

ABSTRACT
Enterococci are naturally tolerant to typically bactericidal cell wall-active antibiotics, meaning that their growth is inhibited but they are not killed even when exposed to a high concentration of the drug. The molecular reasons for this extraordinary tolerance are still incompletely understood. Previous work showed that resistance to killing collapsed specifically in mutants affected in superoxide dismutase (Sod) activity, arguing that bactericidal antibiotic treatment led to induction of a superoxide burst. In the present work, we show that loss of antibiotic tolerance in ΔsodA mutants of pathogenic enterococci is dependent on the energy source present during antibiotic treatment. Hexoses induce greater killing than the pentose ribose, and no killing was observed with glycerol as the energy source. These results point to glycolytic reactions as crucial for antibiotic-mediated killing of ΔsodA mutants. A transposon mutant library was constructed in Enterococcus faecalis ΔsodA mutants and screened for restored tolerance of vancomycin. Partially restored tolerance was observed in mutants with transposon integrations into intergenic regions upstream of regulators implicated in arginine catabolism. In these mutants, the arginine deiminase operon was highly upregulated. A model for the action of cell wall-active antibiotics in tolerant and nontolerant bacteria is proposed.
IMPORTANCE Antibiotic tolerance is a serious clinical concern, since tolerant bacteria have considerably increased abilities to resist killing by bactericidal drugs. Using enterococci as models for highly antibiotic-tolerant pathogens, we showed that tolerance of these bacteria is linked to their superoxide dismutase (Sod), arguing that bactericidal antibiotics induce generation of reactive oxygen species inside cells. Wild-type strains are tolerant because they detoxify these deleterious molecules by the activity of Sod, whereas Sod-deficient strains are killed. This study showed that killing depends on the energy source present during treatment and that an increase in arginine catabolism partially restored tolerance of the Sod mutants. These results are used to propose a mode-of-action model of cell wall-active antibiotics in tolerant and nontolerant bacteria.
INTRODUCTION
Even though enterococci are considered low-virulence pathogens, treatment of enterococcal infections is often difficult and lengthy. International guidelines for treatment of infective enterococcal endocarditis recommend 4 to 6 weeks of administration of penicillin or ampicillin plus an aminoglycoside, with a risk of acute renal failure due to the nephrotoxicity of the latter antibiotics (1–3). Enterococci, which are lactic acid bacteria and part of the human commensal flora, are naturally highly tolerant to antibiotics considered to be bactericidal such as penicillins and glycopeptides (4, 5). The mechanisms that allow these organisms to escape the lethal action of those drugs are still incompletely understood. On the basis of previous studies, it was suggested that the tolerance might be due to alterations in bacterial autolysis, because killing of nontolerant bacteria was often accompanied by massive cell lysis (6). However, penicillin has been shown to kill pathogens such as Streptococcus pyogenes and Enterococcus hirae efficiently without inducing significant lysis of the cells (7–9). Furthermore, suppression of autolysis in Streptococcus pneumoniae and Staphylococcus aureus had no significant effect on killing by penicillin (6, 10–13). These data collectively indicated that a decrease in the autolytic activity could not fully explain tolerance to bactericidal antibiotics for all drug/pathogen combinations.
Characterization of tolerant strains of Streptococcus gordonii that were obtained by cyclic exposure to penicillin at 500 times the MIC showed that two proteins belonging to the arc operon were upregulated in these derivatives (14). The arc operon encodes activities for arginine catabolism. In this pathway, arginine is first deiminated by arginine deiminase (ADI) encoded by arcA. The arginine degradation pathway is therefore also called the ADI pathway. The citrulline formed in the first reaction is then cleaved by phosphorolysis into ornithine and carbamoylphosphate catalyzed by ornithine carbamoyltransferase encoded by arcB. Finally, the high-energy compound carbamoylphosphate is degraded into CO2 and NH3 with the concomitant production of an ATP by carbamate kinase encoded by arcC (15, 16). However, inactivation of the arc locus in drug-tolerant strains of S. gordonii showed that increased tolerance was not a direct consequence of enhanced expression of the ADI pathway (14).
An important discovery for the understanding of the clinically problematic tolerance phenomenon was recently reported for pathogenic enterococci. Tolerance of penicillin and vancomycin was abolished in superoxide dismutase (Sod)-deficient mutants of Enterococcus faecalis and Enterococcus faecium and could be progressively restored by increasing expression of this enzyme from an inducible plasmid (4, 5). Enterococci harbor only one Sod of the manganese type named SodA, and the enzyme catalyzes an electron transfer between two O2− molecules; the first of the two molecules is oxidized to yield oxygen, and the second is reduced to hydrogen peroxide. The above-mentioned finding that ΔsodA mutants of both enterococcal species are between 1,000-fold and 10000-fold more sensitive to penicillin or vancomycin killing demonstrated that SodA is essential for tolerance of bactericidal antibiotics in these pathogenic bacteria (4, 5). The tolerance model proposed from these results foresees that bactericidal antibiotics at concentrations generally equivalent to 20× MIC induce an increased formation of superoxide inside cells. This is not a major problem for wild-type cells, because the O2− molecule is efficiently detoxified by SodA, resulting in tolerance. In contrast, in SodA-deficient strains, O2− accumulates to toxic levels, leading to killing of these mutants. Production of O2− induced by bactericidal antibiotics has been indirectly demonstrated (4). We reasoned that superoxide should also be produced in wild-type cells under conditions of exposure to bactericidal antibiotics. However, the superoxide formed is dismutated to H2O2 by SodA, and the H2O2 molecule is further reduced to H2O by peroxidases. If this cascade takes place in the wild-type cells, then the inactivation of genes encoding peroxidases should lead to an accumulation of H2O2 generated by SodA in the corresponding mutant under conditions of exposure to bactericidal but not bacteriostatic antibiotics. In contrast to superoxide, H2O2 diffuses from the cells and can be detected (38). As predicted, we found an accumulation of H2O2 in cultures of a strain carrying mutations in peroxidase-encoding genes under conditions of exposure to penicillin and vancomycin but not to tetracycline, which is a bacteriostatic antibiotic (4).
Although the model is based on the well-known activity of Sod and has experimental support, issues such as how bactericidal antibiotics induce O2− production and what the targets of O2− are that lead to killing still remain to be addressed. Detailed understanding of these molecular processes might contribute to the development of better therapeutics which would clear infections by these important pathogens more rapidly. In this work, we studied the dependence of tolerance on metabolic activity and utilization of catabolic pathways and screened a transposon mutant library for restored tolerance in a SodA-deficient strain of E. faecalis.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
Strains, plasmids, and oligonucleotides used in this study are listed in Table 1 and Table S1 in the supplemental material. Cultures of E. faecalis and E. faecium strains were grown in M17 medium (23) supplemented with 0.5% (wt/vol) glucose (GM17) or in brain heart infusion (BHI) broth. For experiments performed with specific carbon sources, we used ccM17 MOPS (morpholinepropanesulfonic acid) medium prepared as previously reported (24) (see below) and supplemented with 0.5% glucose (wt/vol) or with lactose, galactose, fructose, mannitol, ribose, or glycerol at a final concentration equimolar to that of glucose (28 mM). Experiments were performed under aerobic conditions in 100-ml Erlenmeyer flasks containing 10 ml medium, and the cultures were incubated at 37°C with gentle agitation (60 rpm) on a rotary shaker. When indicated, catalase from bovine liver (Sigma) (900,000 U/ml) was added to the medium at a final concentration of 500 U/ml. The growth kinetics of E. faecalis cultures were followed both by measurement of optical density at 600 nm (OD600) with a Biophotometer (Eppendorf, Hamburg, Germany) and by counts of viable CFU on agar plates. When necessary, the media were supplemented with 150 μg/ml erythromycin, 10 μg/ml chloramphenicol, or 300 μg/ml gentamicin. Escherichia coli strains were cultivated at 37°C in Luria-Bertani (LB) broth (25) with vigorous shaking (160 rpm). When needed, 100 μg/ml of ampicillin or 13 μg/ml of tetracycline was added to the medium. Bacterial stocks were stored at −80°C in GM17 or LB broth supplemented with glycerol at a final concentration of 15% (vol/vol).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s)a | Reference or source |
|---|---|---|
| Enterococcal strains | ||
| E. faecalis JH2-2 | Fusr Rifr plasmid-free wild-type strain | 17 |
| E. faecalis JH2-2 ΔsodA | JH2-2 sodA deletion mutant | 18 |
| E. faecalis JH2-2/pUCB30:arcA | JH2-2 arcA insertional mutant obtained by SCO event using pUCB30:arcA | This study |
| E. faecalis JH2-2 ΔsodA/pUCB30:arcA | JH2-2 ΔsodA arcA insertional mutant obtained by SCO event using pUCB30:arcA | This study |
| E. faecalis OG1RF | Rifr Fusr wild-type strain | 19 |
| E. faecalis OG1RF ΔsodA | OG1RF sodA deletion mutant | 5 |
| E. faecium Com12 | Healthy volunteer fecal isolate; wild-type strain | 20 |
| E. faecium ΔsodA Com12 | Com12 sodA deletion mutant | 5 |
| E. coli strain Top10F′ | F′ [lacIq Tn1 (Tetr)] mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80dlacZΔM15 ΔlacX74 recA1 araD139 Δ(ara-leu)7697 galU galK rpsL (Strr) endA1 nupG | Invitrogen |
| Plasmids | ||
| pUCB30 | Ori pMB1 lacZ′ Ampr Emr | 21 |
| pUCB30:arcA | pCB30 derivative carrying a 605-bp DNA internal fragment of arcA gene from E. faecalis JH2-2 | This study |
| pZXL5 | Ori(Ts) Cmr Genr Kanr nisR nisKP nisA; mariner transposase; mariner transposon; 2T7 promoters | 22 |
Amp, ampicillin; Em, erythromycin; Fus, fusidic acid; Rif, rifampin; Str, streptomycin; Tet, tetracycline; Cm, chloramphenicol; Gen, gentamicin; Kan, kanamycin; nis, nisin gene; Ts, thermosensitive; SCO, single crossover.
Susceptibility testing and time-kill curves.
The MICs were determined by the Etest method. Time-kill curves were determined with exponentially growing cultures by adding vancomycin to reach a final concentration of 20 μg/ml (20× MIC) at an OD600 of 0.5 (corresponding to ∼4 × 108 CFU/ml). Growth was followed by measuring OD600 at regular intervals, and CFU counts were determined after 24 h of incubation by plating 10-fold serial dilutions of the cultures on GM17 agar plates. The number of survivors giving rise to colonies was determined after 48 h of incubation at 37°C. Killing by antibiotic was assessed as the percentage of surviving colonies relative to unchallenged cells. The sensitization factor is defined as the ratio of survival of a parent strain to that of its derivative mutant strain. For survival experiments in the presence of specific energy sources, cultures were grown on GM17 until an OD600 of 0.5 was reached, harvested by centrifugation, washed with 10 ml of 0.9% NaCl solution, and finally resuspended in ccM17 MOPS medium supplemented with glucose 0.5% (wt/vol) or with lactose, galactose, fructose, mannitol, ribose, or glycerol at a final concentration equimolar to that used for glucose (28 mM). Immediately after, vancomycin or penicillin was added at final concentration of 20 μg/ml.
General molecular methods.
Molecular cloning and other standard techniques were performed as previously described (25). Antibiotics, chemicals, and enzymes were reagent grade. Restriction endonucleases and T4 ligase were obtained from Promega (Madison, WI) and used in accordance with the manufacturer's instructions. Plasmids and PCR products were purified using NucleoSpin kits (Macherey-Nagel, Dünen, Germany). E. coli and E. faecalis competent cells were transformed by electroporation using a Gene Pulser apparatus (Bio-Rad Laboratories, Mares la Coquette, France).
Construction of insertional E. faecalis JH2-2/pUCB30:arcA and JH2-2 ΔsodA/pUCB30:arcA mutants.
Insertional mutants with mutations in the arcA gene were constructed in E. faecalis JH2-2 and its ΔsodA derivative mutant using the pUCB30 suicide vector (21) (Table 1). Briefly, an internal DNA fragment of 605 bp of the arcA gene (EF_0104) was amplified by PCR with specific primers 104ForPst/104RevEco (see Table S1 in the supplemental material), digested by EcoRI and PstI, and ligated into the pUCB30 integrative vector previously treated with the same enzymes. The ligation product was electroporated into E. coli Top10F′ cells (Table 1). The subsequent recombinant plasmid (pUCB30:arcA) (Table 1) was transformed in E. faecalis JH2-2 and its ΔsodA mutant. The generated derivative mutants (JH2-2/pUCB30:arcA and JH2-2 ΔsodA/pUCB30:arcA mutants) (Table 1) were verified by PCR for the insertion of the pUCB30:arcA recombinant plasmid within the chromosome, leading to the inactivation of the arcA gene.
Construction of a high-density transposon mutant library using E. faecalis ΔsodA mutants.
For transposon mutagenesis in the E. faecalis ΔsodA JH2-2 and OG1RF strains, we used the pZXL5 transposon delivery plasmid (GenBank accession number JQ088279) (Table 1) and a method previously described (22). Briefly, after electroporation, gentamicin-resistant transformants were grown overnight in BHI broth supplemented with 300 μg/ml gentamicin and 10 μg/ml chloramphenicol at the permissive temperature of 28°C. A 100-μl volume (approximately 108 viable cells) of this overnight culture was then inoculated into 200 ml of prewarmed BHI broth supplemented with gentamicin and 25 ng/ml nisin and grown overnight at the nonpermissive temperature of 37°C without shaking to induce transposition. Subsequently, 100 μl of this culture was transferred to 200 ml of fresh prewarmed BHI broth and grown similarly overnight without nisin. The mutant library was stored at −80°C in BHI broth containing a 15% (vol/vol) final concentration of glycerol in 1-ml aliquots as mutant library stocks.
Randomness and coverage of transposition were evaluated by PCR analysis and by determination of the integration site of 13 randomly selected colonies. For the first method, genomic DNA was isolated from the library using a NucleoBond RNA/DNA 400 kit (Macherey-Nagel, Dünen, Germany) and then used as the template DNA for PCR using 14 different primers which hybridize to different chromosomal regions. Each of these primers was tested with primer LTn or RTn, corresponding to the outer primers of the transposon. If the transposon integrates randomly, for each primer (Px) specific to a given gene (efx), the PCR carried out with primer pair Px/LTn or Px/RTn should amplify DNA fragments with different sizes, which should result in a smear on agarose gels. This was observed (data not shown). Determination of the integration sites of 13 randomly selected colonies was performed as described for the mapping of the integration sites in mutants with restored tolerance (see next paragraph and Fig. S1 in the supplemental material). Shortly thereafter, the chromosomal DNA of the colonies was extracted and digested with HaeIII and the fragments were circularized by ligation followed by PCR analyses performed using the transposon-specific primer pair LTn/RTn. This showed that 77% of the cells harbored one copy of the transposon, that no multiple insertion sites were found, and that the transposon had integrated in different parts of the chromosome.
Screening for genes involved in tolerance of vancomycin in the E. faecalis OG1RF ΔsodA mutant.
The library of random mutants constructed in the non-vancomycin-tolerant ΔsodA mutant of E. faecalis OG1RF was screened for restored tolerance of vancomycin. For this purpose, the library was grown on GM17 with gentle shaking (60 rpm) until an OD600 of 0.5 was reached, and then 20 μg/ml of vancomycin was added and the culture was incubated for 24 h at 37°C. A second treatment performed under the same conditions was applied again to enrich the number of vancomycin-tolerant mutants. Following this step, the culture was diluted and spread on GM17 agar medium. The survivors were individually tested for tolerance of vancomycin. The integration sites of the transposon of derivatives with increased tolerance of vancomycin were then analyzed. Therefore, genomic DNA was extracted for each tolerant mutant and digested with HaeIII endonuclease, generating DNA fragments containing the transposon with chromosome junctions (see Fig. S1 in the supplemental material). The restriction fragments were then circularized using conditions favoring intramolecular reactions. The loci in which the transposon had inserted were amplified by PCR using the transposon-specific primer pair LTn/RTn with DNA TripleMaster polymerase mix (Eppendorf, Hamburg, Germany) under the following conditions: 94°C for 1 min followed by 32 cycles of 94°C for 18 s, 53°C for 30 s, and 68°C for 7 min. The PCR products were purified and sequenced (Eurofins MWG Operon), and the obtained sequences were compared to the E. faecalis OG1RF genomic sequence (http://blast.ncbi.nlm.nih.gov/) in order to locate the insertion sites.
Analysis of fermentation products.
Glucose, lactate, formate, and acetate concentrations were determined using high-performance liquid chromatography (HPLC). The filtrates resulting from supernatants of 24-h cultures (25 μl) were injected in a Phenomenex PHM-monosaccharide column maintained at 65°C. The flow rate of the mobile phase, 5 mM H2SO4, was 0.5 ml/min. A refractometer detector was used, and the signal was analyzed through the use of Jasco-Borwin software (JMBS Developments, France). The obtained HPLC profiles from the 24-h culture filtrates were compared to the profiles from the control ccM17 MOPS media containing only the specific carbon sources. The concentrations and the peak areas of the carbohydrate substrate and its derivative fermentation products were measured in comparison to those of their corresponding standards.
RNA isolation and RT-qPCR.
To comparatively analyze gene expression, we used the E. faecalis OG1RF ΔsodA strain and its derivative C49, C50, and C56 mutants cultivated on GM17 medium with gentle shaking. Using a Direct-Zol RNA MiniPrep kit (ZymoRecherch), three independent samples of total RNA were isolated from each strain. For reverse transcriptase quantitative PCR (RT-qPCR), specific primers were designed (see Table S1 in the supplemental material) using the E. faecalis OG1RF genome sequence and Primer3 software (http://frodo.wi.mit.edu/primer3/) to produce amplicons of equivalent lengths of 100 bp. Total RNA (2 μg) was reverse transcribed with random hexamer primers and QuantiTect enzyme (Qiagen, Courtaboeuf, France). Quantification of gyrA (encoding the A subunit of DNA gyrase) mRNA provided an internal control. Amplification (using 5 μl of a 1:100 cDNA dilution), detection (with automatic calculation of the threshold value), and real-time analysis were performed twice for each cDNA sample using an iCycleriQ detection system (Bio-Rad Laboratories, Marnes la Coquette, France). Relative mRNA levels for each gene in each sample were calculated using comparative cycle time data, as described elsewhere (26).
Statistics.
Differences among strains in killing by bactericidal antibiotics and gene expression were calculated using the Student t test. P values of less than 0.05 were considered to be significant.
RESULTS
Loss of tolerance of vancomycin or penicillin of Sod-deficient strains is dependent on active cell metabolism.
In reports of a previous study, we showed that Sod is the basis of tolerance to bactericidal antibiotics in pathogenic enterococci (4, 5). We proposed therefore that cell wall-active bactericidal antibiotics induced a superoxide burst responsible for the killing of ΔsodA mutants in which the detoxification of O2− could not be performed. However, the exact molecular mechanisms that explain how these drugs induce O2− formation are still unknown. We also showed that this putative production of O2− is dependent on the primary action of these antibiotics (4, 5) and that no killing occurred under anaerobic conditions (4). We suggested that alterations of the cell wall may trigger modifications in metabolism leading to the formation of O2−. This concept predicts that, during treatment with a bactericidal antibiotic, cells should still be metabolically active. We first studied the effect on the OD600 during treatment of the E. faecalis JH2-2 wild-type strain and its isogenic ΔsodA mutant when vancomycin was added at a final concentration of 20× MIC to cultures at an OD600 of 0.5, which corresponds to the beginning of the exponential phase. After 24 h, those values were nearly unchanged (OD600 = ∼0.6) and were comparable for the two strains.
To test the metabolic activity of the cells after 24 h of vancomycin treatment, we performed HPLC analyses to determine the concentrations of the substrate and final products of fermentation (Fig. 1A and B). The results showed that glucose, used as a substrate in these experiments, was completely consumed by wild-type cultures and that peaks corresponding to the end products lactate, formate, and acetate could be identified. The result obtained with the ΔsodA mutant was different only in that the glucose substrate was consumed only partially (44%). However, since the ΔsodA mutant exhibited a decrease in survival of nearly 4 log10 whereas 80% of the wild-type cells survived (Fig. 1C), it is reasonable to assume that this partial glucose consumption in the ΔsodA mutant reflects cell death. Similar results have been obtained after exposure to 20× MIC of penicillin (see Fig. S2 in the supplemental material). Of note, a kinetic analysis of cell death showed that no killing by vancomycin of the JH2-2 ΔsodA mutant was observed in the first 6 h of treatment. During this period, the mutant cells may have metabolic activity comparable to that of the wild-type cells (Fig. 1C). We concluded from these combined results that cells have active metabolism but do not grow in the presence of 20× MIC of cell wall-active antibiotics.
FIG 1.

Metabolic activity and survival of E. faecalis JH2-2 and the ΔsodA mutant under conditions of vancomycin treatment. Data represent the results of HPLC analysis of the remaining substrate and fermentation products after 24 h of treatment with 20 μg/ml vancomycin in ccM17 MOPS medium supplemented with 0.5% (wt/vol) glucose of the E. faecalis JH2-2 wild-type strain (A) or its ΔsodA mutant (B) and kinetic analysis of killing by vancomycin of the JH2-2 wild-type strain (○) and its isogenic ΔsodA mutant (▵) (C). WT, wild type.
Loss of tolerance of vancomycin or penicillin of Sod-deficient strains is dependent on the energy source.
From the previous observation, we hypothesized that the catabolic activity of the cells is a requirement for antibiotic-induced killing and is the basis of the O2− production. If true, incubation of the ΔsodA mutant with vancomycin in the absence of a carbohydrate would not result in a decrease in viability. As shown in Fig. 2A, this was indeed the case. We then reasoned that if the reactions that are the basis of the O2− formation are linked with particular catabolic pathways, incubation with different substrates during treatment metabolized by different catabolic routes should modify killing of the ΔsodA mutant. Enterococci harbor the three major pathways of carbohydrate catabolism (16), the Embden-Meyerhof-Parnas (glycolysis) pathway, the Entner-Doudoroff (KDPG [2-keto-3-deoxy-6-phosphogluconate]) pathway, and the pentose phosphate pathway (Fig. 2B). The first two pathways are restricted to hexoses, whereas the pentose pathway ferments hexoses and pentoses. First, we tested different hexoses such as glucose, galactose, and fructose but also mannitol and the disaccharide lactose (data not shown). With all these substrates, significant vancomycin killing of the ΔsodA mutant (which was 600-fold to 3,000-fold more sensitive than the parent strain) was observed (Fig. 2A). Using glucose as the substrate, similar results have been obtained with penicillin (see Fig. S2 in the supplemental material). Notably, for the majority of these sugars, the Sod-deficient strain exhibited a slight growth retardation compared to the wild-type parent (see Fig. S3). This difference in growth was most pronounced in the case of the use of galactose as the substrate, probably due to the generation of H2O2 when E. faecalis was aerobically grown on this energy source (24). We then tried the hexose gluconate as the energy source, but neither the JH2-2 strain nor the OG1RF strain grew significantly on this substrate. A similar result has been published for E. faecalis strain V583 (27). In the next step, we tested the pentose ribose, which is metabolized through the pentose phosphate pathway (Fig. 2B). On this sugar as well, growth of the ΔsodA mutant was slightly slower than that of the JH2-2 wild-type strain (see Fig. S3). Interestingly, compared to the hexose results, the use of ribose as a substrate during the antibiotic treatment reduced killing of the ΔsodA mutant by 2 orders of magnitude (Fig. 2A). Comparable results have been obtained with the E. faecium Com12 wild-type strain and its ΔsodA mutant using glucose and ribose as substrates (see Fig. S4).
FIG 2.

Bactericidal activity is dependent on the energy source in the E. faecalis JH2-2 ΔsodA mutant. (A) Ratio of relative survival rates of the E. faecalis ΔsodA mutant and the E. faecalis JH2-2 wild-type strain after 24 h of exposure to 20 μg/ml of vancomycin in ccM17 MOPS medium supplemented with glucose, galactose, fructose, ribose, or glycerol. In the case of glucose, ribose, and glycerol supplementation, similar results in the presence of 20 μg/ml of penicillin have been obtained (see Fig. S2 in the supplemental material). Mean values of the results of at least three different experiments are represented with error bars indicating standard deviations. (B) Integration of the results determined as described for panel A into the metabolic network of E. faecalis. Metabolites are color-coded according to the degree of killing by vancomycin. The following metabolic intermediates are shown: glucose-6-phosphate (Glu-6-P), glucose-1-phosphate (Glu-1-P), fructose-6-phosphate (Fru-6-P), fructose-1,6-bisphosphate (Fru-1,6-P2), galactose-1-phosphate (Gal-1-P), galactose-6-phosphate (Gal-6-P), tagatose-1-6 bisphosphate (Tag-1,6-P2), glycerol-3-phosphate (G-3-P), dihydroxyacetone (DHA), dihydroxyacetone phosphate (DHA-P), glyceraldehyde-3-phosphate (GA-3-P), 1,3-bisphospho-glycerate (G-1,3-P2), 3-phospho-glycerate (3-P-G), 2-phospho-glycerate (2-P-G), phosphoenolpyruvate (PEP), ribulose-5-phosphate (Rbu-5-P), ribose-5-phosphate (Rib-5-P), xylulose-5-phosphate (Xyl-5-P), erythrose-4-phosphate (Ery-4-P), sedoheptulose-7-phosphate (Sed-7-P), 6-phospho-gluconate (6-P-Gln), and 2-keto-3-deoxy-6-phosphogluconate (KDPG).
An interesting substrate to study in the context of this work was glycerol, since the complex catabolism of this sugar alcohol has been recently intensively studied in E. faecalis (28, 29). E. faecalis harbors two pathways of glycerol catabolism, the glycerol kinase (GlpK) pathway and the dihydroxyacetone kinase (DHAK) pathway. Both pathways finally generate dihydroxyacetone phosphate (DHAP), which then enters the lower part of the glycolysis pathway shown in Fig. 2B. The GlpK pathway starts with phosphorylation of glycerol to glycerol-3-phosphate (G-3-P) by the use of glycerol kinase. The G-3-P formed is then oxidized by G-3-P oxidase to DHAP, leading to the generation of hydrogen peroxide (H2O2). The route that proceeds via the DHAK pathway starts with the oxidation of the substrate by glycerol dehydrogenase to dihydroxyacetone (DHA) with the concomitant generation of an NADH. The DHA is then phosphorylated to DHAP by DHAK. We showed recently that glycerol is metabolized simultaneously by the two pathways in the E. faecalis JH2-2 strain (28). When glycerol was used as a substrate during the antibiotic treatment of the ΔsodA mutant, killing by vancomycin was abolished (Fig. 2A). As mentioned above, the metabolism of glycerol via the GlpK pathway leads to the generation of H2O2, and we demonstrated that transient accumulation of this peroxide occurred during growth of the E. faecalis JH2-2 wild-type strain, leading to a growth inhibition which became even more pronounced in mutants affected in peroxidases (24). Since ΔsodA mutants are more sensitive to externally added H2O2 (18, 30, 31), the possibility that the growth on glycerol of the JH2-2 ΔsodA mutant was affected by the intracellular generation of H2O2 during catabolism of this substrate could not be excluded, and we tested this experimentally. While the wild-type JH2-2 strain grew to a final OD600 of 3.5, the isogenic ΔsodA mutant entered stationary phase early after start of growth and reached a final OD600 of only 0.4 (see Fig. S3 in the supplemental material). This growth inhibition was due to the accumulation of H2O2, since addition of active (but not of heat-inactivated) bovine catalase to the growth medium lifted this growth inhibition (see Fig. S3). In the presence of catalase, growth of the ΔsodA mutant was comparable to that of the JH2-2 parental strain. Since we showed that the ΔsodA mutant was not killed by vancomycin in the absence of a carbohydrate source (which led to a lack of growth [see above]), the possibility could not be excluded that the survival of this strain when glycerol was used as a carbon source during antibiotic treatment was due to the absence of significant growth and hence to reduced metabolic activity of the ΔsodA mutant. Therefore, we analyzed killing of the ΔsodA mutant with the concomitant presence of glycerol and catalase during vancomycin treatment. We showed previously that addition of catalase did not affect killing of the JH2-2 ΔsodA mutant in M17 medium containing 0.5% glucose (4). However, in the presence of glycerol and catalase, the SodA-deficient strain survived vancomycin exposure as well as the isogenic parent strain did (Fig. 2A). Therefore, the slow-growth phenotype was not the reason for the absence of killing when glycerol was used as the energy source.
The combined results presented above strongly suggest that the degree of loss of tolerance of the ΔsodA mutant is dependent on the catabolic pathway used to metabolize an energy source. The presence of substrates during the antibiotic treatment upon entering glycolysis led to high-level killing of the ΔsodA mutant. Killing was significantly reduced when ribose, which is metabolized through the pentose phosphate pathway, was the substrate. Finally, in the presence of glycerol, the ΔsodA mutant was as tolerant as the wild type to exposure to 20× MIC of vancomycin. Since glycerol enters the lower part of the glycolysis pathway and no killing of the ΔsodA mutant was observed with this substrate, we speculated that reactions present in the upper part of the glycolysis pathway should be responsible for the increased killing of the ΔsodA mutant. This would also explain the residual killing observed with ribose as the substrate. The pentose phosphate pathway is connected to the upper and lower parts of the glycolytic pathway through fructose-6-phosphate and glyceraldehyde-3-phosphate metabolites that are formed by transketolase and transaldolase reactions (Fig. 2B). To verify this assumption, we tried to construct a mutant defective in the upper part of the glycolysis pathway. We choose to inactivate the pgi gene encoding glucose-6-phosphate isomerase catalyzing isomerization of glucose-6-phosphate (Glu-6-P) to fructose-6-phosphate. We predicted that in the presence of glucose, the Glu-6-P formed would enter the pentose phosphate pathway in this mutant (Fig. 2B). The required enzymes as well as the genes encoding enzymes Glu-6-P dehydrogenase and gluconate-6-P dehydrogenase are present in E. faecalis (32). However, all attempts to obtain this mutant failed, suggesting that the upper part of the glycolysis pathway is essential in E. faecalis. The finding that a 2-fold reduction of phosphofructokinase activity strongly decreased the growth rate of the lactic acid bacterium Lactococcus lactis is in agreement with this conclusion (33).
Construction and screening of a transposon mutant library in E. faecalis.
To gather more insight into the correlation between catabolic pathway utilization and tolerance to bactericidal antibiotics, we created a genome-wide transposon mutant library in the E. faecalis ΔsodA mutant and screened it for variants with increased tolerance of vancomycin in the presence of glucose as the energy source. Recently, construction of a high-density transposon mutant library has been established for E. faecium using a new transposon delivery plasmid named pZXL5 (22). We therefore tested if this plasmid could also be a useful tool for transposon mutagenesis in E. faecalis. We started with the JH2-2 ΔsodA mutant and analyzed the randomness of the transposon insertions by PCR. The results showed that integration was not random in this background but that the transposon integrated preferentially into many regions (hot spots) of the chromosome (data not shown). We then tested a previously constructed ΔsodA mutant of E. faecalis strain OG1RF (5). Construction of the mutant library in this strain also revealed the presence of hot spots, but these were much less frequent than in the JH2-2 background (data not shown). Therefore, we decided to screen the library constructed in the OG1RF ΔsodA mutant background.
To select transposon mutants with increased tolerance of vancomycin, the ΔsodA mutant library was subjected to two rounds of exposure to 20× MIC of vancomycin for 24 h. The survivors of this procedure were plated, and colonies were individually tested for tolerance of the antibiotic. Of 150 colonies analyzed, 10 individual clones showed tolerance of the antibiotic treatment that was increased (between 1 and 2 orders of magnitude) in comparison to that seen with the OG1RF ΔsodA control strain (Fig. 3). The insertion sites of the transposon were determined for the 10 clones, and the results showed that the transposon had integrated in three different intergenic regions (IGR1 to IGR3) (Fig. 4). For the majority (8 of 10) of the clones, the transposon had integrated into IGR1 at three different positions between genes EF_0102 and EF_0103. The two divergent genes surrounding this IGR both encode arginine transcriptional regulators of the ArgR family. We identified two clones in which the transposon had integrated 51 bp upstream of gene EF_0102 and five clones with an integration site 35 bp upstream of gene EF_0103, and in one clone, the transposon was situated 3 bp upstream of EF_0103. In the case of IGR2 and IGR3, the transposon had integrated 1 bp upstream of a gene (EF_0676) encoding a putative ArgR repressor and in the middle of IGR3, respectively. The adjacent genes of IGR3 encode a glycine/betaine ABC transporter (EF_3065) and a deformylase (EF_3066). Since 9 of the 10 clones appear to be involved in the regulation of arginine metabolism, we further focused our study on these transposon mutants.
FIG 3.
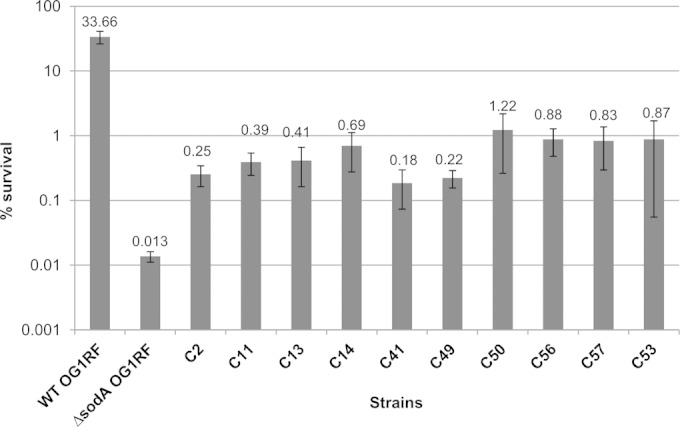
Relative survival rates of E. faecalis OG1RF, ΔsodA, and transposon mutants in the presence of vancomycin. Survival of the OG1RF wild-type strain, its isogenic ΔsodA mutant, and clones screened from the transposon library demonstrating partially restored tolerance (C2, C11, C13, C14, C41, C49, C50, C53, C56, and C57) was determined after 24 h of treatment with 20 μg/ml of vancomycin in GM17 medium. Mean values of the results of at least three different experiments are represented with error bars indicating standard deviations.
FIG 4.

Mapping of insertion sites of the transposon in clones with partially restored tolerance of vancomycin. The transposon (Tn) integrated into three different intergenic regions (IGR1 to IGR3). In IGR1, the Tn integrated at three different positions between genes EF_0102 and EF_0103 encoding both putative arginine regulators (ArgR1and ArgR2). In IGR2, the Tn integrated upstream of gene EF_0676, which encodes a putative arginine repressor (ArgR3). In IGR3, the Tn inserted approximately in the center between genes EF_3065 and EF_3066, encoding a putative glycine/betaine ABC transporter (RpsO) and a putative deformylase (Def), respectively. Numbers above the insertion sites refer to the distances between the ends of the transposon and adjacent genes.
As mentioned above, in the majority of clones the transposon had integrated between two ArgR family regulators (EF_0102 and EF_0103). Directly downstream of this region, genes related to arginine catabolism are present in E. faecalis JH2-2. These genes encode arginine deiminase (arcA; EF_0104), an ornithine carbamoyltransferase (arcF-1; EF_0105), and a carbamate kinase (arcC-1; EF_0106). These three genes seem to be in an operon structure with two other genes encoding a putative Crp/Fnr transcriptional regulator (arcR; EF_0107) and a putative ornithine-arginine antiporter (arcD; EF_0108) (see Fig. S5 in the supplemental material) (15). Due to this context, we hypothesized that one or both ArgR regulators present upstream of the arginine deiminase (ADI) operon are implicated in the regulation of its expression and that insertion of the transposon modified expression of the regulator and hence also that of the ADI operon. This was tested by reverse transcription quantitative PCR (RT-qPCR), which showed that this was indeed the case (Table 2). In clone 50, the transposon integrated 51 bp upstream of gene EF_0102 (Fig. 4). Compared to the ΔsodA mutant, expression of the EF_0102 gene was induced nearly 10-fold in clone 50. Analysis of expression of the EF_0103 gene, which encodes the other ArgR regulator, showed that it was also induced in this Tn mutant, albeit to a (4-fold) lesser extent than EF_0102. Furthermore, genes of the ADI operon were highly (between 8-fold and 23-fold) induced in clone 50 (Table 2). In clone 56, the transposon had integrated 35 bp upstream of EF_0103. The RT-qPCR experiments revealed that, in comparison to the ΔsodA mutant, EF_0103 expression in this clone was strongly repressed but expression of EF_0102 was induced around 8-fold. However, also in this Tn mutant, the ADI operon was highly (between 12-fold and 16-fold) induced. This strongly suggests that induced expression of the ADI operon in the Tn mutants is due to induction of gene EF_0102, whose product acts as a transcriptional activator (see Fig. S5).
TABLE 2.
Ratio of RT-qPCR values of clones 50, 56, and 49 to RT-qPCR values of ΔsodA mutant
| Gene | RT-qPCR value ratioa |
||
|---|---|---|---|
| Clone 50/ΔsodA mutant | Clone 56/ΔsodA mutant | Clone 49/ΔsodA mutanta | |
| EF_0102 | +9.27* | +7.84* | +1.71* |
| EF_0103 | +4.00* | −9.95* | +2.452 |
| EF_0104 | +12.29* | +15.29* | +13.79* |
| EF_0105 | +8.21* | +12.21* | +8.67* |
| EF_0106 | +22.80* | +15.34* | +10.66* |
| EF_0107 | +21.11* | +16.06* | +10.71* |
| EF_0676 | +1.65 | +1.62 | −124.65* |
*, P = <0.05 (considered significant).
In the case of clone 49 of group II, the transposon had integrated 1 bp upstream of gene EF_0676, which encodes a putative ArgR repressor. This integration provoked very strong (125-fold) repression of the expression of this gene (Table 2). Interestingly, expression of the ADI operon was also significantly (between 9 and 14-fold) induced in this Tn mutant, suggesting that the product of gene EF_0676 may act as a transcriptional repressor of the ADI operon (see Fig. S5 in the supplemental material). Of note, the levels of expression of the EF_0102 and EF_0103 genes of clone 49 and the ΔsodA mutant were comparable, and expression of EF_0676 was not modified in the Tn mutants with integration sites between genes EF_0102 and EF_0103 (clones 50 and 56) (Table 2).
Taken together, these results strongly suggest that the increase in expression of the ADI operon is responsible for the increase of tolerance of vancomycin in the Tn mutants. We reasoned therefore that inactivation of arginine catabolism should lead to a decrease of tolerance to this antibiotic, although we could not exclude the possibility that the increased expression of the Crp/Fnr family transcriptional regulator (arcR; EF_0107) which is part of the ADI operon was the basis of the increased tolerance. So we decided to construct mutants with mutations of the first gene of the operon encoding arginine deiminase (arcA; EF_0104) but also, in parallel, to inactivate the arcR (EF_0107) gene by integration of plasmids containing internal fragments of the corresponding genes in both the JH2-2 wild-type strain and the ΔsodA mutant strain. Whereas the arcA mutant was obtained without any difficulty in both backgrounds, all attempts to construct an arcR mutant failed. The JH2-2 arcA mutant and the isogenic ΔsodA arcA double mutant were then tested for tolerance of vancomycin. The results showed that introduction of an arcA mutation into the JH2-2 wild-type strain had no effect on survival in the presence of 20× MIC of vancomycin but that this mutation further decreased survival (20-fold, P = 0.05) of the ΔsodA mutant under these conditions (Fig. 5). From these results, we hypothesized that arginine catabolism may be important in E. faecalis only under oxidative stress conditions, which we postulated to be the case in the SodA-deficient cells due to induction of a superoxide burst upon antibiotic treatment (4, 5).
FIG 5.
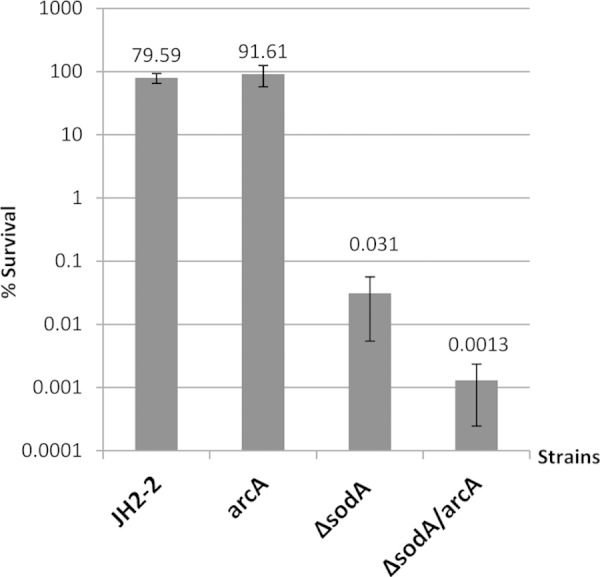
Relative survival rates after vancomycin treatment of E. faecalis JH2-2 and isogenic mutant strains. Survival of the wild-type strain and the arcA, ΔsodA, and ΔsodA arcA mutants was determined after 24 h of treatment with 20 μg/ml of vancomycin in GM17 medium. Mean values of the results of at least three different experiments are represented with error bars indicating standard deviations.
DISCUSSION
The results presented in this communication contribute to the progression of the understanding of the extraordinary tolerance to bactericidal antibiotics of enterococci. As we showed in this and previous work, tolerance is linked to their manganese-cofactored superoxide dismutase, and we postulated that loss of tolerance in Sod-deficient mutants is due to increased production of O2− upon antibiotic treatment (4, 5). The new elements in the present work are that the formation of O2− depends not only on active catabolism but also on the kind of energy source that is present during treatment. To the best of our knowledge, this seems to be the first study demonstrating that, during drug treatment, in our case, treatment with vancomycin or penicillin, antibiotic-susceptible cells continue to consume the substrate but do not grow during exposure to the antibiotic. Furthermore, we showed that this consumption (and, therefore, an active metabolism) is a prerequisite for bactericidal antibiotics to exert their killing effect. This strongly suggests the decoupling of energy production from the cell cycle, so a kind of high-engine-power-while-idling motion may be the basis of the increased production of O2−. However, the molecular basis of this oxidative burst still remains unknown, although our report highlights the importance of the upper part of the glycolysis pathway in this context.
The present work also revealed a link between antibiotic tolerance and arginine catabolism. In Tn mutants with an upregulated ADI operon, tolerance of the Sod-deficient strain was partially restored. These results seem to support the above-mentioned oxidative-stress-based killing model, since a recent study demonstrated the direct implication of the ADI operon in the oxidative stress response of Lactococcus lactis (34). In a spontaneously H2O2-resistant mutant of this species, the ADI operon was found to be upregulated and disruption of the first (arcA) or second (arcB) gene of this operon abolished peroxide resistance. In analogy to these results, it can be suggested that the upregulation of the ADI operon in E. faecalis protects the cells from oxidative stress. As we postulate in the tolerance model, this occurs in ΔsodA mutants treated with bactericidal cell wall-active antibiotics (Fig. 6). This interpretation is further supported by the results obtained with a double mutant affected in Sod activity and the ADI operon. This strain was significantly less tolerant than a ΔsodA single mutant.
FIG 6.

Proposed model for action of cell wall-active antibiotics in tolerant and nontolerant bacteria. Refer to the text for more-detailed information.
Recently, two publications (35, 36) convincingly questioned the hitherto widely accepted model of how bactericidal antibiotics kill bacteria (37). The cell death pathway model proposed that these drugs induce the creation of reactive oxygen species (ROS) which at least contribute to cell killing (37). We have previously published results obtained with E. faecalis (4) which differed in important points from the model proposed by Kohanski et al. (37). Specifically, neither the presumptive hydroxyl radical scavenger thiourea nor iron scavengers protected the SodA-deficient cells from killing. Additionally, peroxidase/catalase mutants as well as DNA repair-deficient recA mutants were not more sensitive to bactericidal antibiotics than their wild-type parents (4). These results are now in line with the new data published in the above-mentioned two studies (35, 36). However, seemingly in conflict with those two studies, which concluded that bactericidal antibiotics do not induce oxidative stress in E. coli, we proposed that cell wall-active antibiotics do induce the creation of superoxide formation in enterococci. This conclusion is based on the specific killing of ΔsodA mutants of these bacteria and on the absence of killing under anaerobic conditions (4, 5). In contrast to enterococci, E. coli is not tolerant of bactericidal antibiotics and is rapidly killed (decrease of 3 to 4 orders of magnitude in 5 h) when exposed to them (35, 37) whereas 10% to 80% of enterococci survive a 24-h exposure to these drugs (4, 5). The results obtained with E. coli and enterococci allowed refining of the tolerance model we previously proposed for enterococci (4). Nontolerant bacteria such as E. coli are quickly killed by the primary actions of bactericidal antibiotics (Fig. 6). Enterococci, however, are tolerant of these primary killing mechanisms. In the long run, cell wall-active antibiotics induce the creation of superoxide radicals in these bacteria in a process which needs active metabolism during exposure to these drugs. In wild-type bacteria, O2− is efficiently detoxified by SodA, leading to long-term tolerance. The accumulation of ROS, and hence their deleterious effects, occurs only in SodA-deficient enterococci. However, killing is not immediate but starts more than 6 h after the exposure to antibiotics. This delay in killing probably reflects the time necessary to accumulate O2− to a threshold concentration, at which point bacterial death occurs. This refined model is now the basis for the design of new experiments which are aimed at further progress in the understanding of bacterial tolerance to cell wall-active antibiotics. In addition, the results obtained from these studies may also be helpful to understand the survival phenotype of the so-called persisters which might be the cause of recurrent infections. Indeed, like enterococci, these specialized cells of a bacterial population are highly tolerant to bactericidal drugs.
Supplementary Material
ACKNOWLEDGMENTS
We thank all members of the EA4655 U2RM-Stress et Virulence laboratory, in particular, Isabelle Rincé, Aurelie Verneuil, Marie-Jeanne Pigny, and Evelyne Marchand, for technical assistance.
We thank the Ministère de l'Enseignement Supérieur et de la Recherche Scientifique Algérien (MESRS) for the doctoral fellowship of R.L. This work was funded by the French Ministry of Education and Science.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00389-15.
REFERENCES
- 1.Baddour LM, Wilson WR, Bayer AS, Fowler VG Jr, Bolger AF, Levison ME, Ferrieri P, Gerber MA, Tani LY, Gewitz MH, Tong DC, Steckelberg JM, Baltimore RS, Shulman ST, Burns JC, Falace DA, Newburger JW, Pallasch TJ, Takahashi M, Taubert KA, Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia, American Heart Association, Infectious Diseases Society of America. 2005. Infective endocarditis: diagnosis, antimicrobial therapy, and management of complications: a statement for healthcare professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, and the Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia, American Heart Association: endorsed by the Infectious Diseases Society of America. Circulation 111:e394–e434. doi: 10.1161/CIRCULATIONAHA.105.165564. [DOI] [PubMed] [Google Scholar]
- 2.Habib G, Hoen B, Tornos P, Thuny F, Prendergast B, Vilacosta I, Moreillon P, de Jesus Antunes M, Thilen U, Lekakis J, Lengyel M, Müller L, Naber CK, Nihoyannopoulos P, Moritz A, Zamorano JL, ESC Committee for Practice Guidelines. 2009. Guidelines on the prevention, diagnosis, and treatment of infective endocarditis (new version 2009): the Task Force on the Prevention, Diagnosis, and Treatment of Infective Endocarditis of the European Society of Cardiology (ESC). Endorsed by the European Society of Clinical Microbiology and Infectious Diseases (ESCMID) and the International Society of Chemotherapy (ISC) for Infection and Cancer. Eur Heart J 30:2369–2413. [DOI] [PubMed] [Google Scholar]
- 3.Fernández-Hidalgo N, Almirante B, Gavaldà J, Gurgui M, Peña C, de Alarcón A, Ruiz J, Vilacosta I, Montejo M, Vallejo N, López-Medrano F, Plata A, López J, Hidalgo-Tenorio C, Gálvez J, Sáez C, Lomas JM, Falcone M, de la Torre J, Martínez-Lacasa X, Pahissa A. 2013. Ampicillin plus ceftriaxone is as effective as ampicillin plus gentamicin for treating Enterococcus faecalis infective endocarditis. Clin Infect Dis 56:1261–1268. doi: 10.1093/cid/cit052. [DOI] [PubMed] [Google Scholar]
- 4.Bizzini A, Zhao C, Auffray Y, Hartke A. 2009. The Enterococcus faecalis superoxide dismutase is essential for its tolerance to vancomycin and penicillin. J Antimicrob Chemother 64:1196–1202. doi: 10.1093/jac/dkp369. [DOI] [PubMed] [Google Scholar]
- 5.Ladjouzi R, Bizzini A, Le Breton F, Sauvageot N, Rincé A, Benachour A, Hartke A. 2013. Analysis of the tolerance of pathogenic enterococci and Staphylococcus aureus to cell wall active antibiotics. J Antimicrob Chemother 68:2083–2091. doi: 10.1093/jac/dkt157. [DOI] [PubMed] [Google Scholar]
- 6.Moreillon P, Markiewicz Z, Nachman S, Tomasz A. 1990. Two bactericidal targets for penicillin in pneumococci: autolysis-dependent and autolysis-independent killing mechanisms. Antimicrob Agents Chemother 34:33–39. doi: 10.1128/AAC.34.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daneo-Moore L, Fletcher SH, Massida O, Pittaluga F. 1987. Penicillin tolerance in Enterococcus hirae ATCC 9790, p 628–635. In Actor P, Daneo-Moore L, Higgins ML, Salton MRJ, Shokman GD (ed), Antibiotic inhibition of bacterial cell surface assembly and function. ASM Press, Washington, DC. [Google Scholar]
- 8.Gutmann L, Tomasz A. 1982. Penicillin-resistant and penicillin-tolerant mutants of group A Streptococci. Antimicrob Agents Chemother 22:128–136. doi: 10.1128/AAC.22.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McDowell TD, Lemanski CL. 1988. Absence of autolytic activity (peptidoglycan nicking) in penicillin-induced nonlytic death in a group A streptococcus. J Bacteriol 170:1783–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujimoto DF, Bayles KW. 1998. Opposing roles of the Staphylococcus aureus virulence regulators, Agr and Sar, in Triton X-100- and penicillin-induced autolysis. J Bacteriol 180:3724–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Novak R, Henriques B, Charpentier E, Normark S, Tuomanen E. 1999. Emergence of vancomycin tolerance in Streptococcus pneumoniae. Nature 399:590–593. doi: 10.1038/21202. [DOI] [PubMed] [Google Scholar]
- 12.Sugai M, Ooku K, Akiyama T, Inoue S, Iseda S, Miyake Y, Suginaka H. 1991. Suppression of penicillin-induced lysis of Staphylococcus aureus by cibacron blue 3G-A. FEMS Microbiol Lett 64:151–154. [DOI] [PubMed] [Google Scholar]
- 13.Sugai M, Yamada S, Nakashima S, Komatsuzawa H, Matsumoto A, Oshida T, Suginaka H. 1997. Localized perforation of the cell wall by a major autolysin: atl gene products and the onset of penicillin-induced lysis of Staphylococcus aureus. J Bacteriol 179:2958–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caldelari I, Loeliger B, Langen H, Glauser MP, Moreillon P. 2000. Deregulation of the arginine deiminase (arc) operon in penicillin-tolerant mutants of Streptococcus gordonii. Antimicrob Agents Chemother 44:2802–2810. doi: 10.1128/AAC.44.10.2802-2810.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barcelona-Andrés B, Marina A, Rubio V. 2002. Gene structure, organization, expression, and potential regulatory mechanisms of arginine catabolism in Enterococcus faecalis. J Bacteriol 184:6289–6300. doi: 10.1128/JB.184.22.6289-6300.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huycke MM. 2002. Physiology of enterococci, 301–354. In Gilmore MS, Clewell DB, Courvalin P, Dunny GM, Murray BE, Rice LB (ed), The enterococci: pathogenesis, molecular biology, antibiotic resistance, and infection control. ASM Press, Washington, DC. [Google Scholar]
- 17.Yagi Y, Clewell DB. 1980. Recombination-deficient mutant of Streptococcus faecalis. J Bacteriol 143:966–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verneuil N, Mazé A, Sanguinetti M, Laplace J-M, Benachour A, Auffray Y, Giard J-C, Hartke A. 2006. Implication of (Mn)superoxide dismutase of Enterococcus faecalis in oxidative stress responses and survival inside macrophages. Microbiology 152:2579–2589. doi: 10.1099/mic.0.28922-0. [DOI] [PubMed] [Google Scholar]
- 19.Murray BE, Singh KV, Ross RP, Heath JD, Dunny GM, Weinstock GM. 1993. Generation of restriction map of Enterococcus faecalis OG1 and investigation of growth requirements and regions encoding biosynthetic function. J Bacteriol 175:5216–5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palmer KL, Carniol K, Manson JM, Heiman D, Shea T, Young S, Zeng Q, Gevers D, Feldgarden M, Birren B, Gilmore MS. 2010. High-quality draft genome sequences of 28 Enterococcus sp. isolates. J Bacteriol 192:2469–2470. doi: 10.1128/JB.00153-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benachour A, Auffray Y, Hartke A. 2007. Construction of plasmid vectors for screening replicons from gram-positive bacteria and their use as shuttle cloning vectors. Curr Microbiol 54:342–347. doi: 10.1007/s00284-006-0358-1. [DOI] [PubMed] [Google Scholar]
- 22.Zhang X, Paganelli FL, Bierschenk D, Kuipers A, Bonten MJM, Willems RJL, van Schaik W. 2012. Genome-wide identification of ampicillin resistance determinants in Enterococcus faecium. PLoS Genet 8:e1002804. doi: 10.1371/journal.pgen.1002804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Terzaghi BE, Sandine WE. 1975. Improved medium for lactic streptococci and their bacteriophages. Appl Microbiol 29:807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.La Carbona S, Sauvageot N, Giard J-C, Benachour A, Posteraro B, Auffray Y, Sanguinetti M, Hartke A. 2007. Comparative study of the physiological roles of three peroxidases (NADH peroxidase, alkyl hydroperoxide reductase and thiol peroxidase) in oxidative stress response, survival inside macrophages and virulence of Enterococcus faecalis. Mol Microbiol 66:1148–1163. doi: 10.1111/j.1365-2958.2007.05987.x. [DOI] [PubMed] [Google Scholar]
- 25.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 26.Meijerink J, Mandigers C, van de Locht L, Tönnissen E, Goodsaid F, Raemaekers J. 2001. A novel method to compensate for different amplification efficiencies between patient DNA samples in quantitative real-time PCR. J Mol Diagn 3:55–61. doi: 10.1016/S1525-1578(10)60652-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fiedler T, Bekker M, Jonsson M, Mehmeti I, Pritzschke A, Siemens N, Nes I, Hugenholtz J, Kreikemeyer B. 2011. Characterization of three lactic acid bacteria and their isogenic ldh deletion mutants shows optimization for YATP (cell mass produced per mole of ATP) at their physiological pHs. Appl Environ Microbiol 77:612–617. doi: 10.1128/AEM.01838-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bizzini A, Zhao C, Budin-Verneuil A, Sauvageot N, Giard J-C, Auffray Y, Hartke A. 2010. Glycerol is metabolized in a complex and strain-dependent manner in Enterococcus faecalis. J Bacteriol 192:779–785. doi: 10.1128/JB.00959-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sauvageot N, Ladjouzi R, Benachour A, Rincé A, Deutscher J, Hartke A. 2012. Aerobic glycerol dissimilation via the Enterococcus faecalis DhaK pathway depends on NADH oxidase and a phosphotransfer reaction from PEP to DhaK via EIIADha. Microbiology 158:2661–2666. doi: 10.1099/mic.0.061663-0. [DOI] [PubMed] [Google Scholar]
- 30.Carlioz A, Touati D. 1986. Isolation of superoxide dismutase mutants in Escherichia coli: is superoxide dismutase necessary for aerobic life? EMBO J 5:623–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poyart C, Pellegrini E, Gaillot O, Boumaila C, Baptista M, Trieu-Cuot P. 2001. Contribution of Mn-cofactored superoxide dismutase (SodA) to the virulence of Streptococcus agalactiae. Infect Immun 69:5098–5106. doi: 10.1128/IAI.69.8.5098-5106.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown AT, Wittenberger CL. 1971. Mechanism for regulating the distribution of glucose carbon between the Embden-Meyerhof and hexose-monophosphate pathways in Streptococcus faecalis. J Bacteriol 106:456–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andersen HW, Solem C, Hammer K, Jensen PR. 2001. Twofold reduction of phosphofructokinase activity in Lactococcus lactis results in strong decreases in growth rate and in glycolytic flux. J Bacteriol 183:3458–3467. doi: 10.1128/JB.183.11.3458-3467.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rochat T, Boudebbouze S, Gratadoux J-J, Blugeon S, Gaudu P, Langella P, Maguin E. 2012. Proteomic analysis of spontaneous mutants of Lactococcus lactis: involvement of GAPDH and arginine deiminase pathway in H2O2 resistance. Proteomics 12:1792–1805. doi: 10.1002/pmic.201100465. [DOI] [PubMed] [Google Scholar]
- 35.Liu Y, Imlay JA. 2013. Cell death from antibiotics without the involvement of reactive oxygen species. Science 339:1210–1213. doi: 10.1126/science.1232751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keren I, Wu Y, Inocencio J, Mulcahy LR, Lewis K. 2013. Killing by bactericidal antibiotics does not depend on reactive oxygen species. Science 339:1213–1216. doi: 10.1126/science.1232688. [DOI] [PubMed] [Google Scholar]
- 37.Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. 2007. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- 38.Seaver LC, Imlay JA. 2001. Hydrogen peroxide fluxes and compartmentalization inside growing Escherichia coli. J Bacteriol 183:7182–7189. doi: 10.1128/JB.183.24.7182-7189.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.