol 3-kinase/Akt stimulates androgen pathway through GSK3β inhibition and nuclear β-catenin accumulation. J. Biol. Chem. 277, 30935–30941 [DOI] [PubMed] [Google Scholar]
- 42. Engelman J. A., Luo J., Cantley L. C. (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 7, 606–619 [DOI] [PubMed] [Google Scholar]
- 43. Vivanco I., Sawyers C. L. (2002) The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2, 489–501 [DOI] [PubMed] [Google Scholar]
- 44. Cross D. A., Alessi D. R., Cohen P., Andjelkovich M., Hemmings B. A. (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785–789 [DOI] [PubMed] [Google Scholar]
- 45. Liu P., Cheng H., Roberts T. M., Zhao J. J. (2009) Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 8, 627–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schultze S. M., Hemmings B. A., Niessen M., Tschopp O. (2012) PI3K/AKT, MAPK and AMPK signalling: protein kinases in glucose homeostasis. Expert Rev. Mol. Med. 14, e1. [DOI] [PubMed] [Google Scholar]
- 47. Song M. S., Salmena L., Pandolfi P. P. (2012) The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 13, 283–296 [DOI] [PubMed] [Google Scholar]
- 48. Alessi D. R., Andjelkovic M., Caudwell B., Cron P., Morrice N., Cohen P., Hemmings B. A. (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 15, 6541–6551 [PMC free article] [PubMed] [Google Scholar]
- 49. Alessi D. R., James S. R., Downes C. P., Holmes A. B., Gaffney P. R., Reese C. B., Cohen P. (1997) Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 7, 261–269 [DOI] [PubMed] [Google Scholar]
- 50. Sarbassov D. D., Guertin D. A., Ali S. M., Sabatini D. M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- 51. Stokoe D., Stephens L. R., Copeland T., Gaffney P. R., Reese C. B., Painter G. F., Holmes A. B., McCormick F., Hawkins P. T. (1997) Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science 277, 567–570 [DOI] [PubMed] [Google Scholar]
- 52. Chan T. O., Rittenhouse S. E., Tsichlis P. N. (1999) AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu. Rev. Biochem. 68, 965–1014 [DOI] [PubMed] [Google Scholar]
- 53. Hemmings B. A., Restuccia D. F. (2012) PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 4, a011189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Iliopoulos D., Hirsch H. A., Struhl K. (2009) An epigenetic switch involving NF-κB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 139, 693–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu L., Lee S., Zhang J., Peters S. B., Hannah J., Zhang Y., Yin Y., Koff A., Ma L., Zhou P. (2009) CUL4A abrogation augments DNA damage response and protection against skin carcinogenesis. Mol. Cell 34, 451–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu L., Yin Y., Li Y., Prevedel L., Lacy E. H., Ma L., Zhou P. (2012) Essential role of the CUL4B ubiquitin ligase in extra-embryonic tissue development during mouse embryogenesis. Cell Res. 22, 1258–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shevchenko A., Wilm M., Vorm O., Mann M. (1996) Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68, 850–858 [DOI] [PubMed] [Google Scholar]
- 58. Fang L., Kaake R. M., Patel V. R., Yang Y., Baldi P., Huang L. (2012) Mapping the protein interaction network of the human COP9 signalosome complex using a label-free QTAX strategy. Mol. Cell. Proteomics 11, 138–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hirsch H. A., Iliopoulos D., Tsichlis P. N., Struhl K. (2009) Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 69, 7507–7511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Iliopoulos D., Jaeger S. A., Hirsch H. A., Bulyk M. L., Struhl K. (2010) STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol. Cell 39, 493–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Deshaies R. J., Joazeiro C. A. (2009) RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434 [DOI] [PubMed] [Google Scholar]
- 62. Schröfelbauer B., Hakata Y., Landau N. R. (2007) HIV-1 Vpr function is mediated by interaction with the damage-specific DNA-binding protein DDB1. Proc. Natl. Acad. Sci. U.S.A. 104, 4130–4135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kirschner R. H., Rusli M., Martin T. E. (1977) Characterization of the nuclear envelope, pore complexes, and dense lamina of mouse liver nuclei by high resolution scanning electron microscopy. J. Cell Biol. 72, 118–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jackson D. A., Yuan J., Cook P. R. (1988) A gentle method for preparing cyto- and nucleo-skeletons and associated chromatin. J. Cell Sci. 90, 365–378 [DOI] [PubMed] [Google Scholar]
- 65. Lee J., Zhou P. (2007) DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase. Mol. Cell 26, 775–780 [DOI] [PubMed] [Google Scholar]
- 66. Angers S., Li T., Yi X., MacCoss M. J., Moon R. T., Zheng N. (2006) Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 443, 590–593 [DOI] [PubMed] [Google Scholar]
- 67. Li T., Chen X., Garbutt K. C., Zhou P., Zheng N. (2006) Structure of DDB1 in complex with a paramyxovirus V protein: viral hijack of a propeller cluster in ubiquitin ligase. Cell 124, 105–117 [DOI] [PubMed] [Google Scholar]
- 68. Ball R. K., Friis R. R., Schoenenberger C. A., Doppler W., Groner B. (1988) Prolactin regulation of β-casein gene expression and of a cytosolic 120-kd protein in a cloned mouse mammary epithelial cell line. EMBO J. 7, 2089–2095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Guenatri M., Bailly D., Maison C., Almouzni G. (2004) Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J. Cell Biol. 166, 493–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kerzendorfer C., Whibley A., Carpenter G., Outwin E., Chiang S. C., Turner G., Schwartz C., El-Khamisy S., Raymond F. L., O'Driscoll M. (2010) Mutations in Cullin 4B result in a human syndrome associated with increased camptothecin-induced topoisomerase I-dependent DNA breaks. Hum. Mol. Genet. 19, 1324–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nakagawa T., Xiong Y. (2011) X-linked mental retardation gene CUL4B targets ubiquitylation of H3K4 methyltransferase component WDR5 and regulates neuronal gene expression. Mol. Cell 43, 381–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Choi D. W., Na W., Kabir M. H., Yi E., Kwon S., Yeom J., Ahn J. W., Choi H. H., Lee Y., Seo K. W., Shin M. K., Park S. H., Yoo H. Y., Isono K., Koseki H., Kim S. T., Lee C., Kwon Y. K., Choi C. Y. (2013) WIP1, a homeostatic regulator of the DNA damage response, is targeted by HIPK2 for phosphorylation and degradation. Mol. Cell 51, 374–385 [DOI] [PubMed] [Google Scholar]
- 73. Klebba J. E., Buster D. W., Nguyen A. L., Swatkoski S., Gucek M., Rusan N. M., Rogers G. C. (2013) Polo-like kinase 4 autodestructs by generating its Slimb-binding phosphodegron. Curr. Biol. 23, 2255–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wang X., Winter D., Ashrafi G., Schlehe J., Wong Y. L., Selkoe D., Rice S., Steen J., LaVoie M. J., Schwarz T. L. (2011) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Imami K., Sugiyama N., Kyono Y., Tomita M., Ishihama Y. (2008) Automated phosphoproteome analysis for cultured cancer cells by two-dimensional nanoLC-MS using a calcined titania/C18 biphasic column. Anal. Sci. 24, 161–166 [DOI] [PubMed] [Google Scholar]
- 76. Townsley F. M., Thompson B., Bienz M. (2004) Pygopus residues required for its binding to Legless are critical for transcription and development. J. Biol. Chem. 279, 5177–5183 [DOI] [PubMed] [Google Scholar]
- 77. Thorne C. A., Hanson A. J., Schneider J., Tahinci E., Orton D., Cselenyi C. S., Jernigan K. K., Meyers K. C., Hang B. I., Waterson A. G., Kim K., Melancon B., Ghidu V. P., Sulikowski G. A., LaFleur B., Salic A., Lee L. A., Miller D. M., 3rd, Lee E. (2010) Small-molecule inhibition of Wnt signaling through activation of casein kinase 1α. Nat. Chem. Biol. 6, 829–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Meijer L., Skaltsounis A. L., Magiatis P., Polychronopoulos P., Knockaert M., Leost M., Ryan X. P., Vonica C. A., Brivanlou A., Dajani R., Crovace C., Tarricone C., Musacchio A., Roe S. M., Pearl L., Greengard P. (2003) GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem. Biol. 10, 1255–1266 [DOI] [PubMed] [Google Scholar]
- 79. Ryves W. J., Harwood A. J. (2001) Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochem. Biophys. Res. Commun. 280, 720–725 [DOI] [PubMed] [Google Scholar]
- 80. Berlato C., Doppler W. (2009) Selective response to insulin versus insulin-like growth factor-I and -II and up-regulation of insulin receptor splice variant B in the differentiated mouse mammary epithelium. Endocrinology 150, 2924–2933 [DOI] [PubMed] [Google Scholar]
- 81. Powis G., Bonjouklian R., Berggren M. M., Gallegos A., Abraham R., Ashendel C., Zalkow L., Matter W. F., Dodge J., Grindey G. (1994) Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res. 54, 2419–2423 [PubMed] [Google Scholar]
- 82. Lydeard J. R., Schulman B. A., Harper J. W. (2013) Building and remodelling Cullin-RING E3 ubiquitin ligases. EMBO Rep. 14, 1050–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Xu C., Kim N. G., Gumbiner B. M. (2009) Regulation of protein stability by GSK3 mediated phosphorylation. Cell Cycle 8, 4032–4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chandrasekaran S., Tan T. X., Hall J. R., Cook J. G. (2011) Stress-stimulated mitogen-activated protein kinases control the stability and activity of the Cdt1 DNA replication licensing factor. Mol. Cell. Biol. 31, 4405–4416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Verma R., Annan R. S., Huddleston M. J., Carr S. A., Reynard G., Deshaies R. J. (1997) Phosphorylation of Sic1p by G1 Cdk required for its degradation and entry into S phase. Science 278, 455–460 [DOI] [PubMed] [Google Scholar]
- 86. Lee J. M., Lee J. S., Kim H., Kim K., Park H., Kim J. Y., Lee S. H., Kim I. S., Kim J., Lee M., Chung C. H., Seo S. B., Yoon J. B., Ko E., Noh D. Y., Kim K. I., Kim K. K., Baek S. H. (2012) EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol. Cell 48, 572–586 [DOI] [PubMed] [Google Scholar]
- 87. Tripathi R., Kota S. K., Srinivas U. K. (2007) Cullin4B/E3-ubiquitin ligase negatively regulates β-catenin. J. Biosci. 32, 1133–1138 [DOI] [PubMed] [Google Scholar]
- 88. Zhang Y., Morrone G., Zhang J., Chen X., Lu X., Ma L., Moore M., Zhou P. (2003) CUL-4A stimulates ubiquitylation and degradation of the HOXA9 homeodomain protein. EMBO J. 22, 6057–6067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. O'Connell B. C., Harper J. W. (2007) Ubiquitin proteasome system (UPS): what can chromatin do for you? Curr. Opin. Cell Biol. 19, 206–214 [DOI] [PubMed] [Google Scholar]
- 90. Tobin A. B. (2002) Are we β-ARKing up the wrong tree? Casein kinase 1 α provides an additional pathway for GPCR phosphorylation. Trends Pharmacol. Sci. 23, 337–343 [DOI] [PubMed] [Google Scholar]
- 91. Watanabe K., Dai X. (2011) A WNTer revisit: new faces of β-catenin and TCFs in pluripotency. Sci. Signal. 4, pe41. [DOI] [PubMed] [Google Scholar]
- 92. Korkaya H., Paulson A., Charafe-Jauffret E., Ginestier C., Brown M., Dutcher J., Clouthier S. G., Wicha M. S. (2009) Regulation of mammary stem/progenitor cells by PTEN/Akt/β-catenin signaling. PLos Biol. 7, e1000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lin Y., Yang Y., Li W., Chen Q., Li J., Pan X., Zhou L., Liu C., Chen C., He J., Cao H., Yao H., Zheng L., Xu X., Xia Z., Ren J., Xiao L., Li L., Shen B., Zhou H., Wang Y. J. (2012) Reciprocal regulation of Akt and Oct4 promotes the self-renewal and survival of embryonal carcinoma cells. Mol. Cell 48, 627–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Reya T., Clevers H. (2005) Wnt signalling in stem cells and cancer. Nature 434, 843–850 [DOI] [PubMed] [Google Scholar]
- 95. Ding V. W., Chen R. H., McCormick F. (2000) Differential regulation of glycogen synthase kinase 3β by insulin and Wnt signaling. J. Biol. Chem. 275, 32475–32481 [DOI] [PubMed] [Google Scholar]
- 96. Ng S. S., Mahmoudi T., Danenberg E., Bejaoui I., de Lau W., Korswagen H. C., Schutte M., Clevers H. (2009) Phosphatidylinositol 3-kinase signaling does not activate the wnt cascade. J. Biol. Chem. 284, 35308–35313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Cajanek L., Adlerz L., Bryja V., Arenas E. (2010) WNT unrelated activities in commercially available preparations of recombinant WNT3a. J. Cell Biochem. 111, 1077–1079 [DOI] [PubMed] [Google Scholar]