

Abstract
Abstract
The literature on arterial mechanics is extensive, but far less is known about mechanisms controlling mechanical properties of veins. We use here a multi-scale approach to identify subcellular sources of venous stiffness. Portal vein tissue displays a severalfold decrease in passive stiffness compared to aortic tissues. The α-adrenergic agonist phenylephrine (PE) increased tissue stress and stiffness, both attenuated by cytochalasin D (CytoD) and PP2, inhibitors of actin polymerization and Src activity, respectively. We quantify, for the first time, cortical cellular stiffness in freshly isolated contractile vascular smooth muscle cells using magnetic microneedle technology. Cortical stiffness is significantly increased by PE and CytoD inhibits this increase but, surprisingly, PP2 does not. No detectable change in focal adhesion size, measured by immunofluorescence of FAK and zyxin, accompanies the PE-induced changes in cortical stiffness. Probing with phospho-specific antibodies confirmed activation of FAK/Src and ERK pathways and caldesmon phosphorylation. Thus, venous tissue stiffness is regulated both at the level of the smooth muscle cell cortex, via cortical actin polymerization, and by downstream smooth muscle effectors of Src/ERK signalling pathways. These findings identify novel potential molecular targets for the modulation of venous capacitance and venous return in health and disease.
Key points
Most cardiovascular research focuses on arterial mechanisms of disease, largely ignoring venous mechanisms.
Here we examine ex vivo venous stiffness, spanning tissue to molecular levels, using biomechanics and magnetic microneedle technology, and show for the first time that venous stiffness is regulated by a molecular actin switch within the vascular smooth muscle cell in the wall of the vein.
This switch connects the contractile apparatus within the cell to adhesion structures and facilitates stiffening of the vessel wall, regulating blood flow return to the heart.
These studies also demonstrate that passive stiffness, the component of total stiffness not attributable to vascular smooth muscle activation, is severalfold lower in venous tissue than in arterial tissue.
We show here that the activity of the smooth muscle cells plays a dominant role in determining total venous stiffness and regulating venous return.
Introduction
In studying the interplay between the heart and the circulatory system, most investigators limit their focus to the left side of the heart and the arterial tree. Accordingly, the venous circulation is considerably under-studied, and its influence on the cardiovascular circuit as a whole is underappreciated. The venous system comprises the major reservoir for blood, holding nearly 70% of the total blood volume in the circulatory system (Guyton & Hall, 2006). The vasoactivity of the veins regulates venous return and the preload on the heart and thereby determines the volume of blood that is pumped into the arterial tree (Rothe, 1983; Tyberg, 2002). As capacitive vessels, the compliance of the veins is essential to their function. Reflecting their specialized function, the veins possess microstructural composition and organization unlike that of arteries (Bohr et al. 1980). Changes in stiffness with age have been documented in veins and are thought to contribute to various disorders, including diastolic dysfunction by impairing venous capacitance, placing excess load on the heart (Safar & London, 1987; Gascho et al. 1989; Monahan et al. 2001; Edwards et al. 2003), and orthostatic hypotension by impairing return of blood to the heart. Overall, however, the mechanisms of regulation of stiffness in veins have received little consideration, and in particular little has been done to study the basic cellular mechanobiology and molecular mechanisms of regulation of venous stiffness (Brecher, 1969; Noordergraaf, 1978; Edwards et al. 2003; Wang et al. 2006).
In the present study, we employed a multi-scale approach to venous biomechanics and examined cytoskeletal regulation of venous cellular stiffness. For the first time, we show that the cortical actin cytoskeleton is an important regulator of venous stiffness and identify differences between the mechanisms regulating cellular stiffness in veins and those reported for arteries. Most notably, we demonstrate that smooth muscle cells exert substantial influence over total venous stiffness due to the low baseline tissue stiffness of passive vessel wall components. These properties have important consequences for venous compliance and venous return.
Methods
Ethical approval
All procedures were performed in accordance with protocols approved by the Boston University Institutional Animal Care and Use Committee (Permit Number: A3316-01). The animals were maintained according to the NIH Guide for the Care and Use of Laboratory Animals, and they were obtained and used in compliance with federal, state, and local laws. Tissue harvest was performed quickly after the animals were killed by isoflurane inhalation.
Portal vein tissue preparation
Ferret portal vein tissue was used because the ferret is recognized as the best non-primate model of the human (Fox & Marini, 2014). Male ferrets (Mustela putorius furo, 12–14 weeks old, n = 24) (Marshall Farms, North Rose, NY, USA) were killed by isoflurane inhalation. The portal vein was promptly excised and rinsed to remove blood inside or around the vessel that can harm the smooth muscle cells and induce contraction (Bulter et al. 1996). Then, the tissue was placed in an oxygenated (95% O2–5% CO2) physiological salt solution (PSS) (in mm: 120 NaCl, 5.9 KCl, 1.2 NaH2PO4, 25 NaHCO3, 11.5 dextrose, 1 CaCl2, and 1.4 MgCl2; pH = 7.4) and carefully dissected to remove connective tissue. Next, the portal vein was cut open along its major axis, and the endothelium removed by gentle abrasion with a rubber policeman.
For tissue stiffness experiments, longitudinal strips were cut (8 mm long × 2 mm wide) and then suspended in vitro in tissue baths containing oxygenated PSS at 37°C. For biochemical analyses, strips in the tissue baths were quick-frozen in a slurry of dry ice and liquid acetone containing 10 mm dithiothreitol and 10% trichloroacetic acid (TCA) (Driska et al. 1981; Bárány, 1996; Marganski et al. 2005).
Venous tissue stiffness measurement with high-frequency, low-amplitude stretches
For in vitro force and stiffness measurements, wire clasps were used to secure portal vein tissue strips on opposite ends to a fixed hook and to a computer-controlled motorized lever arm (Dual-Mode Lever Arm System, Model 300C, Aurora Scientific, Ontario, Canada) capable of setting tissue length while simultaneously measuring force. To minimize slippage and secure the attachment points of the compliant portal vein (PV) tissue to the setup, two small, T-shaped pieces of aluminum foil were wrapped and crimped around either end of the tissue, and mounting wires were threaded through holes that were then punched through the aluminum foil and the enclosed tissue (Brozovich & Morgan, 1989; Rhee & Brozovich, 2000). The strips were stretched uniaxially in the longitudinal direction, as vascular smooth muscle cells in the portal vein wall are oriented primarily in this direction.
Strips were stretched to optimal length LO (1.7 × slack length, or equivalently a strain ε = 70%) for 30 min to allow tensile force to stabilize at a steady state level. The tissue was then tested for viability through 15 min of contraction induced by depolarization with PSS in which 51 mm NaCl had been replaced by KCl, followed by return to PSS for 30 min. Then tissue strips were incubated with vehicle (DMSO, 1:1000), 4-amino-5-(4-chlorophenyl)-7-(dimethylethyl)pyrazolo[3,4-d]pyrimidine (PP2, 10 μm), or CytoD (500 nm), to prevent actin polymerization (Kim et al. 2010) for an additional 30 min. Stiffness measurements at optimal length LO were collected continuously during 10 min of stimulation with the α-agonist phenylephrine (PE, 10 μm) at a maximally effective concentration.
Tissue stiffness was measured by high-frequency, low-amplitude (HFLA) sinusoidal length perturbations as previously described (Brozovich & Morgan, 1989; Rhee & Brozovich, 2000; Saphirstein et al. 2013) (1% length oscillations at 40 Hz). This regime has previously been shown to measure total tissue stiffness without breaking actin–myosin crossbridges (Brozovich & Morgan, 1989). With this protocol, the out-of-phase viscous component of the stiffness is negligible, and the tissue’s elastic modulus, or material stiffness, is calculated as E, the ratio of the stress σ to the strain ε: E = (ΔF/A)/(ΔL/L0), where ΔF is the amplitude of the force response to the cyclic stretches, A is the cross-sectional area, ΔL is the amplitude of the cyclic stretches, and L0 is the optimal length. The cross-sectional area A of the strip is approximated as A = m/(ρ × Lslack), where m is the measured wet weight of the vascular strip, Lslack is the slack length, and ρ is the density of water, which approximates the density of biological tissues.
Cell isolation
Single vascular smooth muscle cells were enzymatically dissociated from ferret portal vein using a modified version of previously published protocols (DeFeo & Morgan, 1985; Lee et al. 2000). Ferret portal vein tissue (wet weight ∼60 mg) was dissected and cleaned, cut open along its longitudinal axis, and then cut into 16 small pieces (2 mm × 2 mm). These pieces were placed in a siliconized flask (Sigmacote, Sigma-Aldrich, Natick, MA, USA) containing oxygenated digestion solution. Digestion solution A contained 18 mg of CLS 2 collagenase (type II, 220 units mg−1; Worthington Biochemical, Lakewood, NJ, USA) and 14 mg of elastase (grade II, 5.41 units mg−1; Roche, Indianapolis, IN, USA) in 7.5 ml of Ca2+,Mg2+-free Hanks buffered salt solution (HBSS) (in mm: 137 NaCl, 5.4 KCl, 0.44 KH2PO4, 0.42 NaH2PO4, 4.17 NaHCO3, 5.55 glucose, and 10 Hepes) with 0.27 mg ml−1 trypsin inhibitor (type II soybean, T-9128, Sigma, Natick, MA, USA) and 0.075% bovine serum albumin (BSA). The tissue pieces were incubated in a shaking water bath at 35°C with 100% O2 for 40 min. The contents of the flask were then poured over a nylon mesh (Spectra/Mesh polypropylene filter, 500 μm pore size, 39% open area, thickness 610 μm; Spectrum Laboratories, Rancho Dominguez, CA, USA) and into a second siliconized flask. The tissue pieces caught on the filter were then rinsed with 10 ml Ca2+,Mg2+-free HBSS, and the rinse was also caught in the second flask. The contents of the second flask were poured onto coverslips (for cell immunofluorescence imaging studies) or 35 mm glass-bottom plates (for magnetic microneedle experiments to measure cortical stiffness) (MatTek, Ashland, MA, USA). This procedure was repeated for two additional batches. All cells were allowed to plate on ice for 1 h, during which Ca2+ and Mg2+ were added back gradually to avoid inducing cell shortening. Isolated cells on one coverslip were tested at room temperature to confirm viability by shortening in response to 51 mm KCl or 10 μm PE.
Bead coating and attachment
For studies using magnetic microneedles, an arginine-glycine-aspartic acid (RGD) peptide (GRGDNP; Enzo Life Sciences, Farmingdale, NY, USA) was covalently coupled to amine-modified superparamagnetic Dynabeads M-270 (2.8 μm; Invitrogen, Grand Island, NY, USA), as previously described, by first suspending the beads in phosphate buffered saline (PBS) at 10% solids, then activating them with 10% glutaraldehyde, reacting them with RGD peptide (0.5 mg ml−1), quenching and blocking them, and finally storing them with blocking agent and preservative in PBS at 4°C (Saphirstein et al. 2013).
Measurement of stiffness with magnetic microneedle technology
The magnetic microtool apparatus, modelled after a system designed and previously described by the Fabry group (Kollmannsberger & Fabry, 2007; Saphirstein et al. 2013), consists of a magnetic microneedle with a finely sharpened tip surrounded by a solenoid. This magnetic microneedle technology allows us to exert very low pulling forces on cell-adherent microbeads (∼50 pN at an operating distance of about 150 μm between the bead and the microneedle tip), as determined by calibration (previously described) which enables probing of the cortical cytoskeleton in the local vicinity of the bead (Saphirstein et al. 2013).
Prior to any measurements with the microneedle, the beads were incubated with the cells for 30 min to allow for their attachment to the cells. Cortical stiffness measurements were made at 37°C first from a set of unstimulated cells on each plate, then repeated on the same set of cells following stimulation with PE for 10 min.
Beads adherent to the cell periphery, distant from the nucleus and on the side of the cell closest to the microneedle tip, were selected for analysis. This minimized the effects of apical topography of the cells. Beads in close proximity to other beads were excluded from analysis. Cortical stiffness was measured using a priming protocol (previously described) consisting of a series of pulls and was calculated as the ratio of the force applied to the displacement of the cell-adherent bead on the final pull (Saphirstein et al. 2013).
Cortical stiffness measurements were performed on an Eclipse TE2000-E inverted microscope at ×20 magnification (NA 0.5) under bright-field illumination (Nikon Instruments, Melville, NY, USA). Video recordings of the bead and microneedle tip were captured with a CCD camera (CoolSNAP HQ2, Photometrics, Tucson, AZ, USA) at 10 frames s−1 with the Nikon NIS Elements imaging software (Nikon Instruments). Bead position was tracked with an intensity-weighted centre-of-mass algorithm using the MTrackJ plug-in for ImageJ (Meijering et al. 2012) and subsequently analysed in MATLAB (Mathworks, Natick, MA, USA).
Immunofluorescence imaging and focal adhesion area analysis
Freshly isolated PV vascular smooth muscle cells (VSMCs) were fixed and stained as previously described (Saphirstein et al. 2013). Cells were examined with an Eclipse TE2000-E fluorescence microscope (Nikon Instruments) equipped with a Nikon Plan Apochromat ×60 (NA 1.4) oil immersion objective and a charge-coupled device camera (CoolSNAP HQ2, Photometrics); NIS-Element Advanced Research software (Nikon Instruments) was used to capture images and for removal of out-of-focus fluorescence blur by deconvolution of Z-sections (Richardson–Lucy algorithm, constrained iterative-maximum likelihood estimation algorithm) as previously described (Min et al. 2012).
Focal adhesion areas (as identified by staining for zyxin and focal adhesion kinase (FAK)) were examined using NIS-Elements Advanced Research software. In a centre-section of the z-stack for a VSMC, punctae within 1 μm of the cell edge around the entire periphery of the cell were randomly selected for analysis.
Western blot analysis
Quick-frozen tissues were homogenized in buffer designed to preserve phosphoproteins and processed at 4°C as previously described (Marganski et al. 2005). Western blot densitometry was performed using an Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE, USA).
Reagents and antibodies
The α-adrenoceptor agonist phenylephrine was purchased from Sigma-Aldrich. PP2 was purchased from EMD Biosciences (La Jolla, CA, USA), and CytoD was purchased from Sigma-Aldrich. Laboratory reagents were of analytical grade or better and purchased from Sigma-Aldrich and Bio-Rad Laboratories (Hercules, CA, USA). The following primary antibodies were used: phospho-tyrosine (mouse, 1:500) from BD Biosciences (San Jose, CA, USA); phospho-FAK Y925 (rabbit, 1:200) and phospho-ERK1/2 (rabbit 9101, 1:2000) from Cell Signaling Technology (Danvers, MA, USA); zyxin (goat, 1:500), FAK (rabbit, 1:400) and α-tubulin (mouse, 1:2000) from Santa Cruz Biotechnology (Dallas, Texas, USA); phospho-caldesmon from Millipore (Darmstadt, Germany) (rabbit, 1:500); and α-tubulin (rabbit, 1:50,000 from Abcam, Cambridge, MA, USA). For immunofluorescence experiments, secondary antibodies used were goat or donkey anti-rabbit and goat or donkey anti-mouse Alexa Fluor 488 and Alexa Fluor 568 (1:1000; Invitrogen). For Western blot experiments, secondary antibodies used were goat Oregon Green 488 or Alexa Fluor 568 labelled anti-rabbit or anti-mouse IgGs (1:1000; LI-COR Biosciences).
Statistical analysis
Data are reported as means ± SEM, with differences between sample populations deemed significant at P < 0.05. For biomechanical observations (venous stress and stiffness), this was evaluated by two-way ANOVA and follow-up multiple comparisons tests: Dunnett’s for tissue stress and stiffness and Tukey–Kramer for cortical stiffness. For measurements of focal adhesion size and quantitative densitometry, two-tailed Student’s t tests were performed.
Results
Smooth muscle cells are the dominant source of total stiffness in portal vein
To examine the biomechanics of the intact venous wall, we measured the stress and stiffness in portal vein strips in vitro (see Methods for details) stretched to optimal length for smooth muscle contraction, or 70% strain, as previously determined (Bradley & Morgan, 1985), at baseline (unstimulated) and during agonist activation (Fig.1). The mean stiffness of portal vein at baseline was measured to be 26.2 ± 2.3 kPa. This value is severalfold lower than the reported baseline stiffness of aorta (Brozovich & Morgan, 1989; Saphirstein et al. 2013).
Figure 1.
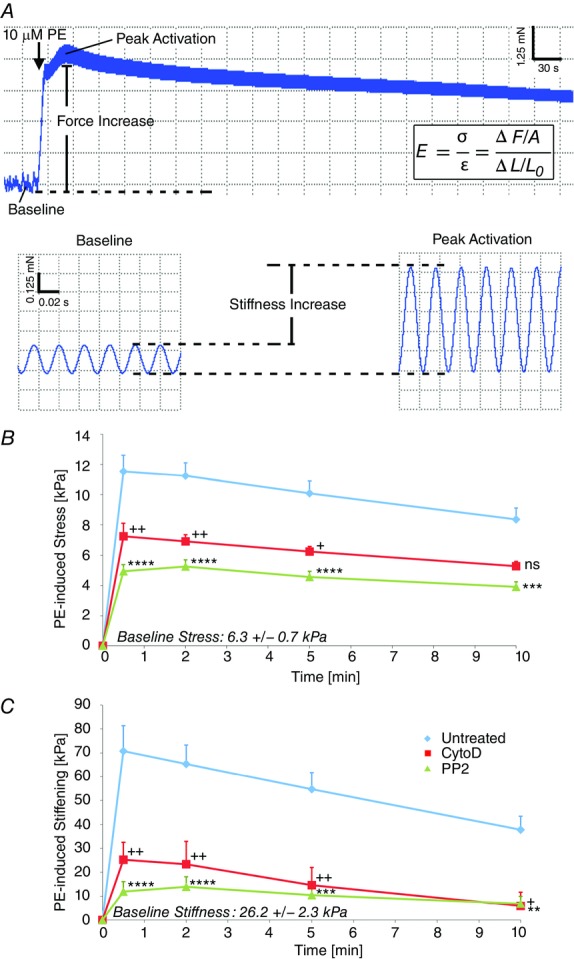
Increases in venous tissue stress and stiffness during contractile stimulation are diminished by inhibition of actin polymerization or Src signalling
A, sample force trace for PV strip during contractile activation with PE illustrating time course of stress and stiffness changes. B, PE-induced stress (in kilopascals) is significantly reduced when longitudinal PV strips are pre-treated with CytoD to inhibit actin polymerization (red) or PP2 to inhibit Src (green). C, PE-induced stiffening (in kilopascals) is significantly reduced when strips are pre-treated with CytoD (red) to inhibit actin polymerization or PP2 to inhibit Src (green). n = 12 PV tissue strips. Two-way ANOVA and Dunnett’s multiple comparison test vs. control +,*P < 0.05, ++, **P < 0.01, ***P < 0.001, ****P < 0.0001. ns, not significant.
When stimulated with the α-adrenergic agonist phenylephrine (PE), both PV stress and stiffness (Fig.1B and C) increased abruptly, peaked within about 30 s, and then declined over the 10 min time course. This is consistent with the phasic nature of these venous smooth muscle cells, which are known to lose tone over the course of stimulation, in contrast to the robust tone maintenance of tonic aortic smooth muscle cells (Somlyo & Somlyo, 1968). The peak agonist-induced increase in stiffness for portal vein is comparable in magnitude to the increase observed in aorta of the same species (Brozovich & Morgan, 1989; Saphirstein et al. 2013). However, since the baseline material stiffness (26.2 ± 2.3 kPa) of PV is much lower than aorta, the increase in stiffness with activation represents a much larger proportion of the total stiffness (∼75%), indicating that venous smooth muscle cells play the dominant role in determining the distensibility of the portal vein and, accordingly, the size of its blood reservoir and its impact on venous return.
Development of venous tissue stress and stiffness during contractile stimulation depends on actin polymerization and Src signalling
It is well accepted that phosphorylation of the 20 kDa light chains of myosin is a major mechanism of regulation of stress and stiffness of portal vein (Jiang & Morgan, 1989; Kitazawa et al. 1991), but recently, focal adhesion signalling and cortical actin polymerization have also been shown to be major regulators of aortic stress and stiffness (Kim et al. 2008; Min et al. 2012; Saphirstein et al. 2013). Thus we investigated the effect of inhibitors of focal adhesion signalling and actin polymerization in this venous preparation. Pre-treatment with the small molecule inhibitor PP2, inhibiting the major focal adhesion kinase Src (Fincham & Frame, 1998; Webb et al. 2004; Bain et al. 2007; Min et al. 2012), significantly decreased PE-induced stress (57 ± 12% at peak) and stiffening (83 ± 20% at peak) (Fig.1B and C, green). Pre-treatment with CytoD (500 nm), inhibiting actin polymerization by capping the barbed ends of filaments (Kim et al. 2010), also significantly reduced PE-induced stress (37 ± 17% at peak) and stiffness (64 ± 28% at peak) (Fig.1B and C, red). Since we have previously shown that PE causes a significant increase in actin polymerization of only the γ isoform of non-muscle actin (Kim et al. 2008), which is present primarily in the cortex of the smooth muscle cell (Gallant et al. 2011), this prompted us to devise a method for the measurement of the stiffness specifically of the cortex of the cell.
Cortical stiffness of portal vein smooth muscle cells is regulated by PE in an actin polymerization-dependent manner but is not affected by Src inhibition
Next, we investigated the regulation of stiffness at a subcellular length scale. We measured and report here for the first time cortical stiffness of individual differentiated vascular smooth muscle cells (dVSMCs) freshly isolated from portal vein. These cells retain normal contractility unlike cultured cells that lose normal contractile responses even on primary culture (Rensen et al. 2007). Cortical stiffness was measured by applying small forces (50 pN) with a magnetic microneedle to cell-adherent RGD-coated magnetic microbeads (Fig.2A). The use of these small forces has been previously shown to restrict measurements to the cortex of the cell (Rotsch et al. 1999; Matthews et al. 2004; Kasas et al. 2005; Sun et al. 2008; Oberleithner et al. 2009; Saphirstein et al. 2013).
Figure 2.
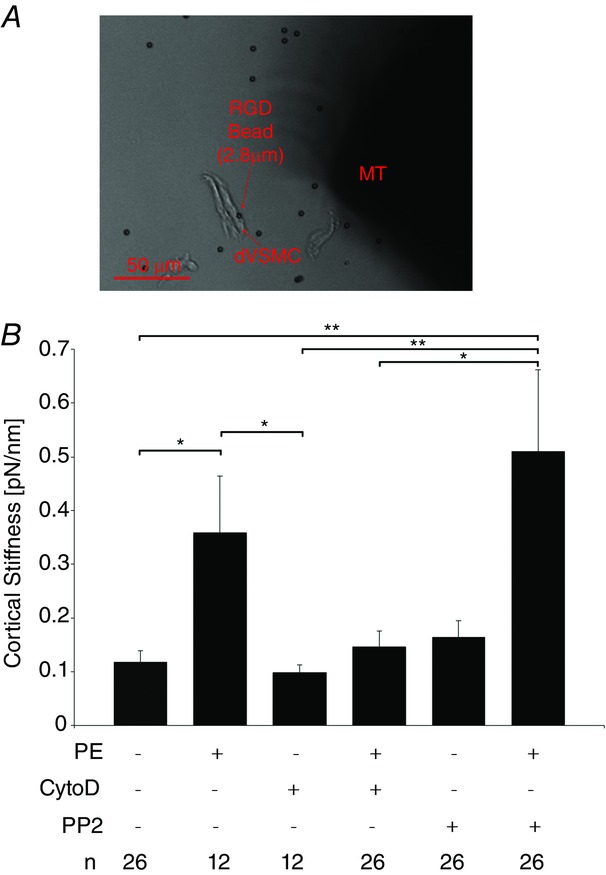
Increase in cortical stiffening in freshly isolated PV VSMCs depend on actin polymerization but not Src signalling
A, brightfield micrograph of magnetic microneedle technology. Using the magnetic microneedle (right, MT), small forces are applied via RGD-coated beads (2.8 μm) (left) adherent to freshly isolated PV VSMCs to probe the stiffness of the underlying cortical cytoskeleton and FA. At an operating distance of 150 μm between bead and microneedle tip, the forces applied are 50 pN. Scale bar, 50 μm. B, mean cortical stiffness + SEM measured with RGD beads increases with vasoconstrictor PE and is inhibited by CytoD but not PP2. n = 12 experiments, 12–26 freshly isolated PV VSMCs per condition. ANOVA and Tukey–Kramer multiple comparison test. *P < 0.05, **P < 0.01.
Mean cortical stiffness (Fig.2B) increased threefold from baseline levels (0.12 ± 0.02 pN nm−1) in cells activated with PE. This increase was significantly diminished by pre-treatment with CytoD, indicating that the addition of actin monomers to the barbed ends of actin filaments are important for cortical reinforcement during agonist-induced contraction of venous smooth muscle cells and consistent with the inhibition of total tissue stiffness by CytoD (Fig.1).
Surprisingly, in comparison to the tissue-level results, PE-induced increases in cortical stiffening were not inhibited by pre-treatment with PP2. This insensitivity to PP2 stands in contrast to our observations at the tissue length scale. Thus, the possibility needs to be considered that the responsiveness to PP2 might be lost on enzymatic isolation of the cells. However, we have previously demonstrated that PP2 is effective in other Src-dependent functions such as endosomal cycling in enzymatically isolated smooth muscle cells (Saphirstein et al. 2013).
Focal adhesion size does not change with contractile stimulation
In order to further investigate the PP2 insensitivity of cortical stiffness in the freshly isolated venous VSMCs, we examined focal adhesion size using deconvolution immunofluorescence microscopy with a resolution down to 140 nm (Parker et al. 1998). We initially hypothesized that focal adhesions (FAs) would grow with contractile stimulation in venous cells based on previous findings in many types of cultured cells (Balaban et al. 2001; Bershadsky et al. 2003) and our previous study of serum-starved aortic smooth muscle cells in culture, where stiffness correlated with FA size (Saphirstein et al. 2013). FAs are reported to contain over 150 different proteins (Zaidel-Bar, 2009). Since different focal adhesions may also be composed of different proteins and the possibility exists that VSMCs may contain more than one type of FA, we monitored two key proteins: zyxin, which is reported to be in the actin regulatory layer of FAs, linking actin to the FAs, and FAK which is an integrin signalling and mechanosensitive protein (Kanchanawong et al. 2010). Surprisingly, no change in focal adhesion size was detectable with PE in venous freshly dissociated dVSMCs as measured by staining for either focal adhesion marker protein. Interestingly, the staining for FAK and zyxin only partially overlap (Fig.3) consistent with either or both of two possibilities: (1) FAK and zyxin label different types of focal adhesions, or (2) FAK and zyxin are targeted to different locations within the individual focal adhesions. At this time, we cannot prove or disprove either possibility. But, as seen in Fig.3, FAK staining was mostly at the cell edge, whereas zyxin staining was punctate throughout the cell reflecting its known association with both focal adhesions and dense bodies (Crawford & Beckerle, 1991). Notably, the punctae measured here averaged 0.20 μm2 in area, much smaller than sizes reported for FAs in cultured cells by similar methods (Balaban et al. 2001; Bershadsky et al. 2003). We have reported in a previous aortic study that individual molecules of zyxin are capable of undergoing Src-dependent endosomal cycling in response to PE (Poythress et al. 2013), but single molecule redistributions would be beyond the resolution of this imaging method and it is unclear as to whether removal of individual molecules would change the stiffness of the focal adhesion. Furthermore, our finding that agonist-induced cortical stiffening is not affected by PP2 inhibition of Src in venous dVSMCs separately also indicates that cortical stiffness is not regulated by changes in focal adhesion size in this cell type but rather by changes in actin linkages.
Figure 3.
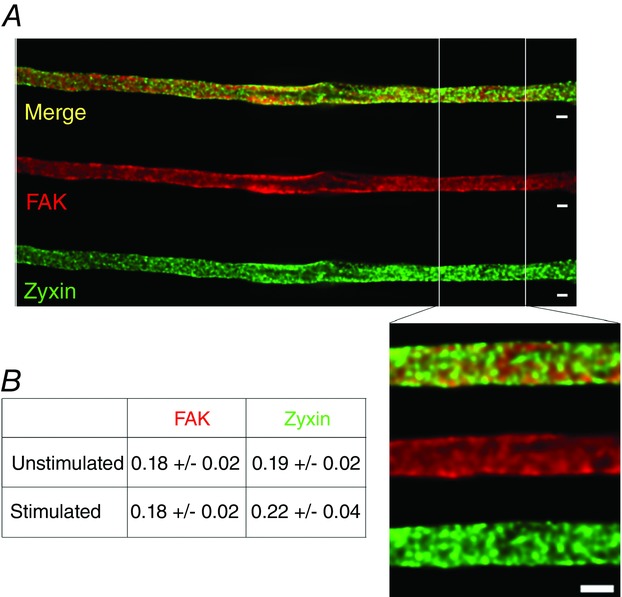
Focal adhesion size in isolated PV VSMCs is unchanged by agonist stimulation
A, deconvolved immunofluorescence images of unstimulated PV dVSMC co-stained for FAK and zyxin as FA markers. Scale bar, 2 μm; inset, 2 μm. B, table of mean FA areas in μm2. There was no detectable difference in FA size with PE stimulation as quantified by the area of the FA punctae with either marker. n = 3–5 experiments with 3–6 cells per condition per experiment. P > 0.05, two-tailed Student’s t test.
Tyrosine phosphorylation of focal adhesion proteins is increased with agonist stimulation in a PP2-dependent manner and signals downstream to directly influence actomyosin
To further investigate the marked difference in PP2 responsiveness between the cellular and tissue levels of venous smooth muscle, we performed phosphotyrosine screening of venous tissue homogenates by Western blot (Fig.4). In smooth muscle tissue, the majority of described phosphorylation events are Ser/Thr phosphorylations (Li et al. 2007; Yamin & Morgan, 2012), with most tyrosine phosphorylations restricted to focal adhesion signalling. PE induced increases in tyrosine phosphorylation of several bands identified as focal adhesion proteins in previous reports for aorta and myometrium (Li et al. 2007; Min et al. 2012) and PP2 was quite effective in inhibiting phosphorylation of those bands. Here we performed phosphotyrosine screening of portal vein homogenates and saw PP2-inhibitable PE-induced increases in tyrosine phosphorylation of several bands previously associated with focal adhesion proteins (Fig.4A). Notably, the 125 kDa band (Fig.4B), which was confirmed to contain FAK by the use of a FAK Y925-specific antibody (Fig.4C), exhibited this phosphorylation profile.
Figure 4.

The vasoconstrictor PE induces FAK/Src-mediated tyrosine phosphorylation of focal adhesion proteins and downstream ERK activation in venous VSMCs
A, typical blot, phosphotyrosine screening of quick-frozen PV tissue homogenates. PE increases tyrosine phosphorylation at several bands, and pre-treatment with Src inhibitor PP2 prevents this increase. B, quantitative densitometry of tyrosine phosphorylation at p125 exhibits a PE-induced increase that is inhibited by PP2 (n = 3). C, phospho-FAK Y925 increases in response to PE in a PP2-attenuated manner (n = 5). D, agonist activation triggers downstream activation of ERK1/2 at Thr202/Tyr204 in a PP2-inhibited manner (n = 6). The densitometry for ERK2 (not shown) follows the same pattern and significance as the ERK1 densitometry. E, phospho-caldesmon 789 increases in response to PE but decreases with the addition of PP2. The brightness of blots has been uniformly altered for visual display, but unaltered blots were used for densitometric quantification. *P < 0.05, **P < 0.01, ***P < 0.001, two-tailed Student’s t test.
Phosphorylation of FAK at 925 in many cell types is known to involve integrin-mediated Src recruitment and signal to extracellular signal-regulated kinases 1 and 2 (ERK1/2) (Mitra et al. 2005). ERK signalling pathways are known to regulate smooth muscle contractility by complex mechanisms. For example, ERK activation has been shown to induce ERK translocation from scaffold complexes in the cell cortex (including focal adhesions; Khalil & Morgan, 1993; Khalil et al. 1995) to the contractile filaments, where it phosphorylates the inhibitory actin filament-binding protein caldesmon (Gangopadhyay et al. 2009), leading to increased force output (Earley et al. 1998; Je et al. 2001; Ishihata et al. 2002; Ishibe et al. 2004; Kim & Hai, 2005). To investigate the involvement of ERK1/2 in venous smooth muscle, we monitored ERK activation via phosphorylation at threonine 202/tyrosine 204 and found that PE stimulation of portal vein tissue leads to phosphorylation of ERK1/2. Furthermore, the activation of ERK (Fig.4D) and the phosphorylation of caldesmon (Fig.4E) is inhibitable by PP2 pre-treatment. These results point to an additional, FAK/Src-induced ERK signalling pathway terminating on non-cortical contractile filaments, leading to the PP2-sensitive stiffening and force increase observed at the tissue level.
Discussion
In this study we assessed the regulation of venous biomechanics using an approach spanning multiple biological levels of scale. The main findings are that agonist-activated smooth muscle represents the major component of total venous stiffness and that the agonist-triggered increases in tissue stiffness and stress are modulated by both (1) a cortical actin switch caused by regulation of cortical actin polymerization, and (2) signalling from the focal adhesion to ERK to the contractile filaments. These two novel mechanisms presumably work in coordination with many other signalling pathways previously reported for vascular smooth muscle.
In the present study, we measured, for the first time, cortical stiffness in freshly isolated, differentiated vascular smooth muscle cells using magnetic microneedle technology. Although it is important to note that we cannot rule out the possibility of some functional changes in isolated cells due to the use of proteases to break connections with extracellular matrix proteins as well as cell-to-cell connections, which are known to alter muscle cells through ‘outside-in signalling’ (Shattil et al. 2010), our low-force measurements made with magnetic microneedle technology are able to target the local cortical stiffness in the vicinity of a cell-adherent microbead (comprising the bead–cell linkage, focal adhesion and cortical cytoskeleton) without breaking crossbridges or provoking a micromyogenic response (Rotsch et al. 1999; Matthews et al. 2004; Kasas et al. 2005; Sun et al. 2008; Oberleithner et al. 2009; Saphirstein et al. 2013). To the best of our knowledge, cortical stiffness in freshly isolated smooth muscle cells has only been examined once before, in airway smooth muscle using magnetic twisting cytometry (Deng et al. 2006). That study did not assess the effects of vasoconstrictors or inhibitors on cortical stiffness. Additionally, to the best of our knowledge, the current study is the first report of stiffness in individual venous contractile VSMCs.
We observed a pronounced effect of CytoD to inhibit both tissue level and cortical stiffness at a concentration that is selective for inhibition of actin filament elongation (Kim et al. 2010). These results, together with our magnetic microneedle technology measurements of cortical stiffness, point to the presence of an active and agonist-regulated cortical actin ‘switch’, where actin polymerization at the barbed ends of the non-muscle actin cytoskeleton can link the contractile filaments to the cortical non-muscle cytoskeleton in which are embedded the focal adhesions, which include integrins. Thus this ‘actin switch’ can promote force and stiffness transmission from the contractile filaments to the cortical actin filaments, through the focal adhesions to the extracellular matrix (Fig.5). Such a non-muscle cortical actin cytoskeleton has been shown by our group to involve γ-actin which remodels in response to an α-agonist (Kim et al. 2008) in contrast to the more stable α smooth muscle-containing contractile filaments. Reinforcement of this cortical switch/bridge by non-muscle cortical actin polymerization connecting to the stable contractile filaments will increase the capacity for force transfer between the inside and the outside of the cell and will regulate stress and stiffness development at the tissue level.
Figure 5.
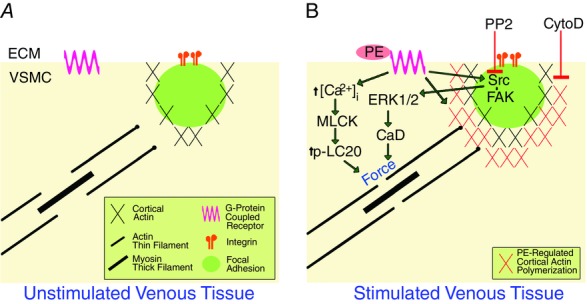
Model: contractile activation of venous smooth muscle alters engagement of the cell–matrix adhesion via cortical actin switch and ERK activation is facilitated by Src
A, diagrammatic view of unstimulated cell showing that the non-muscle cortical actin cytoskeleton containing the FAs is disengaged from a population of the contractile filaments. B, agonist (PE) activation promotes cortical non-muscle actin polymerization which increases the engagement (the ‘actin switch’) of the non-muscle cortical cytoskeleton with the contractile filaments. Conversely, CytoD inhibits agonist-induced cortical actin polymerization and connections to the contractile filaments. Agonist activation also activates Src, facilitating downstream signalling to ERK and caldesmon (CaD) phosphorylation, which enhances agonist-induced contractility and tissue stiffness due to crossbridge attachment. This pathway is inhibited by the Src inhibitor PP2. MLCK, myosin light chain kinase; LC20, 20kDa myosin light chains; ECM, extracellular matrix.
We also observed PP2-induced inhibition of tissue force production and stiffness in portal vein cells, which is not explained by cortical effects alone. We found that PE induced phosphorylation of FAK at Y925 and ERK1/2 at T202/Y204. These results and our model offer an explanation for how PE can increase tissue stiffness in a PP2-attenuated manner, while PE-induced cortical stiffness is PP2 insensitive and FA size is unaffected by agonist stimulation (see model in Fig.5). Agonist-induced force is generated internally by venous VSMC actomyosin crossbridge cycling and transmitted to the non-muscle cytoskeleton and the cell cortex containing the focal adhesions. Since PP2 pre-treatment reduced agonist-induced force generation at the tissue level without affecting cortical stiffness, it would appear that PP2 inhibition of Src is exerting its ultimate effects on stiffness at the level of actomyosin via FAK–ERK signalling. This is further supported by the observation that caldesmon phosphorylation is also inhibited by PP2. Caldesmon is an inhibitory protein found in smooth muscle contractile filaments that is functionally analogous to troponin in skeletal muscle and increases actin availability to augment actomyosin interactions (Katsuyama et al. 1992; Dessy et al. 1998; Earley et al. 1998; Zeidan et al. 2000; Je et al. 2001; Ishihata et al. 2002; Somlyo & Somlyo, 2003; Ishibe et al. 2004; Gerthoffer, 2005; Li et al. 2007, 2009; Huveneers & Danen, 2009; Min et al. 2012). Thus, our results support a pathway linking tyrosine signalling in the focal adhesions to an ERK scaffolding function of the focal adhesions leading to ERK activation and translocation and subsequent ERK-mediated caldesmon phosphorylation to enhance stiffness.
Our results in this study of venous smooth muscle biomechanics highlight several key differences between arteries and veins. The first, that veins are substantially more compliant than arteries, is well known. The basal stiffness of ferret portal vein reported here (26.2 ± 2.3 kPa) is comparable to reports of the stiffness of porcine hepatic portal vein and of veins from other species obtained with various methods (Azuma & Hasegawa, 1973; Wesly et al. 1975; Rhee & Brozovich, 2000; Rossmann, 2010). Ferret portal vein is severalfold less stiff than ferret aorta (∼1400 kPa) (Brozovich & Morgan, 1989) as measured with the HFLA method. The next difference is that smooth muscle contractility represents a much larger proportion of the total activated stiffness in veins (∼75%), as venous stiffness increases up to threefold with agonist stimulation. Accordingly, the matrix stiffness is much less influential to stiffness in activated portal vein. Thus, smooth muscle cells have a far greater relative regulatory impact in portal vein than in aorta and exert extensive control over the venous compliance and the release of the portal blood volume into circulation.
Focal adhesion size in untreated cells differs severalfold between cultured aortic A7r5 cells and freshly isolated PV VSMCs. The difference in FA size is most likely explained on the basis of cellular phenotype. The A7r5s used in our previous study were serum-starved for 24 h to promote a non-spreading, non-migrating more differentiated phenotype, but these differences highlight the limitations of using cell culture models to mimic dVSMCs. Consistent with our current findings in portal vein, another group reported no changes in focal adhesion size in stimulated smooth muscle cells in situ as measured by immunohistochemistry of canine gastrointestinal and airway smooth muscle tissues (Eddinger et al. 2005).
In summary, our results indicate that focal adhesions and cortical actin work cooperatively to support force and stiffness development in venous tissue during contraction. We report that cortical actin polymerization comprises a cortical actin switch that reinforces the focal adhesion linkage between the inside and the outside of the cell and enables venous VSMCs to exert substantial regulatory influence over the biomechanical properties of veins. As a result, large capacitance vessels like the hepatic and portal vein have a large dynamic range that enables significant influence over venous return and cardiovascular function. This study suggests key differences in the mechanisms of cortical regulation of stress and stiffness between arteries and veins and encourages further investigations that may uncover new approaches for selectively combatting cardiovascular disease from the venous side of the circulation.
Acknowledgments
The authors thank Christopher J. Nicholson for critical input regarding the text of the manuscript.
Glossary
- CytoD
cytochalasin D
- dVSMC
differentiated (contractile) vascular smooth muscle cell
- E
modulus of elasticity (Young’s modulus)
- ERK1/2
extracellular signal-regulated kinases 1 and 2
- FA
focal adhesion
- FAK
focal adhesion kinase
- HFLA
high-frequency, low-amplitude
- PE
phenylephrine
- PV
portal vein
- VSMC
vascular smooth muscle cell
Additional information
Competing interests
The authors declare no conflicts of interest.
Author contributions
R.J.S. conceived, designed, and performed the experiments, analysed and interpreted the data, and co-wrote the manuscript. Y.Z.G. contributed to the design of the experiments, analysed and interpreted the data, and revised and critically edited the manuscript. Q.Q.L. performed the experiments and helped interpret the results. K.G.M. conceived and designed the experiments, analysed and interpreted the data, and co-wrote the manuscript. All authors approved the final version of the manuscript.
Funding
This work was supported by NIH P01 HL086655 to K.G.M. and the Aortic Stiffness ARC of the BU Evans Centre.
References
- Azuma T. Hasegawa M. Distensibility of the vein: from the architectural point of view. Biorheology. 1973;10:469–479. doi: 10.3233/bir-1973-10323. [DOI] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR. Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban NQ, Schwarz US, Riveline D, Goichberg P, Tzur G, Sabanay I, Mahalu D, Safran S, Bershadsky A, Addadi L. Geiger B. Force and focal adhesion assembly: a close relationship studied using elastic micropatterned substrates. Nat Cell Biol. 2001;3:466–472. doi: 10.1038/35074532. [DOI] [PubMed] [Google Scholar]
- Bárány M. Biochemistry of Smooth Muscle Contraction. San Diego: Academic Press; 1996. [Google Scholar]
- Bershadsky AD, Balaban NQ. Geiger B. Adhesion-dependent cell mechanosensitivity. Annu Rev Cell Dev Biol. 2003;19:677–695. doi: 10.1146/annurev.cellbio.19.111301.153011. [DOI] [PubMed] [Google Scholar]
- Bohr DF, Somlyo AP. Sparks HV. Handbook of Physiology, section 2, The Cardiovascular System, vol. II, Vascular Smooth Muscle. Vol. 1. Oxford University Press; 1980. pp. 1–31. Rhodin & Johannes A. G., “Architecture of the vessel wall”, Chapter. [Google Scholar]
- Bradley AB. Morgan KG. Cellular Ca2+ monitored by aequorin in adenosine-mediated smooth muscle relaxation. Am J Physiol. 1985;248:H109–H117. doi: 10.1152/ajpheart.1985.248.1.H109. [DOI] [PubMed] [Google Scholar]
- Brecher GA. History of venous research. IEEE Trans Biomed Eng. 1969;16:236–247. doi: 10.1109/tbme.1969.4502655. [DOI] [PubMed] [Google Scholar]
- Brozovich FV. Morgan KG. Stimulus-specific changes in mechanical properties of vascular smooth muscle. Am J Physiol. 1989;257:H1573–H1580. doi: 10.1152/ajpheart.1989.257.5.H1573. [DOI] [PubMed] [Google Scholar]
- Bulter WE, Peterson JW, Zervas NT. Morgan KG. Intracellular calcium, myosin light chain phosphorylation, and contractile force in experimental cerebral vasospasm. Neurosurgery. 1996;38:781–787. doi: 10.1227/00006123-199604000-00029. [DOI] [PubMed] [Google Scholar]
- Crawford AW. Beckerle MC. Purification and characterization of zyxin, an 82,000-dalton component of adherens junctions. J Biol Chem. 1991;266:5847–5853. [PubMed] [Google Scholar]
- DeFeo TT. Morgan KG. Responses of enzymatically isolated mammalian vascular smooth muscle cells to pharmacological and electrical stimuli. Pflugers Arch. 1985;404:100–102. doi: 10.1007/BF00581502. [DOI] [PubMed] [Google Scholar]
- Deng L, Trepat X, Butler JP, Millet E, Morgan KG, Weitz DA. Fredberg JJ. Fast and slow dynamics of the cytoskeleton. Nat Mater. 2006;5:636–640. doi: 10.1038/nmat1685. [DOI] [PubMed] [Google Scholar]
- Dessy C, Kim I, Sougnez CL, Laporte R. Morgan KG. A role for MAP kinase in differentiated smooth muscle contraction evoked by α-adrenoceptor stimulation. Am J Physiol. 1998;275:C1081–C1086. doi: 10.1152/ajpcell.1998.275.4.C1081. [DOI] [PubMed] [Google Scholar]
- Driska SP, Aksoy MO. Murphy RA. Myosin light chain phosphorylation associated with contraction in arterial smooth muscle. Am J Physiol. 1981;240:C222–C233. doi: 10.1152/ajpcell.1981.240.5.C222. [DOI] [PubMed] [Google Scholar]
- Earley JJ, Su X. Moreland RS. Caldesmon inhibits active crossbridges in unstimulated vascular smooth muscle: an antisense oligodeoxynucleotide approach. Circ Res. 1998;83:661–667. doi: 10.1161/01.res.83.6.661. [DOI] [PubMed] [Google Scholar]
- Eddinger TJ, Schiebout JD. Swartz DR. Smooth muscle adherens junctions associated proteins are stable at the cell periphery during relaxation and activation. Am J Physiol Cell Physiol. 2005;289:C1379–C1387. doi: 10.1152/ajpcell.00193.2005. [DOI] [PubMed] [Google Scholar]
- Edwards NM, Maurer MS. Wellner RB. Aging, Heart Disease, and Its Management: Facts and Controversies. Humana Press; 2003. [Google Scholar]
- Fincham VJ. Frame MC. The catalytic activity of Src is dispensable for translocation to focal adhesions but controls the turnover of these structures during cell motility. EMBO J. 1998;17:81–92. doi: 10.1093/emboj/17.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox JG. Marini RP. Biology and Diseases of the Ferret. Wiley-Blackwell; 2014. [Google Scholar]
- Gallant C, Appel S, Graceffa P, Leavis P, Lin JJ, Gunning PW, Schevzov G, Chaponnier C, DeGnore J, Lehman W. Morgan KG. Tropomyosin variants describe distinct functional subcellular domains in differentiated vascular smooth muscle cells. Am J Physiol Cell Physiol. 2011;300:C1356–C1365. doi: 10.1152/ajpcell.00450.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangopadhyay SS, Kengni E, Appel S, Gallant C, Kim HR, Leavis P, DeGnore J. Morgan KG. Smooth muscle archvillin is an ERK scaffolding protein. J Biol Chem. 2009;284:17607–17615. doi: 10.1074/jbc.M109.002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascho JA, Fanelli C. Zelis R. Aging reduces venous distensibility and the venodilatory response to nitroglycerin in normal subjects. Am J Cardiol. 1989;63:1267–1270. doi: 10.1016/0002-9149(89)90188-4. [DOI] [PubMed] [Google Scholar]
- Gerthoffer WT. Signal-transduction pathways that regulate visceral smooth muscle function. III. Coupling of muscarinic receptors to signaling kinases and effector proteins in gastrointestinal smooth muscles. Am J Physiol Gastrointest Liver Physiol. 2005;288:G849–G853. doi: 10.1152/ajpgi.00530.2004. [DOI] [PubMed] [Google Scholar]
- Guyton AC. Hall JE. Textbook of Medical Physiology. Philadelphia: Elsevier Saunders; 2006. [Google Scholar]
- Huveneers S. Danen EH. Adhesion signaling – crosstalk between integrins, Src and Rho. J Cell Sci. 2009;122:1059–1069. doi: 10.1242/jcs.039446. [DOI] [PubMed] [Google Scholar]
- Ishibe S, Joly D, Liu ZX. Cantley LG. Paxillin serves as an ERK-regulated scaffold for coordinating FAK and Rac activation in epithelial morphogenesis. Mol Cell. 2004;16:257–267. doi: 10.1016/j.molcel.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Ishihata A, Tasaki K. Katano Y. Involvement of p44/42 mitogen-activated protein kinases in regulating angiotensin II- and endothelin-1-induced contraction of rat thoracic aorta. Eur J Pharmacol. 2002;445:247–256. doi: 10.1016/s0014-2999(02)01790-9. [DOI] [PubMed] [Google Scholar]
- Je HD, Gangopadhyay SS, Ashworth TD. Morgan KG. Calponin is required for agonist-induced signal transduction – evidence from an antisense approach in ferret smooth muscle. J Physiol. 2001;537:567–577. doi: 10.1111/j.1469-7793.2001.00567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang MJ. Morgan KG. Agonist-specific myosin phosphorylation and intracellular calcium during isometric contractions of arterial smooth muscle. Pflugers Arch. 1989;413:637–643. doi: 10.1007/BF00581814. [DOI] [PubMed] [Google Scholar]
- Kanchanawong P, Shtengel G, Pasapera AM, Ramko EB, Davidson MW, Hess HF. Waterman CM. Nanoscale architecture of integrin-based cell adhesions. Nature. 2010;468:580–584. doi: 10.1038/nature09621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasas S, Wang X, Hirling H, Marsault R, Huni B, Yersin A, Regazzi R, Grenningloh G, Riederer B, Forro L, Dietler G. Catsicas S. Superficial and deep changes of cellular mechanical properties following cytoskeleton disassembly. Cell Motil Cytoskeleton. 2005;62:124–132. doi: 10.1002/cm.20086. [DOI] [PubMed] [Google Scholar]
- Katsuyama H, Wang CL. Morgan KG. Regulation of vascular smooth muscle tone by caldesmon. J Biol Chem. 1992;267:14555–14558. [PubMed] [Google Scholar]
- Khalil RA, Menice CB, Wang CL. Morgan KG. Phosphotyrosine-dependent targeting of mitogen-activated protein kinase in differentiated contractile vascular cells. Circ Res. 1995;76:1101–1108. doi: 10.1161/01.res.76.6.1101. [DOI] [PubMed] [Google Scholar]
- Khalil RA. Morgan KG. PKC-mediated redistribution of mitogen-activated protein kinase during smooth muscle cell activation. Am J Physiol. 1993;265:C406–C411. doi: 10.1152/ajpcell.1993.265.2.C406. [DOI] [PubMed] [Google Scholar]
- Kim HR, Gallant C, Leavis PC, Gunst SJ. Morgan KG. Cytoskeletal remodeling in differentiated vascular smooth muscle is actin isoform dependent and stimulus dependent. Am J Physiol Cell Physiol. 2008;295:C768–C778. doi: 10.1152/ajpcell.00174.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HR, Graceffa P, Ferron F, Gallant C, Boczkowska M, Dominguez R. Morgan KG. Actin polymerization in differentiated vascular smooth muscle cells requires vasodilator-stimulated phosphoprotein. Am J Physiol Cell Physiol. 2010;298:C559–C571. doi: 10.1152/ajpcell.00431.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HR. Hai CM. Mechanisms of mechanical strain memory in airway smooth muscle. Can J Physiol Pharmacol. 2005;83:811–815. doi: 10.1139/y05-060. [DOI] [PubMed] [Google Scholar]
- Kitazawa T, Masuo M. Somlyo AP. G protein-mediated inhibition of myosin light-chain phosphatase in vascular smooth muscle. Proc Natl Acad Sci USA. 1991;88:9307–9310. doi: 10.1073/pnas.88.20.9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollmannsberger P. Fabry B. High-force magnetic tweezers with force feedback for biological applications. Rev Sci Instrum. 2007;78:114301. doi: 10.1063/1.2804771. [DOI] [PubMed] [Google Scholar]
- Lee YH, Gallant C, Guo H, Li Y, Wang CA. Morgan KG. Regulation of vascular smooth muscle tone by N-terminal region of caldesmon. Possible role of tethering actin to myosin. J Biol Chem. 2000;275:3213–3220. doi: 10.1074/jbc.275.5.3213. [DOI] [PubMed] [Google Scholar]
- Li Y, Gallant C, Malek S. Morgan KG. Focal adhesion signaling is required for myometrial ERK activation and contractile phenotype switch before labor. J Cell Biochem. 2007;100:129–140. doi: 10.1002/jcb.21033. [DOI] [PubMed] [Google Scholar]
- Li Y, Reznichenko M, Tribe RM, Hess PE, Taggart M, Kim H, DeGnore JP, Gangopadhyay S. Morgan KG. Stretch activates human myometrium via ERK, caldesmon and focal adhesion signaling. PLoS One. 2009;4:e7489. doi: 10.1371/journal.pone.0007489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marganski WA, Gangopadhyay SS, Je HD, Gallant C. Morgan KG. Targeting of a novel Ca2+/calmodulin-dependent protein kinase II is essential for extracellular signal-regulated kinase-mediated signaling in differentiated smooth muscle cells. Circ Res. 2005;97:541–549. doi: 10.1161/01.RES.0000182630.29093.0d. [DOI] [PubMed] [Google Scholar]
- Matthews BD, Overby DR, Alenghat FJ, Karavitis J, Numaguchi Y, Allen PG. Ingber DE. Mechanical properties of individual focal adhesions probed with a magnetic microneedle. Biochem Biophys Res Commun. 2004;313:758–764. doi: 10.1016/j.bbrc.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Meijering E, Dzyubachyk O. Smal I. Methods for cell and particle tracking. Methods Enzymol. 2012;504:183–200. doi: 10.1016/B978-0-12-391857-4.00009-4. [DOI] [PubMed] [Google Scholar]
- Min J, Reznichenko M, Poythress RH, Gallant CM, Vetterkind S, Li Y. Morgan KG. Src modulates contractile vascular smooth muscle function via regulation of focal adhesions. J Cell Physiol. 2012;227:3585–3592. doi: 10.1002/jcp.24062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA. Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- Monahan KD, Dinenno FA, Seals DR. Halliwill JR. Smaller age-associated reductions in leg venous compliance in endurance exercise-trained men. Am J Physiol Heart Circ Physiol. 2001;281:H1267–H1273. doi: 10.1152/ajpheart.2001.281.3.H1267. [DOI] [PubMed] [Google Scholar]
- Noordergraaf A. Circulatory System Dynamics. New York: Academic Press; 1978. [Google Scholar]
- Oberleithner H, Callies C, Kusche-Vihrog K, Schillers H, Shahin V, Riethmüller C, MacGregor GA. de Wardener HE. Potassium softens vascular endothelium and increases nitric oxide release. Proc Natl Acad Sci U S A. 2009;106:2829–2834. doi: 10.1073/pnas.0813069106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker CA, Takahashi K, Tang JX, Tao T. Morgan KG. Cytoskeletal targeting of calponin in differentiated, contractile smooth muscle cells of the ferret. J Physiol. 1998;508:187–198. doi: 10.1111/j.1469-7793.1998.187br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poythress RH, Gallant C, Vetterkind S. Morgan KG. Vasoconstrictor-induced endocytic recycling regulates focal adhesion protein localization and function in vascular smooth muscle. Am J Physiol Cell Physiol. 2013;305:C215–C227. doi: 10.1152/ajpcell.00103.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rensen SS, Doevendans PA. van Eys GJ. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth Heart J. 2007;15:100–108. doi: 10.1007/BF03085963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee AY. Brozovich FV. The smooth muscle cross-bridge cycle studied using sinusoidal length perturbations. Biophys J. 2000;79:1511–1523. doi: 10.1016/S0006-3495(00)76402-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmann JS. Elastomechanical properties of bovine veins. J Mech Behav Biomed Mater. 2010;3:210–215. doi: 10.1016/j.jmbbm.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Rothe CF. Reflex control of veins and vascular capacitance. Physiol Rev. 1983;63:1281–1342. doi: 10.1152/physrev.1983.63.4.1281. [DOI] [PubMed] [Google Scholar]
- Rotsch C, Jacobson K. Radmacher M. Dimensional and mechanical dynamics of active and stable edges in motile fibroblasts investigated by using atomic force microscopy. Proc Natl Acad Sci U S A. 1999;96:921–926. doi: 10.1073/pnas.96.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safar ME. London GM. Arterial and venous compliance in sustained essential hypertension. Hypertension. 1987;10:133–139. doi: 10.1161/01.hyp.10.2.133. [DOI] [PubMed] [Google Scholar]
- Saphirstein RJ, Gao YZ, Jensen MH, Gallant CM, Vetterkind S, Moore JR. Morgan KG. The focal adhesion: a regulated component of aortic stiffness. PLoS One. 2013;8:e62461. doi: 10.1371/journal.pone.0062461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shattil SJ, Kim C. Ginsberg MH. The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol. 2010;11:288–300. doi: 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somlyo AP. Somlyo AV. Vascular smooth muscle. I. Normal structure, pathology, biochemistry, and biophysics. Pharmacol Rev. 1968;20:197–272. [PubMed] [Google Scholar]
- Somlyo AP. Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- Sun Z, Martinez-Lemus LA, Hill MA. Meininger GA. Extracellular matrix-specific focal adhesions in vascular smooth muscle produce mechanically active adhesion sites. Am J Physiol Cell Physiol. 2008;295:C268–C278. doi: 10.1152/ajpcell.00516.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyberg JV. How changes in venous capacitance modulate cardiac output. Pflugers Arch. 2002;445:10–17. doi: 10.1007/s00424-002-0922-x. [DOI] [PubMed] [Google Scholar]
- Wang JJ, Flewitt JA, Shrive NG, Parker KH. Tyberg JV. Systemic venous circulation. Waves propagating on a windkessel: relation of arterial and venous windkessels to systemic vascular resistance. Am J Physiol Heart Circ Physiol. 2006;290:H154–H162. doi: 10.1152/ajpheart.00494.2005. [DOI] [PubMed] [Google Scholar]
- Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT. Horwitz AF. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell Biol. 2004;6:154–161. doi: 10.1038/ncb1094. [DOI] [PubMed] [Google Scholar]
- Wesly RL, Vaishnav RN, Fuchs JC, Patel DJ. Greenfield JC., Jr Static linear and nonlinear elastic properties of normal and arterialized venous tissue in dog and man. Circ Res. 1975;37:509–520. doi: 10.1161/01.res.37.4.509. [DOI] [PubMed] [Google Scholar]
- Yamin R. Morgan KG. Deciphering actin cytoskeletal function in the contractile vascular smooth muscle cell. J Physiol. 2012;590:4145–4154. doi: 10.1113/jphysiol.2012.232306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidel-Bar R. Evolution of complexity in the integrin adhesome. J Cell Biol. 2009;186:317–321. doi: 10.1083/jcb.200811067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidan A, Nordström I, Dreja K, Malmqvist U. Hellstrand P. Stretch-dependent modulation of contractility and growth in smooth muscle of rat portal vein. Circ Res. 2000;87:228–234. doi: 10.1161/01.res.87.3.228. [DOI] [PubMed] [Google Scholar]