

Background: The GSH-dependent antioxidant system reduces atherosclerosis.
Results: Inhibition of GSH production by BSO enhanced CD36 translational efficiency to induce CD36 protein expression and lipid accumulation that was blocked by antioxidant (enzyme).
Conclusion: Alterations of cellular GSH and GSH/GSSG status regulate macrophage CD36 expression and cellular oxLDL uptake.
Significance: Our study demonstrates an important anti-atherogenic function of the GSH-dependent antioxidant system.
Keywords: antioxidant, atherosclerosis, macrophage, peroxisome proliferator-activated receptor (PPAR), translation regulation, CD36, foam cell, glutamate-cysteine ligase, glutathione
Abstract
The glutathione (GSH)-dependent antioxidant system has been demonstrated to inhibit atherosclerosis. Macrophage CD36 uptakes oxidized low density lipoprotein (oxLDL) thereby facilitating foam cell formation and development of atherosclerosis. It remains unknown if GSH can influence macrophage CD36 expression and cellular oxLDL uptake directly. Herein we report that treatment of macrophages with l-buthionine-S,R-sulfoximine (BSO) decreased cellular GSH production and ratios of GSH to glutathione disulfide (GSH/GSSG) while increasing production of reactive oxygen species. Associated with decreased GSH levels, macrophage CD36 expression was increased, which resulted in enhanced cellular oxLDL uptake. In contrast, N-acetyl cysteine and antioxidant enzyme (catalase or superoxide dismutase) blocked BSO-induced CD36 expression as well as oxLDL uptake. In vivo, administration of mice with BSO increased CD36 expression in peritoneal macrophages and kidneys. BSO had no effect on CD36 mRNA expression and promoter activity but still induced CD36 protein expression in macrophages lacking peroxisome proliferator-activated receptor γ expression, suggesting it induced CD36 expression at the translational level. Indeed, we determined that BSO enhanced CD36 translational efficiency. Taken together, our study demonstrates that cellular GSH levels and GSH/GSSG status can regulate macrophage CD36 expression and cellular oxLDL uptake and demonstrate an important anti-atherogenic function of the GSH-dependent antioxidant system by providing a novel molecular mechanism.
Introduction
Formation of lipid-laden macrophage/foam cells is the initial and critical step in the development of atherosclerosis, which is a major cause of coronary heart disease. Disruption of glutathione (GSH) biosynthesis, metabolism, and functions has been implicated in the development of atherosclerosis (1–4). The decreased cellular GSH levels can result in overproduction of cellular reactive oxygen species (ROS),3 particularly by macrophages, which can cause multiple biological alterations such as increased production of inflammatory molecules and oxidatively modified products like the oxidized low-density lipoprotein (oxLDL). Uptake and internalization of oxLDL by macrophages can enhance cellular lipid accumulation and foam cell formation (5).
CD36 has been defined as a receptor for oxLDL and plays an important role in foam cell formation and development of atherosclerosis (6, 7). The uptake of oxLDL by human monocyte-derived macrophages is blocked by anti-CD36 antibody by 50% (8). Monocytes isolated from humans lacking functional CD36 expression exhibit significantly decreased capacity for oxLDL binding (9). In animal models, genetic deletion of CD36 expression reduces foam cell formation and inhibits the development of atherosclerosis in apoE deficient (apoE−/−) mice (10).
We previously reported that oxLDL induced macrophage CD36 expression, which suggests a positive feedback loop driving foam cell formation by oxLDL (11). Studies on the mechanism(s) responsible for oxLDL-induced CD36 expression have discovered the critical role of peroxisome proliferator-activated receptor γ (PPARγ), a ligand-activated transcription factor, in regulating CD36 transcription (12, 13). Activation of PPARγ by ligand results in formation of a heterodimer of PPARγ with another transcription factor, retinoid X receptor α (RXRα). The complex of PPARγ/RXRα binds to the PPARγ-responsive element in the proximal region of the promoter in target genes including CD36 to initiate their transcription (14). Two of the major oxidized lipid components in oxLDL, 9-hydroxyoctadecadienoic acid (9-HODE) and 13-HODE, have been identified as the endogenous ligands for PPARγ (13), suggesting that oxLDL induces macrophage CD36 expression and foam cell formation in a PPARγ-dependent manner. CD36 expression can also be regulated by cellular glucose levels by a translational mechanism (15).
GSH (glutamate-cysteine-glycine) is a tripeptide thiol and is produced by most mammalian cells at up to millimolar concentrations. Most cellular GSH (80–85%) is present in cytoplasm because of its hydrophilic property (16). In addition to being a co-factor for various enzymatic reactions, GSH can function as an antioxidant to reduce cellular ROS levels. The GSH-dependent antioxidant system has been demonstrated to play an important role in regulating foam cell formation and development of atherosclerosis. Glutamate-cysteine ligase (GCL) is the rate-limiting enzyme in regulating cellular GSH production (17). Genetic deletion of GCL expression enhances atherosclerosis, whereas high expressing GCL inhibits atherosclerosis in apoE deficient (apoE−/−) mice (1). Glutathione peroxidase-1 (GPx-1) is an antioxidant enzyme that uses GSH as the substrate to convert hydrogen peroxide or lipid peroxides into water or the respective alcohols. Deficiency of GPx-1 expression accelerates progression of atherosclerosis (2, 18). Glutathione reductase maintains cellular GSH homeostasis by catalyzing reduction of glutathione disulfide (GSSG) into GSH using NADPH as the reducing cofactor. Increased macrophage glutathione reductase expression also inhibits atherosclerosis in LDL receptor-deficient (LDLR−/−) mice (3).
Despite the above findings, the underlying mechanisms responsible for the anti-atherogenic properties of the GSH-dependent antioxidant system have not been fully elucidated. Because of the importance of CD36 in macrophage oxLDL uptake and foam cell formation, in this study we investigated if inhibition of macrophage GSH production by a potent specific GCL inhibitor, l-buthionine-S,R-sulfoximine (BSO) (19), or regulation of cellular GSH/GSSG status can directly mediate macrophage CD36 expression and cellular oxLDL uptake. We also attempted to determine the involved mechanisms.
Experimental Procedures
Materials
The assay kit for GSH/GSSG was purchased from Beyotime (Nantong, China). The oxLDL ELISA kitwas purchased from Cusabio Biotech (Wuhan, China).LipofectamineTM 2000 was purchased from Invitrogen. The following antibodies were purchased from different sources, respectively: rabbit anti-CD36, Novus Biochemicals (Littleton, CO); mouse anti-puromycin, Millipore (Temecula, CA); rabbit anti-GAPDH and phosphorylated STAT1 (pSTAT1;Tyr-701), Santa Cruz Biotechnology (Dallas, TX); rabbit anti-PPARγ, Abcam (Cambridge, MA); rabbit anti-STAT1, Proteintech Group (Chicago, IL). Low-density lipoprotein (LDL) was purchased from Athens Research and Technology, Inc. (Athens, GA). OxLDL was prepared as described (11). All other reagents were purchased from Sigma except as indicated.
Cell Culture
RAW264.7 cells, a murine macrophage cell line, were purchased from ATCC (Rockville, MD) and cultured in RPMI 1640 medium containing 10% fetal bovine serum and 50 μg/ml penicillin and streptomycin. Peritoneal macrophages were collected from mice as described (20), and purity was determined by immunofluorescent staining with anti-MOMA-2 antibody. Cells received treatment in serum-free medium.
In Vivo Study
The protocol for animal study was approved by the Animal Ethics Committee of Nankai University and conforms to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. C57BL/6 wild type and apoE−/− mice were purchased from the Animal Center of Nanjing University (Nanjing, China). To generate macrophage PPARγ-deficient mice, the homozygous floxed (+/+) PPARγ mice (kindly provide by Dr. S. Duan) crossbred with the transgenic mice containing the CRE gene under the control of the murine M lysozyme promoter (CRE-M lysozyme (+/+)) mice (21). Therefore, the control mice are flox+/+/CRE−/−, and the conditional macrophage PPARγ knock-out mice are flox+/+/CRE+/−. The peritoneal macrophages isolated from these two types of mice are defined as PPARγfl/fl and MacPPARγ KO cells, respectively.
To determine the effect of decreased GSH on CD36 expression in vivo, apoE−/− mice were fed normal chow or normal chow plus BSO (50 mg/100 g of food) for 1 week. Based on the food consumption, the dose of BSO was estimated to be ∼0.25 mmol/kg (bodyweight)/day. At the end of treatment, all of the mice were anesthetized and euthanized in a CO2 chamber followed by collection of peritoneal macrophages and kidneys. The cells were lysed while a piece of kidney was homogenized in a lysis buffer (20 mm Tris, pH 7.5, 137 mm NaCl, 2 mm EDTA, 1% Triton X-100, 25 mm β-glycerophosphate, 2 mm sodium pyrophosphate, 1 mm phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin/leupeptin, 100 mm NaVO4). The cellular lysate or kidney homogenate was centrifuged for 10 min at 16,000 × g at 4 °C. The supernatant was transferred into a new test tube as total protein extract for determination of protein expression by Western blot.
Determination of Cellular GSH and GSSG Levels
Cellular GSH and GSSG levels were determined by enzymatic recycling method (22) using assay kits. The cell samples for GSH/GSSG assay were prepared as follows; after treatment and washing with cold Ca2+/Mg2+-free PBS, macrophages were suspended in ice-cold extraction buffer (1% Triton X-100 and 0.6% sulfosalicylic acid in 0.1 m K3PO4, 5 mm EDTA, pH 7.5) and homogenized. A portion of the homogenate was saved for determination of the cellular protein content, which was used to normalize cellular GSH and GSSG levels. The rest of the homogenate was centrifuged for 5 min at 10,000 × g at 4 °C, and the supernatants were used for GSH/GSSG assay using the assay kit based on the manufacturer's instructions. To prepare kidney samples for GSH determination, a piece of kidney was weighed and homogenized in an ice-cold mixture of 5% metaphosphoric acid and 0.6% sulfosalicylic acid. The supernatant of homogenate was used to determine GSH content, which was normalized by the weight of the piece of kidney and expressed as μmol/g of tissue.
Determination of Cellular ROS Levels
The intracellular ROS levels were determined using a ROS assay kit purchased from Beyotime (Nantong, China) that is based on the principle that oxidation of the non-fluorescent probe 2′,7′-dichlorodihydrofluorescein diacetate by intracellular ROS can generate the highly fluorescent 2′,7′-dichlorofluorescein (23). Briefly, after treatment, 5 μm 2′,7′-dichlorodihydrofluorescein diacetate solution was added to the cells in a 96-well plate and incubated in the dark for 20 min at room temperature followed by washing with PBS 3 times. The 2′,7′-dichlorofluorescein fluorescence generated within the cells was determined at 485-nm (excitation) and 527-nm (emission) wavelengths on a fluorescence microplate reader (EnSpire, PerkinElmer Life Sciences). After determination of fluorescence, the cells remaining in each well of the plate were lysed, and cellular protein content was determined that is used to normalize the 2′,7′-dichlorofluorescein fluorescence. The normalized 2′,7′-dichlorofluorescein fluorescence intensity of control samples was defined as 100%.
Determination of Macrophage oxLDL Uptake
After treatment, macrophages were incubated with rabbit anti-CD36 antibody or normal IgG for 1 h followed by incubation with oxLDL (50 μg/ml) for 3 h. The cells were then fixed in 4% paraformaldehyde for 30 min at room temperature, washed twice with PBS for 5 min, and stained with Oil Red O solution (a mixture of three parts 0.5% Oil Red O in isopropyl alcohol and 2 parts water) for 50 min at room temperature followed by washing twice with water. The cells were counterstained with hematoxylin solution for nuclei for 30 s, kept in water for 5 min, and then photographed. The accumulated Oil Red O dye within macrophages was extracted by added isopropyl alcohol, and the absorbance of the extraction solution was determined at 510 nm. After normalized by cellular protein content, the absorbance of the control group was defined as 1.
Isolation of Total RNA and Determination CD36 mRNA Expression
Total cellular RNA was extracted as described (20). The cDNA was synthesized with 1 μg of total cellular RNA using the reverse transcription kit purchased from New England BioLabs (Ipswich, MA). PCR was performed with the following primers: CD36 forward, (5′-TTTCCTCTGACATTTGCAGGTCTA-3′) and reverse, (5′-AAAGGCATTGGCTGGAAGA-3′) and GAPDH forward (5′-ACCCAGAAGACTGTGGATGG-3′) and reverse, (5′-ACACATTGGGGGTAGGAACA-3′). The RT-PCR product was determined by agarose electrophoresis and photographed. The real time PCR was performed with the above primers and an SYBR Green PCR master mix purchased from Bio-Rad. Expression of CD36 mRNA was normalized by the corresponding GAPDH mRNA.
Western Blot Analysis of CD36, STAT1, and Phosphorylated STAT1 (pSTAT1), FACS, and Immunofluorescent Staining Analysis of CD36
After extraction, an equal amount of cellular protein from each sample was used to determine CD36, STAT1, and pSTAT1 protein levels by Western blot as described (20).
To analyze cell surface CD36 protein levels, ∼1 × 106 cells from each sample were blocked for 30 min at room temperature with PBS containing 5% goat serum. After washing, the cells were incubated with anti-CD36 antibody (1:100) for 1 h at room temperature followed by incubation with goat anti-rabbit FITC-conjugated IgG (1:50) for 45 min and flow cytometric evaluation.
CD36 expression was also determined by immunofluorescent staining as described (20). Images of cells were obtained with a fluorescence microscope (Leica). The mean fluorescence intensity of all the immunofluorescent images was determined as described (24).
Determination of CD36 Protein Translational Efficiency
CD36 protein translational efficiency was determined by two methods: polysomal RNA fractioned-Northern blot analysis and surface sensing of translation (SUnSET) technique. The polysomal RNA fractioned-Northern blot analysis was completed as follows (15, 25). After treatment and washing with PBS, ∼108 cells were resuspended in 0.75 ml of a low salt buffer (20 mm Tris-HCl, pH 7.4, 10 mm NaCl, 3 mm MgCl2) containing Triton X-100 (1.2%, v/v) and sucrose (0.2 m) and homogenized. After centrifugation for 30 s at 27,000 × g at 4 °C, the supernatant was mixed with 0.1 ml of low salt buffer containing heparin (10 mg/ml). After the addition of NaCl (final 0.15 m), the mixture was applied to the top of a 0.5–1.5 m linear sucrose gradient (9.6 ml in low salt buffer) and centrifuged for 3 h at 32,000 × g at 4 °C. The gradients were fractionated into 6 fractions (1 ml/fraction). SDS and proteinase K (final concentration: 0.5% and 0.1 mg/ml, respectively) were added to each fraction and mixed well. The mixture was then incubated for 30 min at 37 °C followed by RNA extraction. Expression of CD36 mRNA in the polysomal RNA fractions was determined by Northern blot (26).
For SUnSET the technique assay (27–29), macrophages were treated with BSO for 12 h followed by treatment with puromycin (10 μg/ml) for 1 h. After whole cellular protein extraction, the input was determined by Western blot with anti-puromycin antibody. Based on the results of input, the same amount of cellular protein from each sample was used to conduct immunoprecipitation by anti-CD36 antibody or normal IgG followed by Western blot analysis with anti-puromycin, anti-CD36, or anti-IgG antibody.
Preparation of Plasmid DNA and Determination of CD36 Promoter Activity
Mouse CD36 promoter (from −1946 to +51) was constructed by PCR with genomic DNA isolated from RAW264.7 cells and the following primers: forward (5′-CCGCTCGAGAAGTGCAGAAGTTTACATGGG-3′) and reverse (5′-CCCAAGCTTAGGAGCTGTCTTCCAGGTG-3′). After the sequence was confirmed, the PCR product was digested with XhoI and HindIII followed by ligation with pGL4 luciferase reporter vector, transformed into Escherichia coli to amplify.
To determine CD36 promoter activity, ∼95% confluent 293T cells were transfected with DNA for CD36 promoter, PPARγ, and RXRα expression vectors and Renilla (for internal normalization) using LipofectamineTM 2000 (Invitrogen). After 20 h of transfection plus treatment, the cells were lysed, and cellular lysate was used to determine firefly and Renilla luciferase activity using the Dual-Luciferase Reporter Assay System from Promega (Madison, WI).
Inhibition of Macrophage GCL or CD36 Expression by siRNA
GCL siRNA was constructed into pSilencer 5.1-H1 Retro vector (Ambion) with the following oligonucleotides: 5′-GATCCGTGGAGGCGATGTTCTTGAGTTCAAGAGACTCAAGAACATCGCCTCCATTTTTTGGAAA-3′ and 5′-AGCTTTTCCAAAAAATGGAGGCGATGTTCTTGAGTCTCTTGAACTCAAGAACATCGCCTCCACG-3′. CD36 siRNA was purchased from Santa Cruz Biotechnology.
Macrophages were transfected with scrambled siRNA, GCL siRNA, or CD36 siRNA with the transfection reagent purchased from Origene (Rockville, MD) in an antibiotics/serum-free RPMI 1640 medium for 24 h. The cells were then switched into complete medium and continued culture or culture or plus treatment for another 48 h. The cells transfected with GCL siRNA were used to extract cellular total RNA and protein followed by determination of GCL mRNA expression by RT-PCR with the forward and reverse primers (5′-CTGCACATCTACCACGCAGT-3′ and 5′-GTCTCAAGAACATCGCCTCC-3′, respectively) and normalized by GAPDH mRNA, CD36 protein expression by Western blot, and cellular GSH levels by assay kit. The cells transfected with CD36 siRNA were treated with BSO or BSO plus antioxidant (enzyme) overnight followed by determination of oxLDL uptake/foam cell formation by incubating cells with oxLDL and ROS production by the ROS assay kit, as described above.
Data Analysis
All experiments were conducted at least three times, and representative results are presented. Data were analyzed by Student's t test (n = 3), and significant difference was considered at p < 0.05.
Results
Cellular GSH/GSSG Status Regulates Macrophage CD36 Protein Expression
Reaction of GSH with ROS generates GSSG, which can be reduced back to GSH by NADPH. Both cellular GSH content and the ratio of GSH to GSSG (GSH/GSSG) play an important role in anti-oxidative stress. GSH is mainly synthesized by kidney in vivo because the highest GCL activity is in this tissue (30). However, other tissues/cell types can also produce GSH. To study if the alterations of cellular GSH levels or GSH/GSSG ratios can influence macrophage CD36 expression, RAW264.7 macrophages were treated with BSO. BSO reduced cellular GSH levels in a concentration-dependent manner (Fig. 1A, left panel). Associated with decreased GSH levels, cellular GSH/GSSG ratios were also reduced (Fig. 1A, right panel), whereas cellular ROS levels were increased (Fig. 1B). The results of a Western blot show that BSO increased CD36 protein expression in a concentration-dependent manner (Fig. 1C, left panel; normally, two bands and one band of CD36 protein were determined in RAW264.7 cells and peritoneal macrophages or kidney, respectively). The time course study indicates that BSO induced CD36 protein expression in a time-dependent fashion (Fig. 1C, right panel).
FIGURE 1.

Inhibition of cellular GSH production by BSO induces macrophage CD36 protein expression. A and B, RAW264.7 macrophages in serum-free medium were treated with BSO at the indicated concentrations for 16 h. Cellular GSH and GSSG concentrations (A) and ROS levels (B) were determined with assay kits, respectively. *, p < 0.05 versus control (n = 3). C, RAW264.7 cells were treated with BSO at the indicated concentrations for 16 h or 5 μm BSO for the indicated times. CD36 protein expression was determined by Western blot. D, after treatment, cell surface CD36 protein levels were determined by FACS. E and F, peritoneal macrophages isolated from C57BL/6 mice received BSO treatment overnight followed by determination of CD36 expression by immunofluorescent staining (E), and the immunofluorescent density (mean fluorescence intensity (MFI)) of the images was quantified (F). *, p < 0.05 versus control (n = 3). G and I, RAW264.7 cells in 6-well plates were transiently transfected with GCL siRNA or scrambled siRNA. After transfection cells were collected for the following assays. G, GCL mRNA expression; H, cellular GSH levels. *, p < 0.05 (n = 3). I, CD36 protein expression.
CD36 is a membrane protein. To determine the effect of BSO on cell surface CD36 protein levels, after treatment cells were subjected to flow cytometric evaluation. Fig. 1D demonstrates that BSO increased cell surface CD36 protein levels. The effect of BSO on CD36 protein expression in primary cells was determined by immunofluorescent staining. Similar to RAW264.7 cells, BSO also induced peritoneal macrophage CD36 protein expression in a concentration dependent manner (Fig. 1, E and F).
To directly link cellular GSH production to macrophage CD36 expression, we selectively inhibited GCL expression by siRNA. Compared with scrambled siRNA, GCL siRNA inhibited GCL mRNA expression (Fig. 1G). Associated with inhibition of GCL expression, cellular GSH levels were decreased (Fig. 1H), whereas expression of macrophage CD36 protein expression was induced (Fig. 1I). Taken together, the results in Fig. 1 suggest that inhibition of cellular GSH production can induce macrophage CD36 protein expression.
Induction of CD36 protein expression by BSO can result in increased cellular oxLDL uptake, which facilitates foam cell formation. To determine it, after BSO treatment macrophages were conducted in Oil Red O staining. The top panel of Fig. 2A shows that BSO treatment led to increased oxLDL uptake, indicating formation of foam cells. The quantitative analysis of accumulated Oil Red O dye within cells (Fig. 2B) also demonstrates that the increased cellular lipid accumulation is in a concentration-dependent manner. However, blocking CD36 function by anti-CD36 antibody substantially attenuated lipid accumulation (Fig. 2A, bottom panel), suggesting that the enhanced macrophage oxLDL uptake and foam cell formation by BSO is mainly completed through induction of CD36 expression, which was also confirmed by a study with CD36 siRNA (Fig. 3F).
FIGURE 2.
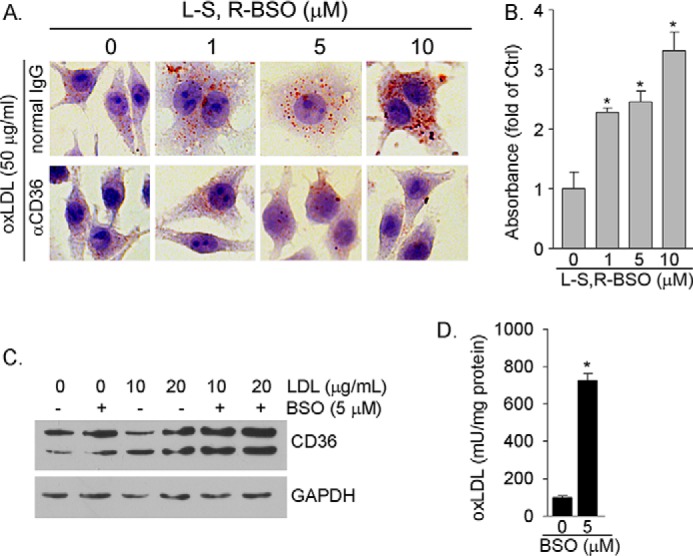
BSO induces macrophage oxLDL uptake by activating CD36 protein expression. A, RAW264.7 cells were treated with BSO for 16 h. After treatment cells were incubated with normal IgG or anti-CD36 antibody (0.3 μg/ml) followed by incubation with oxLDL (50 μg/ml). Cellular oxLDL uptake was determined by Oil Red O staining. B, accumulated Oil Red O dye within cells in each sample of the upper panel in Fig. 2A was extracted individually. The absorbance of extraction solution was determined at 510 nm and normalized by cellular protein content. *, p < 0.05 versus control. C, RAW264.7 cells were treated with LDL or LDL plus BSO as indicated for 16 h. D, RAW264.7 cells were treated with LDL (20 μg/ml) or LDL plus BSO (5 μm) for 16 h. OxLDL concentrations in the cellular lysate were determined by the oxLDL ELISA assay kit. *, p < 0.05 versus LDL alone (n = 3). mU, milliunits.
FIGURE 3.

Cellular GSH/GSSG status regulates macrophage CD36 expression and oxLDL uptake. A, RAW264.7 cells were treated with GSH at the indicated concentrations for 16 h followed by determination of CD36 protein expression. B and C, mouse peritoneal macrophages were treated with CAT, NAC, SOD, and NMMA alone or each plus BSO as indicated for 16 h. After treatment, cells were used to determine GSH/GSSG (B, *, p < 0.05 versus control (n = 3)) and CD36 expression (C). D, peritoneal macrophages were treated with CAT, SOD, NAC, and NMMA or each plus BSO as indicated for 16 h. After treatment, cells were used to determine cellular oxLDL uptake by incubation with oxLDL in the presence of normal IgG (left panel) and anti-CD36 antibody (right panel) followed by Oil Red O staining, extraction, and quantitation of the accumulated Oil Red O dye within cells. *, p < 0.05 (n = 3). E, the cells in Fig. 3D were also determined ROS production by assay kit. *, p < 0.05 (n = 3). DCF, dichlorofluorescein. F, peritoneal macrophages were transfected with scrambled siRNA (Ctrl siRNA) or CD36 siRNA (50 nm of each) and then treated with CAT, SOD, NAC, and NMMA alone or each plus BSO. Inhibition of CD36 expression was determined by real time RT-PCR (right panel). **, p < 0.01 (n = 3). Cellular oxLDL uptake and foam cell formation were determined by incubation with oxLDL followed by Oil Red O staining (left and middle panels) and quantitation of accumulated Oil Red O dye within cells. *, p < 0.05 (n = 3).
The decreased cellular GSH levels and GSH/GSSG ratios by BSO increase cellular ROS production (Fig. 1B), which can enhance production of oxLDL if LDL is present (31). Consequently, the generated oxLDL can activate CD36 expression. To test it, macrophages were treated with LDL or LDL plus BSO. LDL alone had little effect on CD36 expression. However, co-treatment of macrophages with BSO and LDL further enhanced BSO-induced CD36 expression (Fig. 2C), which should be attributed to the oxidation of LDL in response to BSO treatment. In fact, the results of an oxLDL ELISA assay demonstrate that BSO substantially increased cellular oxLDL content in the presence of LDL (Fig. 2D).
We next determined if amelioration of cellular GSH/GSSG status by exogenous GSH or other antioxidant or antioxidant enzyme can attenuate BSO-induced macrophage CD36 protein expression. Fig. 3A demonstrates that treatment of macrophages with exogenous GSH decreased CD36 expression. In the presence of a small molecular antioxidant, such as N-acetyl cysteine (NAC), and antioxidant enzyme (catalase (CAT) or superoxide dismutase (SOD)), the decreased GSH/GSSG ratio was restored to normal (Fig. 3B). Macrophages greatly express inducible nitric-oxide synthase to produce a large amount of nitric oxide when cells receive stimuli. Nitric oxide can also induce cellular oxidative stress. However, treatment of macrophages with NG-monomethyl-l-arginine (l-NMMA, a potent inducible nitric-oxide synthase inhibitor) did not affect the BSO-decreased GSH/GSSG ratio because BSO inhibited inducible nitric-oxide synthase expression (32). Similarly, CAT or NAC or SOD alone slightly inhibited or had no effect on CD36 expression, but each of them blocked CD36 protein expression induced by BSO. Meanwhile, l-NMMA had no effect on CD36 expression in either the absence or presence of BSO (Fig. 3C). Furthermore, amelioration of cellular GSH/GSSG status by CAT, SOD, and NAC attenuated BSO-induced cellular lipid accumulation (Fig. 3D, left half). However, anti-CD36 antibody abolished the effect of BSO or BSO plus antioxidant (enzyme) on oxLDL uptake (Fig. 3D, right half), which suggests that the regulation of cellular oxLDL uptake by BSO or BSO plus antioxidant (enzyme) is contributed by CD36 expression. Correspondingly, BSO increased ROS production, which was blocked by CAT, SOD, and NAC but not NMMA (Fig. 3E).
To further confirm that the BSO-induced cellular oxLDL uptake is mainly contributed by induction of CD36 expression, we transfected macrophages with CD36 siRNA to selectively inhibit CD36 expression (Fig. 3F, right panel) and then determined effect of BSO or BSO plus antioxidant (enzyme) on oxLDL uptake. Similar to normal macrophages (Fig. 3D), BSO treatment substantially induced lipid accumulation in cells transfected with scrambled siRNA (Ctrl siRNA), whereas CAT, SOD, and NAC, but not NMMA, blocked BSO-induced cellular oxLDL uptake (Fig. 3F, left panel). In contrast, cells lacking CD36 expression by CD36 siRNA demonstrate reduced oxLDL uptake at the basal levels, whereas BSO or BSO plus antioxidant (enzyme) had little effect on it (Fig. 3F, middle panel) which further suggest that BSO induced cellular oxLDL uptake is completed through induction of CD36 expression.
Induction of CD36 Expression by GSH Depletion in Vivo
To determine if the decreased GSH levels can induce CD36 expression in vivo, particularly in pro-atherogenic apoE−/− mice, apoE−/− mice were fed normal chow or normal chow plus BSO (50 mg/100 g of food or ∼0.25 mmol/kg (bodyweight)/day) for 1 week. Compared with control mice (chow alone), no difference in food intake, bodyweight gain, and exterior appearance was caused by BSO treatment. Among mouse tissues, the highest GCL expression has been reported in the kidney (30). Therefore, both mouse peritoneal macrophages and kidneys were collected to determine GSH levels and CD36 expression. BSO decreased mouse kidney and peritoneal macrophage GSH levels to ∼15 and ∼25% of controls (Fig. 4, bottom of each image), respectively. Associated with decreased GSH levels, expression of CD36 in both peritoneal macrophages and kidneys was increased (Fig. 4).
FIGURE 4.
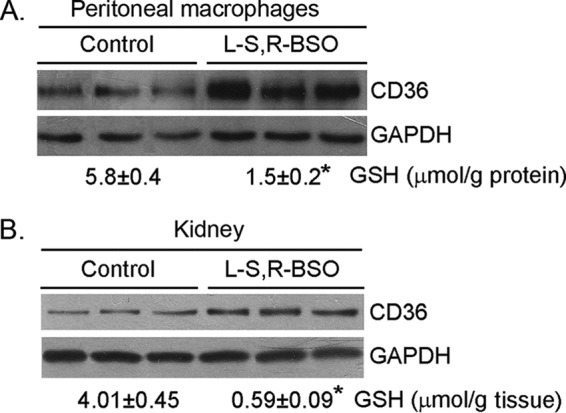
BSO induces peritoneal macrophage and kidney CD36 protein expression in vivo. ApoE−/− mice (3 mice in each group) were fed normal chow or normal chow plus BSO (50 mg/100 g of food) for 1 week. At the end of experiment, peritoneal macrophages and kidneys were collected from each mouse separately and used to extract total cellular protein for determination of CD36 protein expression. A piece of kidney and a portion of peritoneal macrophages were also used to determine GSH contents, and the results were presented at the bottom of each image. *, p < 0.05 (n = 3).
BSO Induces CD36 Protein Expression by Enhancing CD36 Translational Efficiency
To determine if BSO induces CD36 expression at a transcriptional level, we initially determined macrophage CD36 mRNA expression in response to BSO treatment. The results in Fig. 5A show that BSO had little effect on CD36 mRNA levels, which implies that the induction of CD36 protein expression does not occur at the transcriptional level. Furthermore, we determined that antioxidant or antioxidant enzyme alone or in combination with BSO had little effect on CD36 mRNA expression either (Fig. 4B), which indicates that antioxidant (enzyme) blocks BSO-induced CD36 protein expression at a post-transcriptional level also. In contrast to BSO alone, co-treatment of cells with LDL and BSO increased CD36 mRNA expression (Fig. 5C) because the co-treatment generated oxLDL (31) (Fig. 2D), which activates CD36 transcription (11, 13).
FIGURE 5.

BSO does not activate CD36 transcription. RAW264.7 cells received the following treatment. A, BSO at the indicated concentrations for 16 h or with 5 μm BSO for the indicated times. B, CAT (10 units/ml), SOD (500 units/ml), NAC (10 mm), and NMMA (0.1 mm) or each plus BSO (5 μm) overnight. C, LDL or LDL plus BSO overnight. Expression of CD36 mRNA was determined by real time RT-PCR. *, p < 0.05 versus control (n = 3). D, RAW264.7 cells were treated with actinomycin D (ActD, 5 μg/ml) or actinomycin D plus BSO (5 μm) for the indicated times. Expression of CD36 and GAPDH mRNA was determined by RT-PCR. E, ∼90% confluent 293T cells were transfected with DNA for the CD36 promoter, PPARγ and RXRα expression vectors, and Renilla luciferase for 4 h followed by treatment with BSO or BSO plus pioglitazone overnight. After transfection and treatment, the cellular lysate was extracted for determination of the activity of firefly and Renilla luciferases. F, peritoneal macrophages isolated from the PPARγfl/fl and the conditional macrophage PPARγ knock-out (MacPPARγ KO) mice were treated with BSO at the indicated concentrations overnight followed by determination of PPARγ and CD36 protein expression. G, RAW264.7 cells were treated with BSO at the indicated concentrations (left panel) or BSO plus CAT as indicated (right panel) overnight. Expression of total STAT1 and pSTAT1 was determined by Western blot.
To determine the effect of BSO on CD36 mRNA stability, macrophages were treated with actinomycin D or actinomycin D plus BSO for different times followed by determination of CD36 and GAPDH mRNA levels. Actinomycin D can potently inhibit cellular RNA transcription. Thus, mRNA levels in the presence of actinomycin D demonstrate the rate of mRNA degradation and mRNA stability indirectly. Fig. 5D shows similar CD36 mRNA levels between the group receiving actinomycin D alone treatment and the group receiving actinomycin D plus BSO treatment, suggesting that BSO has no effect on CD36 mRNA stability.
CD36 transcription is mainly controlled by transcription factor of PPARγ. To further confirm that BSO has no effect on CD36 transcription, we constructed a CD36 promoter (from −1946 to +51), which includes the PPARγ-responsive element motif. As shown in Fig. 5E, high expression of PPARγ and RXRα induced CD36 promoter activity, and the induction was further enhanced by a PPARγ ligand, pioglitazone. However, BSO had little effect on CD36 promoter activity either in the absence or presence of pioglitazone (Fig. 5E).
To further confirm that the induction of CD36 protein expression by BSO is independent of PPARγ activity, we isolated peritoneal macrophages from the conditional macrophage PPARγ knock-out mice and the corresponding control mice and then treated the cells with BSO. As expected, deficiency of PPARγ expression reduced CD36 expression at the basal levels (Fig. 5F), which suggests the importance of PPARγ in CD36 expression. Treatment of control cells with BSO did not affect PPARγ expression. Furthermore, BSO induced CD36 protein expression in both control macrophages and PPARγ-deficient macrophages (Fig. 5F, left panel) at a similar degree (Fig. 5F, right panel), indicating that PPARγ is not involved in BSO-induced CD36 protein expression.
15(S)-Hydroxyeicosatetraenoic acid, the major 15-lipoxygenase 1/2 metabolite of arachidonic acid, has been reported to induce macrophage CD36 expression and foam cell formation through xanthine oxidase and NADPH oxidase-dependent ROS production and STAT1 activation at both transcriptional and translational levels (33). To exclude the induction of CD36 expression by BSO is related to STAT1 activation, we determined expression of total STAT1 (STAT1) and pSTAT1 in response to BSO treatment. The results in Fig. 5G, left panel, demonstrate that BSO has little effect on both STAT1 and pSTAT1 levels. In addition, the presence of antioxidant enzyme, such as CAT, had no effect on STAT1 and pSTAT1 levels either (Fig. 5G, right panel), which suggests that STAT1 activation is not involved in BSO-induced CD36 expression. Taken together, the results in Fig. 5 demonstrate that BSO does not affect CD36 transcription, and neither PPARγ nor STAT1 is involved in BSO induced CD36 protein expression. It also implies that the induction of CD36 protein expression by BSO should occur at the translational level.
To determine the mechanisms by which BSO induced CD36 protein expression, we initially determined if BSO can increase CD36 protein stability. Cycloheximide is a potent inhibitor of cellular protein biosynthesis. Similar to actinomycin D, CD36 protein levels in the presence of cycloheximide demonstrate the rate of CD36 protein degradation and CD36 protein stability indirectly in response BSO treatment. As shown in Fig. 6A, the similar reduction of CD36 protein levels was determined in the presence of cycloheximide alone and cycloheximide plus BSO, which suggests that BSO had no effect on CD36 protein stability.
FIGURE 6.

BSO treatment enhances macrophage CD36 translational efficiency. A, RAW264.7 cells were treated with 5 μm cycloheximide (CHX) or cycloheximide plus 5 μm BSO for the indicated times followed by determination of CD36 protein expression. B, RAW264.7 cells were treated with 5 μm BSO for 16 h. The cells were then used to prepare polysomal RNA fractions followed by determination of CD36 and GAPDH mRNA expression by Northern blot analysis. C, RAW264.7 cells were treated with BSO at the indicated concentrations for 12 h followed by treatment with puromycin (10 μg/ml) for 1 h. After treatment, the cellular protein was extracted and used to conduct input assay by Western blot (IB) with anti-puromycin antibody (left panel). After input assay, the protein samples were used to conduct immunoprecipitation (IP) with anti-CD36 antibody or normal IgG followed by Western blot with anti-puromycin antibody for newly synthesized CD36 protein or with anti-CD36 antibody for whole CD36 protein expression (right panel).
The above results imply that induction of CD36 protein expression by BSO should occur through enhanced CD36 translation. To determine it, we performed the following two experiments. We initially conducted the polysomal RNA fractioned-Northern blot analysis. After BSO treatment, polysomal fractions were prepared from cells by a sucrose gradient centrifugation, and RNA extracted from each fraction was then subjected to Northern blot analysis. The results of Northern blot demonstrate that distribution of GAPDH mRNA in each of the sucrose gradient fractions was similar between control and BSO-treated macrophages. In contrast, more CD36 mRNA derived from BSO-treated macrophages than control cells was found in the heaviest fractions (Fig. 6B, fractions 5 and 6), indicating that BSO increases the number of ribosomes/transcript and enhances CD36 translational efficiency.
The effect of BSO on CD36 protein translation was further determined by SUnSET analysis (27–29). Puromycin can effectively end label newly synthesized proteins (peptides), which can be detected by Western blot with anti-puromycin antibody. Specifically, after immunoprecipitation by anti-CD36 antibody, the newly synthesized CD36 protein can be determined by Western blot with anti-puromycin antibody. Fig. 6C, left panel, demonstrates that BSO had little effect on the global protein translation. However, compared with control sample (Fig. 6C, lane 2, right panel), after immunoprecipitated by anti-CD36 antibody, more puromycin-labeled CD36 protein was determined in samples with BSO treatment (Fig. 6C, lanes 3 and 4, right panel) by Western blot with anti-puromycin antibody. Therefore, by using two different assays, we demonstrate that BSO can enhance CD36 protein translational efficiency.
Discussion
Although antioxidant vitamins, such as vitamins C and E, can reduce ROS production and suppress oxidative stress, the role played by these vitamins in atherosclerosis is still controversial. Recent large clinical trials do not show the favorable effects of antioxidant vitamins on the established atherosclerosis (34, 35). In animal models, selective macrophage ascorbic acid (vitamin C) deficiency surprisingly suppresses early atherosclerosis (36). In contrast, combined vitamin C and E deficiency enhances the development of atherosclerosis in apoE−/− mice, in particular the mice also lacking vitamin C biosynthesis by genetic deleting gulonolactone oxidase (gulo) expression (37). Vitamin E, in combination with coenzyme Q10 (CoQ10) or selenium, also reduces atherosclerosis in apoE−/− mice or rabbits (38, 39).
GSH is an abundant endogenous antioxidant and protects cells against the oxidative stress-induced cell injuries. The anti-atherogenic properties of the GSH-dependent antioxidant system have been determined at different levels. Clinically, an inverse correlation between the activity of red blood cell GPx-1 and the risk of cardiovascular events in patients has been reported (4). Polymorphisms in the 5′ flanking region of the GCL catalytic subunit (GCLC) and GCL modifier subunit (GCLM) genes in patients results in decreased plasma GSH levels and increased risk of myocardial infarction and endothelial dysfunction (40, 41). In animal models alterations of expression of molecules involved in GSH production, utilization, and cycles, such as GCL, GPx-1, and glutathione reductase, influence atherosclerosis in both LDLR−/− and apoE−/− mice (3, 18). Several actions can make contributions to the anti-atherogenic properties of GSH-dependent antioxidant system. For instance, GCL protects macrophages from mitochondrial hyperpolarization induced by oxLDL (3). Deficiency of GPx-1 results in activation of pro-inflammatory and pro-fibrotic pathways (2). Administration of LDLR−/− mice with streptozotocin decreases GSH/GSSG ratios in macrophages and enhances macrophage recruitment into aortic wall, thereby accelerating the development of atherosclerosis (42).
The formation of foam cells is the initial and critical step in the development of atherosclerosis, and it is completed by uptake of modified LDL (mainly oxLDL) by macrophage scavenger receptors including CD36. Formation of foam cells is also influenced by the balance between pro-oxidants and antioxidants. Antioxidants can prevent LDL oxidation as well as cellular lipid accumulation (43). In contrast, the overproduction of ROS can enhance oxLDL generation. It may also result in activation of molecules, which are responsible for oxLDL uptake. For instance, activation of Toll-like receptor 9 by CpG oligodeoxynucleotide increases ROS production and activates expression of lectin-like oxLDL receptor-1 (Lox1) at the transcriptional level, another macrophage scavenger receptor for oxLDL, thereby enhancing oxLDL uptake and formation of foam cells (44).
CD36 facilitates foam cell formation by binding and internalizing oxLDL. Expression of macrophage CD36 can also be regulated by cellular oxidative status. Treatment of macrophages with 15(S)-hydroxyeicosatetraenoic acid enhances ROS production, which results in increased oxLDL uptake and foam cell formation (33). In contrast, vitamin E or α-tocopherol decrease macrophage CD36 expression and oxLDL uptake (45, 46). Similarly, in our study treatment of macrophage with BSO decreased cellular GSH levels and GSH/GSSG ratios while increasing ROS production, CD36 expression, and cellular oxLDL uptake (Figs. 1 and 2). In contrast, amelioration of cellular GSH/GSSG status by antioxidant (NAC) and antioxidant enzymes (CAT and SOD) restored BSO-decreased GSH/GSSG ratios thereby blocking BSO-induced CD36 expression and oxLDL uptake (Fig. 3, B–D). Induction of CD36 expression by GCL siRNA (Fig. 1, G and I) indicates the direct effect of cellular GSH/GSSG status on cellular oxLDL uptake. Lack of CD36 expression abolished BSO-induced oxLDL uptake (Fig. 3F), demonstrating the direct contribution by BSO-induced CD36 expression.
Expression of CD36 is transcriptionally activated by PPARγ. Regulation of CD36 expression by vitamin E and 15(S)-hydroxyeicosatetraenoic acid is also completed at the transcriptional level (33, 45, 46). In contrast, in our study, although we determined that BSO substantially induced macrophage CD36 protein expression and cellular oxLDL uptake in a CD36-dependent manner while the induction was blocked by antioxidant (enzyme), we observed that BSO or BSO plus antioxidant (enzyme) had little effect on CD36 mRNA levels and promoter activity (Fig. 5, A–D). In addition, we excluded the involvement of PPARγ and STAT1 in BSO-induced CD36 expression (Fig. 5, E–G). These results suggest that the post-transcriptional regulation should play a critical role in BSO-induced CD36 expression. In fact, we demonstrated that BSO enhanced CD36 translational efficiency (Fig. 6).
The translational regulation of CD36 expression in different cell types has been reported by a few studies. In macrophages, glucose increased CD36 translation due to the existence of the specific features of CD36 transcript. A 5′-UTR length of >200 bp that contains 3 upstream open reading frames (uORFs) and several stable predicted secondary structures exists in CD36 transcript. The first ORF plays a critical role for induction of CD36 translation in response to glucose treatment by increasing translation reinitiation (15). In hepatocytes, activation of mTOR signaling pathway is involved in regulation of CD36 translation. Induction of inflammatory stress in HepG2 cells by TNF-α and IL-6 treatment or in mice by administration of casein increases cellular lipid accumulation and CD36 protein expression through enhancement of CD36 translational efficiency, which is blocked by rapamycin, an mTOR-specific inhibitor (47). The induction of hepatic CD36 translation by palmitate is also completed through activation of the mTOR signaling pathway (48). However, the interaction of ROS and mTOR is still unclear. Cadmium induces ROS production to activate mTOR pathway, leading to neuronal cell death (49). In contrast, mTOR pathway is inhibited by hydrogen peroxide (49). Salvianolic acid A activates mTOR by reducing ROS production (50). In our study inhibition of GSH production by BSO or GCL siRNA induced macrophage CD36 expression and enhanced oxLDL uptake, which was associated with increased ROS production and blocked by antioxidant (enzyme). Although we determined the induction of CD36 expression occurred at the translational level, it needs more investigation to unveil if this induction is related to enhanced reinitiation of CD36 translation or activation of mTOR signaling pathway.
In summary, our study not only demonstrates that macrophage CD36 expression and oxLDL uptake can be regulated by multiple mechanisms including the translational regulation but also suggests an anti-atherogenic property of GSH-dependent antioxidant system by providing cellular and molecular evidence.
Author Contributions
J. H. and Y. D. designed the study, X. Y., H. Y., Y. C., L. S., Y. L., X. M., S. D., X. L., and R. X. performed experiments, and J. H., Y. D., and X. Y. wrote the manuscript.
This work was supported by National Science Foundation of China (NSFC) Grants 81272460 and 81473204 (to J. H.) and 31400694 (to Y. C.), Tianjin Municipal Science and Technology Commission of China Grants 14JCYBJC25100 (to Y. D.) and 13JCYBJC24600 (to X. L.), respectively, and the Ph.D. Candidate Research Innovation Fund of Nankai University (to X. Y.). The authors declare that they have no conflicts of interest with the contents of this article.
- ROS
- reactive oxygen species
- oxLDL
- oxidized low-density lipoprotein
- PPAR
- peroxisome proliferator-activated receptor
- RXRα
- retinoid X receptor α
- GCL
- glutamate-cysteine ligase
- GPx-1
- glutathione peroxidase-1
- BSO
- l-buthionine-S,R-sulfoximine
- NAC
- N-acetyl cysteine
- CAT
- catalase
- SOD
- superoxide dismutase
- l-NMMA
- NG-monomethyl-l-arginine
- pSTAT1
- phosphorylated STAT1.
References
- 1. Callegari A., Liu Y., White C. C., Chait A., Gough P., Raines E. W., Cox D., Kavanagh T. J., Rosenfeld M. E. (2011) Gain and loss of function for glutathione synthesis: impact on advanced atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 31, 2473–2482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lewis P., Stefanovic N., Pete J., Calkin A. C., Giunti S., Thallas-Bonke V., Jandeleit-Dahm K. A., Allen T. J., Kola I., Cooper M. E., de Haan J. B. (2007) Lack of the antioxidant enzyme glutathione peroxidase-1 accelerates atherosclerosis in diabetic apolipoprotein E-deficient mice. Circulation 115, 2178–2187 [DOI] [PubMed] [Google Scholar]
- 3. Qiao M., Kisgati M., Cholewa J. M., Zhu W., Smart E. J., Sulistio M. S., Asmis R. (2007) Increased expression of glutathione reductase in macrophages decreases atherosclerotic lesion formation in low-density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 27, 1375–1382 [DOI] [PubMed] [Google Scholar]
- 4. Blankenberg S., Rupprecht H. J., Bickel C., Torzewski M., Hafner G., Tiret L., Smieja M., Cambien F., Meyer J., Lackner K. J. (2003) Glutathione peroxidase 1 activity and cardiovascular events in patients with coronary artery disease. N. Engl. J. Med. 349, 1605–1613 [DOI] [PubMed] [Google Scholar]
- 5. Silverstein R. L., Li W., Park Y. M., Rahaman S. O. (2010) Mechanisms of cell signaling by the scavenger receptor CD36: implications in atherosclerosis and thrombosis. Trans. Am. Clin. Climatol. Assoc. 121, 206–220 [PMC free article] [PubMed] [Google Scholar]
- 6. Endemann G., Stanton L. W., Madden K. S., Bryant C. M., White R. T., Protter A. A. (1993) CD36 is a receptor for oxidized low density lipoprotein. J. Biol. Chem. 268, 11811–11816 [PubMed] [Google Scholar]
- 7. Rahaman S. O., Lennon D. J., Febbraio M., Podrez E. A., Hazen S. L., Silverstein R. L. (2006) A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell. Metab. 4, 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nicholson A. C., Frieda S., Pearce A., Silverstein R. L. (1995) Oxidized LDL binds to CD36 on human monocyte-derived macrophages and transfected cell lines. Evidence implicating the lipid moiety of the lipoprotein as the binding site. Arterioscler. Thromb. Vasc. Biol. 15, 269–275 [DOI] [PubMed] [Google Scholar]
- 9. Nozaki S., Kashiwagi H., Yamashita S., Nakagawa T., Kostner B., Tomiyama Y., Nakata A., Ishigami M., Miyagawa J., Kameda-Takemura K. (1995) Reduced uptake of oxidized low density lipoproteins in monocyte-derived macrophages from CD36-deficient subjects. J. Clin. Invest. 96, 1859–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Febbraio M., Podrez E. A., Smith J. D., Hajjar D. P., Hazen S. L., Hoff H. F., Sharma K., Silverstein R. L. (2000) Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J. Clin. Invest. 105, 1049–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Han J., Hajjar D. P., Febbraio M., Nicholson A. C. (1997) Native and modified low density lipoproteins increase the functional expression of the macrophage class B scavenger receptor, CD36. J. Biol. Chem. 272, 21654–21659 [DOI] [PubMed] [Google Scholar]
- 12. Tontonoz P., Nagy L., Alvarez J. G., Thomazy V. A., Evans R. M. (1998) PPARγ promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell 93, 241–252 [DOI] [PubMed] [Google Scholar]
- 13. Nagy L., Tontonoz P., Alvarez J. G., Chen H., Evans R. M. (1998) Oxidized LDL regulates macrophage gene expression through ligand activation of PPARγ. Cell 93, 229–240 [DOI] [PubMed] [Google Scholar]
- 14. Kersten S., Desvergne B., Wahli W. (2000) Roles of PPARs in health and disease. Nature 405, 421–424 [DOI] [PubMed] [Google Scholar]
- 15. Griffin E., Re A., Hamel N., Fu C., Bush H., McCaffrey T., Asch A. S. (2001) A link between diabetes and atherosclerosis: glucose regulates expression of CD36 at the level of translation. Nat. Med. 7, 840–846 [DOI] [PubMed] [Google Scholar]
- 16. Griffith O. W., Meister A. (1985) Origin and turnover of mitochondrial glutathione. Proc. Natl. Acad. Sci. U.S.A. 82, 4668–4672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meister A., Anderson M. E. (1983) Glutathione. Annu. Rev. Biochem. 52, 711–760 [DOI] [PubMed] [Google Scholar]
- 18. Torzewski M., Ochsenhirt V., Kleschyov A. L., Oelze M., Daiber A., Li H., Rossmann H., Tsimikas S., Reifenberg K., Cheng F., Lehr H. A., Blankenberg S., Förstermann U., Münzel T., Lackner K. J. (2007) Deficiency of glutathione peroxidase-1 accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 27, 850–857 [DOI] [PubMed] [Google Scholar]
- 19. Griffith O. W., Meister A. (1979) Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine). J. Biol. Chem. 254, 7558–7560 [PubMed] [Google Scholar]
- 20. Chen Y., Duan Y., Kang Y., Yang X., Jiang M., Zhang L., Li G., Yin Z., Hu W., Dong P., Li X., Hajjar D. P., Han J. (2012) Activation of liver X receptor induces macrophage interleukin-5 expression. J. Biol. Chem. 287, 43340–43350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Babaev V. R., Yancey P. G., Ryzhov S. V., Kon V., Breyer M. D., Magnuson M. A., Fazio S., Linton M. F. (2005) Conditional knockout of macrophage PPAR increases atherosclerosis in C57BL/6 and low-density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 25, 1647–1653 [DOI] [PubMed] [Google Scholar]
- 22. Rahman I., Kode A., Biswas S. K. (2006) Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 1, 3159–3165 [DOI] [PubMed] [Google Scholar]
- 23. Aranda A., Sequedo L., Tolosa L., Quintas G., Burello E., Castell J. V., Gombau L. (2013) Dichloro-dihydro-fluorescein diacetate (DCFH-DA) assay: a quantitative method for oxidative stress assessment of nanoparticle-treated cells. Toxicol. In Vitro 27, 954–963 [DOI] [PubMed] [Google Scholar]
- 24. Zhang B., Zhang Z., Xia S., Xing C., Ci X., Li X., Zhao R., Tian S., Ma G., Zhu Z., Fu L., Dong J. T. (2013) KLF5 activates microRNA 200 transcription to maintain epithelial characteristics and prevent induced epithelial-mesenchymal transition in epithelial cells. Mol. Cell. Biol. 33, 4919–4935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mach M., White M. W., Neubauer M., Degen J. L., Morris D. R. (1986) Isolation of a cDNA clone encoding S-adenosylmethionine decarboxylase: expression of the gene in mitogen-activated lymphocytes. J. Biol. Chem. 261, 11697–11703 [PubMed] [Google Scholar]
- 26. Sun L., Nicholson A. C., Hajjar D. P., Gotto A. M. Jr., Han J. (2003) Adipogenic differentiating agents regulate expression of fatty acid binding protein and CD36 in the J744 macrophage cell line. J. Lipid Res. 44, 1877–1886 [DOI] [PubMed] [Google Scholar]
- 27. Goodman C. A., Mabrey D. M., Frey J. W., Miu M. H., Schmidt E. K., Pierre P., Hornberger T. A. (2011) Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. FASEB J. 25, 1028–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schmidt E. K., Clavarino G., Ceppi M., Pierre P. (2009) SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 6, 275–277 [DOI] [PubMed] [Google Scholar]
- 29. David A., Dolan B. P., Hickman H. D., Knowlton J. J., Clavarino G., Pierre P., Bennink J. R., Yewdell J. W. (2012) Nuclear translation visualized by ribosome-bound nascent chain puromycylation. J. Cell Biol. 197, 45–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen Y., Shertzer H. G., Schneider S. N., Nebert D. W., Dalton T. P. (2005) Glutamate cysteine ligase catalysis: dependence on ATP and modifier subunit for regulation of tissue glutathione levels. J. Biol. Chem. 280, 33766–33774 [DOI] [PubMed] [Google Scholar]
- 31. Rosenblat M., Aviram M. (1998) Macrophage glutathione content and glutathione peroxidase activity are inversely related to cell-mediated oxidation of LDL: in vitro and in vivo studies. Free. Radic. Biol. Med. 24, 305–317 [DOI] [PubMed] [Google Scholar]
- 32. Nikulina M. A., Andersen H. U., Karlsen A. E., Darville M. I., Eizirik D. L., Mandrup-Poulsen T. (2000) Glutathione depletion inhibits IL-1 β-stimulated nitric oxide production by reducing inducible nitric oxide synthase gene expression. Cytokine 12, 1391–1394 [DOI] [PubMed] [Google Scholar]
- 33. Kotla S., Singh N. K., Traylor J. G. Jr., Orr A. W., Rao G. N. (2014) ROS-dependent Syk and Pyk2-mediated STAT1 activation is required for 15(S)-hydroxyeicosatetraenoic acid-induced CD36 expression and foam cell formation. Free. Radic. Biol. Med. 76, 147–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sesso H. D., Buring J. E., Christen W. G., Kurth T., Belanger C., MacFadyen J., Bubes V., Manson J. E., Glynn R. J., Gaziano J. M. (2008) Vitamins E and C in the prevention of cardiovascular disease in men: the Physicians' Health Study II randomized controlled trial. JAMA 300, 2123–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cook N. R., Albert C. M., Gaziano J. M., Zaharris E., MacFadyen J., Danielson E., Buring J. E., Manson J. E. (2007) A randomized factorial trial of vitamins C and E and β carotene in the secondary prevention of cardiovascular events in women: results from the Women's Antioxidant Cardiovascular Study. Arch. Intern. Med. 167, 1610–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Babaev V. R., Whitesell R. R., Li L., Linton M. F., Fazio S., May J. M. (2011) Selective macrophage ascorbate deficiency suppresses early atherosclerosis. Free. Radic. Biol. Med. 50, 27–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Babaev V. R., Li L., Shah S., Fazio S., Linton M. F., May J. M. (2010) Combined vitamin C and vitamin E deficiency worsens early atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 30, 1751–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thomas S. R., Leichtweis S. B., Pettersson K., Croft K. D., Mori T. A., Brown A. J., Stocker R. (2001) Dietary cosupplementation with vitamin E and coenzyme Q10 inhibits atherosclerosis in apolipoprotein E gene knockout mice. Arterioscler. Thromb. Vasc. Biol. 21, 585–593 [DOI] [PubMed] [Google Scholar]
- 39. Schwenke D. C., Behr S. R. (1998) Vitamin E combined with selenium inhibits atherosclerosis in hypercholesterolemic rabbits independently of effects on plasma cholesterol concentrations. Circ. Res. 83, 366–377 [DOI] [PubMed] [Google Scholar]
- 40. Koide S., Kugiyama K., Sugiyama S., Nakamura S., Fukushima H., Honda O., Yoshimura M., Ogawa H. (2003) Association of polymorphism in glutamate-cysteine ligase catalytic subunit gene with coronary vasomotor dysfunction and myocardial infarction. J. Am. Coll. Cardiol. 41, 539–545 [DOI] [PubMed] [Google Scholar]
- 41. Nakamura S., Kugiyama K., Sugiyama S., Miyamoto S., Koide S., Fukushima H., Honda O., Yoshimura M., Ogawa H. (2002) Polymorphism in the 5′-flanking region of human glutamate-cysteine ligase modifier subunit gene is associated with myocardial infarction. Circulation 105, 2968–2973 [DOI] [PubMed] [Google Scholar]
- 42. Qiao M., Zhao Q., Lee C. F., Tannock L. R., Smart E. J., LeBaron R. G., Phelix C. F., Rangel Y., Asmis R. (2009) Thiol oxidative stress induced by metabolic disorders amplifies macrophage chemotactic responses and accelerates atherogenesis and kidney injury in LDL receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 29, 1779–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Aviram M. (1999) Macrophage foam cell formation during early atherogenesis is determined by the balance between pro-oxidants and anti-oxidants in arterial cells and blood lipoproteins. Antioxid. Redox. Signal. 1, 585–594 [DOI] [PubMed] [Google Scholar]
- 44. Lee J. G., Lim E. J., Park D. W., Lee S. H., Kim J. R., Baek S. H. (2008) A combination of Lox-1 and Nox1 regulates TLR9-mediated foam cell formation. Cell. Signal. 20, 2266–2275 [DOI] [PubMed] [Google Scholar]
- 45. Ricciarelli R., Zingg J. M., Azzi A. (2000) Vitamin E reduces the uptake of oxidized LDL by inhibiting CD36 scavenger receptor expression in cultured aortic smooth muscle cells. Circulation 102, 82–87 [DOI] [PubMed] [Google Scholar]
- 46. Devaraj S., Hugou I., Jialal I. (2001) α-Tocopherol decreases CD36 expression in human monocyte-derived macrophages. J. Lipid Res. 42, 521–527 [PubMed] [Google Scholar]
- 47. Wang C., Hu L., Zhao L., Yang P., Moorhead J. F., Varghese Z., Chen Y., Ruan X. Z. (2014) Inflammatory stress increases hepatic CD36 translational efficiency via activation of the mTOR signalling pathway. PloS ONE 9, e103071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang C., Yan Y., Hu L., Zhao L., Yang P., Moorhead J. F., Varghese Z., Chen Y., Ruan X. Z. (2014) Rapamycin-mediated CD36 translational suppression contributes to alleviation of hepatic steatosis. Biochem. Biophys. Res. Commun. 447, 57–63 [DOI] [PubMed] [Google Scholar]
- 49. Chen L., Xu B., Liu L., Luo Y., Yin J., Zhou H., Chen W., Shen T., Han X., Huang S. (2010) Hydrogen peroxide inhibits mTOR signaling by activation of AMPKα leading to apoptosis of neuronal cells. Lab. Invest. 90, 762–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang H., Liu Y. Y., Jiang Q., Li K. R., Zhao Y. X., Cao C., Yao J. (2014) Salvianolic acid A protects RPE cells against oxidative stress through activation of Nrf2/HO-1 signaling. Free Radic. Biol. Med. 69, 219–228 [DOI] [PubMed] [Google Scholar]