

Summary
Dimethyl fumarate and synthetic oleanane triterpenoids are effective inducers of the cytoprotective Nrf2 transcription factor. However, we show that they have very different effects on experimental lung carcinogenesis.
Abstract
Lung cancer accounts for the highest number of cancer-related deaths in the USA, highlighting the need for better prevention and therapy. Activation of the Nrf2 pathway detoxifies harmful insults and reduces oxidative stress, thus preventing carcinogenesis in various preclinical models. However, constitutive activation of the Nrf2 pathway has been detected in numerous cancers, which confers a survival advantage to tumor cells and a poor prognosis. In our study, we compared the effects of two clinically relevant classes of Nrf2 activators, dimethyl fumarate (DMF) and the synthetic oleanane triterpenoids, CDDO-imidazolide (CDDO-Im) and CDDO-methyl ester (CDDO-Me) in RAW 264.7 mouse macrophage-like cells, in VC1 lung cancer cells and in the A/J model of lung cancer. Although the triterpenoids and DMF both activated the Nrf2 pathway, CDDO-Im and CDDO-Me were markedly more potent than DMF. All of these drugs reduced the production of reactive oxygen species and inhibited nitric oxide production in RAW264.7 cells, but the triterpenoids were 100 times more potent than DMF in these assays. Microarray analysis revealed that only 52 of 99 Nrf2-target genes were induced by all three compounds, and each drug regulated a unique subset of Nrf2 genes. These drugs also altered the expression of other genes important in lung cancer independent of Nrf2. Although all three compounds enhanced the phosphorylation of CREB, only DMF increased the phosphorylation of Akt. CDDO-Me, at either 12.5 or 50mg/kg of diet, was the most effective drug in our lung cancer mouse model. Specifically, CDDO-Me significantly reduced the average tumor number, size and burden compared with the control group (P < 0.05). Additionally, 52% of the tumors in the control group were high-grade tumors compared with only 14% in the CDDO-Me group. Though less potent, CDDO-Im had similar activity as CDDO-Me. In contrast, 61–63% of the tumors in the DMF groups (400–1200mg/kg diet) were high-grade tumors compared with 52% for the controls (P < 0.05). Additionally, DMF significantly increased the average number of tumors compared with the controls (P < 0.05). Thus, in contrast to the triterpenoids, which effectively reduced pathogenesis in A/J mice, DMF enhanced the severity of lung carcinogenesis in these mice. Collectively, these results suggest that although CDDO-Im, CDDO-Me and DMF all activate the Nrf2 pathway, they target distinct genes and signaling pathways, resulting in opposite effects for the prevention of experimental lung cancer.
Introduction
Despite intensive research efforts aimed at finding an effective treatment for lung cancer, this disease is still the primary cause of cancer-related mortality in the USA. Lung cancer accounts for >220000 deaths each year or 25% of all cancer-related deaths (1). The 5 year survival for lung cancer is <18% (1), which is considerably lower than the survival rates for most other types of cancer. Hence, the search for effective therapeutic agents to treat lung cancer as well as methods to prevent this disease continues.
The Nrf2 pathway has been explored as a potential target for both the prevention and treatment of a variety of cancers. NFE2-related factor 2 is a transcription factor that serves as a defense system in response to damaging stimuli that disrupt homeostasis. Under basal conditions, Nrf2 is targeted for ubiquitination and proteasomal degradation through its interaction with its repressor, Kelch-like erythroid-associated protein 1 (KEAP1). However, in the presence of oxidative stress or electrophiles, numerous reactive cysteines in KEAP1 can be modified, resulting in a conformational change that releases the Nrf2 protein. Activated Nrf2 then enters the nucleus and dimerizes with members of the masculoaponeurotic fibrosarcoma protein family, which facilitates the binding of Nrf2 to the antioxidant response element on target genes. The binding of Nrf2 to the antioxidant response element induces the transcription of a wide array of cytoprotective phase II enzymes, such as NAD(P)H:quinone oxidoreductase 1 (NQO1) and heme oxygenase 1 (HO-1), which are responsible for detoxifying insults and reducing oxidative stress (2–4).
Because DNA damage leads to elevated levels of reactive oxygen species (ROS) and inflammation, which contribute to tumor development, the activation of the Nrf2 pathway is an important target for the prevention of cancer. The activation of Nrf2 by numerous natural products or by genetic manipulation can inhibit carcinogenesis, especially at its earliest stages, in a wide variety of experimental cancer models (5,6). However, the antitumorigenic properties of Nrf2 are context dependent. Studies have shown that the activation of Nrf2 can have either a positive or a negative effect in the progression of lung cancer (7). Constitutive activation of Nrf2 caused by mutations in the Nrf2 pathway has been found in lung, breast, stomach, pancreas and colorectal cancer (8). Moreover, elevated Nrf2 levels can protect tumor cells from radiation or chemotherapy, causing them to be resistant to treatment. Consequently, activation of Nrf2 in cancer cells can provide a survival advantage that allows them to proliferate and promote carcinogenesis (2,4,9,10). Because Nrf2 can play a beneficial or detrimental role in cancer, careful considerations are essential for optimizing the use of drugs that target the Nrf2 pathway in lung cancer.
The synthetic oleanane triterpenoids are a class of multifunctional drugs that have been used successfully to delay tumor progression or treat established tumors in a variety of preclinical cancer models (11). One of these triterpenoids, CDDO-methyl ester (CDDO-Me, bardoxolone methyl), displayed promising anticancer activity in patients in early clinical trials (12,13). Nanomolar concentrations of CDDO-Me and CDDO-imidazolide (CDDO-Im) inhibit inducible nitric oxide (NO) synthase and ROS production in cells stimulated with inflammatory cytokines (14), and these triterpenoids are among the most potent known inducers of the Nrf2 pathway and the NQO1 and HO-1 proteins (15). However, Nrf2 is not the only target of the triterpenoids, as these drugs interact with other proteins containing reactive cysteine residues, including IkappaB (IKK), STAT3 (signal transducer and activation of transcription 3), HER2 (human epidermal growth factor receptor 2) and mammalian target of rapamycin (mTOR), albeit at higher doses than required for Nrf2 activation (11). CDDO-Me markedly reduced the number, size and severity of tumors in a vinyl carbamate-induced model of lung cancer (16,17); but only a single moderate dose was tested, and this concentration probably targeted other pathways in addition to Nrf2. The effect of CDDO-Im has not been tested in this lung carcinogenesis model.
In 2013, the Food and Drug Administration approved dimethyl fumarate (DMF; Tecfidera®) for the treatment of relapsing-remitting multiple sclerosis, a disease which affects 350000 people in the USA alone. DMF was originally approved in Germany for the treatment of psoriasis due to its immunomodulatory activity (18), but the anti-inflammatory properties of this drug and its ability to activate the Nrf2 pathway also contribute to its mechanism of action (19–23). DMF inhibits proliferation and induces apoptosis in a variety of human cancer cells (24–28), blocks invasion and angiogenesis (26) and reduces tumor growth and metastasis in vitro and in vivo (29,30). Moreover, fumaric acid can suppress the development of chemically induced cancer in the liver, lung and stomach (31,32). The ability of DMF to prevent lung adenocarcinomas induced by vinyl carbamate is not known.
Because of the emerging importance and apparently conflicting roles of the Nrf2 pathway in cancer, we studied three clinically relevant drugs with similar biological activities. The triterpenoids, CDDO-Im and CDDO-Me, are among the most potent activators of the Nrf2 pathway reported in the literature, and DMF is the first Food and Drug Administration-approved drug known to activate this cytoprotective pathway. In these experiments, we compared the ability of these three drugs to inhibit inflammation, induce the Nrf2-target gene NQO1, differentially regulate gene expression and suppress lung carcinogenesis.
Materials and methods
Reagents, cell culture and western blotting
CDDO-Im and CDDO-Me were synthesized as described previously (33,34). DMF and tert-butyl hydroperoxide (tBHP) were purchased from Sigma (St. Louis, MO), MG132 from EMD Millipore (Billerica, MA) and 2′,7′-dichlorodihydrofluorescein diacetate from Molecular Probes (Grand Island, NY). VC1 cells were derived from a lung tumor in an A/J mouse (16). Both VC1 and RAW 264.7 mouse macrophage-like cells (American Type Culture Collection) were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. Nrf2 wild-type (WT) (+/+) and Nrf2 knockout (KO) (−/−) mouse embryonic fibroblasts (MEFs) were generously provided by Dr Jefferson Chan (UC Irvine). These cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 15% fetal bovine serum, 0.05mM of 2-mercaptoethanol, 1mM of non-essential amino acids and 2mM of glutamine. RAW264.7 cells from American Type Culture Collection are tested and authenticated using morphology, karyotyping and PCR-based approaches, and VC1 cells and MEFs have been characterized as described previously (16,35,36). Vials frozen from the 2nd through 5th passage (RAW 264.7 cells) or 3rd through 12th passage (VC1 cells and MEFs) in culture were used for these experiments. For western blotting, VC1 cells or MEFs were treated with compounds, lysed in TNTE (50mM of Tris-HCl, 150mM of NaCl, 0.5% of TX-100, 1mM of ethylenediaminetetraacetic acid) buffer with protease inhibitors and processed for western blotting using the following antibodies: NQO1 (Abcam, Cambridge, MA); HO-1 (Enzo Life Sciences, Farmingdale, NY); p85 (R & D Systems, Minneapolis, MN); Akt, phospho-Akt, cAMP-response element-binding (CREB) protein and phospho-CREB (Cell Signaling Technology, Danvers, MA); and Sox9, GAPDH, α-tubulin, Nrf2 and all secondary antibodies (Santa Cruz Biotechnologies, Dallas, TX).
NQO1, HO-1 and AKR1B8 induction by qPCR
To examine NQO1 and HO-1 gene induction, VC1 cells and RAW 264.7 cells were treated with dimethyl sulfoxide (DMSO, control) or drugs for 6h. RNA was isolated using the Qiagen RNAasy kit (Valencia, CA). Two micrograms of RNA was used for reverse transcription, and 1 μl of complementary DNA from this reaction was added to 12.5 μl of Bio-Rad iQ SYBRGREEN Supermix (Hercules, CA), 1 μl of validated RT2 quantitative PCR (qPCR) primers from Qiagen (Valencia, CA) and DNAse-free water for real-time qPCR. For in vivo studies, vehicle, CDDO-Im, CDDO-Me or DMF were administered by intraperitoneal (i. p.) injection or by gavage to mice. Lung and liver tissues were collected after 4h. RNA was extracted by the Trizol method and qPCR was performed as described previously. All expression data were normalized using actin as the housekeeping control.
Inducible NO synthase assay
RAW 264.7 cells were plated in 96-well plates and incubated with various concentrations of drugs and stimulated with either 10ng/ml of interferon-γ (IFNγ) or 3ng/ml of lipopolysaccharide (LPS) for 24h. NO levels in media were measured in the form of nitrite by the Griess reaction (37).
Detection of ROS
Cells were treated with compounds for 16h and then incubated with 10 μM of the indicator 2′,7′-dichlorodihydrofluorescein diacetate for 30min. To induce oxidative stress, cells were challenged with 250 μM of tBHP for 15min. Samples were washed with phosphate-buffered saline, trypsinized and analyzed by flow cytometry. To measure the intracellular production of ROS, an excitation wavelength of 480nm and an emission wavelength of 525nm were used. Data were analyzed using Flowjo software.
Microarray studies
VC1 cells were treated with DMSO, 0.05 μM of CDDO-Im, 0.1 μM of CDDO-Me or 15 μM of DMF in triplicate wells for 6h, and RNA was extracted using the Qiagen RNAeasy kit. Two hundred and fifty nanograms of RNA was amplified and labeled using the TargetAmp™-Nano Labeling Kit from Epicentre (Madison, WI) according to the manufacturer’s protocol. One and a half micrograms of labeled RNA was then hybridized onto the Illumina Mouse WG-6 v2 Expression BeadChip (San Diego, CA) and read using the Illumina BeadArray reader. The microarray analyses were performed using BRB-Array Tools Version 4.2.1 (Biometric Research Branch at the National Cancer Institute). Variance stabilizing transformation was applied to raw intensity data that were then normalized using robust spline normalization and filtered to remove non-detected spots as determined by Illumina BeadStudio Software. We identified genes that were differentially expressed among two classes using a random-variance t-test (or F-test among multiple classes) with a P value cutoff of 0.01 (38). Hierarchical or K-means clustering was employed using a Euclidean distance measure to generate heat maps for subsets of significant genes using the open source software Cluster/Treeview (39). The commercial software Pathway Studio software (Ariadne Genomics/Elsevier) was used to identify ontology groups and pathways statistically enriched in the gene set. The pathways (both canonical and non-canonical) are based on a curated database created using information extracted from published literature.
In vivo experiments
All animal studies were done in accordance with protocols approved by the Institutional Animal Care and Use Committee at Dartmouth. Six- to seven-week-old female A/J mice (Jackson Laboratory, Bar Harbor, ME) were given two i. p. injections, 1 week apart, of 0.32mg vinyl carbamate (Toronto Research Chemicals, Ontario, Canada) in acidified saline (pH 5). One week later, mice were randomized and fed either control AIN-93G diet or diet containing CDDO-Im (50, 100 or 200mg/kg of diet), CDDO-Me (12.5 or 50mg/kg of diet) or DMF (400, 800 or 1200mg/kg of diet). After 15–16 weeks on diet, lungs were removed and inflated with formalin. The left lung of each mouse was step sectioned (200 μm between sections) starting at the medial hilar surface, and sections were stained with hematoxylin and eosin. The number of tumors, size and histopathology were evaluated on two sections from each lung. All analyses of the lung were done in a blinded manner by two independent investigators. The tumors were assigned a grade of high, medium or low according to published criteria (17,40). Induction of the Nrf2-target gene AKR1B8 was analyzed from the livers of these mice by qPCR.
Statistical analyses
Results are described as mean ± standard error of the mean. In vitro data were analyzed by one-way analysis of variance followed by a Sidak’s multiple comparisons test or the Tukey test (Prism 6). Results from the in vivo studies were analyzed by one-way analysis of variance and the Tukey test or by one-way analysis of variance on ranks followed by Dunn’s test (SigmaStat3.5).
Results
CDDO-Im, CDDO-Me and DMF activate the Nrf2 pathway and reduce inflammatory and oxidative stress
The pathways that regulate inflammation and oxidative stress work intricately together to maintain homeostasis in the body. The deregulation of these processes has long been known to contribute to tumor development (11). As such, the synthetic triterpenoids were initially developed as anticancer agents and screened for their ability to inhibit inflammation. In order to compare the antioxidative and anti-inflammatory properties of the triterpenoids (CDDO-Im and CDDO-Me) and DMF, we first examined the concentration ranges at which these drugs activate the Nrf2 pathway in RAW 264.7 mouse macrophage-like cells (11). The activation of the Nrf2 transcription factor induces numerous cytoprotective proteins including NQO1 and HO-1. When RAW 264.7 cells were incubated with DMSO (control) or various concentrations of drugs for 6h, all compounds significantly (P < 0.05) induced both NQO1 and HO-1 gene expression, but the triterpenoids were much more potent than DMF (Figure 1A). NQO1 messenger RNA (mRNA) levels increased nearly 100-fold above control levels when cells were incubated with concentrations as low as 0.025 μM of CDDO-Im and 0.05 μM CDDO-Me. However, 10 μM of DMF was necessary to elicit similar effects in these cells (Figure 1A, left panel). The robust induction of NQO1 mRNA was not dose-dependent, suggesting that a maximum threshold was reached. Conversely, the induction of HO-1 was dose-dependent with all three compounds (Figure 1A, right panel), but the triterpenoids again were active at much lower concentrations (0.025–0.1 µM) than DMF (>10 µM). Surprisingly, the induction of HO-1 mRNA by CDDO-Im was significantly (P < 0.05) higher not only when compared with controls but also when compared with cells treated with CDDO-Me or DMF.
Figure 1.

CDDO-Im (Im), CDDO-Me (Me) and DMF induce NQO1 and HO-1 gene expression and inhibit NO and ROS production in RAW 264.7 cells. RAW 264.7 macrophage-like cells were treated with DMSO or various concentrations of Im, Me or DMF. In (A), cells were lysed after 6h and RNA was extracted to measure NQO1 (left panel) and HO-1 (right panel) mRNA induction by qPCR; *P < 0.05 versus DMSO; #P < 0.05 versus 0.05 μM Im or 0.1 μM Im. In (B), cells were treated with drugs and stimulated with 10ng/ml of IFNγ (left panel) or 3ng/ml of LPS (right panel) for 24h before the NO level in media was measured by the Griess reaction; *P < 0.05 versus IFNγ or LPS-stimulated control. In (C), cells were treated with compounds for 24h, incubated with 10 μM of 2′,7′-dichlorodihydrofluorescein diacetate for 30min, and then challenged with 250 μM of tBHP for 15min. Cells were harvested and fluorescence of ROS-positive cells was measured by flow cytometry. *P < 0.05 versus tBHP-stimulated controls.
The induction of phase II enzymes such as NQO1 is strongly correlated with the protection against oxidative and inflammatory stress (14). To investigate the physiological consequences of Nrf2 pathway activation, we compared the ability of DMF and the triterpenoids to inhibit the production of NO and reactive oxygen species (ROS). Consistent with previous studies, CDDO-Im and CDDO-Me reduced NO production in a dose-dependent manner at low nanomolar concentrations when stimulated with either IFNγ (Figure 1B, left panel) or LPS (Figure1B, right panel). However, even though DMF decreased NO production in a dose-dependent manner, its effects were only observed at micromolar concentrations. To assess ROS production, RAW 264.7 cells were treated with various concentrations of drugs for 24h. Cells were then challenged with 250 μM of tBHP for 15min to induce ROS production, and ROS-positive fluorescent cells were detected by flow cytometry. All three compounds significantly (P < 0.05) inhibited tBHP-induced ROS production, and DMF was less potent than the triterpenoids. Specifically, 10 μM of DMF reduced ROS production to 56% of the levels in control cells treated with tBHP, whereas 0.01 μM of CDDO-Im and CDDO-Me reduced ROS production to 32% and 41%, respectively, of control levels (Figure 1C).
Because mutations in KEAP1, and less frequently in NRF2, have been detected in tumors and altered expression of Keap1 or Nrf2 may be a negative prognostic marker in lung cancer (41–44), we examined Nrf2 activation in lung cancer cells. For these studies, VC1 cells, derived from an adenocarcinoma of the lung in an A/J mouse treated with vinyl carbamate (16), were used. CDDO-Im, CDDO-Me and DMF induced NQO1 and HO-1 mRNA in a dose-dependent manner in these cells, as confirmed by qPCR (data not shown). To examine the stabilization of Nrf2 and the induction of NQO1 and HO-1 at the protein level, VC1 cells were treated with compounds for 0–24h (Figure 2A) before they were lysed and analyzed by western blotting. Unless activated and released from Keap1, Nrf2 is targeted for proteasomal degradation. In control cells, no Nrf2 protein was detected at any time point in the absence of MG132 (Figure 2A), a proteasome inhibitor used as a positive control. In cells treated with triterpenoids or DMF, Nrf2 protein was detected as early as 30min after treatment, and this protein stabilization was maintained for 8h. As a result, NQO1 and HO-1 proteins increased in a dose-dependent manner with all three drugs. The kinetics differed between the 2 proteins, as the peak of NQO1 induction was at 24h whereas the peak of HO-1 was at 8h (Figure 2B). The induction of HO-1 was most robust with 0.5 µM of CDDO-Im, although CDDO-Me and DMF also increased HO-1 protein levels.
Figure 2.
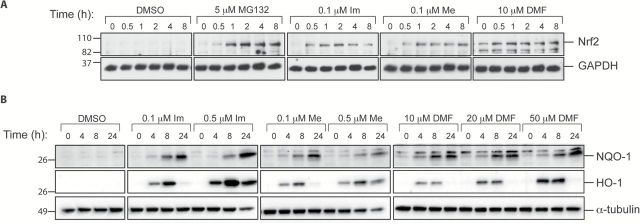
Triterpenoids and DMF stabilize Nrf2 proteins and induce NQO1 and HO-1 protein expression in vitro. VC1 lung cancer cells were treated with DMSO or various concentrations of CDDO-Im (Im), CDDO-Me (Me) or DMF for 0–8h (A) or for 0–24h (B) before cells were lysed and processed by western blotting; MG132 is a proteasome inhibitor used as a positive control.
Microarray analysis of VC1 cells treated with triterpenoids or DMF
Our studies have confirmed that the synthetic triterpenoids and DMF are Nrf2 activators that induce NQO1 and HO-1 expression both at the gene and protein level. To determine whether the triterpenoids and DMF differentially regulate target genes, we performed a microarray analysis. Based on our results in Figures 1 and 2, we chose concentrations of CDDO-Im, CDDO-Me and DMF of 0.05, 0.1 and 15 μM, respectively, as these concentrations induced the prototypical Nrf2-target gene NQO1 to a similar extent (Figure 3A). VC1 cells were incubated with these concentrations of drugs for 6h, and RNA was extracted and processed for microarray analysis using an Illumina mouse WG-6 BeadChip. The full microarray data (GSE65623) are available on the NCBI’s Gene Expression Omnibus website.
Figure 3.
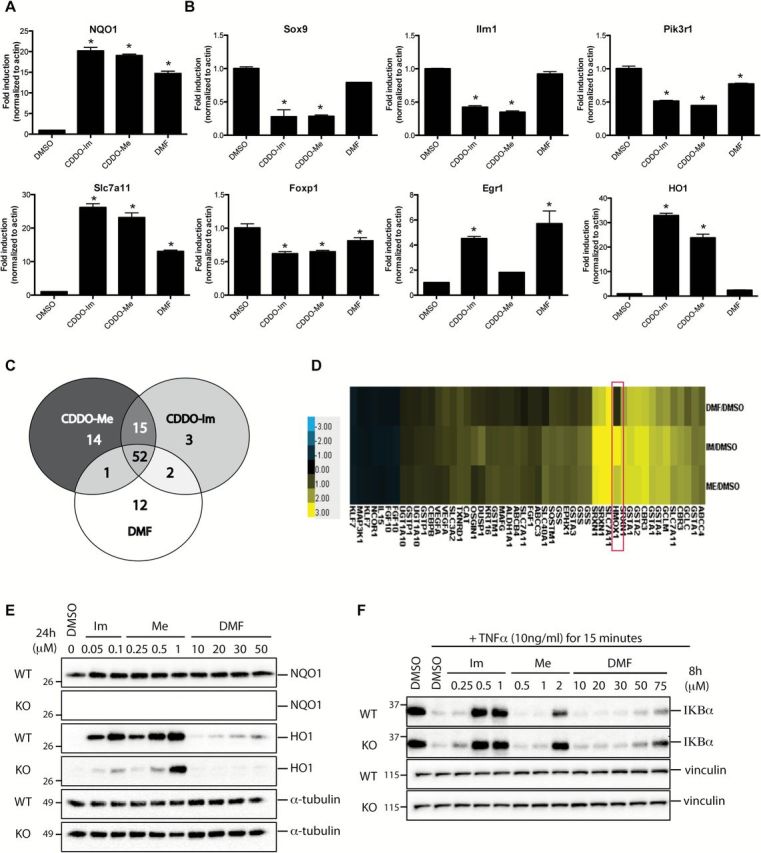
CDDO-Im (Im), CDDO-Me (Me) and DMF activate overlapping but non-identical subsets of Nrf2 genes. VC1 cells were treated in triplicates with DMSO, 0.05 μM of CDDO-Im, 0.1 μM of CDDO-Me or 15 μM of DMF for 6h before RNA was extracted and processed for microarray analysis. Expression patterns of Nrf2 genes (A) and non-Nrf2 genes (B) that were detected in the microarray were confirmed by qPCR. *P < 0.05 versus DMSO. (C) Venn diagram depicting the subsets of Nrf2 genes that were targeted by the compounds. (D) Heat map showing the Nrf2 genes that were significantly up-regulated (yellow) or down-regulated (blue) by 1.5-fold or 0.75-fold, respectively by drug treatment compared with control (DMSO) in the microarray. The fold change of Hmox1 was the greatest of all the genes examined (red box).WT Nrf2 mouse embryonic fibroblasts and Nrf2 KO mouse embryonic fibroblasts were treated with DMSO or various concentrations of Im, Me or DMF for 24h (E) or 8h (F) before cells were lysed and processed for western blotting.
To validate the microarray data, we used qPCR to measure the expression patterns of seven genes identified as being significantly altered by the drugs, including NQO1, Slc7a11, Sox9, Foxp1, Egr1, Pik3r1 and Ilrn1. The expression patterns for all 7 genes tested by qPCR were comparable with the results obtained from the microarray. Besides NQO1, Slc7a11 is a well-known Nrf2-target gene. Specifically, Slc7a11 is a cystine–glutamate carrier important for intracellular glutathione synthesis and is often expressed at high levels in lung cancer and thus is involved in chemoresistance (45). Similar to what was observed with NQO1, Slc7a11 expression was induced by all treatments with less robust induction with DMF (Figure 3A). The Sox9, Foxp1, Pik3r1, Egr1 and Ilrn1 genes are also important in lung cancer. Specifically, Sox9 and Pik3r1, a gene that encodes for the p85 PI3K regulatory subunit, are often overexpressed in lung adenocarcinoma, which in turn, promotes tumor cell proliferation and survival (46–48). Conversely, both Egr1 and Foxp1 proteins have an inhibitory role in tumor development and improve the survival rate of patients with non-small cell lung cancer (49–51). Ilrn1 serves as a natural inhibitor for interleukin-1 (IL1), a cytokine that plays a critical role in tumorigenesis by regulating genes involved in metastasis, angiogenesis and growth factor expression. IL1 mRNA has been shown to be highly expressed in numerous human cancer xenografts, including non-small cell lung cancer samples, and several studies found that the IL1 antagonist, Ilrn1, could reverse IL1-mediated pathological effects in cancer (52,53). Our results with these genes not only confirmed the microarray data but also showed that each of these drugs differentially regulated the expression of specific genes (Figure 3B). For instance, all three compounds reduced Foxp1 and Pik3r1 expression. In contrast, the expression of Sox9 and Il1rn was reduced in the presence of triterpenoids, but the expression of these genes was unchanged when cells were treated with DMF. Interestingly, the expression pattern of Egr1 revealed differences between the triterpenoids. Specifically, CDDO-Im and DMF significantly (P < 0.05) increased the expression of Egr1, whereas CDDO-Me had no effect. Notably, the triterpenoids increased HO-1 expression 23- to 34-fold over the control, whereas DMF only increased gene expression by 2.4-fold.
To examine whether the triterpenoids and DMF regulate the same subset of Nrf2-target genes, we utilized Pathway Analysis software to search for all the genes directly related to Nrf2 based on published literature. Using this list, we then performed a comparative analysis to examine whether these genes were targeted by CDDO-Im, CDDO-Me or DMF. The Venn diagram generated from this analysis revealed that these drugs regulated 99 genes involved in the Nrf2 pathway (Figure 3C). Interestingly, only 52 of those 99 genes were targeted by all three compounds, and each of the drugs displayed an affinity for unique subsets of Nrf2 genes. For example, Prkcd, Tlr4 and Txnrd3 were specifically targeted by CDDO-Im; expression of Bach1, Cyb5r1, Hsp90AA1, Edn1, Meis1, Gnai2, Apex1, Rb1, Mapk14, Pmf1, Nfe2l2, Hbegf, Junb and Klf2 was significantly regulated by CDDO-Me; and Fos, Ddit3, Pparg, Rarg, Prdx5, Cul3, Cycs, Optn, Jun, Mterf, Dusp16 and Chd6 were altered by DMF. The details of these changes are provided in Supplementary Table 1, available at Carcinogenesis Online. In addition, when we examined the Nrf2-target genes that were significantly up-regulated by any of our three compounds by >1.5-fold or <0.75-fold (both P < 0.01), HO-1 was the most highly induced, as it was up-regulated by 9.1-, 5.3- and 1.6-fold compared with the control when treated with CDDO-Im, CDDO-Me or DMF, respectively (Figure 3D).
Although the triterpenoids and DMF activate the Nrf2 pathway, they also regulate other signaling pathways (11), which might account for the difference we observed in HO-1 expression. Even at concentrations in which NQO1 induction was similar with all three drugs, the induction of HO-1 protein and mRNA expression by the triterpenoids were much greater than for DMF (Figures 2B and 3A). In order to understand the difference in HO-1 induction between treatment groups, we used Nrf2 wildtype (WT) and knockout (KO) mouse embryonic fibroblasts (MEFs). After 24h of treatment, all three compounds increased NQO1 protein expression in WT cells but not in KO cells (Figure 3E). HO-1 protein also was induced in WT cells by all three drugs in a dose-dependent manner. However, in the KO cells, no HO-1 induction was detectable following treatment with DMF. In contrast, CDDO-Im and CDDO-Me both induced HO-1 protein expression in the KO cells, albeit at a lower level than in the WT cells. These results confirmed that although HO-1 is partially regulated by Nrf2, it is also regulated by other genes that are targeted by the triterpenoids (54,55).
The differential regulation of HO-1 is just one example of the importance of other targets beyond Nrf2 that explain the biological effects of the triterpenoids and DMF. These drugs also down-regulate the pro-inflammatory nuclear factor-kappaB (NF-κB) pathway (56–59), which crosstalks with the Nrf2 pathway (60,61). Therefore, we explored the effect of these compounds on the NF-κB pathway and tested whether these effects were dependent on Nrf2. As shown in Figure 3F, stimulation with tumor necrosis factor α caused degradation of the IKBα protein. Pretreatment with 0.5 µM of CDDO-Im, 2 µM of CDDO-Me or 50–75 µM of DMF prevented this degradation in both Nrf2 WT and KO cells, suggesting that the effects of these drugs on the NF-κB pathway are independent of Nrf2. Notably, higher concentrations of drugs are required to inhibit IKBα degradation (Figure 3F) than to increase NQO1 or HO-1 protein expression (Figure 3E).
In addition to HO-1 and IKBα, we examined two other cancer relevant, non-Nrf2-target genes detected in the microarray studies. As noted previously, all three drugs reduced mRNA expression of Sox9 and Pik3r1 (Figure 3B). To investigate changes at the protein level, VC1 cells were treated with various concentrations of compounds, and all three drugs decreased Sox9 expression in a dose-dependent manner. Surprisingly, Sox9 expression increased at higher concentrations of CDDO-Im (Figure 4A). We next assessed p85 protein expression, which is encoded by the Pik3r1 gene, and examined the activation of Akt and CREB, as these proteins play an intricate role downstream of PI3K signaling in tumor development. In these experiments, there was no change in total p85 protein expression, but all three drugs increased CREB/ATF (activating transcription factor) phosphorylation in a dose-dependent manner (Figure 4B). At higher concentrations, CDDO-Im alone decreased total CREB levels. Notably, a dose-dependent increase in Akt phosphorylation was evident only in DMF-treated cells, suggesting differential regulation compared with the triterpenoids.
Figure 4.
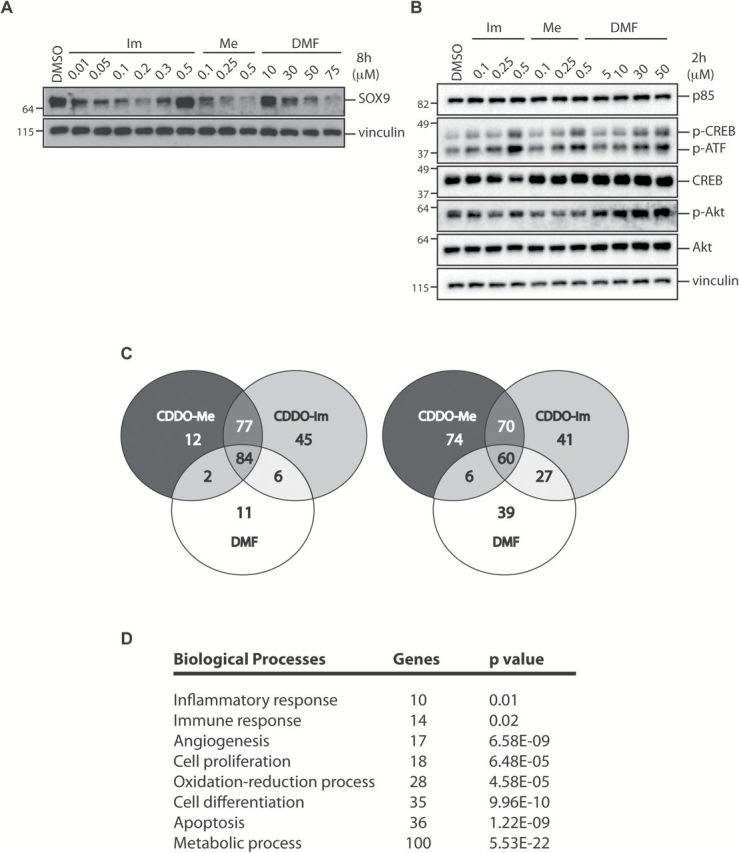
Triterpenoids and DMF regulate non-Nrf2-target genes that are involved in biological processes related to cancer progression. VC1 cells were treated with DMSO or various concentrations of CDDO-Im (Im), CDDO-Me (Me) or DMF for 8h (A) or 2h (B) before cells were lysed and processed by western blotting. (C) Venn diagram showing the subsets of genes that were significantly up-regulated by 1.5-fold (left panel) or down-regulated by 0.75-fold (right panel) in the presence of drugs (P < 0.01). (D) Gene Ontology analysis of the number of genes that were targeted by the compounds in eight major biological processes involved in carcinogenesis with respective P values.
To further explore other Nrf2-independent pathways regulated by these drugs, we utilized the microarray data to examine global transcription changes between groups. Of the 45000 transcripts that were present on the microarray chip, a total of 237 genes were significantly up-regulated by CDDO-Im, CDDO-Me or DMF by at least 1.5-fold (Figure 4C, left panel; P < 0.01), and 317 genes were significantly down-regulated by these compounds by at least 0.75-fold (Figure 4C, right panel; P < 0.01). Although there were 84 common genes that were up-regulated and 60 genes that were down-regulated by all three drugs, there were numerous genes that were specifically targeted by only CDDO-Im, or CDDO-Me or DMF. Moreover, the microarray data indicated that CDDO-Im and CDDO-Me had their own unique targets. The detailed information for these differentially regulated genes is listed in Supplementary Table 2, available at Carcinogenesis Online.
We next performed a Gene Ontology analysis to examine whether genes that were significantly up-regulated by 1.5-fold or down-regulated by 0.75-fold in our microarray studies were enriched in biological processes independent of Nrf2. Our results showed that these compounds targeted genes that are involved in various pathways related to cancer progression (Figure 4D). For instance, Mapk9 and Cyclin D are important players in cell proliferation (62,63). Mapk9 was targeted by all three drugs, and gene expression increased by 1.77-, 2.08- and 2.13-fold, respectively (P < 0.01), in the presence of CDDO-Im, CDDO-Me and DMF. Interestingly, Cyclin D1 was targeted by the triterpenoids only; CDDO-Im and CDDO-Me significantly (P < 0.01) reduced gene expression to 0.69- and 0.72-fold of the controls, respectively. Similarly, Gadd45γ, a gene intricately involved in inhibiting cell growth and inducing apoptosis, was also targeted only by the triterpenoids. Specifically, CDDO-Im and CDDO-Me increased the mRNA expression of Gadd45γ by 1.82 and 1.64, respectively (P < 0.01). Moreover, we found that IL15 mRNA expression was reduced by 0.71-fold in the presence of CDDO-Im only, further highlighting differences between the triterpenoids.
Other cancer-related biological processes that were targeted by these compounds included the inflammatory response, immune response, angiogenesis, oxidation–reduction, cell differentiation, apoptosis and metabolism. Expression of 17 genes involved in angiogenesis was altered on the microarray, and 16 of the 17 genes were regulated in the same manner by all three compounds. Tnfaip2, a pro-angiogenic factor (64), was down-regulated by CDDO-Me, but DMF had no effect. Thirty apoptosis-related genes were regulated on the microarray. Five genes were targeted only by the triterpenoids and not DMF. In addition to the up-regulation of Gadd45γ mentioned previously, the triterpenoids increased the expression of Sgk1 but decreased the expression of Tnfaip3 and Tns4. Sgk1 and Tnfaip3 inhibit apoptosis, whereas Tns4 enhances apoptosis via caspase3-mediated cleavage (65–67). Intriguingly, our Gene Ontology analysis revealed that metabolic-related genes were the largest category altered by these compounds. Of 89 genes, 72 of them were targeted by all three drugs, whereas 12 were targeted by the triterpenoids. Of note, the expression of Acsl3 and Ampd3, the key enzymes for fatty acid and adenosine monophosphate metabolism, respectively, were increased by the triterpenoids. Increased expression of ACSLs is evident in numerous cancers including lung cancer and the changes in expression in cancer cells are associated with survival, proliferation and chemoresistance (68), whereas Ampd3 is up-regulated in LKB-mutated lung adenocarcinoma (69). Although Nrf2 genes are vital in a plethora of cellular functions, it is evident that the Nrf2 pathway is not the only target of CDDO-Im, CDDO-Me or DMF. Moreover, there are a substantial number of non-Nrf2 genes and pathways that these compounds regulate, which may play an important role in carcinogenesis.
Triterpenoids and DMF have opposite effects in vivo in a preclinical model of lung cancer
We have successfully used the NO assay (Figure 1B) as a screen to predict for the anticancer activity of triterpenoids and other drugs (70). Before initiating in vivo carcinogenesis studies, we first tested whether these compounds could activate the Nrf2 pathway in vivo. It is difficult to translate concentrations used for in vitro experiments directly to appropriate in vivo doses so several pilot experiments were performed. For many compounds, it is easier to detect changes in biomarkers when drugs are administered as a bolus instead of with chronic feeding. I. P. injections of DMF (100–200mg/kg) and the triterpenoids (12.5mg/kg) both significantly (P < 0.05) increased NQO1 and HO-1 mRNA levels in the lungs and liver of mice. Notably, the magnitude of NQO1 induction was higher in the lungs (4- to 10-fold versus controls) than in the liver (1.5- to 2-fold) when treated with these compounds (Figure 5A). In contrast, HO-1 mRNA was more robustly induced in the liver (2.5- to 10.8-fold) than in the lungs (1.9- to 3.8-fold) of the mice injected with triterpenoids or DMF. When administered by gavage, DMF (200mg/kg) and CDDO-Me (12.5mg/kg) increased mRNA expression of NQO1 and the Nrf2-target gene AKR1B8 in the lungs and livers of mice but, as expected, the induction was lower than with i. p. injection (Figure 5B).
Figure 5.
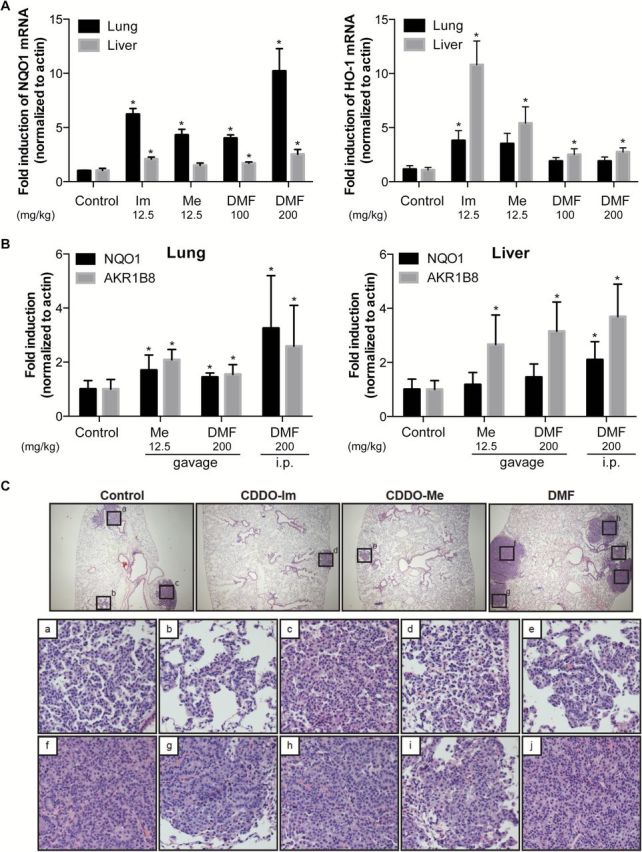
Triterpenoids and DMF both induce NQO1 and HO-1 mRNA expression in vivo but have opposing effects in the severity of lung cancer in A/J mice. Mice (n = 5–6 per group) were injected i. p. (A) with vehicle, 12.5mg/kg of CDDO-Im (Im) and CDDO-Me (Me) or 100mg/kg of DMF or gavaged with 12.5mg/kg of Me and/or injected i. p. with 200mg/kg of DMF (B). Four hours later, tissues were harvested, and RNA from lungs and liver were extracted to assess NQO1 (left panel) and HO-1 (right panel) (A) or NQO1 and AKR1B8 (B) mRNA induction by qPCR. *P < 0.05 versus control. (C) Representative images of the lungs of mice fed with control, 50mg/kg of CDDO-Im, 50mg/kg of CDDO-Me or 1200mg/kg of DMF in diet for 15 weeks (magnification, 40×). Histopathology of tumors from control (insets a–c), CDDO-Im (inset d), CDDO-Me (inset e) and DMF (insets f–j) are shown (magnification, 200×).
A/J mice are commonly used for preclinical lung cancer studies because of their susceptibility to form tumors in the lungs in response to tobacco smoke and other carcinogens. For our studies, female A/J mice were injected with vinyl carbamate, which induces adenocarcinomas in a time-dependent manner (40,71). Beginning 1 week after initiation, A/J mice were fed with control diet, or various doses of CDDO-Im (50, 100 or 200mg/kg diet, ~12.5, 25 and 50mg/kg body wt), CDDO-Me (12.5 or 50mg/kg diet, ~3 and 12.5mg/kg body wt) or DMF (400, 800 or 1200mg/kg diet, ~ 100, 200 and 300mg/kg body wt). Notably, the concentrations of DMF used in these studies are comparable with the levels of DMF or fumaric acid fed in diet (500–1000mg/kg diet) and shown to induce NQO1 in rodent tissue and inhibit carcinogenesis in the forestomach of mice (32,72). These compounds were well tolerated by the mice at all doses tested, and there were no significant changes in the average weight of each group during the study (Supplementary Figure 1, available at Carcinogenesis Online). After 15 weeks on diet, lungs were harvested; the number of lung tumors was counted, the size of the tumors was measured and the histopathology of the tumors and nuclei was graded using previously published criteria (17,40). Because the entire lungs are harvested en bloc and inflated with formalin to optimize the evaluation of the histopathology, it is not possible to measure drug levels or induction of mRNA in the lungs. However, we did examine expression of the Nrf2-target gene AKR1B8 in the liver of these mice, as induction of this gene in the liver was more robust than for NQO1 in the preliminary pharmacokinetic studies (Figure 5B). Both DMF and the triterpenoids enhanced expression of this gene (Supplementary Figure 2, available at Carcinogenesis Online), confirming activation of the Nrf2 pathway in the A/J mice, but the induction was higher with the triterpenoids.
Surprisingly, DMF and the synthetic triterpenoids had very different effects on lung tumorigenesis in this model (Table 1). Even though CDDO-Im was the most potent compound in vitro, CDDO-Me was the most effective for significantly (P < 0.05) reducing tumor number, size and severity. The average number of tumors was decreased by 32% (2.2±0.3) compared with the controls (3.2±0.15), and the size of tumors was decreased by 76% to an average of 0.09±0.01mm3 compared with the control group (0.37±0.04mm3) when mice were fed with the low dose of 12.5mg/kg CDDO-Me diet. Average tumor burden was also reduced by 83% (0.2±0.03mm3) compared with the control group (1.2±0.14mm3). Moreover, the tumors in the CDDO-Me group were less malignant than in the control group. Specifically, the percentage of low-grade tumors increased from 8% in the control group to 19% in mice fed with 12.5mg/kg of CDDO-Me diet (P < 0.05). Importantly, the percentage of high-grade tumors significantly decreased from 52% in the control group to 31% with this low dose of CDDO-Me diet. The higher dose of CDDO-Me (50mg/kg diet) was even more effective, as the average tumor burden (0.08±0.02mm3) was reduced by 93% compared with the control group (P < 0.05), and 35% of these tumors were low grade and only 14% were high grade (P < 0.01 versus controls).
Table 1.
Triterpenoids (CDDO-Im and CDDO-Me) reduce the total burden and severity of lung tumors whereas DMF increases the number and severity of lung tumors in A/J mice injected with vinyl carbamate.
| Treatment (diet in mg/kg) | Control | CDDO-Im | CDDO-Me | DMF | |||||
|---|---|---|---|---|---|---|---|---|---|
| 50 | 100 | 200 | 12.5 | 50 | 400 | 800 | 1200 | ||
| Tumor number, size, burden | |||||||||
| n | 104 | 24 | 24 | 24 | 24 | 24 | 44 | 48 | 24 |
| Average number of tumors (% control) | 3.21±0.15 (100) | 2.54±0.34 (79) | 3±0.49 (94) | 2.17±0.29*(68) | 2.17±0.27* (68) | 2.13±0.24* (66) | 4.09±0.25* (127) | 3.25±0.25 (101) | 4.13±0.44* (129) |
| Average tumor size, mm3 (% control) | 0.37±0.04 (100) | 0.16±0.02* (43) | 0.16±0.02* (43) | 0.05±0.008*(14) | 0.09±0.01* (24) | 0.037±0.007* (11) | 0.25±0.02 (68) | 0.25±0.02 (68) | 0.23±0.03* (62) |
| Average tumor burden, mm3 (% control) | 1.19±0.14 (100) | 0.39±0.07* (33) | 0.49±0.11* (41) | 0.12±0.02*(10) | 0.2±0.03* (17) | 0.08±0.02* (7) | 1.02±0.08 (86) | 0.82±0.09 (69) | 0.95±0.17 (80) |
| Tumor histopathology | |||||||||
| Number of tumors | 334 | 61 | 73 | 52 | 52 | 51 | 180 | 156 | 99 |
| Total number of medium-grade tumors (% total) | 134 (40) | 27 (44) | 36 (49) | 30* (58) | 26 (50) | 26 (51) | 61 (34) | 53 (34) | 34 (34) |
| Total number of high-grade tumors (% total) | 172 (52) | 27 (44) | 26* (36) | 11* (21) | 16* (31) | 7** (14) | 110* (61) | 98* (63) | 59 (60) |
Female mice were injected i. p. with two doses of vinyl carbamate (0.32mg per mouse) 1 week apart and fed with control, CDDO-Im (50, 100 and 200mg/kg), CDDO-Me (12.5 and 50mg/kg) or DMF (400, 800 and 1200mg/kg) diets, beginning 1 week after the last injection of carcinogen, for 15 weeks. Number, size of tumors and histopathology of tumors were evaluated by two independent investigators. Values represent mean ± standard error.
*P < 0.05, **P < 0.001.
CDDO-Im was less potent than CDDO-Me, but the lowest dose of CDDO-Im (50mg/kg diet) significantly (P < 0.05) reduced the average tumor size (0.16±0.02mm3) and tumor burden (0.39±0.07mm3) by 57% and 67%, respectively compared with the controls (0.37±0.04mm3 and 1.19±0.14mm3). The number of tumors was also significantly (P < 0.05) lower than the controls when mice were fed with a higher dose of CDDO-Im (200mg/kg diet), and the average tumor burden was reduced by 90% at this higher dose. High-dose CDDO-Im also significantly increased the percentages of low- (21%) and medium-grade (58%) tumors and significantly reduced the percentage of high-grade tumors (21%) compared with their respective controls (8%, 40% and 52%; P < 0.05).
However, these parameters were markedly different in mice fed DMF. The number of tumors increased by 27–29% versus controls when mice were fed with 400 and 1200mg/kg of DMF diets (P < 0.05). Notably, DMF significantly (P < 0.05) increased the severity of tumors in this mouse model, as 61–63% of the tumors were high-grade tumors in mice fed with 400 and 800mg/kg of DMF diets compared with 51% in the control group. Despite a trend toward a reduction in tumor size, the tumors were only significantly (P < 0.05) smaller (0.23±0.03mm3 versus 0.37±0.04mm3) when mice were fed with the highest dose (1200mg/kg diet) of DMF. Representative images showing an increase in tumor number and severity with DMF compared with the controls versus a decrease in tumor number and size with the triterpenoids are shown in Figure 5C.
Discussion
In this study, we showed that CDDO-Im, CDDO-Me and DMF induced phase II cytoprotective enzymes, including NQO1 and HO-1, and inhibited inflammation by reducing the production of NO and ROS. However, the drug concentrations required for activating Nrf2 and other pathways varies with the pharmacological agent. Specifically, the triterpenoids elicited their effects at considerably lower (nanomolar) concentrations than required for DMF (high micromolar). Our microarray analysis confirmed that these drugs (i) targeted different subsets of genes in the Nrf2 pathway and (ii) these transcriptional alterations were not limited to the Nrf2 pathway. When tested in a carcinogen-induced model of lung cancer, the triterpenoids significantly reduced the number, size and severity of the tumors at all doses tested. Surprisingly, DMF treatment had the opposite effect, as it increased the number and severity of the tumors.
Under physiological conditions, fumarate is metabolized by the fumarate hydratase (FH) enzyme to malate for energy production via the Krebs cycle. When administered as a pharmacological agent, fumaric acid inhibited chemically induced tumors in the liver, lung and stomach (31,32). However, a homozygous loss of function mutation in FH leads to elevated levels of fumarate in the kidney. Fumarate then forms adducts with KEAP1, resulting in constitutive activation of Nrf2 and induction of genes containing an antioxidant response element (73,74). The dysregulation of Nrf2 is thought to be the cause of renal carcinoma in patients with a FH mutation. Constitutive activation of the Nrf2 pathway in cancer cells, the result of either spontaneous mutations or genetic manipulation in animal models, is known to be oncogenic and cause drug resistance. However, to the best of our knowledge, the use of DMF or another pharmacological activator of the Nrf2 pathway has not been shown to enhance carcinogenesis in vivo.
In many carcinogenesis studies, the pharmacological agent is given prior to initiation with a carcinogen. If the compound activates the Nrf2 pathway and thus enhances detoxification of electrophiles or reduces DNA damage, tumor burden will be reduced. In our lung studies, triterpenoids and DMF treatments were started 1 week after initiation with vinyl carbamate. This experimental paradigm is more challenging for developing effective interventions, as a drug must interrupt tumor progression and growth, but is more relevant to carcinogenesis in humans. Although previous studies suggested that DMF has antitumorigenic effects, these reports utilized concentrations of DMF up to 300 μM, which exceed physiologically and even pharmacologically relevant doses (26,29,75). The concentrations of DMF used in the lung cancer studies (400–1200mg/kg diet) activated the Nrf2 pathway in vivo (Figure 5A and B) and are comparable with doses (500–1000mg/kg diet) shown to induce NQO1 in rodent tissue and inhibit forestomach carcinogenesis in mice when fed in diet (32,72).
In our lung cancer model, it is possible that (i) DMF activated Nrf2, which improved survival of transformed cells and thus enhanced tumor progression or (ii) these observations are the result of an off-target effect, rather than the activation of the Nrf2 pathway. Similarly, it is not known if the pronounced reduction in the number, size and histopathology of lung tumors in the A/J model by the multifunctional triterpenoids, at all doses tested, is the result of Nrf2 activation or signaling through other pathways. Of note, the microarray studies revealed that the three drugs induced different subsets of genes in the Nrf2 pathway and differentially regulated numerous genes not connected to the Nrf2 pathway (Figures 3 and 4).
One of the most striking differences between these compounds was the markedly higher induction of Hmox1 with the triterpenoids than with DMF. Hmox1 encodes for the HO-1 protein, which is responsible for heme degradation and the production of carbon monoxide, iron and biliverdin, which is converted into bilirubin. These metabolites, in turn, activate various downstream pathways that lead to an anti-inflammatory response and cytoprotection. Although HO-1 is regulated by Nrf2, HO-1 expression can also be induced by other transcription factors and signaling cascades (54). The triterpenoids target multiple signaling pathways, and the induction of HO-1 was still detectable in Nrf2 KO cells when treated with CDDO-Im and CDDO-Me, but not with DMF (Figure 3E).
Interestingly, even within the same class of compounds, CDDO-Im and CDDO-Me activated unique subsets of target genes. This phenomenon was observed for the expression of Sox9, a gene often overexpressed in lung adenocarcinoma (46,47) and Egr1. Sox9 protein expression was reduced by both CDDO-Im and CDDO-Me (Figure 4A); however, at higher concentrations of CDDO-Im, Sox9 expression increased. Similarly, CDDO-Im increased the expression of Egr1, which inhibits tumor growth, whereas CDDO-Me had no effect (Figure 3B). These data further support the notion that derivatives of CDDO can affect diverse signaling pathways via different mechanisms (11). Moreover, the Sox9 results confirm that the biological effect of a triterpenoid is dependent on dose.
Aberrant PI3K signaling is often observed in cancer, but the protein expression of p85 was not affected by these drugs (Figure 4B), even though there was a decrease in Pik3r1 expression at the mRNA level (Figure 3B). However, when we assessed the phosphorylation of CREB and Akt, important downstream proteins in this pathway, they were affected by all three compounds. Although the CREB transcription factor is involved in a plethora of processes including cell proliferation, survival and differentiation, it also has emerged as an important player in inflammation (76). Specifically, phosphorylated CREB can inhibit NF-κB activation and its downstream responses by competing with NF-κB for the binding of CBP/p300, an important component needed for the activation of the NF-κB pathway (77,78). CREB was phosphorylated following treatment with all three drugs, which are also anti-inflammatory agents (Figure 1). Notably, the phosphorylation of Akt was increased only in cells treated with DMF, but not the triterpenoids. Akt is an important player in cell survival, proliferation, metabolism, migration and angiogenesis, all of which are classic hallmarks of cancer. As such, aberrant Akt signaling is evident in numerous cancers (79–81); suggesting that this pathway could contribute to our in vivo results, and future studies will explore the role of DMF and the Akt pathway in the A/J model.
Although we were hopeful that additional conclusions would emerge from the microarray data to help explain the tumor data, we have been unable to identify a specific gene, pathway or process that explains our results. Differential regulation of angiogenesis or apoptosis pathways by the triterpenoids and DMF would be informative, but except for the changes described previously, many of the differences between these drugs were changes in magnitude and thus quantitative rather than qualitative. Because the α,β-unsaturated carbonyl group in DMF and the triterpenoids are Michael acceptor sites, these compounds can interact with reactive cysteines on target proteins such as KEAP1, IkappaB (IKK) and PI3K/Akt (reviewed in ref. 11). The effects of these drugs on target proteins and downstream signaling pathways and networks may not be detectable by microarray studies, as differential effects on other target proteins might be needed to explain the results in the lung cancer model. In order to explore this possibility, proteomic analysis of VC1 cells treated with DMF and a triterpenoid should also be explored.
In summary, our data collectively demonstrate that although DMF and triterpenoids both activate the Nrf2 pathway, they also regulate different subsets of Nrf2-target genes as well as a number of Nrf2-independent genes. These drugs had opposite effects in a preclinical model of lung cancer, but it is not possible to predict whether the promotion or suppression of carcinogenesis by either DMF or CDDO-Me, have any direct implications in the current clinical use of these drugs for conditions other than cancer. Although both agents activate the Nrf2 pathway, whether such enhanced Nrf2 activity is advantageous or disadvantageous with respect to human cancer is known to depend on context (9), and the complexity of the microarray results further complicates the overall benefit versus risk analysis. Future studies will utilize Nrf2 KO mice to study the role of DMF and triterpenoids for prevention and treatment of cancer at different stages of progression and use proteomic studies to help identify the mechanisms responsible for these results.
Supplementary material
Supplementary Figures 1 and 2 and Tables 1 and 2 can be found at http://carcin.oxfordjournals.org/
Funding
National Foundation for Cancer Research and Reata Pharmaceuticals, Inc. Microarray studies were carried out at Dartmouth Medical School in the Genomics Shared Resource, which was established by equipment grants from the National Institutes of Health and National Science Foundation and is supported in part by a Cancer Center Core Grant (P30CA023108) from the National Cancer Institute.
Supplementary Material
Acknowledgements
The authors thank E.York for his expertise in preparing the slides for histology studies.
Conflict of Interest Statement: M.B.S. and K.T.L. have a commercial research grant from Reata Pharmaceuticals Inc. and have patent interests in synthetic triterpenoids. The other authors have no potential conflict of interests.
Glossary
Abbreviations
- CDDO-Im
CDDO-imidazolide
- CDDO-Me
CDDO-methyl ester
- CREB
cAMP response element-binding protein
- DMF
dimethyl fumarate
- HO-1
heme oxygenase 1
- IFNγ
interferon-γ
- IL1
interleukin-1
- KEAP1
Kelch-like erythroid-associated protein 1
- KO
knockout
- LPS
lipopolysaccharide
- MEFs
mouse embryonic fibroblasts
- mRNA
messenger RNA
- NO
nitric oxide
- NF-κB
nuclear factor-kappaB
- NQO1
NAD(P)H:quinone oxidoreductase 1
- qPCR
quantitative PCR
- ROS
reactive oxygen species
- tBHP
tert-butyl hydroperoxide
- WT
wild-type
References
- 1. Siegel R., et al. (2014) Cancer statistics, 2014. CA Cancer J. Clin., 64, 9–29. [DOI] [PubMed] [Google Scholar]
- 2. Jaramillo M.C., et al. (2013) The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev., 27, 2179–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kensler T.W., et al. (2010) Nrf2: friend or foe for chemoprevention? Carcinogenesis, 31, 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Suzuki T., et al. (2013) Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol. Sci., 34, 340–346. [DOI] [PubMed] [Google Scholar]
- 5. Hayes J.D., et al. (2010) Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid. Redox Signal., 13, 1713–1748. [DOI] [PubMed] [Google Scholar]
- 6. Hu R., et al. (2010) Regulation of NF-E2-related factor 2 signaling for cancer chemoprevention: antioxidant coupled with antiinflammatory. Antioxid. Redox Signal., 13, 1679–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bauer A.K., et al. (2013) The involvement of NRF2 in lung cancer. Oxid. Med. Cell. Longev., 2013, 746432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moon E.J., et al. (2015) Dual roles of NRF2 in tumor prevention and progression: possible implications in cancer treatment. Free Radic. Biol. Med., 79, 292–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sporn M.B., et al. (2012) NRF2 and cancer: the good, the bad and the importance of context. Nat. Rev. Cancer, 12, 564–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. DeNicola G.M., et al. (2011) Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature, 475, 106–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liby K.T., et al. (2012) Synthetic oleanane triterpenoids: multifunctional drugs with a broad range of applications for prevention and treatment of chronic disease. Pharmacol. Rev., 64, 972–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hong D.S., et al. (2012) A phase I first-in-human trial of bardoxolone methyl in patients with advanced solid tumors and lymphomas. Clin. Cancer Res., 18, 3396–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nagaraj S., et al. (2010) Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin. Cancer Res., 16, 1812–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dinkova-Kostova A.T., et al. (2005) Extremely potent triterpenoid inducers of the phase 2 response: correlations of protection against oxidant and inflammatory stress. Proc. Natl Acad. Sci. USA, 102, 4584–4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yates M.S., et al. (2007) Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol. Cancer Ther., 6, 154–162. [DOI] [PubMed] [Google Scholar]
- 16. Liby K., et al. (2009) Triterpenoids CDDO-methyl ester or CDDO-ethyl amide and rexinoids LG100268 or NRX194204 for prevention and treatment of lung cancer in mice. Cancer Prev. Res. (Phila.), 2, 1050–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liby K., et al. (2007) The synthetic triterpenoids CDDO-methyl ester and CDDO-ethyl amide prevent lung cancer induced by vinyl carbamate in A/J mice. Cancer Res., 67, 2414–2419. [DOI] [PubMed] [Google Scholar]
- 18. Meissner M., et al. (2012) Dimethyl fumarate—only an anti-psoriatic medication? J. Dtsch. Dermatol. Ges., 10, 793–801. [DOI] [PubMed] [Google Scholar]
- 19. Damal K., et al. (2013) Optimizing therapeutics in the management of patients with multiple sclerosis: a review of drug efficacy, dosing, and mechanisms of action. Biologics, 7, 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lin S.X., et al. (2011) The anti-inflammatory effects of dimethyl fumarate in astrocytes involve glutathione and haem oxygenase-1. ASN Neuro, 3, 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Linker R.A., et al. (2013) Dimethyl fumarate for treatment of multiple sclerosis: mechanism of action, effectiveness, and side effects. Curr. Neurol. Neurosci. Rep., 13, 394. [DOI] [PubMed] [Google Scholar]
- 22. Scannevin R.H., et al. (2012) Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J. Pharmacol. Exp. Ther., 341, 274–284. [DOI] [PubMed] [Google Scholar]
- 23. Stangel M., et al. (2013) Dimethyl fumarate (BG-12) for the treatment of multiple sclerosis. Expert Rev. Clin. Pharmacol., 6, 355–362. [DOI] [PubMed] [Google Scholar]
- 24. Boivin A., et al. (2011) Transient alteration of cellular redox buffering before irradiation triggers apoptosis in head and neck carcinoma stem and non-stem cells. PLoS One, 6, e14558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dethlefsen L.A., et al. (1988) Toxic effects of acute glutathione depletion by buthionine sulfoximine and dimethylfumarate on murine mammary carcinoma cells. Radiat. Res., 114, 215–224. [PubMed] [Google Scholar]
- 26. García-Caballero M., et al. (2011) Dimethylfumarate inhibits angiogenesis in vitro and in vivo: a possible role for its antipsoriatic effect? J. Invest. Dermatol., 131, 1347–1355. [DOI] [PubMed] [Google Scholar]
- 27. Gu B., et al. (2005) Enhanced cytotoxicity of bioreductive antitumor agents with dimethyl fumarate in human glioblastoma cells. Anticancer Drugs, 16, 167–174. [DOI] [PubMed] [Google Scholar]
- 28. Kirlin W.G., et al. (1999) Dietary compounds that induce cancer preventive phase 2 enzymes activate apoptosis at comparable doses in HT29 colon carcinoma cells. J. Nutr., 129, 1827–1835. [DOI] [PubMed] [Google Scholar]
- 29. Loewe R., et al. (2006) Dimethylfumarate impairs melanoma growth and metastasis. Cancer Res., 66, 11888–11896. [DOI] [PubMed] [Google Scholar]
- 30. Yamazoe Y., et al. (2009) Dimethylfumarate inhibits tumor cell invasion and metastasis by suppressing the expression and activities of matrix metalloproteinases in melanoma cells. Cell Biol. Int., 33, 1087–1094. [DOI] [PubMed] [Google Scholar]
- 31. Kuroda K., et al. (1989) Inhibitory effect of fumaric acid on 3′-methyl-4-(dimethylamino)azobenzene-induced hepatocarcinogenesis in rats. Chem. Pharm. Bull. (Tokyo), 37, 1345–1346. [DOI] [PubMed] [Google Scholar]
- 32. Kuroda K., et al. (1982) Inhibitory effect of fumaric acid on forestomach and lung carcinogenesis by a 5-nitrofuran naphthyridine derivative in mice. J. Natl. Cancer Inst., 69, 1317–1320. [PubMed] [Google Scholar]
- 33. Honda T., et al. (2000) Synthetic oleanane and ursane triterpenoids with modified rings A and C: a series of highly active inhibitors of nitric oxide production in mouse macrophages. J. Med. Chem., 43, 4233–4246. [DOI] [PubMed] [Google Scholar]
- 34. Liby K., et al. (2006) The synthetic triterpenoid CDDO-imidazolide suppresses STAT phosphorylation and induces apoptosis in myeloma and lung cancer cells. Clin. Cancer Res., 12(14 Pt 1), 4288–4293. [DOI] [PubMed] [Google Scholar]
- 35. Chan K., et al. (1996) NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl Acad. Sci. USA, 93, 13943–13948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Leung L., et al. (2003) Deficiency of the Nrf1 and Nrf2 transcription factors results in early embryonic lethality and severe oxidative stress. J. Biol. Chem., 278, 48021–48029. [DOI] [PubMed] [Google Scholar]
- 37. Suh N., et al. (1998) Novel triterpenoids suppress inducible nitric oxide synthase (iNOS) and inducible cyclooxygenase (COX-2) in mouse macrophages. Cancer Res., 58, 717–723. [PubMed] [Google Scholar]
- 38. Wright G.W., et al. (2003) A random variance model for detection of differential gene expression in small microarray experiments. Bioinformatics, 19, 2448–2455. [DOI] [PubMed] [Google Scholar]
- 39. Eisen M.B., et al. (1998) Cluster analysis and display of genome-wide expression patterns. Proc. Natl Acad. Sci. USA, 95, 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liby K., et al. (2008) The rexinoid LG100268 and the synthetic triterpenoid CDDO-methyl amide are more potent than erlotinib for prevention of mouse lung carcinogenesis. Mol. Cancer Ther., 7, 1251–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ohta T., et al. (2008) Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res., 68, 1303–1309. [DOI] [PubMed] [Google Scholar]
- 42. Shibata T., et al. (2008) Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc. Natl Acad. Sci. USA, 105, 13568–13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Singh A., et al. (2006) Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med., 3, e420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Solis L.M., et al. (2010) Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin. Cancer Res., 16, 3743–3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huang Y., et al. (2005) Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res., 65, 7446–7454. [DOI] [PubMed] [Google Scholar]
- 46. Jiang S.S., et al. (2010) Upregulation of SOX9 in lung adenocarcinoma and its involvement in the regulation of cell growth and tumorigenicity. Clin. Cancer Res., 16, 4363–4373. [DOI] [PubMed] [Google Scholar]
- 47. Zhou C.H., et al. (2012) Clinical significance of SOX9 in human non-small cell lung cancer progression and overall patient survival. J. Exp. Clin. Cancer Res., 31, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zito C.R., et al. (2012) Multi-level targeting of the phosphatidylinositol-3-kinase pathway in non-small cell lung cancer cells. PLoS One, 7, e31331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang H., et al. (2014) EGR1 decreases the malignancy of human non-small cell lung carcinoma by regulating KRT18 expression. Sci. Rep., 4, 5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ferraro B., et al. (2005) EGR1 predicts PTEN and survival in patients with non-small-cell lung cancer. J. Clin. Oncol., 23, 1921–1926. [DOI] [PubMed] [Google Scholar]
- 51. Feng J., et al. (2012) High expression of FoxP1 is associated with improved survival in patients with non-small cell lung cancer. Am. J. Clin. Pathol., 138, 230–235. [DOI] [PubMed] [Google Scholar]
- 52. Elaraj D.M., et al. (2006) The role of interleukin 1 in growth and metastasis of human cancer xenografts. Clin. Cancer Res., 12, 1088–1096. [DOI] [PubMed] [Google Scholar]
- 53. Lewis A.M., et al. (2006) Interleukin-1 and cancer progression: the emerging role of interleukin-1 receptor antagonist as a novel therapeutic agent in cancer treatment. J. Transl. Med., 4, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Paine A., et al. (2010) Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem. Pharmacol., 80, 1895–1903. [DOI] [PubMed] [Google Scholar]
- 55. Liby K., et al. (2005) The synthetic triterpenoids, CDDO and CDDO-imidazolide, are potent inducers of heme oxygenase-1 and Nrf2/ARE signaling. Cancer Res., 65, 4789–4798. [DOI] [PubMed] [Google Scholar]
- 56. Ahmad R., et al. (2006) Triterpenoid CDDO-Me blocks the NF-kappaB pathway by direct inhibition of IKKbeta on Cys-179. J. Biol. Chem., 281, 35764–35769. [DOI] [PubMed] [Google Scholar]
- 57. Yore M.M., et al. (2006) The synthetic triterpenoid 1-[2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole blocks nuclear factor-kappaB activation through direct inhibition of IkappaB kinase beta. Mol. Cancer Ther., 5, 3232–3239. [DOI] [PubMed] [Google Scholar]
- 58. Loewe R., et al. (2002) Dimethylfumarate inhibits TNF-induced nuclear entry of NF-kappa B/p65 in human endothelial cells. J. Immunol., 168, 4781–4787. [DOI] [PubMed] [Google Scholar]
- 59. Seidel P., et al. (2009) Dimethylfumarate inhibits NF-{kappa}B function at multiple levels to limit airway smooth muscle cell cytokine secretion. Am. J. Physiol. Lung Cell. Mol. Physiol., 297, L326–L339. [DOI] [PubMed] [Google Scholar]
- 60. Buelna-Chontal M., et al. (2013) Redox activation of Nrf2 & NF-κB: a double end sword? Cell. Signal., 25, 2548–2557. [DOI] [PubMed] [Google Scholar]
- 61. Wakabayashi N., et al. (2010) When NRF2 talks, who’s listening? Antioxid. Redox Signal., 13, 1649–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wagner E.F., et al. (2009) Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer, 9, 537–549. [DOI] [PubMed] [Google Scholar]
- 63. Musgrove E.A., et al. (2011) Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer, 11, 558–572. [DOI] [PubMed] [Google Scholar]
- 64. Sarma V., et al. (1992) Cloning of a novel tumor necrosis factor-alpha-inducible primary response gene that is differentially expressed in development and capillary tube-like formation in vitro . J. Immunol., 148, 3302–3312. [PubMed] [Google Scholar]
- 65. Herr I., et al. (2007) Regulation of differential pro- and anti-apoptotic signaling by glucocorticoids. Apoptosis, 12, 271–291. [DOI] [PubMed] [Google Scholar]
- 66. Lo S.S., et al. (2005) Cleavage of cten by caspase-3 during apoptosis. Oncogene, 24, 4311–4314. [DOI] [PubMed] [Google Scholar]
- 67. Won M., et al. (2010) Novel anti-apoptotic mechanism of A20 through targeting ASK1 to suppress TNF-induced JNK activation. Cell Death Differ., 17, 1830–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Balaban S., et al. (2015) Obesity and cancer progression: is there a role of fatty acid metabolism? Biomed Res. Int., 2015, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fernandez P., et al. (2004) Distinctive gene expression of human lung adenocarcinomas carrying LKB1 mutations. Oncogene, 23, 5084–5091. [DOI] [PubMed] [Google Scholar]
- 70. Tran K., et al. (2013) The combination of the histone deacetylase inhibitor vorinostat and synthetic triterpenoids reduces tumorigenesis in mouse models of cancer. Carcinogenesis, 34, 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gunning W.T., et al. (2000) Chemoprevention of vinyl carbamate-induced lung tumors in strain A mice. Exp. Lung Res., 26, 757–772. [DOI] [PubMed] [Google Scholar]
- 72. Spencer S.R., et al. (1990) Induction of glutathione transferases and NAD(P)H:quinone reductase by fumaric acid derivatives in rodent cells and tissues. Cancer Res., 50, 7871–7875. [PubMed] [Google Scholar]
- 73. Adam J., et al. (2011) Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell, 20, 524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ooi A., et al. (2011) An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell, 20, 511–523. [DOI] [PubMed] [Google Scholar]
- 75. Ghods A.J., et al. (2013) Beneficial actions of the anti-inflammatory dimethyl fumarate in glioblastomas. Surg. Neurol. Int., 4, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wen A.Y., et al. (2010) The role of the transcription factor CREB in immune function. J. Immunol., 185, 6413–6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Parry G.C., et al. (1997) Role of cyclic AMP response element-binding protein in cyclic AMP inhibition of NF-kappaB-mediated transcription. J. Immunol., 159, 5450–5456. [PubMed] [Google Scholar]
- 78. Shenkar R., et al. (2001) Interactions between CBP, NF-kappaB, and CREB in the lungs after hemorrhage and endotoxemia. Am. J. Physiol. Lung Cell. Mol. Physiol., 281, L418–L426. [DOI] [PubMed] [Google Scholar]
- 79. Altomare D.A., et al. (2005) Perturbations of the AKT signaling pathway in human cancer. Oncogene, 24, 7455–7464. [DOI] [PubMed] [Google Scholar]
- 80. Crowell J.A., et al. (2007) Targeting the AKT protein kinase for cancer chemoprevention. Mol. Cancer Ther., 6, 2139–2148. [DOI] [PubMed] [Google Scholar]
- 81. Romano G. (2013) The role of the dysfunctional akt-related pathway in cancer: establishment and maintenance of a malignant cell phenotype, resistance to therapy, and future strategies for drug development. Scientifica (Cairo), 2013, 317186. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.