

Abstract
Adaptation lies at the heart of Darwinian evolution. Accordingly, numerous studies have tried to provide a formal framework for the description of the adaptive process. Of these, two complementary modeling approaches have emerged: While so-called adaptive-walk models consider adaptation from the successive fixation of de novo mutations only, quantitative genetic models assume that adaptation proceeds exclusively from preexisting standing genetic variation. The latter approach, however, has focused on short-term evolution of population means and variances rather than on the statistical properties of adaptive substitutions. Our aim is to combine these two approaches by describing the ecological and genetic factors that determine the genetic basis of adaptation from standing genetic variation in terms of the effect-size distribution of individual alleles. Specifically, we consider the evolution of a quantitative trait to a gradually changing environment. By means of analytical approximations, we derive the distribution of adaptive substitutions from standing genetic variation, that is, the distribution of the phenotypic effects of those alleles from the standing variation that become fixed during adaptation. Our results are checked against individual-based simulations. We find that, compared to adaptation from de novo mutations, (i) adaptation from standing variation proceeds by the fixation of more alleles of small effect and (ii) populations that adapt from standing genetic variation can traverse larger distances in phenotype space and, thus, have a higher potential for adaptation if the rate of environmental change is fast rather than slow.
Keywords: adaptation, standing genetic variation, natural selection, models/simulations, population genetics
ONE of the biggest surprises that has emerged from evolutionary research in the past few decades is that, in contrast to what has been claimed by the neutral theory (Kimura 1983), adaptive evolution at the molecular level is widespread. In fact, some empirical studies concluded that up to of all amino acid changes between Drosophila simulans and D. yakuba are adaptive (Smith and Eyre-Walker 2002; Orr 2005b). Along the same line, Wichman et al. (1999) evolved the single-stranded DNA bacteriophage ΦX174 to high temperature and a novel host and found that of the observed nucleotide substitutions had an adaptive effect. These and other results have led to an increased interest in providing a formal framework for the adaptive process that goes beyond traditional population- and quantitative-genetic approaches and considers the statistical properties of suites of substitutions in terms of “individual mutations that have individual effects” (Orr 2005a, p. 121). In general, selection following a change in the environmental conditions may act either on de novo mutations or on alleles already present in the population, also known as standing genetic variation. Consequently, from the numerous studies that have attempted to address this subject, two complementary modeling approaches have emerged.
So-called adaptive-walk models (Gillespie 1984; Kauffman and Levin 1987; Orr 2002, 2005b) typically assume that selection is strong compared to mutation, such that the population can be considered monomorphic all the time and all observed evolutionary change is the result of de novo mutations. These models have produced several robust predictions (Orr 1998, 2000; Martin and Lenormand 2006a,b), which are supported by growing empirical evidence (Cooper et al. 2007; Rockman 2012; Hietpas et al. 2013; but see Bell 2009), and have provided a statistical framework for the fundamental event during adaptation, that is, the substitution of a resident allele by a beneficial mutation. Specifically, the majority of models (e.g., Gillespie 1984; Orr 1998; Martin and Lenormand 2006a) consider the effect-size distribution of adaptive substitutions following a sudden change in the environment. Recently, Kopp and Hermisson (2009b) and Matuszewski et al. (2014) extended this framework to gradual environmental change.
In contrast, most quantitative-genetic models consider an essentially inexhaustible pool of preexisting standing genetic variants as the sole source for adaptation (Lande 1976). Evolving traits are assumed to have a polygenic basis, where many loci contribute small individual effects, such that the distribution of trait values approximately follows a Gaussian distribution (Bulmer 1980; Barton and Turelli 1991; Kirkpatrick et al. 2002). Since the origins of quantitative genetics lie in the design of plant and animal breeding schemes (Wricke and Weber 1986; Tobin et al. 2006; Hallauer et al. 2010), the traditional focus of these models was on predicting short-term changes in the population mean phenotype (often assuming constant genetic variances and covariances) and not on the fate and effect of individual alleles. The same is true for the relatively small number of models that have studied the contribution of new mutations in the response to artificial selection (e.g., Hill and Rasbash 1986a) and the shape and stability of the G matrix [i.e., the additive variance–covariance matrix of genotypes (Jones et al. 2004, 2012a)].
It is only in the past decade that population geneticists have thoroughly addressed adaptation from standing genetic variation at the level of individual substitutions (Orr and Betancourt 2001; Hermisson and Pennings 2005; Chevin and Hospital 2008). Hermisson and Pennings (2005) calculated the probability of adaptation from standing genetic variation following a sudden change in the selection regime. They found that, for small-effect alleles, the fixation probability is considerably increased relative to that from new mutations. Furthermore, Chevin and Hospital (2008) showed that the selective dynamics at a focal locus are substantially affected by genetic background variation. These results were experimentally confirmed by Lang et al. (2011), who followed beneficial mutations in hundreds of evolving yeast populations and showed that the selective advantage of a mutation plays only a limited role in determining its ultimate fate. Instead, fixation or loss is largely determined by variation in the genetic background—which need not to be preexisting, but could quickly be generated by a large number of new mutations. Still, predictions beyond these single-locus results have been verbal at best, stating that “compared with new mutations, adaptation from standing genetic variation is likely to lead to faster evolution [and] the fixation of more alleles of small effect […]” (Barrett and Schluter 2008, p. 38). Thus, despite recent progress, one of the central questions still remains unanswered: From the multitude of standing genetic variants segregating in a population, which are the ones that ultimately become fixed and contribute to adaptation, and how does their distribution differ from that of (fixed) de novo mutations?
The aim of this article is to contribute to overcoming what has been referred to as “the most obvious limitation” (Orr 2005b, p. 6) of adaptive-walk models and to study the ecological and genetic factors that determine the genetic basis of adaptation from standing genetic variation. Specifically, we consider the evolution of a quantitative trait in a gradually changing environment. We develop an analytical framework that accurately describes the distribution of adaptive substitutions from standing genetic variation and discuss its dependence on the effective population size, the strength of selection, and the rate of environmental change. In line with Barrett and Schluter (2008), we find that, compared to adaptation from de novo mutations, adaptation from standing genetic variation proceeds, on average, by the fixation of more alleles of small effect. Furthermore, when standing genetic variation is the sole source for adaptation, faster environmental change can enable the population to remain better adapted and to traverse larger distances in phenotype space.
Model and Methods
Phenotype, selection, and mutation
We consider the evolution of a diploid population of N individuals with discrete and nonoverlapping generations characterized by a single phenotypic trait z, which is under Gaussian stabilizing selection with regard to a time-dependent optimum ,
| (1) |
where describes the width of the fitness landscape (see Table 1 for a summary of our notation and Figure 1 for a graphical abstract of the model). Throughout this article we choose the linearly moving optimum,
| (2) |
where v is the rate of environmental change.
Table 1. A summary of notation and definitions.
| Notation | Definition |
|---|---|
| α | Phenotypic effect of mutation |
| (Gaussian) distribution of new mutations | |
| z | Phenotype |
| Mean genetic background phenotype | |
| ν | Rate of environmental change |
| (Gaussian) fitness function | |
| Width of Gaussian fitness function | |
| Variance of new mutations | |
| (Background) genetic variance | |
| Time-dependent selection coefficient for allele with phenotypic effect α | |
| x | Frequency of mutant allele |
| Effective population size | |
| θ | Per-locus mutation rate |
| Θ | Population-wide mutation rate (per trait) |
| Fixation probability | |
| Distribution of mutant allele frequency at a single locus with phenotypic effect α | |
| Probability to adapt from standing genetic variation | |
| Distribution of adaptive substitutions from standing genetic variation | |
| Equilibrium lag |
Figure 1.

Illustration of our main model assumptions. (A) Phenotypic evolution in the moving-optimum model: The black curve and colored circles give the distribution of phenotypes for three different snapshots in time for a population that adapts to a time-dependent phenotypic optimum (green circles) under Gaussian stabilizing selection (see equation). Note that the population mean phenotype typically lags behind the optimum. (B) The biallelic infinite-sites model: Each mutation creates a new allele at a unique site. (C) The focal-locus genetic-background model used in our analytical approximation (Lande 1983).
Mutations enter the population at rate Θ/2 (with , where u is the per-haplotype mutation rate), and we assume that their phenotypic effect size α follows a Gaussian distribution with mean 0 and variance (which we refer to as the distribution of new mutations); that is,
| (3) |
All mutations that are segregating in the population at the time the environment starts changing are considered as standing genetic variants, whereas all mutations introduced after that point are considered as de novo mutations.
Throughout this article we equate genotypic with phenotypic values and, thus, neglect any environmental variance. Note that this model is, so far, identical to the moving-optimum model proposed by Kopp and Hermisson (2009b) (see also Bürger 2000).
Genetic assumptions and simulation model
To study the distribution of adaptive substitutions from standing genetic variation, we conducted individual-based simulations (IBS) (available from the corresponding author upon request; see Bürger 2000; Kopp and Hermisson 2009b) that explicitly model the simultaneous evolution at multiple loci, while making additional assumptions about the genetic architecture of the selected trait, the life cycle of individuals, and the regulation of population size. This serves as our main model.
Genome:
Individuals are characterized by a linear (continuous) genome of diploid loci, which determine the phenotype z additively (i.e., there is no phenotypic epistasis; note, however, that there is epistasis for fitness). Mutations occur at constant rate u per haplotype. In contrast to the majority of individual-based models (e.g., Jones et al. 2004; Kopp and Hermisson 2009b; Matuszewski et al. 2014), we do not fix the number of loci a priori, but instead assume that each mutation creates a unique polymorphic locus, whose position is drawn randomly from a uniform distribution over the entire genome (where genome length is determined by the recombination parameter r described below). Consequently, each locus contains only a wild-type allele with phenotypic effect 0 and a mutant allele with phenotypic effect α, which is drawn from Equation 3. Thus, we effectively design a biallelic infinite-sites model with a continuum of alleles (see also Figure 1B).
To monitor adaptive substitutions, we introduce a population-consensus genome that keeps track of all polymorphic loci, that is, of all mutant alleles that are segregating in the population. If a mutant allele becomes fixed in the population, it is declared the new wild-type allele and its phenotypic effect is reset to 0. The phenotypic effects of all fixed mutations are taken into account by a variable , which can be interpreted as a phenotypic baseline effect. Thus, the phenotype z of an individual i is given by
| (4a) |
where
| (4b) |
Life cycle:
Each generation, the following steps are performed:
Viability selection: Individuals are removed with probability (see Equation 1).
Population regulation: If, after selection, the population size N exceeds the carrying capacity K, randomly chosen individuals are removed.
Reproduction: The surviving individuals are randomly assigned to mating pairs, and each mating pair produces exactly offspring. Note that under this scheme, the effective population size equals times the census size (Bürger 2000, p. 274). To account for this difference, Θ in the analytical approximations needs to be calculated on the basis of this effective size; i.e., . The offspring genotypes are derived from the parent genotypes by taking into account segregation, recombination, and mutation.
Recombination:
For each reproducing individual, the number of crossing-over events during gamete formation (i.e., the number of recombination breakpoints) is drawn from a Poisson distribution with (genome-wide recombination) parameter r (i.e., the total genome length is cM; see Supporting Information, File S1). The genomic position of each recombination breakpoint is then drawn from a uniform distribution over the entire genome, and the offspring haplotype is created by alternating between the maternal and the paternal haplotype, depending on the recombination breakpoints. Free recombination (where all loci are assumed to be unlinked) corresponds to . In this case, for each locus a Bernoulli-distributed random number is drawn to determine whether the offspring haplotype will receive the maternal or the paternal allele at that locus.
Simulation initialization and termination:
Starting from a population of K wild-type individuals with phenotype (i.e., the population was perfectly adapted at ), we allowed for the establishment of genetic variation, , by letting the population evolve for generations under stabilizing selection with a constant optimum. Increasing the number of generations had no effect on the average for the population sizes considered in this study. Following this equilibration time, the optimum started moving under ongoing mutational input, and the simulation was stopped once all alleles from the standing genetic variation were either fixed or lost (i.e., when ). Simulations were replicated until a total number of 5000 adaptive substitutions from standing genetic variation were recorded.
The individual-based simulation program is written in C++ and makes use of the GNU Scientific Library (Galassi et al. 2009). Mathematica (Wolfram Research, Champaign, IL) was used for the numerical evaluation of integrals and to create plots and graphics, making use of the LevelScheme package (Caprio 2005).
Analytical approximations: Evolution of a focal locus in the presence of genetic background variation
To obtain an analytically tractable model, we need to approximate the multilocus dynamics. Clearly, simple interpolation of single-locus theory will fail, because when alleles at different loci influencing the same trait segregate as standing genetic variation, the selective dynamics of any individual allele are critically affected by the collective evolutionary response at other loci. In particular, any allele that brings the mean phenotype closer to the optimum simultaneously decreases the selective advantage of other such alleles (epistasis for fitness). Thus, if simultaneous evolution at many loci allows the population to closely follow the optimum, large-effect alleles at any given locus are likely to remain deleterious (as their carriers would overshoot the optimum). To account for these effects, we adopt a quantitative-genetics approach originally developed by Lande (1983) and introduce a genetic background whose mean phenotype evolves according to Lande’s equation
| (5a) |
where is the change between generations, is the (additive) genetic variance, and
| (5b) |
denotes the selection gradient, which measures the change in log mean fitness per unit change of the mean phenotype (Lande 1976). Furthermore, under the simplifying assumption that remains constant over time, the mean background phenotype evolves according to
| (6a) |
with
| (6b) |
Given the dynamics of the genetic background, we then choose a single focal locus and derive the time-dependent selection coefficient for a mutant allele with phenotypic effect α (for details see below; see also Figure 1C). We then use theory for adaptation from standing genetic variation (Hermisson and Pennings 2005) and for fixation under time-inhomogeneous selection (Uecker and Hermisson 2011) (summarized in Appendix B) to estimate the probability that this allele contributes to adaptation. If there is no linkage (i.e., there is free recombination between all loci), each locus can be viewed as the focal locus (with a specific phenotypic effect α), allowing us to obtain an estimate for the overall distribution of adaptive substitutions from standing genetic variation. Thus, in these approximations, our multilocus model is effectively treated within a single-locus framework. Note that a similar focal-locus approach has recently been used to analyze the effect of genetic background variation on the trajectory of an allele sweeping to fixation (Chevin and Hospital 2008) and to study the probability of adaptation to novel environments (Gomulkiewicz et al. 2010), with both studies stressing the fact that genetic background variation cannot be neglected and critically affects the adaptive outcome.
Results
In the following, we calculate, first, the probability that a focal allele from the standing genetic variation becomes fixed when the population adapts to a moving phenotypic optimum. Based on this intermediate result, we then derive the effect-size distribution of such alleles and discuss the overall potential for adaptation from standing genetic variation and its importance relative to de novo mutations.
Note that, for the first result, we assume the focal allele to arise from a wild-type allele by recurrent mutation with the per-locus (population-wide) mutation rate θ. For all subsequent results, we let , in accordance with the infinite-sites model described above.
The probability for adaptation from standing genetic variation
The probability that a focal mutant allele from the standing genetic variation contributes to adaptation depends on the dynamics of its selection coefficient in the presence of genetic background variation. For an allele with phenotypic effect α and a genetic background with mean and (constant) variance , the selection coefficient can be calculated as
| (7) |
Note that the genetic background variance has the effect of broadening the fitness landscape experienced by the focal allele (the term ). Plugging Equation 6a into Equation 7 then yields the time-dependent selection coefficient,
| (8) |
Assuming that the population is perfectly adapted at (), the initial (deleterious) selection coefficient is given by
Unlike in the model without genetic background variation (Kopp and Hermisson 2009b), does not increase linearly, but instead depends on the evolution of the phenotypic lag δ between the optimum and the mean background phenotype. In particular, the population will reach a dynamic equilibrium with , where it follows the optimum with a constant lag
| (9) |
(Bürger and Lynch 1995). Consequently, the selection coefficient for α approaches
| (10) |
Note that the right-hand side can be written as , where is the equilibrium selection gradient (Kopp and Matuszewski 2014). In this case, the largest obtainable selection coefficient is for and evaluates to
| (11) |
The range of allelic effects α that can reach a positive selection coefficient is bounded by and . Note that in previous adaptive-walk models (e.g., Kopp and Hermisson 2009b; Matuszewski et al. 2014) there was no strict , since the population followed the optimum by stochastic jumps, whereas in the present model, the genetic background evolves deterministically and establishes a constant equilibrium lag.
Assuming the mutant allele was initially deleterious and arises recurrently at rate , its allele frequency spectrum prior to the onset of environmental change, , is given by Equation B5. The fixation probability for a mutation starting with n initial copies, , is then given by Equation B7a, where Equation B7b is given by
| (12) |
(using Equation 8). In the general case, this expression needs to be evaluated numerically. Only in the limit is there an explicit solution
| (13) |
where denotes the Gaussian complementary error function.
Finally, combining Equations B5, B7, and 12, the probability that an allele with allelic effect α is present in the standing variation and contributes to adaptation can then be calculated as
| (14) |
where .
The analytical approximation (Equation 14) represents an important intermediate result toward our goal of deriving the distribution of adaptive substitutions from standing genetic variation. Yet, unlike the main model, it assumes that the focal locus is subject to recurrent mutation (see Equation B5). To test its accuracy, we therefore used an alternative simulation approach based on the Wright–Fisher model (see Appendix A for details). As shown in Figure 2 and Figure S3_1 (File S3), Equation B5 performs generally very well. The only exception occurs when the background variation is high (large ) and stabilizing selection is weak (i.e., if is large). In this case, Equation 14 underestimates for small . The reason is that, under a constant optimum (i.e., before the environmental change), the genetic background compensates for the deleterious effect of α (i.e., , in violation of our assumption that ), effectively reducing the selection strength against the deleterious mutant allele. Consequently, α is, on average, present at higher initial frequencies than predicted by Equation B5.
Figure 2.

The probability for a mutant allele to adapt from standing genetic variation as a function of the rate of environmental change v. Solid lines correspond to the analytical prediction (Equation 14), the gray dashed line shows the probability for a neutral allele ( Equation 15), and symbols give results from Wright–Fisher simulations (see Appendix A). The phenotypic effect size α of the mutant allele ranges from (top line; black) to (bottom line; purple) with increments of . The numbers in each parameter box (per locus mutation rate θ, width of fitness landscape ) correspond to different values of the genetic background variation with (no background variation; top left), (top right), (bottom left), and (bottom right). Other parameters: , ,
Note that, if α is small compared to the genetic background variation (i.e., in the limit of ), selection is weak ( large), and environmental change is slow (i.e., ), will approach the probability of fixation from standing genetic variation for a neutral allele [i.e., see Figure S3_1, bottom right (File S3)]. This probability can be calculated as
| (15) |
where is given by Equation B3, denotes the harmonic number, is Euler’s gamma, and is the polygamma function [see dashed lines in Figure 2 and Figure S3_1 (File S3)].
Figure 2 and Figure S3_1 (File S3) show some general trends: First, the probability for a mutant allele to become fixed increases with the rate of environmental change, v (irrespective of its effect size α, the per-locus mutation rate θ and the width of the fitness landscape ), since only the positive term in Equation 8 depends (linearly) on v. Second, is proportional to θ as long as θ is small [compare and in Figure S3_1 (File S3)], simply because the probability that α is present in the population is linear in θ. Thus, Figure 2 is representative for the limit , which is used below. Indeed, only if the per-locus mutation rate is fairly large (), does the shape of the distribution of allele frequencies become important, and the increase in with θ becomes less than linear (Figure S3_1 (File S3)). Third, changes in the width of the fitness landscape, , have a dual effect: While increasing promotes the initial frequency of the focal allele in the standing genetic variation (because stabilizing selection is weaker), the selection coefficient increases more slowly after the onset of environmental change (such that the allele is less likely to be picked up by selection; see Equation 7). Our results, however, show that the former effect always outweighs the latter (as increases with ). Finally, if the genetic background variation is below a threshold value (e.g., the exact threshold should depend on θ and ), it only marginally affects the fixation probability of the focal allele α. Once surpasses this value, however, it critically affects (in accordance with the results by Chevin and Hospital 2008). In particular, as increases decreases, because most large-effect alleles remain deleterious even if environmental change is fast. Thus, enlarged background variation acts as if reducing the rate of environmental change v.
The distribution of adaptive substitutions from standing genetic variation
We now derive the distribution of adaptive substitutions from standing genetic variation over all mutant effects α. In the previous section, we derived the probability of adaptation at a focal locus (with a given effect α) by treating the genetic background variance as an independent model parameter. In the full model (as in the individual-based simulations), this variance emerges from a balance of mutation, selection, and drift at all background loci. As such, it is a function of the basic model parameters for these forces. Since we use an infinite-sites model, there is no recurrent mutation and each allele originates from a single mutation. In this situation, the background variance is accurately predicted by the stochastic house-of-cards (SHC) approximation (results not shown; Bürger and Lynch 1995)
| (16) |
where mutation is parametrized by the total (per trait) mutation rate Θ and the variance of de novo mutations , the width of the fitness landscape is given by , and the effective population size is a measure for genetic drift.
To derive the probability that an allele with a given phenotypic effect α contributes to adaptation, we first need to calculate the probability that such an allele segregates in the population at time 0. Following Hermisson and Pennings (2005), the probability that the allele is not present can be approximated by integrating over the distribution of allele frequencies (Equation B5) from 0 to , yielding
| (17) |
(equation 7 and appendix of Hermisson and Pennings 2005). The fixation probability can then be calculated by conditioning on segregation of the allele in the limit (due to the infinite-sites assumption). Using Equation 14, this probability reads
| (18) |
where (see also Equation B5) and is given by Equation 12. The limit in Equation 18 can be approximated numerically by setting θ to a very small, but positive value.
Multiplying by the rate of mutations with effect α [i.e., ], the distribution of adaptive substitutions from standing genetic variation is given by
| (19) |
where is a normalization constant [black line in Figure 3, Figure 4, Figure 5, and Figure S3_2 and Figure S3_3 (File S3)]. Note that Equation 19 still depends on Θ through its effect on the background variance [which affects ]. In particular, in the SHC approximation (Equation 16), scales linearly with Θ. Furthermore, Equation 19 should be valid for any distribution of mutational effects .
Figure 3.

The distribution of adaptive substitutions from standing genetic variation. Histograms show results from individual-based simulations. The black line corresponds to the analytical prediction (Equation 19), with the genetic background variation determined by the SHC approximation (Equation 16). The red line gives the analytical prediction for the limiting case where the equilibrium lag is reached fast (Equation 21). The blue line is based on the analytical prediction (Equation 25), which assumes a neutral fixation probability, but has been shifted so that it is centered around the empirical mean. The gray-shaded area gives the analytical prediction for substitutions from de novo mutations under the assumption that the phenotypic lag has reached its equilibrium (Equation 23). The asterisks indicate where Fixed parameter:
Figure 4.

The distribution of adaptive substitutions from standing genetic variation for various rates of environmental change. For further details see Figure 3. Fixed parameters: , ,
Figure 5.

The mean size of adaptive substitutions from standing genetic variation, measured in units of mutational standard deviations () as a function of the rate of environmental change ν (A, C, and E) and for various ν as a function of the population-wide mutation rate Θ (B), the width of the fitness landscape (D), and the population size N (F). Lines show the analytical prediction (the mean of the distribution, Equation 19), and symbols give results from individual-based simulations. Error bars for standard errors are contained within the symbols. For , no simulation results are shown, as these constitute a degenerate case (for details see The accuracy of the approximation). Fixed parameter:
In the limit where the equilibrium lag is reached fast (i.e., when γ is large; Equation 6b), the moving-optimum model reduces to a model with constant selection for any focal allele (i.e., as in Hermisson and Pennings 2005). Using Equations B6 and 17, the fixation probability for a segregating allele can be calculated as
| (20) |
Plugging Equation 20 into Equation 19, the distribution of adaptive substitutions from standing genetic variation can be approximated by
| (21) |
where is a normalization constant [red line in Figure 3, Figure 4, and Figure S3_2 and Figure S3_3 (File S3)].
Similarly, the fixation probability of de novo mutations under the equilibrium lag can be derived [using Equation 10 and Equation B2 with an initial frequency of ] as
| (22) |
yielding the distribution of adaptive substitutions
| (23) |
where is a normalization constant [gray-shaded area in Figure 3, Figure 4, and Figure S3_2 and Figure S3_3 (File S3)].
In contrast, if the environment changes very slowly, we can calculate the limit distribution of adaptive substitutions from standing genetic variation by approximating the fixation probability by that of a neutral allele, which is equal to its initial allele frequency x (even though the frequency spectrum, i.e., the distribution of x, is not that of a neutral allele). In this case,
| (24a) |
with
| (24b) |
where is given by Equation B4 and the right-hand side is a ratio of hypergeometric functions.
Using Equation 24a, the distribution of substitutions from standing genetic variation reads
| (25) |
where again denotes a normalization constant (blue line in Figure 3; Figure 4; and Figure S3_2, Figure S3_3 and Figure S3_4 (File S3)).
The accuracy of the approximation:
When compared to individual-based simulations, our analytical approximation for the distribution of adaptive substitutions from standing genetic variation (Equation 19) performs, in general, very well as long as selection is strong; that is, the rate of environmental change v is high and/or the width of the fitness landscape is not too large [Figure 3 and Figure S3_2 (File S3)]. Under weak selection, however, Equation 19 fails to capture the fixation of alleles with neutral or negative effects (“backward fixations”; ). The reason is that Equation B7 considers only the fixation of alleles whose selection coefficient becomes positive in the long term. But if the rate of environmental change is slow (or is very large), most alleles get fixed or lost simply by chance, that is, genetic drift. In particular, if genetic drift is the main driver of phenotypic evolution [i.e., ], the distribution of adaptive substitutions is almost symmetric around 0 [see Figure S3_4 (File S3)]. This distribution is described very well by Equation 25, which assumes that the fixation probability of an allele is proportional to its initial frequency in the standing variation. In addition, even for cases where environmental change imposes modest directional selection, Equation 25 still captures the shape of the distribution of adaptive substitutions reasonably well, when centered around the empirical mean (blue line in Figure 3, Figure 4, and Figure S3_2 and Figure S3_3 (File S3)).
With a moving phenotypic optimum, the selection coefficient (Equation 8) is initially very small. Accordingly, there is always a phase during the adaptive process where genetic drift dominates, that is, where for all mutant alleles. The length of this phase (i.e., the time it takes until selection becomes the main force of evolution) depends on the interplay of multiple parameters, notably , , and A good heuristic to determine whether evolution will ultimately become dominated by selection is to calculate (Equation 11), which gives the maximal population-scaled selection coefficient. Since the selection coefficient of most mutations will be smaller than this value, one can consider as a rule of thumb that selection is the main driver of evolution as long as In this case, Equation 19 matches the individual-based simulations very well [see asterisks in Figure 3, Figure 4, and Figure S3_2 and Figure S3_3 (File S3)], and this holds true even when the mutation rate Θ is an order of magnitude larger than the range used in the majority of our individual-based simulations [Figure S3_5 (File S3)]. In summary, the accuracy of our approximation crucially depends on the efficacy of selection.
While our analytical approximations assume that the genetic background variation remains constant, in our IBS is an emergent property of the model parameters (notably ) and, thus, is expected to fluctuate (e.g., due to genetic drift). Furthermore, with a moving phenotypic optimum, is expected to increase—the larger v is, the stronger the increase in —because of initially rare large-effect alleles increasing in frequency (Bürger 1999). In general, however, our approximations seem to be robust to these effects, suggesting that these temporal changes in do not affect the adaptive process strongly.
The effects of linkage on the distribution of adaptive substitutions from standing genetic variation are discussed in File S1. The main result is that only tight linkage has a noticeable effect, namely to reduce the efficacy of selection and increase the proportion of backward fixations (moving the distribution closer to the prediction from Equation 25).
Biological interpretation:
As shown in Figure 3 and Figure 4 [see also Figure S3_2, Figure S3_3 and Figure S3_5 (File S3)], adaptive substitutions from standing genetic variation have, on average, smaller phenotypic effects than those from de novo mutations. There are two reasons for this result. First, in the standing genetic variation, small-effect alleles are more frequent than large-effect alleles and might already segregate at appreciable frequency (increasing their fixation probability). Second, substitutions from standing variation occur in the initial phase of the adaptive process, where the phenotypic lag is small, whereas our approximation for de novo mutations (Equation 23) assumes that the phenotypic lag has reached its maximal (equilibrium) value (which need not be large, depending on the amount of genetic background variation). The relative importance of these two effects can be seen in Figure 3 and Figure 4 [see also Figure S3_2 and Figure S3_3 (File S3)]: Comparing the gray-shaded area (Equation 23; de novo mutations under the equilibrium lag) with the red line (Equation 21; standing genetic variation under the equilibrium lag) shows the effect of larger starting frequencies of small-effect mutations from the standing genetic variation. The difference of the black (Equation 19; standing genetic variation) and red (Equation 21; standing genetic variation under the equilibrium lag) lines shows the effects of the initially smaller lag (i.e., the effect of the dynamical selection coefficient). Note that the first effect is always important (even if Θ and are large and v is small, where the red line and the gray-shaded area almost coincide—although this is only because the approximation is bad). The second effect, however, becomes particularly important if is small (i.e., if the time to reach the equilibrium lag is large), such that selection coefficients are dynamic and small-effect alleles are selected earlier than large-effect alleles, explaining the relative lack of large-effect alleles in the distribution of adaptive substitutions.
Generally, the distribution of adaptive substitutions is unimodal and resembles a log-normal distribution [Figure 3, Figure 4, and Figure S3_5 (File S3)]. Only if selection is very weak (i.e., when is large and/or v is small), does it contain a significant proportion of backward fixations [with negative α; Figure 4 and Figure S3_3 (File S3); see The accuracy of the approximation]. As the rate of environmental change v increases, the mean phenotypic effect of substitutions increases (Figure 5, top row), too, but the mode may actually decrease (Figure 4); that is, the distribution becomes more asymmetric and skewed, resembling the “almost exponential” distribution of substitutions from de novo mutations in the sudden-change scenario (Orr 1998). A likely explanation is that small-effect alleles, which are common in the standing variation, are under stronger selection and have an increased fixation probability if v is large (see Figure 2).
Interestingly, if the environment changes very fast, the simulated distribution of adaptive substitutions from standing genetic variation almost exactly matches the one predicted by Equation 23 for de novo mutations [Figure 6; see also Figure 3, Figure 4, and Figure S3_2 and Figure S3_3 (File S3)]. However, this seems to be an artifact rather than a relevant biological phenomenon. The reason is that the environment changes so fast that the population quickly dies out. Thus, the resulting distribution of adaptive substitutions is that for a dying population and might not necessarily reflect the adaptive process. In an experimental setup, though, where populations evolve until they go extinct, the distribution of adaptive substitutions from standing genetic variation might truly be indistinguishable from that from de novo mutations.
Figure 6.

The distribution of adaptive substitutions from standing genetic variation in the case of fast environmental change. For further details see Figure 3. Fixed parameters: , , , ,
In the following, we discuss the influence of the other model parameters (Θ, , and N) on the distribution of adaptive substitutions from standing genetic variation and, in particular, its mean (Figure 5).
Increasing the rate of mutational supply Θ generally decreases via its effect on the equilibrium lag . More precisely, increasing Θ increases the background genetic variance (Equation 16) and hence γ, which in turn decreases and reduces the likelihood for the fixation of large-effect alleles. This effect is strongest if is small relative to the mutational standard deviation , but weakens if is large. Note that, as long as , is approximately proportional to . Consequently, in Figure 5, A and B, decreases with Θ for small but not large values of the rate of environmental change v, but, when increasing Θ to very large values, decreases even for comparably large v [Figure S3_5 (File S3)]. In summary, the effect of the rate of mutational supply on the mean size and the distribution of adaptive substitutions from standing genetic variation depends strongly on the rate of environmental change.
The width of the fitness landscape affects different aspects of the adaptive process, but its net effect is an increase of the mean effect size of fixed alleles as increases (i.e., as stabilizing selection gets weaker), especially if the rate of environmental change is intermediate (Figure 5D). The reason is that weak stabilizing selection increases the frequency of large-effect alleles in the standing variation. In addition, weak selection also increases the phenotypic lag (Equation 9; see also Kopp and Matuszewski 2014), again favoring large-effect alleles. Note that the latter point holds true even though weak selection increases the background variance . Finally, the effect of is strongest for intermediate v, because for small v, large-effect alleles are never favored, whereas for large v, all alleles with positive effect have a high fixation probability.
Similar arguments hold for (when the rate of mutational supply, Θ, is held constant). First, increasing will always increase the efficacy of selection, resulting in lower initial frequencies of mutant alleles (Equation B4) and decreased (Equation 16). If the environment changes slowly, increases with , because the equilibrium lag increases (caused by the decrease in ). In contrast, if the rate of environmental change is fast, slightly decreases with due to the lower starting frequency of large-effect alleles and because small-effect alleles are selected more efficiently (i.e., they are less prone to get lost by genetic drift; Figure 5F).
The potential for adaptation from standing genetic variation and the rate of environmental change
So far, we have focused on the distribution of adaptive substitutions for individual fixation events. We now address what can be said about the total progress that can be made from standing genetic variation following a moving phenotypic optimum. The overall potential for adaptation from standing genetic variation depends on the mean number of alleles segregating as standing genetic variation, which can be accurately approximated as
| (26) |
(results not shown; Foley 1992). The mean number of alleles that become fixed can then be calculated as
| (27) |
where the integral equals the normalization constant in Equation 19 (i.e., the proportion of fixed alleles). Finally, using Equation 27, the average distance traveled in phenotype space before standing variation is exhausted is given by
| (28) |
where is the mean phenotypic effect size of adaptive substitutions from standing genetic variation, and the factor 2 in Equation 28 comes from the fact that we are considering diploids (and α denotes the phenotypic effect per haplotype). Note that, once the shift of the optimum considerably exceeds , the population will inevitably go extinct without the input of new mutations.
Figure 7 [see also Figure S3_6, Figure S3_7, Figure S3_8, Figure S3_9, Figure S3_10 and Figure S3_11 (File S3)] illustrates these predictions and compares them to results from individual-based simulations (where, unlike in the rest of this article, new mutational input was turned off after the onset of the environmental change).
Figure 7.

The average distance traversed in phenotype space, , as a function of the rate of environmental change ν, when standing genetic variation is the sole source for adaptation. Symbols show results from individual-based simulations (averaged over 100 replicate runs). The solid line gives the analytical prediction (Equation 28), with taken from Equation 16. The shaded dashed line gives the critical rate of environmental change (Equation 29). Error bars for standard errors are contained within the symbols. Fixed parameters: ,
Both the mean number of fixations and the mean phenotypic distance traveled increase with the rate of environmental change, reflecting the fact that more and larger-effect alleles become fixed if the environment changes fast.
Only for very large v do and decrease sharply, because the population goes extinct before fixations can be completed. Note that in these cases the rate of environmental change exceeds the “critical rate of environmental change” (Bürger and Lynch 1995, p. 151) [shaded dashed line in Figure 7 and Figure S3_9, Figure S3_10 and Figure S3_11 (File S3)], which for our choice for the number of offspring equals
| (29) |
At small values of v, matches the “neutral” prediction (gray dashed line in Figure S3_6, Figure S3_7 and Figure S3_8 (File S3)). Note that these fixations have almost no effect on , because their average effect is zero. At intermediate v, Equation 28 slightly underestimates for parameter values leading to large background variance (i.e., high Θ and ). The likely reason is that the analytical approximation assumes to be constant, while it obviously decreases in the simulations (since there are no de novo mutations). All these results are qualitatively consistent across different values of and Θ [Figure 7 and Figure S3_9 and Figure S3_11 (File S3)].
The relative importance of standing genetic variation and de novo mutations over the course of adaptation
Until now we have compared adaptation from standing genetic variation to that from de novo mutations in terms of their distribution of fixed phenotypic effects. We now turn to investigating their relative importance over the course of adaptation. For this purpose, we recorded (in individual-based simulations) the contributions of both sources of variation to the phenotypic mean and variance. An average time series for both measures is shown in Figure 8. As expected, the initial response to selection is almost entirely based on standing variation, but the contribution of de novo mutations increases over time. As a quantitative measure for this transition, we define as the point in time where the cumulative contribution of de novo mutations has reached Indeed, we find that, beyond this time, adaptation almost exclusively proceeds by the fixation of de novo mutations (Figure 8A). As expected, decreases with v [Figure 9 and Figure S3_12 (File S3), first row], while the total phenotypic response increases (Figure 9 and Figure S3_12 (File S3), second row). The reason is that faster environmental change induces stronger directional selection and increases the phenotypic lag, such that standing variation is depleted more quickly and de novo mutations contribute earlier. Note that, as in Figure 7, the total phenotypic response at time decreases once v exceeds the maximal sustainable rate of environmental change, for the same reasons as discussed above. Furthermore, increases with both Θ and (due to the increased standing variation; see Equation 16). Interestingly, the relative contribution of original standing genetic variation to the total genetic variance at time remains largely constant (at ∼) over a large range of v and does not show any dependence on Θ or [Figure 9 and Figure S3_12 (File S3), third row]. Deviations occur only if v is either very small or very large. In particular, if v is small, standing variation is almost completely depleted before new mutations play a significant role. Conversely, if v is very large, standing genetic variation still forms the majority of the total genetic variance. As mentioned above, this is most likely because the population goes extinct before fixations can be completed, that is, before the entire (standing) adaptive potential is exhausted. All these results remain qualitatively unchanged if, instead of , we define as the point in time where of the current genetic variance goes back to de novo mutations [Figure S3_13 and Figure S3_14 (File S3)].
Figure 8.
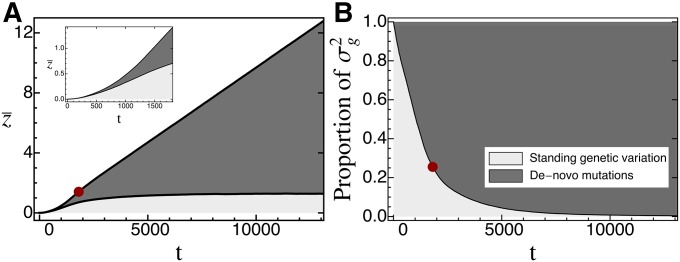
The contributions of standing genetic variation (light gray) and de novo mutations (dark gray) to the cumulative phenotypic response to selection (A) and the current genetic variance (B) over time. Plots show average trajectories over 1000 replicate simulations. The red circle marks the point in time where of the total phenotypic responses were due to de novo mutations. The inset in A shows a more detailed plot of the dynamics of up to this point. Fixed parameters: , , , ,
Figure 9.

(Top row) The point in time where of the phenotypic responses to moving-optimum selection have been contributed by de novo mutations as a function of the rate of environmental change for various values of Θ (left) and (right). Insets show the results for large ν on a log scale. (Middle row) The mean total phenotypic response at this time. (Bottom row) The relative contribution of original standing genetic variation to the total genetic variance at time . Data are means (and standard deviations) from 1000 replicate simulation runs. Fixed parameters (if not stated otherwise): , , ,
Discussion
When studying the genetic basis of adaptation to changing environments, most theoretical work has focused on adaptation from new mutations (e.g., Gillespie 1984; Orr 1998, 2000; Collins et al. 2007; Kopp and Hermisson 2007, 2009a,b; Matuszewski et al. 2014). Consequently, very little is known about the details of adaptation from standing genetic variation (but see Orr and Betancourt 2001; Hermisson and Pennings 2005), that is, which of the alleles segregating in a population will become fixed and contribute to the evolutionary response. Here, we have used analytical approximations and stochastic simulations to study the effects of standing genetic variation on the genetic basis of adaptation in gradually changing environments. Supporting a verbal hypothesis by Barrett and Schluter (2008), we show that, when comparing adaptation from standing genetic variation to that from de novo mutations, the former proceeds, on average, by the fixation of more alleles of small effect. In both cases, however, the genetic basis of adaptation crucially depends on the efficacy of selection, which in turn is determined by the population size, the strength of (stabilizing) selection, and the rate of environmental change. When standing genetic variation is the sole source for adaptation, we find that fast environmental change enables the population to traverse larger distances in phenotype space than under slow environmental change, in contrast to studies that consider adaptation from new mutations only (Perron et al. 2008; Bell and Gonzalez 2011; Bell 2013; Lindsey et al. 2013). We now discuss these results in greater detail.
The genetic basis of adaptation in the moving-optimum model
Introduced as a model for sustained environmental change, such as global warming (Lynch et al. 1991; Lynch and Lande 1993), the moving-optimum model describes the evolution of a quantitative trait under stabilizing selection toward a time-dependent optimum (Bürger 2000). A large number of studies have analyzed both the basic model and several modifications, for example, models with a periodic or fluctuating optimum or models for multiple traits (Slatkin and Lande 1976; Charlesworth 1993; Bürger and Lynch 1995; Lande and Shannon 1996; Kopp and Hermisson 2007, 2009a,b; Gomulkiewicz and Houle 2009; Zhang 2012; Chevin 2013; Matuszewski et al. 2014). Following traditional quantitative-genetic approaches, the majority of these studies assumed that the distribution of genotypes (and phenotypes) is Gaussian with constant (time-invariant) genetic variance, and they have mostly focused on the evolution of the population mean phenotype and on the conditions for population persistence (Bürger and Lynch 1995; Lande and Shannon 1996; Gomulkiewicz and Houle 2009). None of these models, however, allows one to address the fate of individual alleles (i.e., whether they become fixed or not). In a recent series of articles on the moving-optimum model, Kopp and Hermisson (2007, 2009a,b) studied the genetic basis of adaptation from new mutations and derived the distribution of adaptive substitutions (i.e., the distribution of the phenotypic effects of those mutations that arise and become fixed in a population); this approach has recently been generalized to multiple phenotypic traits by Matuszewski et al. (2014). The shape of this distribution resembles a gamma distribution with an intermediate mode. Thus, most substitutions are of intermediate effect with only a few large-effect alleles contributing to adaptation. The reason is that small-effect alleles—despite appearing more frequently than large-effect alleles—have only small effects on fitness (and are, hence, often lost due to genetic drift), while large-effect alleles might be removed because they “overshoot” the optimum (Kopp and Hermisson 2009b). A detailed comparison and discussion of the distribution of adaptive substitutions from de novo mutations with (Equation 23) and without (Kopp and Hermisson 2009b) genetic background variation are given in File S2.
Here, we have studied the genetic basis of adaptation from standing genetic variation. We find that the distribution of substitutions from standing genetic variation depends on the distribution of standing genetic variants (i.e., distribution of alleles segregating in the population prior to the environmental change) and the intensity of selection. The former is shaped primarily by the distribution of new mutations and the strength of stabilizing selection, which removes large-effect alleles. Depending on the speed of change v, we find two regimes that are characterized by separate distributions of standing substitutions. If the environment changes sufficiently fast, the distribution of adaptive substitutions resembles a lognormal distribution with a strong contribution of small-effect alleles [Equation 19; Figure 3; and Figure S3_2 (File S3)]. The reason is that, in the standing genetic variation, small-effect alleles are more frequent than large-effect alleles and might already segregate at appreciable frequency (so that they are not lost by genetic drift). With a moving optimum, they furthermore are the first to become positively selected, hence reducing the time they are under purifying selection. Finally, epistatic interactions between cosegregating alleles (or between a focal allele and the genetic background) also favor alleles of small effect. Consequently, when adapting from standing genetic variation, most substitutions are of small phenotypic effect.
The second regime occurs if the rate of environmental change v is very small. In this case, allele-frequency dynamics are dominated by genetic drift, and the distribution of adaptive substitutions reflects the approximately Gaussian distribution of standing genetic variants [Equation 25; Figure S3_4 (File S3)]. It should be noted, however, that fixations under this regime take a very long time, similar to that of purely neutral substitutions (i.e., on the timescale of ).
Finally, we have studied the relative importance of standing genetic variation and de novo mutations over the course of adaptation. As shown in Figure 4 and Figure 5, the initial response to selection is almost entirely based on standing variation, with de novo mutations becoming gradually more important. The timescale of this transition strongly depends on the rate of environmental change, but for slow or moderately fast change, it typically occurs over at least hundreds of generations [Figure 9 and Figure S3_12, Figure S3_13 and Figure S3_14 (File S3)]. This observation is in contrast to results by Hill and Rasbash (1986b), who found that under strong artificial (i.e., truncation) selection in small populations (), new mutations might contribute up to one-third of the total response after as little as 20 generations. Our results show that the situation is very different for large populations under natural selection in gradually changing environments. The likely reason for this difference is that truncation selection induces strong directional selection (corresponding to large v) and only extreme phenotypes reproduce. Thus, truncation selection is much more efficient in maintaining large-effect de novo mutations, while eroding genetic variation more quickly (because it introduces a large skew in the offspring distribution). However, the similarities and differences in the genetic basis of responses to artificial vs. natural selection are an interesting topic—in particular, for the interpretation of the large amount of genetic data available from breeding programs (Stern and Orgogozo 2009)—that should be addressed in future studies.
Throughout this study, we have focused on adaptation to a moving optimum, that is, a scenario of gradual environmental change. An obvious question is how our results would change under the alternative scenario of a one-time sudden shift in the optimum (as assumed in numerous studies, e.g., Orr 1998; Hermisson and Pennings 2005; Chevin and Hospital 2008). While beyond the scope of this article, our approach should, in principle, still be applicable. In particular, each focal allele still experiences a gradual change in its selection coefficient, due to the evolution of the genetic background. Unlike in the moving-optimum model, however, the selection coefficient decreases, as the mean phenotype gradually approaches the new optimum. Hence, a suitably modified version of Equation 12 would give the probability that a focal allele establishes in the population (i.e., escapes stochastic loss), but in the absence of continued environmental change, establishment does not guarantee fixation. In other words, alleles need to “race for fixation” before other competing alleles get fixed and they become deleterious (Kopp and Hermisson 2007, 2009a). The dynamics of a mutation along its trajectory should therefore be even more complex than in the moving-optimum model and show an even stronger dependence on the genetic background (Chevin and Hospital 2008).
Extinction and the rate of environmental change
Recently, several experimental studies have explored how the rate of environmental change affects the persistence of populations that rely on new mutations for adapting to a gradually changing environment (Perron et al. 2008; Bell and Gonzalez 2011; Lindsey et al. 2013). In line with theoretical predictions (Bell 2013), all studies found that “evolutionary rescue” is contingent on a small rate of environmental change. In particular, Lindsey et al. (2013, p. 463) evolved replicate populations of Escherichia coli under different rates of increase in antibiotic concentration and found that certain genotypes were evolutionarily inaccessible under rapid environmental change, suggesting that “rapidly deteriorating environments not only limit mutational opportunities by lowering population size, but […] also eliminate sets of mutations as evolutionary options”. This is in stark contrast to our prediction that faster environmental change can enable the population to remain better adapted and to traverse larger distances in phenotype space when standing genetic variation is the sole source for adaptation [Figure 7 and Figure S3_9, Figure S3_10 and Figure S3_11 (File S3)]; in line with recent experimental observations; H. Teotonio, private communication). The difference between these results arises from the availability of the “adaptive material.” While de novo mutations first need to appear and survive stochastic loss before becoming fixed, standing genetic variants are available right away and might already be segregating at appreciable frequency. Thus, in both cases, the rate of environmental change plays a critical, although antagonistic, role in determining the evolutionary options. While fast environmental change eliminates sets of new mutations, it simultaneously helps to preserve standing genetic variation until it can be picked up by selection. Under slow change, in contrast, most large-effect alleles from the standing variation, by the time they are needed, are already eliminated by drift or stabilizing selection.
Our results also mean that, if the optimum stops moving at a given value , populations will achieve a higher degree of adaptation (higher ) if the final optimum is reached fast rather than slowly (see also Uecker et al. 2014), at least if standing genetic variation is the sole source for adaptation. While this assumption is an obvious simplification, it may often be approximately true in natural populations. The same holds true in experimental populations, where selection is usually strong and the duration of the experiment short, such that de novo mutations can frequently be neglected (see Figure 9).
Testing the predictions
The predictions made by our model can in principle be tested empirically, even though suitable data might be sparse and experiments challenging. There is, of course, ample evidence for adaptation from standing genetic variation (e.g., Teotónio et al. 2009; Jerome et al. 2011; Jones et al. 2012b; Sheng et al. 2015). For example, Domingues et al. (2012) showed that camouflaging pigmentation of oldfield mice (Peromyscus polionotus) that have colonized Florida’s Gulf Coast has evolved quite rapidly from a preexisting mutation in the Mc1r gene, Limborg et al. (2014) investigated selection in two allochronic but sympatric lineages of pink salmon (Oncorhynchus gorbuscha) and identified 24 divergent loci that had arisen from different pools of standing genetic variation, and Turchin et al. (2012) showed that height-associated alleles in humans display a clear signal for widespread selection on standing genetic variation.
However, testing the predictions of our model requires, in addition, detailed knowledge of the genotype–phenotype relation. Currently, there are only a small (yet increasing) number of systems for which both a set of functionally validated beneficial mutations and their selection coefficients under different environmental conditions are available (Jensen 2014). Thus, estimating the distribution of standing substitutions will be challenging, because of the often unknown phenotypic and fitness effects of beneficial mutations and the large number of replicate experiments needed to obtain a reliable empirical distribution. Furthermore, from an experimental point of view it is often difficult to discriminate between phenotypic (or fitness) effects of individual mutations and phenotypic changes induced by phenotypic plasticity and environmental variance. However, even if these problems were solved, small-effect alleles might not be detectable due to statistical limitations (Otto and Jones 2000), and in certain limiting cases where the population quickly goes extinct (i.e., when the environment changes very fast), the distribution of adaptive substitutions from standing genetic variation might be indistinguishable from that from de novo substitutions (Figure 6).
Recent developments in laboratory systems (Morran et al. 2009; Parts et al. 2011), however, have created opportunities for experimental evolution studies in which population size, the selective regime, and the duration of selection can be manipulated and adaptation from de novo mutations and standing genetic variation can be recorded (Burke 2012; Schlotterer et al. 2015; see also Teotonio et al. 2012). Applying these techniques in experiments in the vein of Lindsey et al. (2013), but starting from a polymorphic population, should make it possible to test the relation between the rate of environmental change and population persistence and to assess the probability of adaptation from standing genetic variation. First experiments along these lines are currently being carried out in populations of Caenorhabditis elegans, with the aim of determining the limits of adaptation to different rates of increase in sodium chloride concentration (H. Teotonio, private communication; see also Theologidis et al. 2014). Furthermore, Pennings (2012) recently applied the Hermisson and Pennings (2005) framework to show that standing genetic variation plays an important role in the evolution of drug resistance in human immunodeficiency virus, affecting up to of patients (depending on treatment) and explaining why resistance mutations in patients who interrupt treatment are likely to become established within the first year. A similar approach should also be applicable to scenarios of gradual environmental change (e.g., evolution of resistance mutations under gradually increasing antibiotic concentrations).
Conclusion
As global climate change continues to force populations to respond to the altered environmental conditions, studying adaptation to changing environments—both empirically and theoretically—has become one of the main topics in evolutionary biology. Despite increased efforts, however, very little is known about the genetic basis of adaptation from standing genetic variation. Our analysis of the moving-optimum model shows that this process has, indeed, a very different genetic basis than that of adaptation from de novo mutations. In particular, adaptation proceeds via the fixation many small-effect alleles (and just a few large ones). In accordance with previous studies, the adaptive process critically depends on the tempo of environmental change. Specifically, when populations adapt from standing genetic variation only, the potential for adaptation increases as the environment changes faster.
Supplementary Material
Acknowledgments
We thank S. Aeschbacher, R. Bürger, L. M. Chevin, H. Teotonio, C. Vogl, and two anonymous reviewers for constructive comments on this manuscript. This study was supported by the Austrian Science Fund (grant P 22581-B17 to M.K. and grant P22188 to Reinhard Bürger), the Austrian Agency for International Cooperation in Education and Research (grant FR06/2014 to J.H.), Campus France (grant PHC AMADEUS 31642SJ to M.K.), and a Writing-Up Fellowship from the Konrad Lorenz Institute for Evolution and Cognition Research (to S.M.).
Appendix
Appendix A: Wright–Fisher Simulations: A Focal Locus with Recurrent Mutations
In Appendix A, we describe the Wright–Fisher (WF) simulations used (solely) for checking our analytical approximations regarding the probability for adaptation from standing genetic variation (Equation 14). To simulate evolution at a focal locus, we followed Hermisson and Pennings (2005) and implemented a multinomial WF sampling approach (available from the corresponding author upon request). These simulations serve as an additional analysis tool that has been adjusted to the approximation method and allows the adaptive process to be simulated fast and efficiently. In addition, they go beyond the individual-based model in one aspect, as they do not make the infinite-sites assumption but allow for recurrent mutation at the focal locus (but still with at most two alleles).
Genome
At the focal locus, mutations with a fixed allelic effect α appear recurrently at rate θ and convert ancestral alleles into derived mutant alleles. Accordingly, despite a genetic background with normally distributed genotypic values, there are at most two types of (focal) alleles in the population, where each type “feels” only the mean background , which evolves according to Lande’s equation (Equation 5, see above). The genetic background variation is assumed to be constant and serves as a free parameter that is independent of θ, , and . Note that the evolutionary response at the focal locus is influenced by that of the genetic background, and vice versa, meaning that the two are interdependent.
Procedure
We follow the evolution of alleles at the focal locus. Each generation is generated by multinomial sampling, where the probability of choosing an allele of a given type (ancestral or derived) is weighted by its respective (marginal) fitness. Furthermore, the mean phenotype of the genetic background evolves deterministically according to Equation 5 with constant . To let the population reach mutation–selection–drift equilibrium, each simulation is started generations before the environment starts changing. Initially, the population consists of only ancestral alleles “0”; the derived allele “1” is created by mutation. If the derived allele reaches fixation before the environmental change (by drift), it is itself declared “ancestral”; i.e., the population is set back to the initial state. After generations, the optimum starts moving, such that the selection coefficient of the derived allele, which is initially deleterious [i.e., ], increases and may eventually become beneficial [i.e., ], depending on the response at the genetic background. Simulations continue until the derived allele is either fixed or lost. Fixation probabilities are estimated from simulation runs.
Appendix B: Theoretical Background
In Appendix B, we briefly recapitulate results from previous studies that form the basis for our analytical derivations.
The probability of adaptation from standing genetic variation for a single biallelic locus after a sudden environmental change
Hermisson and Pennings (2005) studied the situation where the selection scheme at a single biallelic locus changes following a sudden environmental change. In particular, they derived the probability for a mutant allele to reach fixation that was neutral or deleterious prior to the change but has become beneficial in the new environment. In the continuum limit for allele frequencies this probability is given by
| (B1) |
where is the density function for the allele frequency x of the mutant allele in mutation–selection–drift balance and denotes its fixation probability.
For a mutant allele present at frequency x in a population with effective size and with selective advantage in the new environment, the fixation probability is given by
| (B2) |
(Kimura 1957). There are two points to make here. First, mutational effects in the Hermisson and Pennings (2005) model are directly proportional to fitness, whereas mutations in our model affect a phenotype under selection. Second, in our framework, denotes the (beneficial) selection coefficient for heterozygotes.
Approximations for can be derived from standard diffusion theory (Ewens 2004; for details see Hermisson and Pennings 2005). If the mutant allele was neutral prior to the change in the selection scheme,
| (B3) |
Here, θ is the per-locus mutation rate, denotes a normalization constant where is Euler’s gamma, and is the polygamma function. Similarly, if the mutant allele was deleterious before the environmental change (with negative selection coefficient ), the allele-frequency distribution is given by
| (B4) |
where denotes a normalization constant and is the hypergeometric function. If the allele was sufficiently deleterious (), Equation B4 can further be approximated as
| (B5) |
where again denotes a normalization constant with denoting the lower incomplete gamma function.
Finally, the probability that a population successfully adapts from standing genetic variation can be derived as
| (B6) |
Fixation probabilities under time-inhomogeneous selection
In gradually changing environments, the selection coefficient of a given (mutant) allele is not fixed but changes over time (i.e., as the position of the optimum changes). Uecker and Hermisson (2011) recently developed a mathematical framework based on branching-process theory to describe the fixation process of a beneficial allele under temporal variation in population size and selection pressures. They showed that the probability of fixation of a mutation starting with n initial copies is given by
| (B7a) |
where
| (B7b) |
Assuming that the population size remains constant and that the selection coefficient increases linearly in time, , Equation B7a becomes
| (B8) |
where denotes the complementary Gaussian error function.
Footnotes
Communicating editor: J. Wakeley
Supporting information is available online at www.genetics.org/lookup/suppl/doi:10.1534/genetics.115.178574/-/DC1.
Literature Cited
- Barrett R. D., Schluter D., 2008. Adaptation from standing genetic variation. Trends Ecol. Evol. 23: 38–44. [DOI] [PubMed] [Google Scholar]
- Barton N. H., Turelli M., 1991. Natural and sexual selection on many loci. Genetics 127: 229–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell G., 2009. The oligogenic view of adaptation. Cold Spring Harb. Symp. Quant. Biol. 74: 139–144. [DOI] [PubMed] [Google Scholar]
- Bell G., 2013. Evolutionary rescue and the limits of adaptation. Philos. Trans. R. Soc. Lond., B 368: 20120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell G., Gonzalez A., 2011. Adaptation and evolutionary rescue in metapopulations experiencing environmental deterioration. Science 332: 1327–1330. [DOI] [PubMed] [Google Scholar]
- Bulmer M. G., 1980. The Mathematical Theory of Quantitative Genetics. Oxford University Press, Oxford. [Google Scholar]
- Bürger R., 1999. Evolution of genetic variability and the advantage of sex and recombination in a changing environment. Genetics 153: 1055–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bürger R., 2000. The Mathematical Theory of Selection, Recombination, and Mutation. Wiley, Chichester, UK. [Google Scholar]
- Bürger R., Lynch M., 1995. Evolution and extinction in a changing environment: a quantitative genetic analysis. Evolution 49: 151–163. [DOI] [PubMed] [Google Scholar]
- Burke M. K., 2012. How does adaptation sweep through the genome? Insights from long-term selection experiments. Proc. Biol. Sci. 279: 5029–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caprio M., 2005. Levelscheme: a level scheme drawing and scientific figure preparation system for Mathematica. Comput. Phys. Commun. 171: 107–118. [Google Scholar]
- Charlesworth B., 1993. Directional selection and the evolution of sex and recombination. Genet. Res. 61: 205–224. [DOI] [PubMed] [Google Scholar]
- Chevin L. M., 2013. Genetic constraints on adaptation to a changing environment. Evolution 67: 708–721. [DOI] [PubMed] [Google Scholar]
- Chevin L. M., Hospital F., 2008. Selective sweep at a quantitative trait locus in the presence of background genetic variation. Genetics 180: 1645–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins S., de Meaux J., Acquisti C., 2007. Adaptive walks toward a moving optimum. Genetics 176: 1089–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper T. F., Ostrowski E. A., Travisano M., 2007. A negative relationship between mutation pleiotropy and fitness effect in yeast. Evolution 61: 1495–1499. [DOI] [PubMed] [Google Scholar]
- Domingues V. S., Poh Y.-P., Peterson B. K., Pennings P. S., Jensen J. D., et al. , 2012. Evidence of adaptation from ancestral variation in young populations of beach mice. Evolution 66: 3209–3223. [DOI] [PubMed] [Google Scholar]
- Ewens W. J., 2004. Mathematical Population Genetics, Ed. 2 Springer-Verlag, Berlin. [Google Scholar]
- Foley P., 1992. Small population genetic variability at loci under stabilizing selection. Evolution 46: 763–774. [DOI] [PubMed] [Google Scholar]
- Galassi, M., J. Davies, J. Theiler, B. Gough, and G. Jungman, 2009 GNU Scientific Library - Reference Manual, for GSL Version 1.12, Ed. 3. Network Theory Ltd, Bristol, UK. [Google Scholar]
- Gillespie J. H., 1984. Molecular evolution over the mutational landscape. Evolution 38: 1116–1129. [DOI] [PubMed] [Google Scholar]
- Gomulkiewicz R., Houle D., 2009. Demographic and genetic constraints on evolution. Am. Nat. 174: E218–E229. [DOI] [PubMed] [Google Scholar]
- Gomulkiewicz R., Holt R. D., Barfield M., Nuismer S. L., 2010. Genetics, adaptation, and invasion in harsh environments. Evol. Appl. 3: 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallauer, A., M. Carena, and J. Filho, 2010 Quantitative Genetics in Maize Breeding (Handbook of Plant Breeding). Springer-Verlag, New York. [Google Scholar]
- Hermisson J., Pennings P. S., 2005. Soft sweeps: molecular population genetics of adaptation from standing genetic variation. Genetics 169: 2335–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hietpas R. T., Bank C., Jensen J. D., Bolon D. N. A., 2013. Shifting fitness landscapes in response to altered environments. Evolution 67: 3512–3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill W. G., Rasbash J., 1986a Models of long-term artificial selection in finite population. Genet. Res. 48: 41–50. [DOI] [PubMed] [Google Scholar]
- Hill W. G., Rasbash J., 1986b Models of long-term artificial selection in finite population with recurrent mutation. Genet. Res. 48: 125–131. [DOI] [PubMed] [Google Scholar]
- Jensen J. D., 2014. On the unfounded enthusiasm for soft selective sweeps. Nat. Commun. 5: 5281. [DOI] [PubMed] [Google Scholar]
- Jerome J. P., Bell J. A., Plovanich-Jones A. E., Barrick J. E., Brown C. T., et al. , 2011. Standing genetic variation in contingency loci drives the rapid adaptation of Campylobacter jejuni to a novel host. PLoS ONE 6: e16399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones A. G., Arnold S. J., Bürger R., 2004. Evolution and stability of the G-matrix on a landscape with a moving optimum. Evolution 58: 1636–1654. [DOI] [PubMed] [Google Scholar]
- Jones A. G., Bürger R., Arnold S. J., Hohenlohe P. A., Uyeda J. C., 2012a The effects of stochastic and episodic movement of the optimum on the evolution of the G-matrix and the response of the trait mean to selection. J. Evol. Biol. 25: 2010–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones F. C., Grabherr M. G., Chan Y. F., Russell P., Mauceli E., et al. , 2012b The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484: 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman S. A. A., Levin S., 1987. Towards a general theory of adaptive walks on rugged landscapes. J. Theor. Biol. 128: 11–45. [DOI] [PubMed] [Google Scholar]
- Kimura M., 1957. Some problems of stochastic processes in genetics. Ann. Math. Stat. 28: 882–901. [Google Scholar]
- Kimura M., 1983. The Neutral Theory of Molecular Evolution. Cambridge University Press, Cambridge, UK. [Google Scholar]
- Kirkpatrick M., Johnson T., Barton N., 2002. General models of multilocus evolution. Genetics 161: 1727–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp M., Hermisson J., 2007. Adaptation of a quantitative trait to a moving optimum. Genetics 176: 715–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp M., Hermisson J., 2009a The genetic basis of phenotypic adaptation I: fixation of beneficial mutations in the moving optimum model. Genetics 182: 233–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp M., Hermisson J., 2009b The genetic basis of phenotypic adaptation II: the distribution of adaptive substitutions in the moving optimum model. Genetics 183: 1453–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp M., Matuszewski S., 2014. Rapid evolution of quantitative traits: theoretical perspectives. Evol. Appl. 7: 169–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lande R., 1976. The maintenance of genetic variability by mutation in a polygenic character with linked loci. Genet. Res. 26: 221–235. [DOI] [PubMed] [Google Scholar]
- Lande R., 1983. The response to selection on major and minor mutations affecting a metrical trait. Heredity 50: 47–65. [Google Scholar]
- Lande R., Shannon S., 1996. The role of genetic variation in adaptation and population persistence changing environment. Evolution 50: 434–437. [DOI] [PubMed] [Google Scholar]
- Lang G. I., Botstein D., Desai M. M., 2011. Genetic variation and the fate of beneficial mutations in asexual populations. Genetics 188: 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limborg M. T., Waples R. K., Seeb J. E., Seeb L. W., 2014. Temporally isolated lineages of pink salmon reveal unique signatures of selection on distinct pools of standing genetic variation. Heredity 105: 741–751. [DOI] [PubMed] [Google Scholar]
- Lindsey H. A., Gallie J., Taylor S., Kerr B., 2013. Evolutionary rescue from extinction is contingent on a lower rate of environmental change. Nature 494: 463–467. [DOI] [PubMed] [Google Scholar]
- Lynch M., Lande R., 1993. Evolution and extinction in response to environmental change, pp. 43–53 in Biotic Interactions and Global Change, edited by Kareiva P., Kingsolver J. G., Juey R. B. Sinauer Associates, Sunderland, MA. [Google Scholar]
- Lynch M., Gabriel W., Wood A. M., 1991. Adaptive and demographic responses of plankton populations to environmental change. Limnol. Oceanogr. 36: 1301–1312. [Google Scholar]
- Martin G., Lenormand T., 2006a A general multivariate extension of Fisher’s geometrical model and the distribution of mutation fitness effects across species. Evolution 60: 893–907. [PubMed] [Google Scholar]
- Martin G., Lenormand T., 2006b The fitness effect of mutations in stressful environments: a survey in the light of fitness landscape models. Evolution 60: 2413–2427. [PubMed] [Google Scholar]
- Matuszewski S., Hermisson J., Kopp M., 2014. Fisher’s geometric model with a moving optimum. Evolution 68: 2571–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morran L. T., Parmenter M. D., Phillips P. C., 2009. Mutation load and rapid adaptation favour outcrossing over self-fertilization. Nature 462: 350–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr H. A., 1998. The population genetics of adaptation: the distribution of factors fixed during adaptive evolution. Evolution 52: 935–949. [DOI] [PubMed] [Google Scholar]
- Orr H. A., 2000. Adaptation and the cost of complexity. Evolution 54: 13–20. [DOI] [PubMed] [Google Scholar]
- Orr H. A., 2002. The population genetics of adaptation: the adaptation of DNA sequences. Evolution 56: 1317–1330. [DOI] [PubMed] [Google Scholar]
- Orr H. A., 2005a The genetic theory of adaptation: a brief history. Nat. Rev. Genet. 6: 119–127. [DOI] [PubMed] [Google Scholar]
- Orr H. A., 2005b Theories of adaptation: what they do and don’t say. Genetica 123: 3–13. [DOI] [PubMed] [Google Scholar]
- Orr H. A., Betancourt A. J., 2001. Haldane’s sieve and adaptation from standing genetic variation. Genetics 157: 875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto S. P., Jones C. D., 2000. Detecting the undetected: estimating the total number of loci underlying a quantitative trait. Genetics 156: 2093–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parts L., Cubillos F. A., Warringer J., Jain K., Salinas F., et al. , 2011. Revealing the genetic structure of a trait by sequencing a population under selection. Genome Res. 21: 1131–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennings P. S., 2012. Standing genetic variation and the evolution of drug resistance in HIV. PLoS Comput. Biol. 8: e1002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perron G. G., Gonzalez A., Buckling A., 2008. The rate of environmental change drives adaptation to an antibiotic sink. J. Evol. Biol. 21: 1724–1731. [DOI] [PubMed] [Google Scholar]
- Rockman M. V., 2012. The QTN program and the alleles that matter for evolution: all that’s gold does not glitter. Evolution 66: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlotterer C., Kofler R., Versace E., Tobler R., Franssen S. U., 2015. Combining experimental evolution with next-generation sequencing: a powerful tool to study adaptation from standing genetic variation. Heredity 114: 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng, Z., M. E. Pettersson, C. F. Honaker, P. B. Siegel, and O. Carlborg, 2015 Standing genetic variation as a major contributor to adaptation in the Virginia chicken lines selection experiment. bioRxiv. http://dx.doi.org/10.1101/018721. [DOI] [PMC free article] [PubMed]
- Slatkin M., Lande R., 1976. Niche width in a fluctuating environment-density independent model. Am. Nat. 110: 31–55. [Google Scholar]
- Smith N. G. C., Eyre-Walker A., 2002. Adaptive protein evolution in Drosophila. Nature 415: 1022–1024. [DOI] [PubMed] [Google Scholar]
- Stern D. L., Orgogozo V., 2009. Is genetic evolution predictable? Science 323: 746–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teotónio H., Chelo I. M., Bradic M., Rose M. R., Long A. D., 2009. Experimental evolution reveals natural selection on standing genetic variation. Nat. Genet. 41: 251–257. [DOI] [PubMed] [Google Scholar]
- Teotonio H., Carvalho S., Manoel D., Roque M., Chelo I. M., 2012. Evolution of outcrossing in experimental populations of Caenorhabditis elegans. PLoS ONE 7: e35811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theologidis I., Chelo I. M., Goy C., Teotonio H., 2014. Reproductive assurance drives transitions to self-fertilization in experimental Caenorhabditis elegans. BMC Biol. 12: 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin D., Kause A., Mäntysaari E. A., Martin S. A., Houlihan D. F., et al. , 2006. Fat or lean? The quantitative genetic basis for selection strategies of muscle and body composition traits in breeding schemes of rainbow trout (Oncorhynchus mykiss). Aquaculture 261: 510–521. [Google Scholar]
- Turchin M. C., Chiang C. W., Palmer C. D., Sankararaman S., Reich D., et al. , 2012. Evidence of widespread selection on standing variation in Europe at height-associated SNPs. Nat. Genet. 44: 1015–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uecker H., Hermisson J., 2011. On the fixation process of a beneficial mutation in a variable environment. Genetics 188: 915–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uecker H., Otto S. P., Hermisson J., 2014. Evolutionary rescue in structured populations. Am. Nat. 183: E17–E35. [DOI] [PubMed] [Google Scholar]
- Wichman H. A., Badgett M. R., Scott L. A., Boulianne C. M., Bull J. J., 1999. Different trajectories of parallel evolution during viral adaptation. Science 285: 422–424. [DOI] [PubMed] [Google Scholar]
- Wricke G., Weber E., 1986. Quantitative Genetics and Selection in Plant Breeding. Walter de Gruyter, Berlin, Boston. [Google Scholar]
- Zhang X. S., 2012. Fisher’s geometrical model of fitness landscape and variance in fitness within a changing environment. Evolution 66: 2350–2368. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.