

Abstract
Streptolysin S (SLS) is a post-translationally modified peptide cytolysin that is produced by the human pathogen Streptococcus pyogenes. SLS belongs to a large family of azole-containing natural products that are biosynthesized via an evolutionarily conserved pathway. SLS is an important virulence factor during S. pyogenes infections, but despite an extensive history of study, further investigations are needed to clarify several steps of its biosynthesis. To this end, chemical inhibitors of SLS biosynthesis would be valuable tools to interrogate the various maturation steps of both SLS and biosynthetically-related natural products. Such chemical inhibitors could also potentially serve as anti-virulence therapeutics, which in theory may alleviate the spread of antibiotic resistance. In this work, we demonstrate that FDA-approved HIV protease inhibitors, especially nelfinavir, block a key proteolytic processing step during SLS production. This inhibition was demonstrated in live S. pyogenes cells and through in vitro protease inhibition assays. A panel of 57 nelfinavir analogs was synthesized, leading to a series of compounds with improved anti-SLS activity while illuminating structure-activity relationships. Nelfinavir was also found to inhibit the maturation of other azole-containing natural products, namely those involved in listeriolysin S, clostridiolysin S, and plantazolicin production. The use of nelfinavir analogs as inhibitors of SLS production has allowed us to begin examining the proteolysis event in SLS maturation and will aid in further investigations of the biosynthesis of SLS and related natural products.
Introduction
The ribosomally synthesized and post-translationally modified peptides (RiPPs) comprise a rapidly expanding class of natural products that includes a wide variety of structural modifications.1 These modifications impart RiPPs with diverse activities, giving rise to a range of products from antibacterials2–4 to anticancer agents.5 The installation of azole and/or azoline heterocycles is one such modification common to many RiPPs, forming a sub-class of natural products called the thiazole/oxazole-modified microcins (TOMMs).6 The azoles are biosynthesized by the cyclodehydration and subsequent dehydrogenation of cysteine, serine, and threonine residues to form thiazole and (methyl)oxazole rings on the C-terminal portion, or “core”, of a ribosomally produced precursor peptide.6 The azole-containing peptides will often undergo further processing, including the proteolytic removal of the N-terminal “leader” portion of the peptide and export of the mature product.7 Although recent discoveries have shed light on the mechanism of azole formation,8–10 the proteolytic processing step of most TOMMs has yet to be investigated.
Streptolysin S (SLS), a key virulence factor of Streptococcus pyogenes, is one such TOMM whose biosynthesis is incompletely understood.11 S. pyogenes is the causative agent of diseases ranging in severity from pharyngitis to necrotizing fasciitis12 and is a major global health burden, causing over 600 million infections and 500,000 deaths annually.13 SLS is the cytolytic toxin responsible for the classic β-hemolytic phenotype when S. pyogenes is grown on blood agar14 and has been shown to be critical to pathogenesis in mammalian infection models.15–17 Although a few strains of non-β-hemolytic, pathogenic S. pyogenes have been described, such as the Lowry strain,18 the vast majority of S. pyogenes isolates produce SLS.19 The toxin is biosynthesized by a 9-gene biosynthetic operon that encodes the precursor peptide (sagA), cyclodehydratase and dehydrogenase enzymes (sagBCD), a putative leader peptidase (sagE), a multicomponent ABC-transporter (sagGHI), and a protein of unknown function (sagF) (Figure 1A).14 The SagBCD heterocycle synthetase is known to install azol(in)e rings on the core region of the precursor peptide,20, 21 which is followed by proteolytic removal of the leader peptide prior to export of the mature, bioactive natural product (Figure 1B,C). The final molecular weight of SLS has been inferred from classic gel filtration studies to be 2.8 kDa,22 which is consistent with the bioinformatic prediction of the scissile bond being C-terminal to a Gly-Gly motif based on the similarity to bacteriocins from other Gram-positive bacteria.23 Additionally, proteolysis following small residues is common in RiPPs with known cleavage sites.7 Although SLS was first defined nearly 80 years ago,24 with the β-hemolytic phenotype being known since the late 1800s,25 a detailed mechanism of SLS biosynthesis and the structure of the mature toxin remain elusive. Thus, effective SLS biosynthetic inhibitors could serve as powerful chemical tools to shed more light on the biochemistry of SLS and the infection biology of S. pyogenes.
Figure 1.

TOMM gene clusters and the biosynthetic pathway. (A) Open reading frame diagram showing the organization of several TOMM gene clusters, grouped by function of the mature TOMM. The letters over each gene correspond to the name of the sag genes in S. pyogenes. The function of each gene is color-coded in the legend. (B) Sequences of the precursor peptides from the clusters shown in panel A. The putative (*) or known (−) cleavage sites are shown. The residues substituted in SagA to generate SagA-VLPLL are shown in red. SagA, from S. pyogenes; ClosA, from C. botulinum; StaphA, from S. aureus; LlsA, from L. monocytogenes; BamA, from B. amyloliquefaciens. (C) Generalized mechanism of TOMM maturation with SLS as the example. The proteins putatively responsible for each step are shown above each arrow.
Gaining a better understanding of how pathogens employ their various virulence factors also aids in the development of selective treatment strategies that could help to increase the lifespan of clinically important antibiotics. Unlike traditional broad-spectrum antibiotic treatment, specifically targeting virulence would retain the human microbiota, helping to eradicate secondary infections and, in some cases, could theoretically reduce the evolutionary pressure for the development of resistance.26 Previous approaches to targeting virulence have included disruption of quorum sensing, which often regulates virulence factor expression, blocking toxin delivery or function, and inhibition of bacterial adhesion.26 Several compounds designed around these approaches have been efficacious in vivo and have prompted further study,26, 27 but the selective nature of targeting virulence requires a tailored therapy for each pathogen, which when developed, would stimulate vast improvements in clinical diagnostics. SLS is an interesting anti-virulence target, as it plays a major role in paracellular invasion, immune evasion, and host-metabolism manipulation.11, 28 Furthermore, additional roles of SLS in iron acquisition have been suggested but require further confirmation.11 Due to the importance of SLS in a multitude of pathogenic processes, chemical inhibitors developed to probe the biosynthesis of SLS may find future roles in virulence attenuation strategies that protect the vital microbiota and limit the spread of antibiotic resistance in other pathogens that employ SLS-like toxins (i.e. S. pyogenes and specific strains of Listeria monocytogenes, Clostridium botulinum, and Staphylococcus aureus).
In this study, we identified inhibitors SLS biosynthesis in S. pyogenes by searching for compounds that block an essential proteolytic maturation step. This proteolysis event has been proposed to be performed by SagE, a putative peptidase with homology to a large family of proteases referred to as the CaaX proteases and bacteriocin-processing enzymes (CPBP; often confusingly annotated as abortive infection proteins, Abi), which includes the eukaryotic type II CaaX proteases as well as prokaryotic proteins with putative bacteriocin-related functions.29, 30 The type II CaaX proteases are involved in the processing of a number of C-terminally prenylated proteins in eukaryotes and have been much more thoroughly studied than their prokaryotic counterparts.31–33 Type II CaaX proteases were initially believed to be cysteine proteases,34 but the presumed catalytic cysteine was later proven to be unnecessary for activity.33 In contrast, conserved glutamate and histidine residues were shown to be important for activity, leading to the hypothesis that type II CaaX proteases were metalloproteases.33 Many of the prokaryotic members of the CPBP family, including SagE, have been annotated as immunity proteins due to the role in bacteriocin self-immunity of the family members in Lactobacillus plantarum and L. sakei.35 However, the production of viable allelic exchange mutants of sagE and the homolog in Listeria monocytogenes (llsP) indicates that SagE may not be serving an immunity role or may be redundant with other uncharacterized immunity mechanisms.14, 36 Thus, it is plausible that the prokaryotic CPBPs actually act as proteases and that this function is exploited to provide self-immunity in certain cases. The results presented herein support a role for SagE as a protease through the discovery and characterization of a family of small molecule inhibitors of SLS biosynthesis. Appealingly, these inhibitors were identified through the repurposing of existing drugs by an examination of known off-target effects. This approach facilitated the rapid identification of lead compounds without the need to perform expensive and laborious high-throughput screens, as well as aiding in the synthesis of analogs by leveraging previous work on the compounds.37–39
Results
Evidence for the role of SagE as a protease
Reconstitution of SagE in vitro would be the most direct means of testing for the predicted protease activity and for the screening of inhibitors. Unfortunately, numerous attempts to heterologously express SagE proved unsuccessful and alternative assays were developed to address this issue. We attribute much of the difficulty in expressing SagE to the predicted transmembrane nature of the protein; topology modeling of SagE with SPOCTOPUS40 predicted five transmembrane helixes and an N-terminal signal peptide (Figure S1). With this in mind, crude membranes from S. pyogenes were prepared from total cellular lysates by ultracentrifugation. The resultant samples were then assessed for proteolytic activity towards 35S-labeled SagA, generated through in vitro transcription/translation. Robust proteolysis of SagA to the predicted molecular weight22 was observed after treatment with the membrane fraction (Figure 2A). SagA processing in the whole cell lysate and supernatant fractions was much less extensive and often not observable (Figure S2). To test the substrate specificity of proteolysis, a mutant version of SagA with residues Ala20, Gly22, and Gly23 mutated to leucines (SagA-VLPLL) was generated (Figure 1B). These residues are directly N-terminal to the predicted leader peptide cleavage site and were expected to be important for protease recognition. Pro21 was left intact to avoid inducing a drastic structural change on the peptide. When treated with isolated S. pyogenes membranes, SagA-VLPLL was processed at a rate slower than wild-type SagA (Figure 2A), suggesting that the cleavage was performed by a membrane protease specifically recognizing the SagA cleavage site (other membrane-bound proteases may also be minor contributors to the observed proteolysis).
Figure 2.

The role of SagE in the processing of pre-SLS. (A) Purified S. pyogenes membranes display proteolytic activity towards SagA. Both wild type (WT) SagA, and to a reduced extent, the predicted cut site mutant SagA-VLPLL, function as cleavage substrates. (B) Lytic activity of E. coli expressing the SLS biosynthetic machinery after a 2 h induction. The hemolytic activity of extracted SLS on erythrocytes is measured by a colorimetric readout of hemoglobin release.
To provide additional evidence for the role of SagE, a previously reported multi-plasmid-based expression system for the generation of SLS in Escherichia coli was used. In that study, a lytic entity was generated by the expression of a maltose-binding protein (MBP)-tagged SagA in conjunction with SagB-D from pETDuet vectors after extended induction times.41 SLS produced in this system could be extracted with bovine serum albumin (BSA) and applied to blood using procedures long established for S. pyogenes.42 The heterologously produced SLS was presumably exported by generalized transporters after non-specific proteolysis of the MBP tag or was released via E. coli cell death following buildup of the toxin. Using this system, no lytic activity was observable after a considerably shorter induction time (2 h) from a strain expressing only SagA-D; however, E. coli expressing SagA-F was highly lytic after 2 h (Figure 2B), indicating that SagEF expedite SLS maturation. The additional inclusion of SagF was required for this effect, although its functional role is unknown. SagF shares no homology to any known proteins but has previously been demonstrated to be necessary for SLS biosynthesis14 and may aid in the folding or localization of the other modification machinery. To support the role of SagE as a protease involved in SLS maturation, two highly conserved glutamine residues (Glu131 and Glu132) that are critical for activity in mammalian CPBP family members30, 33 were mutated to alanine, resulting in the complete loss of the observed lytic activity.
Aspartyl protease inhibitors block SagA proteolysis
A small panel of general mechanism-based protease inhibitors was screened for inhibition of SagA leader proteolysis using the membrane cleavage assay with S. pyogenes membranes. This assay was preferred over the E. coli multi-plasmid system to avoid potential issues with membrane penetration or general toxicity of the inhibitors. From the panel, only the aspartyl protease inhibitor pepstatin displayed inhibitory activity (Figure S3), which was unexpected given that SagE bears no similarity to known aspartyl proteases and that members of the type II CaaX protease family were previously hypothesized to function as zinc-dependent metalloproteases.33 However, inhibition of a metalloprotease by aspartyl protease inhibitors has literature precedent, as several inhibitors of HIV protease were found to also inhibit the type I CaaX protease ZMPSTE24, which is a known zinc metalloprotease.43–45 Lipodystrophy is a possible side effect of treatment with certain HIV protease inhibitors and also occurs from genetic deficiencies in ZMPSTE24, leading to the discovery of this off-target effect for these drugs.43 Although type I and II CaaX proteases do not share sequence similarity, they redundantly process some of the same substrates and share similar substrate-binding site architectures.46, 47 Given this, we reasoned that HIV protease inhibitors might be repurposed as inhibitors of SLS production.
HIV protease inhibitors block SLS production
A whole-cell assay in S. pyogenes based on the extraction of SLS with BSA was used to screen a panel of nine FDA-approved HIV protease inhibitors (Figure S4). Nelfinavir, ritonavir, saquinavir, and lopinavir were found to inhibit the production of SLS when tested at 50 µM, while indinavir, amprenavir, atazanavir, and darunavir did not inhibit SLS production (Figure 3A). Interestingly, tipranavir caused significant growth suppression in S. pyogenes, and treated cultures never reached late exponential phase (Figure S5), which is when SLS becomes detectable in vitro.48 The efficacies of the HIV protease inhibitors shown to inhibit SLS production in the initial screen were evaluated by determining 50% inhibition concentration (IC50) values. The IC50 of nelfinavir (6 µM) was much lower than those of ritonavir (35 µM), saquinavir (25 µM), and lopinavir (25 µM). Owing to its greater potency, nelfinavir (Figure 3B) was selected for further study.
Figure 3.

Inhibition of SLS production. (A) Hemolytic activity of SLS extracts from S. pyogenes treated with HIV protease inhibitors. Tipranavir is shown separated by a dashed line due to significant growth effects during treatment (Figure S5). Asterisks indicate a P-value < 0.01 relative to the DMSO control. (B) The structure of nelfinavir (1). (C) The proteolytic effect of S. pyogenes membranes treated with nelfinavir on MBP-SagA, relative to a DMSO-treated control.
Nelfinavir was evaluated in the membrane proteolysis assay to determine if the observed loss of β-hemolysis was due to inhibition of the proteolytic processing step of SLS maturation. As expected, treatment with nelfinavir greatly reduced the proteolytic activity toward SagA contained within S. pyogenes membranes (Figure 3C, the membrane fraction used in this experiment was prepared on a different day than the fraction used in Figure 2 and displayed much higher activity). Evidence that nelfinavir was not drastically perturbing normal cellular function came from the observation that the growth rates of S. pyogenes treated with nelfinavir were identical to the DMSO control (Figure S5). Additionally, minimum inhibitory concentration (MIC) testing revealed no growth inhibition up to the highest concentration tested (64 µM) for a range of bacterial species (Table S1). Transmission electron microscopy (TEM) was used to examine the morphology of S. pyogenes treated with nelfinavir, with no apparent changes compared to the control sample (Figure S6). The transcription levels of a panel of virulence factor genes, as assessed by qRT-PCR, were also not significantly impacted (Table S2). Notably, the levels of sagA and sagB expression were unchanged. These data indicate that nelfinavir inhibited peptide processing directly, rather than through transcriptional regulation or by significantly perturbing other cellular processes.
Structure-activity relationships
A series of nelfinavir analogs was synthesized to gain a better understanding of the structure-activity relationships (SAR) for inhibition of SLS maturation. If nelfinavir were to interact with the SLS leader peptidase in a manner analogous to HIV protease, the secondary hydroxyl group would function as a tetrahedral intermediate mimic, as is the case for many aspartyl and metalloprotease inhibitors (Figure S7). Synthetic routes to nelfinavir have been thoroughly explored and a route that allowed facile derivatization was adapted for this study (Scheme 1).37 The efficacy of each analog at 25 µM was evaluated using the hemolysis assay (Table 1 and Table S3).
Scheme 1.

Synthesis of nelfinavir. a, i-BuOCOCl, Et3N, THF; CH2N2, Et2O (62%); b, HCl, Et2O; c, NaBH4, THF (62% over two steps); d, KOH, EtOH (91%); e, 6, KOH, IPA, 80 °C (84%); f, 3-hydroxy-2-methylbenzoic acid, EDC, HOBt, THF (66%).
Table 1.
Relative activity of nelfinavir analogs for SLS inhibition.
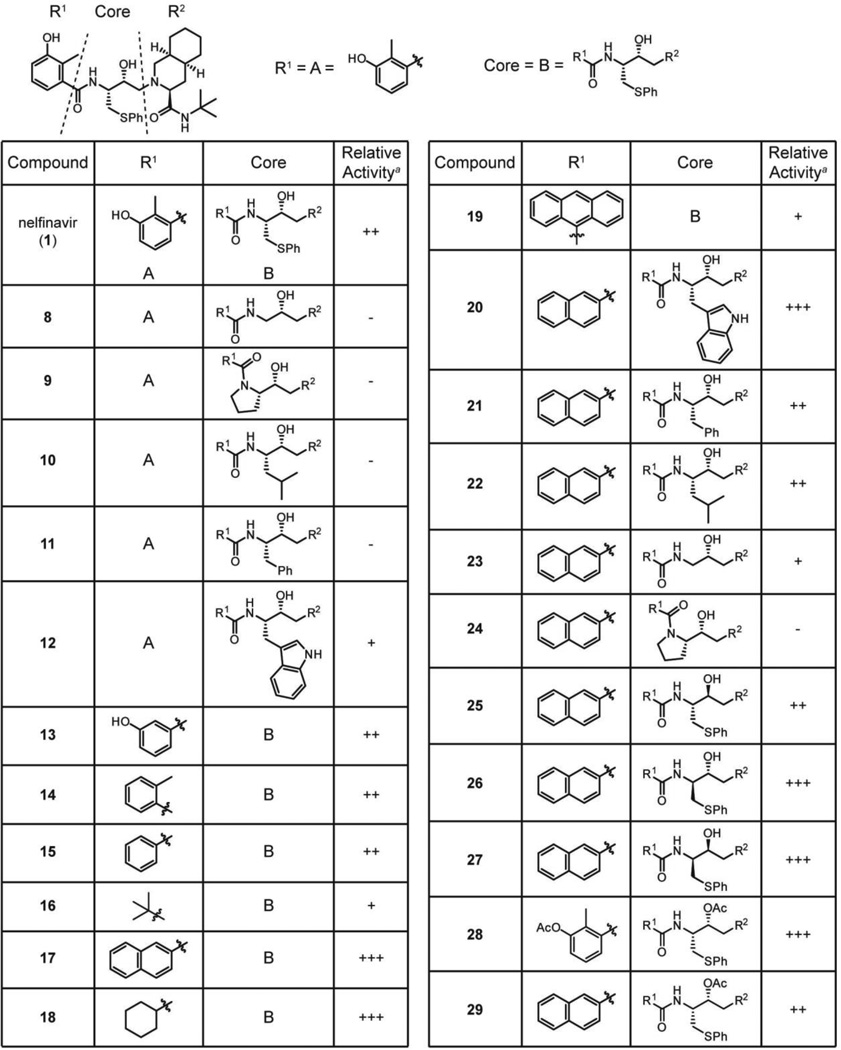 |
The activity of each analog is reported qualitatively relative to nelfinavir due to variability in commercial blood lots and extraction effectiveness. Inhibitory activity that is >3-fold than nelfinavir is designated as (+++); activity that is equal to nelfinavir is (++); detectable activity that is <3-fold than nelfinavir is (+); non-detectable activity is denoted (−).
Nelfinavir has peptidomimetic features, including an S-phenyl-ethyl group intended to replace the side chain of phenylalanine commonly found in the P1 position of the HIV protease substrate. Since the putative P1 position of SagA is a much smaller glycine residue (Figure 1B, Gly23), we surmised that smaller substituents at this location on nelfinavir might improve the observed anti-SLS activity. Instead, removal of the side chain (8) abolished detectable activity. It is possible that the cleavage site is closer to the N-terminus, however, and that Pro21 or an aliphatic residue preceding it resides in the P1 position. Accordingly, analogs mimicking these amino acids (9, 10) were synthesized, but these compounds did not exhibit detectable inhibitory activity. Even retention of the phenyl ring in a phenylalanine mimic (11) was insufficient to maintain activity. Conversely, a tryptophan mimic (12) was inhibitory, albeit weakly, suggesting that a relatively large group in that position may be necessary for activity.
The lack of detectable activity with the series of P1 position analogs prevented the rigorous establishment of SAR. Modifications to other portions of the molecule were prepared in order to enhance the inhibitory activity to address this pitfall. As installation of the benzamide is the final step of the synthesis (Scheme 1), preparation of analogs at this location was convenient. Initial analogs revealed that neither the hydroxyl nor the methyl groups (13–15) were important for activity but that the ring itself was necessary (16). Replacement of the ring with bulkier naphthyl and cyclohexyl groups (17, 18) provided >5-fold increases in potency, indicating that this part of the pharmacophore may reside in a hydrophobic pocket not fully occupied by the single planar ring. However, increasing the size of this moiety to an anthracene (19) greatly reduced activity. In general, electron-deficient rings had improved activity relative to electron-rich rings (Table S3), although hydrophobicity appeared to be a more significant contributing factor towards potency.
Given the enhanced activity of the naphthylamide-bearing compound (17), this group was incorporated into the collection of P1 position analogs (Table 1; Table S3), increasing the activity of these analogs to detectable levels. The tryptophan mimic with the naphthylamide (20) displayed considerably increased potency relative to the 3-hydroxy-2-methylbenzamide analog (12). Within the naphthylamide series, the phenylalanine and leucine mimics (21, 22) were equivalently active to nelfinavir, while the glycine mimic (23) displayed weak SLS-inhibitory activity. The proline mimic (24) remained devoid of activity, possibly due to structural perturbations enforced by the ring. These data support a trend of larger substituents imparting higher activity that was foreshadowed by the initial SAR.
To further probe the pseudo-P1 position of nelfinavir, the stereochemical configurations of both the side chain and the secondary hydroxyl group were varied. Surprisingly, inverting stereocenters with this series of analogs (25–27) did not result in large changes in activity. Many of the derivatives were still highly potent, suggesting that nelfinavir may not inhibit SagE through mimicking the proteolytic tetrahedral intermediate (Figure S7). This is further supported by the retention of activity in acetylated derivatives (28, 29). Overall, the sum of the SAR analysis resulted in the development of an analog with significantly improved potency (17, IC50 = 1 µM). Additionally, the activity trends led us to believe that nelfinavir might serve as an inhibitor for the protease in other TOMM natural product biosynthetic clusters in which the precursor peptides do not always contain a predicted Gly-Gly cleavage motif (Figure 1B).
Biosynthesis inhibition of other TOMMs
SLS is the best-studied member of a group of related cytolysins produced by a number of bacteria, including pathogens such as L. monocytogenes and C. botulinum.11, 20 The SLS-like biosynthetic gene clusters in these strains are highly similar to that of S. pyogenes and include orthologs of sagE (Figure 1A, Figure S8). The SLS-like toxin from L. monocytogenes, listeriolysin S (LLS), is known to be expressed during oxidative stress; therefore, a strain with LLS under the control of a constitutive promoter was used in the blood lysis assay to determine if nelfinavir could also inhibit LLS production.49 The strain was deficient in production of an unrelated cytolysin, listeriolysin O (LLO), to ensure any hemolysis observed derived from LLS production. When treated with nelfinavir, this strain produced significantly less LLS (Figure 4A). The LLO−/LLS− strain of L. monocytogenes was included as a negative control to demonstrate the observed hemolysis was indeed LLS-dependent (Figure 4A).
Figure 4.

Inhibition of the biosynthesis of other TOMMs by nelfinavir. (A) Lytic activity of extracts from an LLS-producing strain (LLS+) of L. monocytogenes treated with nelfinavir relative to a DMSO control (n = 3). Extracts from a separate strain with llsA deleted (LLS−) were also used as a control. (B) Lytic activity of extracts from a CLS-producing strain of C. sporogenes treated with nelfinavir relative to a DMSO control (n = 3). (C) Production of PZN by B. amyloliquefaciens treated with nelfinavir relative to a DMSO control (n = 4). Nelfinavir was used at 50 µM in all cases. Excluding LLS- negative control, P-values < 0.05 were obtained for all nelfinavir-treated samples relative to the corresponding DMSO positive controls.
Similar to SLS and LLS, clostridiolysin S (CLS) is a hemolysin from C. botulinum as well as from certain strains of C. sporogenes, which are nearly identical to C. botulinum but do not produce botulinum toxin.21 The presence of the CLS cluster in an unsequenced strain of C. sporogenes (ATCC 19404) known to be hemolytic on blood agar was confirmed by PCR amplification of closC and closD (the cyclodehydratase genes). When C. sporogenes was grown in the presence of nelfinavir, the production of CLS was significantly reduced (Figure 4B). The inhibition of not only SLS but also LLS and CLS production suggests that nelfinavir would likely inhibit the production of additional TOMM cytolysins.
TOMM biosynthetic gene clusters are wide-spread among bacteria and archaea, and the products of the vast majority of these clusters have not been structurally or functional characterized.6 A bioinformatic analysis of TOMM clusters revealed that 22% (328 out of 1520 as of October 2014) contained a CPBP family member within the cluster, many of which are predicted or known to have non-cytolytic products. The presence of a CPBP member in these clusters led us to postulate that nelfinavir would also inhibit TOMM production in these cases. Plantazolicin (PZN) is a TOMM produced by Bacillus amyloliquefaciens FZB42 with highly selective antibacterial activity against Bacillus anthracis and has a sagE-like gene (bamE) in its biosynthetic gene cluster (Figure 1A, Figure S8).50 The structure of PZN has been determined; thus the precise cleavage site is known (after Ala27, Figure 1B), although no biochemical studies have directly linked BamE to leader peptide cleavage. Methanolic surface extracts from B. amyloliquefaciens were analyzed by liquid chromatography-mass spectrometry and the nelfinavir-treated cultures were found to produce significantly less PZN than a DMSO control (Figure 4C). Unlike the cytolysins, PZN can be readily observed and quantified by mass spectrometry, which permitted confirmation of the nelfinavir dose-dependent inhibition (Figure S9). The nelfinavir-dependent inhibition in divergent organisms of additional TOMM cytolysins, as well as a functionally distinct antimicrobial TOMM, not only supports the assignment of the CPBP protein being responsible for leader peptide removal during maturation, but also suggests that nelfinavir, and analogs thereof, could be generally useful for inhibition of TOMM production in a large number of hitherto uncharacterized clusters.
Discussion
In this work, the FDA-approved HIV protease inhibitor nelfinavir was repurposed as the first small molecule inhibitor of SLS production in S. pyogenes, displaying low micromolar activity. Nelfinavir was identified as a lead compound by leveraging the extensive basic and clinical research data accumulated on the effects of the drug. Lipodystrophy, a known side effect of nelfinavir and several other HIV protease inhibitors, had been previously linked to the off-target inhibition of the CaaX protease ZMPSTE24. We surmised that the HIV protease inhibitors would also inhibit SagE due to its homology with CaaX proteases, allowing us to rapidly identify a lead compound for the inhibition of SLS production. This strategy for lead identification negated the need for high-throughput screening or for a crystal structure of the target for in silico and structure-based design. Utilizing a drug with synthetic routes that have been thoroughly explored also greatly accelerated the creation of analogs for SAR efforts that yielded compound 17, with an improved IC50 value of 1 µM.
Nelfinavir and related compounds are inhibitors of SLS biosynthesis, most likely through inhibition of proteolytic cleavage of the protoxin by the CPBP family member SagE. Although in vitro reconstitution was unsuccessful, the necessity for SagE during SLS production in the multi-plasmid expression system in E. coli provides considerable evidence for its role in proteolytic processing. Like many CPBP members, SagE is commonly referred to as an immunity protein in the literature,14, 15 but inhibition by nelfinavir did not have any effect on the growth of S. pyogenes, providing evidence that SagE is not involved in self-immunity. Alternatively, compensatory mutations that abolish SLS production may arise when SagE is inactivated, as has been previously suggested.15 The original annotation of SagE as an immunity protein stems from its similarity to PlnP from Lactobacillus plantarum. PlnP and several related proteins are found downstream of bacteriocin structural genes in L. plantarum and have been shown to provide immunity to the antibacterial effect of the respective bacteriocins.35 Yet, unlike these bacteriocins, SLS has not been demonstrated to possess any antibacterial activity against intact S. pyogenes cells. A large buildup of intracellular SLS might result in toxicity, but this would be expected to affect any S. pyogenes strains in which the transport machinery is inactivated as well, which has not been observed.14 Furthermore, treatment of SagA with the cyclization machinery (SagBCD) in vitro results in a lytic entity without cleavage of the leader peptide, indicating that while proteolysis is required for cellular export, it is unnecessary for lytic activity.20 These observations lead us to conclude that the principal function of SagE is to proteolytically mature SLS.
In addition to experimentally supporting a biochemical role for SagE, we have also addressed the mechanism of proteolysis and the probable inhibition by nelfinavir. CPBP family members have been postulated to function through a zinc metalloprotease mechanism based on the presence of two glutamates, two histidines, and an asparagine residue that are conserved across the family (Figure S8).33 Mutation of these residues in the eukaryotic type II CaaX protease Ras-converting enzyme (Rce1) has also demonstrated that these residues are critical for activity.33 We found that Glu131 and Glu132 were critical for the activity of SagE. Thus, it was initially unexpected that proteolysis was inhibited by pepstatin, a general aspartyl protease inhibitor, but not by bestatin, a metalloprotease inhibitor (Figure 2B). However, a recent report detailing the crystal structure of Rce1 from Methanococcus maripaludis provides compelling evidence that this family of proteases actually functions through a novel glutamate-dependent mechanism (Figure S10).47 The authors hypothesized that a glutamate residue extending into the active site is responsible for deprotonating a water molecule, activating it for nucleophilic attack. Given the similarities between this proposed mechanism and the mechanism of aspartyl proteases, it is perhaps unsurprising that a CaaX protease homolog would be inhibited by aspartyl protease inhibitors, including nelfinavir.
In addition to increasing the potency of inhibition with compound 17, our synthetic effort also yielded information on how nelfinavir may interact with SagE. The SAR analysis revealed that a rather large side chain in the pseudo P1 position of the structure was required for potent activity, with the original S-phenyl-ethyl group displaying the greatest inhibition. This result was not anticipated, given that the P1 residue in the SagA substrate is putatively glycine. One possible explanation for this discrepancy is that the catalytic site architecture of SagE is highly conserved with the type II CaaX proteases, which normally cleave substrates with a prenylated cysteine in the P1 position. In this case, nelfinavir would be an ideal fit for the active site, as the core of the molecule closely resembles a cysteine with a hydrophobic group appended. An alternative explanation is that nelfinavir does not bind in the active site in the expected fashion (with the secondary hydroxyl interacting with the catalytic residues, Figure S7) or does not bind in the active site at all. These possibilities are supported by the potent activity of several analogs with different stereochemical configurations, as these molecules would likely be forced into different conformations that do not allow the same favorable interactions with active site residues. In this scenario, binding of nelfinavir to the membrane protease may be driven by hydrophobic interactions. This explanation would also account for the attenuated activity of saquinavir, which is nearly identical to nelfinavir except for the presence of a more hydrophilic group at the benzamide position (Figure S4, XLogP3 values from PubChem for nelfinavir and saquinavir are 5.7 and 4.2, respectively). Comparisons to the other HIV protease inhibitors do not yield much additional information due to their low similarity to nelfinavir, as reflected in the Tanimoto similarity coefficients (Table S4).
Unequivocal confirmation of the bacterial target of nelfinavir (and analogs) was unfortunately not possible in the present study, which can be attributed to the technical challenges inherent to the study of integral membrane proteins. Nelfinavir also likely interacts with multiple targets in S. pyogenes, as the drug is known to have multiple off-target effects in humans.43 A significant body of work in mammalian cell lines has demonstrated that nelfinavir displays promiscuous activity, such as interruption of Akt signaling and inhibition of the proteasome.51 Target promiscuity likely also exists in bacteria, so possible interactions of nelfinavir with additional targets cannot be ruled out. Thus, nelfinavir may be inhibiting additional participants that indirectly result in the inhibition of SLS production in a mechanism unrelated to proteolysis. Further targets of nelfinavir may also exist that do not result in observable phenotypes. However, nelfinavir also inhibited the biosynthesis of other natural products that include a CPBP family member in the gene cluster (i.e. LLS, CLS, and PZN). The fact that this inhibition occurred in a range of disparate bacterial species decreases the probability that another protease is responsible and provides substantial, albeit indirect, support that SagE is the primary target of nelfinavir in S. pyogenes. Finally, the HIV protease inhibitors found to inhibit SLS production parallel those capable of inhibiting the human CaaX protease ZMPSTE24 (Figure S4),44, 45 providing additional evidence that nelfinavir and its analogs inhibit SLS production by blocking the action of SagE.
Conclusion
Despite their prevalence, prokaryotic members of the CPBP family have not yet been thoroughly investigated. Many of the family members are incorrectly annotated or have a predicted function based solely on distant homology. The discovery of nelfinavir as an inhibitor of CPBPs has provided evidence that SagE functions as a protease and will aid in the assignment of functions to other family members, including other human pathogens such as S. aureus. Additionally, nelfinavir is the first reported inhibitor of the production of SLS and related toxins. Nelfinavir and improved analogs will provide a new tool to investigate toxin function without the need to create genetic deletions while also allowing for temporal control over toxin production. Reversible control of SLS production with nelfinavir analogs will also help to clarify the precise contribution of SLS to virulence in in vivo models of infection and may open the door to the development of virulence-targeting strategies for the control of S. pyogenes infections. Finally, the chemical knockdown effect of nelfinavir can be utilized for the discovery of natural products from the 22% of TOMM gene clusters that contain a CPBP family member, potentially accelerating the structural and functional characterization of these compounds.
Supplementary Material
Acknowledgements
We would like to thank P. Cotter (Teagasc, Moorepark Food Research Center, Fermoy, Co. Cork, Ireland) for the gift of engineered L. monocytogenes strains. We would like to thank B. White (University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA) for the use of his anaerobic chamber. All FDA-approved HIV protease inhibitors were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS (NIAID). Electron microscopy was carried out in part in the Frederick Seitz Materials Research Laboratory Central Research Facilities, University of Illinois. This research was supported in part by the NIH Director's New Innovator Award Program (DP2 OD008463, to D.A.M.). T.M. and C.D.D. were supported in part by fellowships from the Department of Chemistry at the University of Illinois at Urbana-Champaign and the NIH Chemical Biology Interface Training Program (T32 GM070421). E.M.M. was formerly funded by the Irish Research Council for Science, Engineering and Technology through University College Cork, Ireland, and received travel-related funding from the UK Society for General Microbiology.
Footnotes
Supporting Information Available: This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, Cotter PD, Craik DJ, Dawson M, Dittmann E, Donadio S, Dorrestein PC, Entian KD, Fischbach MA, Garavelli JS, Goransson U, Gruber CW, Haft DH, Hemscheidt TK, Hertweck C, Hill C, Horswill AR, Jaspars M, Kelly WL, Klinman JP, Kuipers OP, Link AJ, Liu W, Marahiel MA, Mitchell DA, Moll GN, Moore BS, Muller R, Nair SK, Nes IF, Norris GE, Olivera BM, Onaka H, Patchett ML, Piel J, Reaney MJ, Rebuffat S, Ross RP, Sahl HG, Schmidt EW, Selsted ME, Severinov K, Shen B, Sivonen K, Smith L, Stein T, Sussmuth RD, Tagg JR, Tang GL, Truman AW, Vederas JC, Walsh CT, Walton JD, Wenzel SC, Willey JM, van der Donk WA. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013;30:108–160. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bagley MC, Dale JW, Merritt EA, Xiong X. Thiopeptide antibiotics. Chem. Rev. 2005;105:685–714. doi: 10.1021/cr0300441. [DOI] [PubMed] [Google Scholar]
- 3.Chatterjee C, Paul M, Xie L, van der Donk WA. Biosynthesis and mode of action of lantibiotics. Chem. Rev. 2005;105:633–684. doi: 10.1021/cr030105v. [DOI] [PubMed] [Google Scholar]
- 4.Maksimov MO, Pan SJ, James Link A. Lasso peptides: structure, function, biosynthesis, and engineering. Nat. Prod. Rep. 2012;29:996–1006. doi: 10.1039/c2np20070h. [DOI] [PubMed] [Google Scholar]
- 5.Sivonen K, Leikoski N, Fewer DP, Jokela J. Cyanobactins-ribosomal cyclic peptides produced by cyanobacteria. Appl. Microbiol. Biot. 2010;86:1213–1225. doi: 10.1007/s00253-010-2482-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Melby JO, Nard NJ, Mitchell DA. Thiazole/oxazole-modified microcins: complex natural products from ribosomal templates. Curr. Opin. Chem. Biol. 2011;15:369–378. doi: 10.1016/j.cbpa.2011.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oman TJ, van der Donk WA. Follow the leader: the use of leader peptides to guide natural product biosynthesis. Nat. Chem. Biol. 2010;6:9–18. doi: 10.1038/nchembio.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dunbar KL, Melby JO, Mitchell DA. YcaO domains use ATP to activate amide backbones during peptide cyclodehydrations. Nat. Chem. Biol. 2012;8:569–575. doi: 10.1038/nchembio.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunbar KL, Mitchell DA. Insights into the mechanism of peptide cyclodehydrations achieved through the chemoenzymatic generation of amide derivatives. J. Am. Chem. Soc. 2013;135:8692–8701. doi: 10.1021/ja4029507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melby JO, Li X, Mitchell DA. Orchestration of enzymatic processing by thiazole/oxazole-modified microcin dehydrogenases. Biochemistry. 2014;53:413–422. doi: 10.1021/bi401529y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molloy EM, Cotter PD, Hill C, Mitchell DA, Ross RP. Streptolysin S-like virulence factors: the continuing sagA. Nat. Rev. Microbiol. 2011;9:670–681. doi: 10.1038/nrmicro2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cunningham MW. Pathogenesis of group A streptococcal infections. Clin. Microbiol. Rev. 2000;13:470–511. doi: 10.1128/cmr.13.3.470-511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect. Dis. 2005;5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 14.Nizet V, Beall B, Bast DJ, Datta V, Kilburn L, Low DE, De Azavedo JC. Genetic locus for streptolysin S production by group A streptococcus. Infect. Immun. 2000;68:4245–4254. doi: 10.1128/iai.68.7.4245-4254.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Datta V, Myskowski SM, Kwinn LA, Chiem DN, Varki N, Kansal RG, Kotb M, Nizet V. Mutational analysis of the group A streptococcal operon encoding streptolysin S and its virulence role in invasive infection. Mol. Microbiol. 2005;56:681–695. doi: 10.1111/j.1365-2958.2005.04583.x. [DOI] [PubMed] [Google Scholar]
- 16.Betschel SD, Borgia SM, Barg NL, Low DE, De Azavedo JC. Reduced virulence of group A streptococcal Tn916 mutants that do not produce streptolysin S. Infect. Immun. 1998;66:1671–1679. doi: 10.1128/iai.66.4.1671-1679.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fontaine MC, Lee JJ, Kehoe MA. Combined contributions of streptolysin O and streptolysin S to virulence of serotype M5 Streptococcus pyogenes strain Manfredo. Infect. Immun. 2003;71:3857–3865. doi: 10.1128/IAI.71.7.3857-3865.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.James L, McFarland RB. An epidemic of pharyngitis due to a nonhemolytic group A streptococcus at lowry air force base. N. Engl. J. Med. 1971;284:750–752. doi: 10.1056/NEJM197104082841403. [DOI] [PubMed] [Google Scholar]
- 19.Yoshino M, Murayama SY, Sunaoshi K, Wajima T, Takahashi M, Masaki J, Kurokawa I, Ubukata K. Nonhemolytic Streptococcus pyogenes isolates that lack large regions of the sag operon mediating streptolysin S production. J. Clin. Microbiol. 2010;48:635–638. doi: 10.1128/JCM.01362-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee SW, Mitchell DA, Markley AL, Hensler ME, Gonzalez D, Wohlrab A, Dorrestein PC, Nizet V, Dixon JE. Discovery of a widely distributed toxin biosynthetic gene cluster. Proc. Natl. Acad. Sci. U.S.A. 2008;105:5879–5884. doi: 10.1073/pnas.0801338105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gonzalez DJ, Lee SW, Hensler ME, Markley AL, Dahesh S, Mitchell DA, Bandeira N, Nizet V, Dixon JE, Dorrestein PC. Clostridiolysin S, a post-translationally modified biotoxin from Clostridium botulinum. J. Biol. Chem. 2010;285:28220–28228. doi: 10.1074/jbc.M110.118554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bernheimer AW. Physical Behavior of Streptolysin S. J. Bacteriol. 1967;93:2024–2025. doi: 10.1128/jb.93.6.2024-2025.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jack RW, Tagg JR, Ray B. Bacteriocins of gram-positive bacteria. Microbiol. Rev. 1995;59:171–200. doi: 10.1128/mr.59.2.171-200.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Todd EW. The differentiation of two distinct serological varieties of streptolysin, streptolysin O and streptolysin S. J. Pathol. Bacteriol. 1938;47:423–445. [Google Scholar]
- 25.Marmorek A. Le streptocoque et le sérum antistreptococcique. Ann. Inst. Pasteur. 1895;9:593–620. [Google Scholar]
- 26.Rasko DA, Sperandio V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 2010;9:117–128. doi: 10.1038/nrd3013. [DOI] [PubMed] [Google Scholar]
- 27.Cegelski L, Marshall GR, Eldridge GR, Hultgren SJ. The biology and future prospects of antivirulence therapies. Nat. Rev. Microbiol. 2008;6:17–27. doi: 10.1038/nrmicro1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baruch M, Belotserkovsky I, Hertzog BB, Ravins M, Dov E, McIver KS, Le Breton YS, Zhou Y, Cheng CY, Hanski E. An extracellular bacterial pathogen modulates host metabolism to regulate its own sensing and proliferation. Cell. 2014;156:97–108. doi: 10.1016/j.cell.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pei J, Grishin NV. Type II CAAX prenyl endopeptidases belong to a novel superfamily of putative membrane-bound metalloproteases. Trends Biochem. Sci. 2001;26:275–277. doi: 10.1016/s0968-0004(01)01813-8. [DOI] [PubMed] [Google Scholar]
- 30.Pei J, Mitchell DA, Dixon JE, Grishin NV. Expansion of type II CAAX proteases reveals evolutionary origin of gamma-secretase subunit APH-1. J. Mol. Biol. 2011;410:18–26. doi: 10.1016/j.jmb.2011.04.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bergo MO, Ambroziak P, Gregory C, George A, Otto JC, Kim E, Nagase H, Casey PJ, Balmain A, Young SG. Absence of the CAAX endoprotease Rce1: effects on cell growth and transformation. Mol. Cell. Biol. 2002;22:171–181. doi: 10.1128/MCB.22.1.171-181.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bergo MO, Wahlstrom AM, Fong LG, Young SG. Genetic analyses of the role of RCE1 in RAS membrane association and transformation. Methods Enzymol. 2008;438:367–389. doi: 10.1016/S0076-6879(07)38026-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Plummer LJ, Hildebrandt ER, Porter SB, Rogers VA, McCracken J, Schmidt WK. Mutational analysis of the ras converting enzyme reveals a requirement for glutamate and histidine residues. J. Biol. Chem. 2006;281:4596–4605. doi: 10.1074/jbc.M506284200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dolence JM, Steward LE, Dolence EK, Wong DH, Poulter CD. Studies with recombinant Saccharomyces cerevisiae CaaX prenyl protease Rce1p. Biochemistry. 2000;39:4096–4104. doi: 10.1021/bi9923611. [DOI] [PubMed] [Google Scholar]
- 35.Kjos M, Snipen L, Salehian Z, Nes IF, Diep DB. The Abi Proteins and Their Involvement in Bacteriocin Self-Immunity. J. Bacteriol. 2010;192:2068–2076. doi: 10.1128/JB.01553-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clayton EM, Hill C, Cotter PD, Ross RP. Real-time PCR assay to differentiate Listeriolysin S-positive and -negative strains of Listeria monocytogenes. Appl. Environ. Microb. 2011;77:163–171. doi: 10.1128/AEM.01673-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaldor SW, Kalish VJ, Davies JF, 2nd, Shetty BV, Fritz JE, Appelt K, Burgess JA, Campanale KM, Chirgadze NY, Clawson DK, Dressman BA, Hatch SD, Khalil DA, Kosa MB, Lubbehusen PP, Muesing MA, Patick AK, Reich SH, Su KS, Tatlock JH. Viracept (nelfinavir mesylate, AG1343): a potent, orally bioavailable inhibitor of HIV-1 protease. J. Med. Chem. 1997;40:3979–3985. doi: 10.1021/jm9704098. [DOI] [PubMed] [Google Scholar]
- 38.Albizati KF, Babu S, Birchler A, Busse JK, Fugett M, Grubbs A, Haddach A, Pagan M, Potts B, Remarchuk T, Rieger D, Rodriguez R, Shanley J, Szendroi R, Tibbetts T, Whitten K, Borer BC. A synthesis of the HIV-protease inhibitor nelfinavir from D-tartaric acid. Tetrahedron Lett. 2001;42:6481–6485. [Google Scholar]
- 39.Ma D, Zou B, Zhu W, Xu HD. A short synthesis of the HIV-protease inhibitor nelfinavir via a diastereoselective addition of ammonia to the alpha, beta-unsaturated sulfoxide derived from (R)-glyceraldehyde acetonide. Tetrahedron Lett. 2002;43:8511–8513. [Google Scholar]
- 40.Viklund H, Bernsel A, Skwark M, Elofsson A. SPOCTOPUS: a combined predictor of signal peptides and membrane protein topology. Bioinformatics. 2008;24:2928–2929. doi: 10.1093/bioinformatics/btn550. [DOI] [PubMed] [Google Scholar]
- 41.Markley AL, Jensen ER, Lee SW. An Escherichia coli-based bioengineering strategy to study streptolysin S biosynthesis. Anal. Biochem. 2012;420:191–193. doi: 10.1016/j.ab.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alouf JE. Streptococcal toxins (streptolysin O, streptolysin S, erythrogenic toxin) Pharmacol. Ther. 1980;11:661–717. doi: 10.1016/0163-7258(80)90045-5. [DOI] [PubMed] [Google Scholar]
- 43.Caron M, Auclair M, Sterlingot H, Kornprobst M, Capeau J. Some HIV protease inhibitors alter lamin A/C maturation and stability, SREBP-1 nuclear localization and adipocyte differentiation. Aids. 2003;17:2437–2444. doi: 10.1097/00002030-200311210-00005. [DOI] [PubMed] [Google Scholar]
- 44.Coffinier C, Hudon SE, Farber EA, Chang SY, Hrycyna CA, Young SG, Fong LG. HIV protease inhibitors block the zinc metalloproteinase ZMPSTE24 and lead to an accumulation of prelamin A in cells. Proc. Natl. Acad. Sci. U.S.A. 2007;104:13432–13437. doi: 10.1073/pnas.0704212104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coffinier C, Hudon SE, Lee R, Farber EA, Nobumori C, Miner JH, Andres DA, Spielmann HP, Hrycyna CA, Fong LG, Young SG. A potent HIV protease inhibitor, darunavir, does not inhibit ZMPSTE24 or lead to an accumulation of farnesyl-prelamin A in cells. J. Biol. Chem. 2008;283:9797–9804. doi: 10.1074/jbc.M709629200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quigley A, Dong YY, Pike AC, Dong L, Shrestha L, Berridge G, Stansfeld PJ, Sansom MS, Edwards AM, Bountra C, von Delft F, Bullock AN, Burgess-Brown NA, Carpenter EP. The structural basis of ZMPSTE24-dependent laminopathies. Science. 2013;339:1604–1607. doi: 10.1126/science.1231513. [DOI] [PubMed] [Google Scholar]
- 47.Manolaridis I, Kulkarni K, Dodd RB, Ogasawara S, Zhang Z, Bineva G, O'Reilly N, Hanrahan SJ, Thompson AJ, Cronin N, Iwata S, Barford D. Mechanism of farnesylated CAAX protein processing by the intramembrane protease Rce1. Nature. 2013;504:301–305. doi: 10.1038/nature12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bernheimer AW. Formation of a bacterial toxin (streptolysin S) by resting cells. J. Exp. Med. 1949;90:373–392. doi: 10.1084/jem.90.5.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cotter PD, Draper LA, Lawton EM, Daly KM, Groeger DS, Casey PG, Ross RP, Hill C. Listeriolysin S, a novel peptide haemolysin associated with a subset of lineage I Listeria monocytogenes. PLoS Pathog. 2008;4:e1000144. doi: 10.1371/journal.ppat.1000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Molohon KJ, Melby JO, Lee J, Evans BS, Dunbar KL, Bumpus SB, Kelleher NL, Mitchell DA. Structure determination and interception of biosynthetic intermediates for the plantazolicin class of highly discriminating antibiotics. ACS Chem. Biol. 2011;6:1307–1313. doi: 10.1021/cb200339d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gantt S, Casper C, Ambinder RF. Insights into the broad cellular effects of nelfinavir and the HIV protease inhibitors supporting their role in cancer treatment and prevention. Curr. Opin. Oncol. 2013;25:495–502. doi: 10.1097/CCO.0b013e328363dfee. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.