

Abstract
The search for drugs that can kill replicating and nonreplicating Mycobacterium tuberculosis faces practical bottlenecks. Measurement of CFU and discrimination of bacteriostatic from bactericidal activity are costly in compounds, supplies, labor, and time. Testing compounds against M. tuberculosis under conditions that prevent the replication of M. tuberculosis often involves a second phase of the test in which conditions are altered to permit the replication of bacteria that survived the first phase. False-positive determinations of activity against nonreplicating M. tuberculosis may arise from carryover of compounds from the nonreplicating stage of the assay that act in the replicating stage. We mitigate these problems by carrying out a 96-well microplate liquid MIC assay and then transferring an aliquot of each well to a second set of plates in which each well contains agar supplemented with activated charcoal. After 7 to 10 days—about 2 weeks sooner than required to count CFU—fluorometry reveals whether M. tuberculosis bacilli in each well have replicated extensively enough to reduce a resazurin dye added for the final hour. This charcoal agar resazurin assay (CARA) distinguishes between bacterial biomasses in any two wells that differ by 2 to 3 log10 CFU. The CARA thus serves as a pretest and semiquantitative surrogate for longer, more laborious, and expensive CFU-based assays, helps distinguish bactericidal from bacteriostatic activity, and identifies compounds that are active under replicating conditions, nonreplicating conditions, or both. Results for 14 antimycobacterial compounds, including tuberculosis (TB) drugs, revealed that PA-824 (pretomanid) and TMC207 (bedaquiline) are largely bacteriostatic.
INTRODUCTION
Most antibiotics were discovered by identifying compounds with activity against replicating (R) bacteria (1). This paradigm is changing, as treatment of persistent bacterial infections, such as tuberculosis (TB), may benefit from the additional targeting of nonreplicating (NR) populations of bacteria that are phenotypically tolerant to antibiotics active against replicating bacteria (2, 3). Recent studies have begun to address this need through target-based and whole-cell screens to identify compounds that kill Mycobacterium tuberculosis under conditions that prevent its replication (4–11).
Phenotypic tolerance gives rise to bacterial persistence, that is, survival of some bacteria during exposure to drugs to which bacteria with the same genomic sequence are killed under standard conditions. The diverse mechanisms of phenotypic tolerance and persistence have been broadly categorized as class I and class II (3). Class I persisters are rare cells in otherwise replicating bacterial populations. Their phenotypic tolerance is intrinsic and may or may not be associated with a nonreplicating state prior to antibiotic exposure (12–14). In contrast, class II persisters are the majority in a population exposed to an extrinsic stress that prevents them from replicating. Host immunity can generate several such stresses. For this reason, while immunity contributes to the control of M. tuberculosis, immunity can, at the same time, frustrate the ability of drugs to eradicate it (15–17).
To identify molecules that target M. tuberculosis class II persisters, we undertook a screening campaign using nonreplicating conditions designed to mimic four physiological stresses encountered by M. tuberculosis in one or more of its microenvironments in the host: acidity, reactive nitrogen intermediates, hypoxia, and a fatty acid as a carbon source (6, 16, 18–26). The detection of killing of nonreplicating bacteria poses a challenge for high-throughput assays in which the readout is based on failure to resume growth in replicating conditions after removal of the compounds. Washing cells to remove extracellular- and membrane-associated antibiotics requires multiple steps of centrifuging a bacterial culture and resuspending the bacterial pellet. This is not a feasible option in high-throughput testing of molecules. Instead, we added a stage to the screening assay in which an aliquot of a culture in nonreplicating conditions is diluted into replicating conditions to permit outgrowth of surviving cells (6). The price to pay for the convenience of this approach is the false-positive rate that results when potent molecules active only in replicating conditions register as active candidates in nonreplicating conditions by virtue of their carryover from the nonreplicating phase to the outgrowth phase. Comparing the MICs in replicating conditions with those in the two-stage nonreplicating assay can suggest whether a compound is likely to be selectively active against replicating M. tuberculosis, selectively active against nonreplicating M. tuberculosis, or active against both (selectively replicating-active, selectively nonreplicating-active, and dual-active compounds, respectively) (Fig. 1). However, a definitive distinction has thus far required a classical CFU-based assay that dilutes the compound to extinction. In the CFU technique, an aliquot from each replicate with a different concentration of compound is itself serially diluted, and small aliquots taken from the latter dilutions are then spread on much larger volumes of agar.
FIG 1.

Flow chart demonstrating the placement of the CARA (gray shaded area) in a workflow between high-throughput MIC-type concentration-response assays and low-throughput assays of CFU. As a semiquantitative, medium-throughput assay for relative bacterial numbers, the CARA allows the rapid screening of many test agents to identify those that are inactive, active under replicating conditions, active under nonreplicating conditions, or both (dual-active compounds). Unlike a standard broth MIC assay, the CARA also distinguishes bacteriostatic and bactericidal activity for replicating-active compounds. Dotted lines indicate unanticipated, and often very weak, drug activities. Bacteriostatic activity is not a pertinent concept under nonreplicating conditions, because the assay conditions themselves are bacteriostatic by design.
High-throughput screening that employs a nonreplicating stage, followed by a replicating outgrowth of survivors, has identified thousands of candidate nonreplicating-active molecules and many candidate dual-active molecules (6, 26, 27). Characterization of these screening hits by a CFU-based readout presents a bottleneck in terms of labor, time, and cost (Fig. 1). To resolve this problem, we sought to couple a standard MIC broth dilution assay with a new assay that is faster than determining the CFU remaining at each concentration of compound, allows for the testing of compounds in a large number of concentrations in replicate, provides a value similar to the minimal bactericidal concentration (MBC), that is, the concentration that kills 99% of cells (MBC99) as determined from reduction of CFU on solid bacteriologic agar, eliminates the serial dilutions of the samples at each test concentration that are involved in CFU assays, reduces the activity of compounds after they are carried over into the outgrowth phase, accurately indicates whether compounds are selectively replicating-active, selectively nonreplicating-active, or dual-active, and identifies whether compounds are bactericidal or bacteriostatic under the conditions tested. The charcoal agar resazurin assay (CARA) described here fulfills those criteria.
MATERIALS AND METHODS
Strains and growth conditions.
All experiments were performed with wild-type Mycobacterium tuberculosis H37Rv strains and Mycobacterium smegmatis MC2155. M. tuberculosis strains were obtained from the ATCC (PA-824 experiments) or from R. North (the Trudeau Institute, Saranac, NY) (all other experiments). M. tuberculosis was passaged in Middlebrook 7H9-albumin-dextrose-NaCl-Tyl (0.2% glycerol, 0.5% albumin, 0.2% dextrose, 0.085% NaCl, and 0.02% tyloxapol) or Middlebrook 7H9-oleic acid-albumin-dextrose-catalase (OADC)-Tyl (0.2% glycerol, oleic acid, albumin, dextrose, catalase) and plated on solid 7H11-OADC at 5% CO2 and 20% O2. The replicating and nonreplicating high-throughput screening methods were as described previously (6, 28). The nonreplicating conditions consist of a modified Sauton's medium-based mixture (per liter: 0.5 g KH2PO4, 0.5 g MgSO4, 0.05 g ferric ammonium citrate, 0.5% bovine serum albumin (BSA), 0.085% NaCl, and 0.02% tyloxapol) at pH 5.0 with 0.5 mM NaNO2, 0.05% butyrate in place of dextrose, and incubated at 1% O2 and 5% CO2. M. tuberculosis in mid-log phase was washed twice in Dulbecco's phosphate-buffered saline (PBS) containing 0.02% tyloxapol prior to resuspension in the nonreplicating medium at an optical density at 580 nm (OD580) of 0.1 for MIC and high-throughput screening (HTS) assays. Replicating M. tuberculosis was used at an OD580 of 0.01 for all MIC assays. Single cell suspensions were used for CFU-based assays and prepared by removing clumped bacilli using a 10-min centrifugation at 123 × g with no brake.
Charcoal agar microplates (CARA plates).
The 7H11 Middlebrook agar containing 10% OADC supplement was prepared with additional activated carbon (0.4%, wt/vol). The activated charcoal was autoclaved with 1 liter of 7H11 agar in a 2-liter Erlenmeyer flask containing a large magnetic stir bar. The autoclave tray holding the flask was filled with 1 to 2 in. of water to prevent the medium from boiling out of the Erlenmeyer flask. After autoclaving, the medium was cooled to 55°C with slow stirring. OADC supplement was added after the medium had cooled to the touch. The 2-liter Erlenmeyer flask containing the medium was kept on a magnetic stir plate at 55 to 60°C at the slowest speed (1 to 1.5, using a VWR hot plate stirrer) to maintain the charcoal in suspension and prevent particles from settling. Two hundred microliters of 7H11-OADC-charcoal were added to 96-well Corning microplate wells using a Rainin 12-channel multipipettor. Care was taken to avoid dripping liquid agar between wells because the agar may support growth of mold upon incubation. Tips were changed frequently because the medium rapidly solidifies and blocks the tips. Upon solidifying, agar plates were placed in large Ziploc bags to prevent drying and stored at 4°C. Before use, plates were equilibrated to room temperature (RT) and their lids were dried to remove condensation. Overdried CARA plates might immediately absorb the resazurin (7-hydroxy-3H-phenoxazin-3-one-10-oxide) solution, leading to variation or inability to determine fluorescence units.
CARA for Mycobacterium tuberculosis.
Figure 2 presents an overview. A single-cell suspension of M. tuberculosis was exposed to a test agent or a dimethyl sulfoxide (DMSO) vehicle control for 7 days in 96-well microplates containing 200 μl liquid medium specific to replicating or nonreplicating conditions. DMSO was maintained at a 1% final concentration. Compound dilutions were placed in columns 3 to 11, and the vehicle control was in column 2. Columns 1 and 12 did not contain test agents but were filled with 200 μl sterile Dulbecco's PBS to minimize the evaporation of interior wells. Rows A to H were used. Dose-response assays were prepared with 3 replicates per concentration. Results are presented as mean fluorescence ± standard deviation (SD). At day 7, cells were mixed with a P200 multichannel pipette set at 40 μl (pipetting 10 times up and down while stirring in a circular motion to ensure cells on the edges of the well bottoms were mixed); then, using a P20 multichannel pipette, 10 μl was spotted onto the surface of the 7H11-OADC-charcoal agar plates (CARA microplates). On the same day that test wells were transferred to CARA microplates for the outgrowth, eleven 2-fold dilutions of a single-cell suspension of wild-type M. tuberculosis culture, starting at an OD580 of 0.1, were spotted in 8 replicates across a CARA microplate from wells 1 to 11. This plate served as a standard curve. Inoculated microplates were stacked, placed in Ziploc bags with the zipper cracked open, and incubated at 37°C at 20% O2 and 5% CO2 for 7 days. On day 7, one may observe masses of microcolonies growing in the control wells. Forty microliters of a 1:1 solution of alamarBlue (AB) stock solution and 10% polyethylene sorbitol ester (Tween 80 [TW80], 10% vol/vol in double-distilled water [ddH2O]) was added directly to the surface of the microplate and on top of M. tuberculosis microcolony growth. The developing reagent is AB-TW80. Premoistening all wells with 40 μl 7H9-OADC prior to addition of the AB-TW80 developing solution was recommended if the CARA plates appeared that they became dry during their incubation at 37°C. This technique can help prevent the CARA developing reagent from absorbing into the agar.
FIG 2.

Schematic of the CARA illustrating results for 8 hypothetical compounds with distinct replicating and nonreplicating activities as labeled, manifested by the pink color of bacterially reduced resazurin in microtest plate wells in the lower part of the schematic diagram. Microplates used for OD580 readings are light-colored: white wells depict no growth, and brown wells indicate turbidity due to growth. The CARA microplate wells are black due to the charcoal, which is in suspension, and remains black where there are insufficient bacteria to reduce the resazurin but turn bright pink if there is bacterial growth. Detailed descriptions of these 8 combinations of MIC/CARA results, and how to interpret them, can be found in Fig. S5 and S6 in the supplemental material. PAE*, postantibiotic effect.
The plates were slowly rocked back and forth to ensure the AB-TW80 mixture coated the surface of the wells. Plates were bagged again and incubated at 37°C for 1 h. If the M. tuberculosis colony mass floated to the liquid surface, it was gently pushed down to ensure it was moistened by the AB-TW80 solution. At 1 h post-AB-TW80 addition, CARA microplates were placed in a biosafety hood, and lids were removed for 15 min for temperature equilibration, which was critical to avoid accumulation of condensation on the lids or stickers that would affect the fluorescence reading. An optical-quality PCR sticker (Denville) was placed over the plate's surface and pressed onto the plate using a Kimwipe to create a tight seal. Fluorescence was determined via top read with an excitation of 530 nm and emission at 590 nm (SpectraMax M5; Molecular Devices). Resazurin powder is a less expensive alternative to commercial alamarBlue and can be used to prepare the CARA developing solution: 0.01% resazurin (wt/vol) in sterile Dulbecco's PBS (calcium chloride and magnesium chloride free; Gibco) containing 5% Tween 80.
CARA for Mycobacterium smegmatis.
The M. smegmatis CARA was identical to the M. tuberculosis CARA but with the following differences: CARA microplates were prepared with 7H11 agar without the OADC supplement, the length of drug exposure was 24 h; the length of bacterial recovery on CARA plates was 24 or 48 h; and Tween 80 was dissolved in Dulbecco's PBS (calcium chloride and magnesium chloride free; Gibco) instead of water.
Source of chemicals.
Unless otherwise indicated, all chemicals were from Sigma at the maximum purity available. We are grateful to H. Boshoff and the TB Alliance for kindly providing TMC207 and PA-824, respectively.
Drug binding assays.
Test agents were added from DMSO stocks to Dulbecco's PBS (calcium chloride and magnesium chloride free; Gibco) containing or not containing 0.5% BSA or 0.4% activated charcoal (wt/vol). After 20 min at room temperature, samples were passed across 10-kDa molecular weight (MW) cutoff filters (Amicon) to remove residual charcoal- and BSA-drug complexes. The flowthrough was analyzed for any unbound drug by liquid chromatography-mass spectrometry (LC-MS). To address binding to charcoal at later time points, microplates containing compounds in PBS containing or not containing 0.4% activated charcoal (wt/vol) were incubated 18 to 24 h at room temperature. Microplates were then centrifuged to pellet the charcoal and supernatants analyzed by LC-MS.
Data analysis.
Data were archived and analyzed using Collaborative Drug Discovery (Burlingame, CA) (29). When there was obvious M. tuberculosis growth in DMSO control wells or wells in which a compound was inactive, we observed significant variation in maximal fluorescence (sometimes >2-fold). In such cases, it may be easier to observe trends by increasing the number of experimental replicates and plotting data as means ± standard error. Curve fit plots (shown as red lines) for inhibitor versus fluorescence plots were generated in Prism 6 for Mac OS X using the analysis parameters of “log inhibitor versus response—variable slope (4 parameters)” to help illustrate fluorescence trends. Given the variability in DMSO-treated values, including bleaching in wells with high fluorescence values, 50% inhibitory concentrations (IC50s) were not calculated due to the difficulty normalizing data as percent inhibition compared to that of controls. Examples of how to determine the CARA MBC≥99 are shown in Fig. S5 in the supplemental material, and examples of how to determine the inactive, bactericidal, or bacteriostatic activities of small molecules are shown in Fig. S6 in the supplemental material and summarized in Table 1. The JChem add-on (ChemAxon) was used with Microsoft Excel to visualize structures with data.
TABLE 1.
Data analysis of MIC90 and CARA data derived from a 96-well microplate formata
| Assay type | Parameter | Example A | Example B | Example C | Example D | Example E | Example F | Example G | Example H | Example I |
|---|---|---|---|---|---|---|---|---|---|---|
| Example(s) in Fig. 2 | 1, 2 | 3 | 4 | 4 | 5 | 6 | 5 | 6 | 7 | |
| Nonreplicating assay | Inactive | Inactive | Inactive | Inactive | Cidal | Cidal | Potent cidal | Potent cidal | Cidal | |
| Replicating assay | Cidal | Static + no PAEb | Static + weak PAE | Static + potent PAE | Cidal | Static (±PAE) | Cidal | Static (± PAE) | Inactive | |
| Activity classification | R-cidal | R-static | R-static | R-static | Dual-active | Dual-active | Dual-active | Dual-active | NR-active | |
| Ratio of MIC90sc: R/NR | <0.2 | <0.2 | <0.2 | <0.2 | 0.2–5 | 0.2–5 | >5 | >5 | >5 | |
| CARA: nonreplicating | No fluorescence decrease | Yes | Yes | Yes | Yes | |||||
| Dose-dependent decrease to background at MIC90 | Maybe | Maybe | Yes | Yes | Yes | |||||
| Dose-dependent decrease to background at ≥4× MIC90 | Maybe | Maybe | Maybe | |||||||
| CARA: replicating | Static window | Yes | Yes | Maybe | Yes | Yes | ||||
| No fluorescence decrease | Yes | Maybe | Maybe | Yes | ||||||
| Dose-dependent decrease to background at MIC90 | Yes | Maybe | Yes | Maybe | Yes | Maybe | ||||
| Dose-dependent decrease to background at ≥4× MIC90 | Yes | Maybe | Maybe | Maybe | ||||||
| Expand axis to view SW? | Yes | Maybe | Maybe | |||||||
| Alternative explanation | Charcoal may not bind drug effectively | Charcoal may not bind drug effectively | Charcoal may not bind drug effectively | Charcoal may not bind drug effectively |
This table summarizes Fig. 2 and Fig. S5 and S6 in the supplemental material. Blank cells represent no impact or a lack of relevance.
PAE, postantibiotic effect.
Derived from data obtained using a 384-well format.
RESULTS
Limitations of broth-based MIC determinations for assessing activity under replicating and nonreplicating conditions.
Our replicating and nonreplicating conditions for screening compounds against M. tuberculosis differ in inoculum (OD580 of 0.01 versus that of 0.1), gas phase (20% O2 versus 1% O2, each with 5% CO2), base medium (Middlebrook 7H9 versus Sauton's), and duration of exposure to compound (7 days in the replicating assay versus 3 days in the nonreplicating assay at the original concentration, followed by 7 more days after a 1:5 dilution into the outgrowth conditions) (6, 28). Nevertheless, we can infer from the ratio of MIC90 values in the two conditions (R-MIC90/NR-MIC90) whether a compound is likely to be selectively replicating-active or nonreplicating-active (Fig. 1). In the 384-well format of the replicating and nonreplicating assays, for an R-MIC90/NR-MIC90 of <0.2, the compound is probably selectively replicating-active; if the R-MIC90/NR-MIC90 is between 0.2 and 5, the compound has potential to have dual activity; and if R-MIC90/NR-MIC90 is >5, the compound is probably selectively nonreplicating-active or dual-active with more potent nonreplicating activity than that of replicating activity. These predictive ratios are different for a 96-well, 2-plate assay format that allows for a 21-fold dilution into outgrowth conditions (R/NR): <0.05, replicating active; 0.05 to 21, candidate dual; >21, nonreplicating active or dual active with more potent nonreplicating activity.
Some compounds, such as moxifloxacin and rifampin, confound the prediction of dual activity based on ratios of ∼0.25 to 0.5. A 7-day CFU-based assay revealed that they only exerted bactericidal activity in the multistress nonreplicating conditions at concentrations—25 and 50 μM, respectively—far above their R-MIC90 (≤0.16 μM), and even then, the extent of killing was relatively modest: 1.5 to 3 log10 CFU (26). We speculated that the apparent activity of moxifloxacin and rifampin in the nonreplicating screening assay might have reflected carryover of the drug from the nonreplicating phase to the outgrowth phase.
Such observations prompted us to test the impact of including activated charcoal in liquid medium and solid bacteriologic agar, first with untreated M. tuberculosis as a control and then with M. tuberculosis exposed to drugs under replicating conditions. Finally, we compared the results of MICs and CARAs for 13 antimycobacterial compounds, including TB drugs, under replicating and nonreplicating conditions.
The impact of activated charcoal on assays with untreated M. tuberculosis.
In the single-plate, 384-well format of our multistress nonreplicating assay (6), compounds are carried over into the replicating outgrowth phase of the nonreplicating assay at 20% of their original screening concentration (Fig. 1). As noted, potent replicating-active compounds may register as potential nonreplicating-active compounds by killing in the outgrowth phase of the assay. To reduce such false-positive results, we supplemented liquid 7H9-glycerol bacteriologic medium with 0.4% (wt/vol) activated charcoal. Constant stirring was needed to keep the charcoal powder in suspension. Moreover, activated charcoal decolorized the 7H9 medium (see Fig. S1a in the supplemental material), suggesting that it bound small molecule components, and the charcoal quenched the fluorescence of resazurin (see Fig. S1b in the supplemental material), whose reduction to resorufin is a useful quantitative reflection of relative mycobacterial numbers (30, 31). To bypass these problems, we used an agar-plate-based method to replace the liquid-based outgrowth (Fig. 2). Activated charcoal is a common supplement in bacteriologic agar, where it remains homogeneously suspended. Addition of 0.4% (wt/vol) activated charcoal to 7H11-OADC agar had no detectable effect on the number of M. tuberculosis CFU or the time of their appearance, although the colonies were often slightly smaller (see Fig. S1c and d in the supplemental material). When untreated M. tuberculosis was taken from replicating conditions and dispensed onto charcoal agar, microcolonies were visible by day 5. When the M. tuberculosis came from nonreplicating conditions, microcolonies were detectable by days 7 to 10. Microcolonies were too small to count reliably, and microwells were too small to support statistically useful numbers of discrete macrocolonies. Therefore, to semiquantify the M. tuberculosis biomass on the agar surface, we added alamarBlue and Tween 80 as a 1:1 mixture (vol/vol, 100% alamarBlue and 10% TW80) on top of the microcolonies in each well of the CARA plate and recorded fluorescence by top reading. Fluorescence generated by untreated M. tuberculosis was readily detectable after 1 h of incubation at 37°C (Fig. 2). Longer incubation times were confounded by the agar absorbing the AB-TW80 developing solution and the charcoal-mediated quenching of fluorescence (see Fig. S1b in the supplemental material).
There was a linear-log relationship between fluorescence (linear) and the starting number of untreated M. tuberculosis bacilli (log) over a 2 to 3 log10 range of M. tuberculosis bacilli spotted prior to incubating the plate (Fig. 3). Thus, we can infer that complete loss of CARA fluorescence indicates ≥99% reduction in bacterial numbers. Since the lowest concentration that affords a 2 log10 kill of M. tuberculosis bacilli commonly defines the minimum bactericidal concentration of antibiotics (MBC99), we reasoned that in the CARA, the lowest drug concentration giving rise to no fluorescence above background might serve as a surrogate for the MBC (see Fig. S5 in the supplemental material). This concentration will here be termed the “CARA-MBC≥99.”
FIG 3.

Correlation of the number of M. tuberculosis bacilli initially plated and alamarBlue fluorescence upon developing the CARA microplate at day 7 or day 10 postinoculation. Ten microliters of 10-fold serial dilutions of a culture of M. tuberculosis H37Rv, beginning at an OD580 of 0.1 (corresponding to ∼5 × 107 CFU/ml), was spotted onto wells of either NARA (a and c) or CARA (b and d) microplates. The M. tuberculosis was a single-cell suspension prepared from an exponentially growing culture in mid-log phase and was not exposed to test agents. Results are means ± SD from 8 replicates in one of 3 similar experiments.
The actual number of initially plated M. tuberculosis bacilli that generated a signal within the 2 to 3 log10 dynamic range and whether the range extended over 2 log10 or 3 log10 depended on how long M. tuberculosis was allowed to grow on the CARA plates before the addition of resazurin (Fig. 3a to d) and, to a lesser extent, on how long the cells were incubated with resazurin before fluorescence was recorded. A 10-day growth of M. tuberculosis on a CARA plate increased sensitivity and dynamic range compared to those of a 7-day growth. Nevertheless, we used 7-day incubation because it was faster, and its sensitivity was adequate.
Inclusion of charcoal did not affect the relative relationship between fluorescence intensity and initial bacterial number, as shown in Fig. 3 for the comparison of CARA plates with normal agar resazurin assay (NARA) plates. However, inclusion of charcoal did reduce the maximum fluorescence intensity attained and increased the standard deviations of replicates at higher fluorescence intensities (Fig. 3).
Similar results were obtained for the fast-growing Mycobacterium smegmatis (see Fig. S2 in the supplemental material) with minor adjustments to the protocol. We reasoned that the faster doubling time of M. smegmatis (3 h compared to ∼18 to 24 h for M. tuberculosis) would require shorter CARA incubation times. Incubations of 24 h (see Fig. S2a in the supplemental material) or 48 h (see Fig. S2b in the supplemental material) prior to developing with AB-Tw80 showed that, similar to the CARA for M. tuberculosis, the CARA for M. smegmatis had a 2 to 3 log10 CFU window. Although the 48-h CARA incubation provided a larger dynamic range than that of the 24-h CARA incubation for M. smegmatis, we routinely employed 24-h CARA incubations to decrease the overall assay time. For nonreplicating M. smegmatis assays, we decided to develop the CARA plate at 24 or 48 h based on the presence or absence of visible growth from DSMO-treated control wells.
Operational terms and data analysis to distinguish bactericidal and bacteriostatic drug actions.
Based on the results presented below as well as those obtained in testing over 100 replicating-, nonreplicating-, or dual-active compounds from academic and industrial collaborations, we developed a guide to interpreting CARA data (Table 1 and Fig. 2; see also Fig. S5 and S6 in the supplemental material). For the sake of clarity, we present the guide here before completing the description of how the CARA was validated. To illustrate the interpretive concepts, reference will be made to figures that will be discussed again later in the context of their specific results.
Although classifying drugs as replicating- and/or nonreplicating-active compounds is often straightforward (Fig. 2), for some drugs, further classification of a replicating-active compound as bacteriostatic or bactericidal requires careful analysis of CARA results. By definition, all bacteriostatic drugs abrogate bacterial replication. However, there is no standardized definition for the amount of bacterial kill tolerated for a drug to remain classified as bacteriostatic and the point at which further CFU reduction changes the classification to bactericidal. Drugs active against fast-growing bacteria such as Staphylococcus aureus are bacteriostatic if they decrease CFU by <99.9% in 24 h (32), whereas drugs active against slow-growing mycobacteria are conventionally considered bacteriostatic if they result in a <99% CFU reduction in 15 days (33). Such bacteriostatic-bactericidal distinctions are arbitrary, depending on the time of drug exposure, the bacterial strain and species, the type of liquid medium used during the drug exposure, and the type of agar medium used for recovery (33). Below, we illustrate 99% and 99.9% CFU reduction for CFU data (Fig. 7n, 8h, and 9a, c, and e). In the interest of speed, we chose to limit the replicating drug exposure in the CARA to 7 days rather than 15 days. The 7-day drug exposure was also necessary for nonreplicating conditions in which we have observed that untreated M. tuberculosis bacilli decrease in CFU after 7 days (26).
FIG 7.

Replicating-bacteriostatic molecules. (a) Results expected for bacteriostatic compounds that possess a postantibiotic effect or not. Compounds whose R-MIC90 and R-CARA data for PAS (b, c), fenamisal (d, e), TMC207 (f, g), ethambutol (h, i), linezolid (j, k), and ethionamide (l, m) were consistent with bacteriostatic activity. The insets for PAS (b) and fenamisal (d) have an expanded right y axis to allow visualization of the correct CARA-MBC≥99 (this method of interpretation is described in Fig. S5 in the supplemental material). CFU for wild-type replicating (n) and nonreplicating (o) M. tuberculosis after a 7-day exposure to linezolid confirmed CARA results that predicted it was replicating-bacteriostatic and nonreplicating-inactive. To help determine if any given compound concentration is inactive (grow), bacteriostatic (static), or bactericidal (cidal), the CFU graphs have been annotated to the right of data points with respect to the starting inoculum at time zero. Two definitions of bacteriostatic compounds are shown, in which a compound results in 99% or 99.9% kill relative to the starting inoculum. CFU data for replicating M. tuberculosis exposed to PAS and fenamisal are shown in Fig. S4 in the supplemental material. Results are means ± SD from 3 replicates in one of two similar experiments.
FIG 8.

Dual-active molecules. (a) Expected activity profiles of molecules with activity against replicating and nonreplicating bacilli. Drugs that were identified as dual-active compounds are PA-824 (b, c), moxifloxacin (d, e), and rifampin (f, g). CFU assays for replicating (h) and nonreplicating (i) wild-type M. tuberculosis treated with rifampin for 7 days corroborate CARA results that indicated rifampin is replicating-bactericidal with modest nonreplicating-bactericidal activity at concentrations manyfold higher than its R-MIC90 and R-CARA. CFU data show means ± SD from 3 replicates in 3 similar experiments. CFU data for PA-824 are shown in Fig. 9 and, for moxifloxacin, in Fig. S3c and d in the supplemental material.
FIG 9.
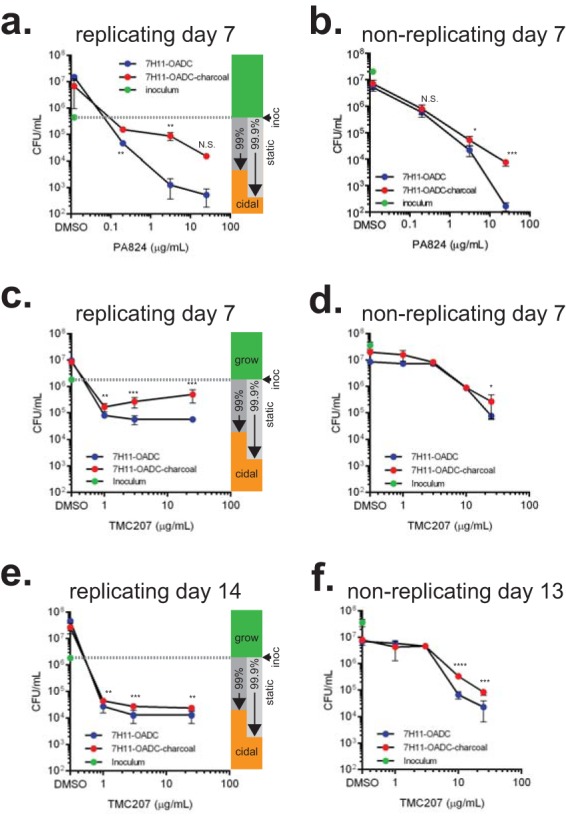
PA-824 and TMC207 display less killing when M. tuberculosis bacilli are enumerated on 7H11-OADC plates supplemented with activated charcoal. CARA results indicated that PA-824 and TMC207 were replicating bacteriostatic drugs. PA-824 and TMC207 display hallmarks of bacteriostatic drugs after 7 days' exposure when tested by the CARA under replicating conditions (Fig. 7f and 8b) and nonreplicating conditions (Fig. 7g and 8c). The impact of 0.4% (wt/vol) activated charcoal on drug carryover was determined by a CFU assay in which aliquots of drug-treated replicating (a, c, e) or nonreplicating (b, d, f) cells were plated on 7H11-OADC with or without 0.4% activated charcoal. P values (unpaired Student's t test) compare plating ± charcoal for each drug concentration. N.S., not significant; *, P < 0.2; **, P < 0.1; ***, P < 0.05. Results are means ± SD from 3 replicates in one of at least two similar experiments.
We anticipated that direct comparison of the CARA-MBC≥99 with the standard liquid broth MIC90 might distinguish bacteriostatic from bactericidal compounds (Fig. 2). Because liquid broth MIC90 dose response curves often have a 2-fold variation between biological replicates, significant differences in MIC90 are generally considered to be those that are >4-fold. Thus, a test agent would be considered bacteriostatic if it failed to reduce fluorescence in the CARA to the background level when tested at its liquid broth MIC90 and at concentrations of ≥4-fold higher (Fig. 2; see also Fig. S5 in the supplemental material).
While the CARA accurately predicts bactericidal activity when the CARA curve mirrors the curve used to determine the MIC90 (Fig. 2; see also S6a in the supplemental material), CARA results for a bacteriostatic drug can be difficult to interpret (Fig. 2; see also Fig. S6b to d in the supplemental material). Although some compounds are obviously bacteriostatic because they have minimal impact on CARA fluorescence relative to DMSO controls (Fig. 2; see also Fig. S6b and c in the supplemental material), in some cases, the CARA for replicating M. tuberculosis fails to afford a clear-cut distinction between bacteriostatic and bactericidal activity (see Fig. S6d in the supplemental material). This can result from bacteriostatic compounds exhibiting a postantibiotic effect (PAE) that strongly suppresses colony growth on the CARA plates (Fig. 2), such that bacteriostatic compounds with a postantibiotic effect give results similar to those for bactericidal compounds.
For these reasons, we have introduced a replicating-assay-specific operational term, “static window” (SW), to describe a dose response shift (the window) of ≥4-fold between an MIC90 and the CARA-MBC≥99 (see Fig. S6d in the supplemental material). A static window is a hallmark of a subset of bacteriostatic compounds such as linezolid (described later). Key features of static windows are 2-fold: the CARA fluorescence must decrease to background levels in a dose-dependent manner, and the CARA-MBC≥99 is ≥4-fold higher than the broth MIC90. In some cases, a static window is discernible from visual inspection of graphed results only after expanding the y axis to pinpoint the concentration of drug at which the CARA fluorescence rises above the background (see Fig. S5b and c in the supplemental material). Static windows are predictive of bacteriostatic activity; however, they do not rule out the possibility that some bactericidal activity might occur at some drug concentrations or longer drug exposures as has been observed for fluoroquinolone antibiotics (34, 35).
Because the nonreplicating assay is bacteriostatic and coupled to a replicating outgrowth stage to permit surviving cells to increase in biomass, a replicating-inactive drug must be nonreplicating-bactericidal to give an NR-MIC90 (Fig. 1). For a nonreplicating-specific compound, decreases in CARA fluorescence can be attributed to CFU reduction, and the concept of a static window does not apply.
For compounds that are inferred to be candidate dual-active based on the R-MIC90 to NR-MIC90 ratio (Fig. 1), a dose-dependent NR-CARA fluorescence decrease to background levels suggests bona fide nonreplicating-bactericidal activity. However, when the R/NR-MIC90 ratio indicates that a compound is replicating-active (Fig. 1), sometimes the NR-CARA fluorescence decreases at concentrations significantly greater than the R-MIC90 and R-CARA-MBC≥99. Such a compound might be a replicating compound whose NR-CARA fluorescence decrease results from the failure of the charcoal to bind a carried over compound, a compound with weak bactericidal activity against nonreplicating bacteria, or a combination. An example of this phenomenon is described below for kanamycin.
Activated charcoal binds antimycobacterial compounds.
We first compared the rapid binding of antimycobacterial compounds to activated charcoal or bovine serum albumin fraction V (BSA) and then tested the impact of incubation time on the binding of the compounds to activated charcoal. In the first experiment, we compared 0.4% activated charcoal (wt/vol) to 0.5% BSA (wt/vol), the concentration of BSA found in 7H11-OADC agar. We used 30 μg/ml and 100 μg/ml of drugs to model concentrations at the high end of MIC90 assays, in which carryover might play a significant role. Compounds were diluted into phosphate-buffered saline (PBS), mixed with PBS containing charcoal or BSA for 20 min at room temperature, and then passed over a 10-kDa filter to retain charcoal or BSA drug complexes. Unbound drug (the flowthrough) was quantified by LC-MS. For ethambutol, ethionamide, fenamisal, isoniazid (INH), linezolid, moxifloxacin, para-aminosalicylate (PAS), streptomycin, and PA-824, <25% of the compound was retained in the BSA fraction. For nitazoxanide, oxyphenbutazone, and rifampin, 25% to 75% of the compound was retained in the BSA fraction. Put differently, BSA failed to rapidly bind >75% of any compound tested. In contrast, after 20 min, activated charcoal had removed 95% to 100% of the ethambutol, ethionamide, fenamisal, linezolid, nitazoxanide, oxyphenbutazone, PA-824, PAS, and rifampin, leaving ∼1% to 12% of moxifloxacin and nitazoxanide and ∼55% to 80% of isoniazid and streptomycin unbound. When we extended the incubation time to 18 to 24 h, activated charcoal bound >99.9% of all compounds tested except ethambutol (∼3% unbound) and streptomycin (58% unbound) (Table 2).
TABLE 2.
Impact of activated charcoal on binding antimycobacterial compounds in aqueous solution and in agar plates
| Compound | Replicating MIC90 (μg/ml) | Replicating NARA-MBC≥99 (μg/ml) | Replicating CARA-MBC≥99 (μg/ml) | Nonreplicating MIC90 (μg/ml) | Nonreplicating NARA-MBC≥99 (μg/ml) | Nonreplicating CARA-MBC≥99 (μg/ml) | Replicating CARA-MBC≥99/NARA-MBC≥99 (fold shift)a | Nonreplicating CARA-MBC≥99/NARA-MBC≥99 (fold shift)a | Percentage removed by 0.4% (wt/vol) activated charcoalb |
|---|---|---|---|---|---|---|---|---|---|
| Streptomycin | 2.5 | 2.5 | 2.5 | 5–>10 | >10 | >10 | 1 | ≥2 | 41.7 |
| Ethambutol | 3.13–6.25 | 3.13–12.5 | 50–100 | 25–50 | 50–100 | >100 | 8 | ≥2 | 97.2 |
| PA-824c | 0.2–0.4 | 0.78 | ≥25 | 0.4–0.78 | 0.78 | 12.5–25 | ≥32 | 16–32 | ≥99.9 |
| Rifampin | 0.08–0.16 | 0.16–0.31 | 0.16–0.31 | 0.31–1.25 | 0.63–1.25 | 5–10 | ≤2 | 4–16 | ≥99.9 |
| Moxifloxacin | 0.08–0.2 | <0.4 | <0.4 | 0.78–3.13 | 1.56–3.13 | ≥100 | NDd | ≥32 | ≥99.9 |
| Oxyphenbutazone | >100 | >100 | >100 | 3.13–12.5 | 12.5 | 12.5–25 | ND | ≤2 | ≥99.9 |
| Ethionamide | 1.56–3.13 | 6.25–12.5 | 25 | 25 | 25 | >100 | 2–4 | ≥4 | ≥99.9 |
| Isoniazid | <0.4 | ND | ND | 12.5 | 12.5 | >100 | ND | ≥4 | ≥99.9 |
| TMC207 | 0.2 | 0.78–1.56 | 12.5–>25 | 1.56–3.13 | 1.56 | 12.5–≥25 | ≥8 | ≥8 | ≥99.9 |
| Linezolid | 1.03–2.03 | NTe | 33 | 16.5 | NT | >33 | ND | ND | ≥99.9 |
| PAS | <0.4 | <0.4–1.56 | 12.5–100 | 25 | 25–50 | 50–100 | ≥32–64 | 2–4 | ≥99.9 |
| Fenamisal | <0.4 | <0.4 | 100 | 0.78–1.56 | 0.78 | 100 | ≥256 | 128 | ≥99.9 |
| Nitazoxanidec | 25 | 100 | 100 | 3.13 | 6.25 | 6.25 | ≤2 | ≤2 | ≥99.9 |
Range of two independent experiments measuring impact of 0.4% (wt/vol) in CARA microplate. Ratios of ≥4 indicate significant loss of activity when plated on activated charcoal.
At 24 h, average of two replicates.
In the NARA, n = 1.
ND, not determined because the compound was inactive or active at concentrations tested, had an indeterminate MBC≥99 in the replicating or nonreplicating assay, or was not tested in the NARA.
NT, not tested.
Thus, for every compound tested, activated charcoal sequestered the drug faster and more extensively than BSA. In many cases, 100% of compound was absorbed by activated charcoal within 20 min, and by 24 h, activated charcoal had absorbed almost all compounds to >99.9%. The notable exception was streptomycin and, to a lesser extent, ethambutol.
Impact of activated charcoal on assays with drug-treated M. tuberculosis.
Next, we tested the impact of including activated charcoal in the agar to allow recovery of viable wild-type replicating or nonreplicating M. tuberculosis bacilli from liquid MIC90 assays (Table 2). To do this, we compared the results of NARA versus those of CARA plates for a set of antimycobacterial compounds. Inclusion of charcoal had a major effect on the apparent activity of INH, rifampin, moxifloxacin, PAS, fenamisal, ethionamide, PA-824, TMC207, and ethambutol but little impact for oxyphenbutazone, nitazoxanide, and streptomycin (Table 2) for head-to-head comparisons with and without charcoal; instead, results are shown below for replicate experiments with charcoal. Because inclusion of activated charcoal never increased a drug's apparent potency and only decreased the apparent potency of some drugs, its effect did not appear to be directly exerted on M. tuberculosis but on drugs with the potential to inhibit growth during the agar outgrowth phase. Thus, as a routine, we included activated charcoal in the microplate agar assay and used the resulting CARA to study the impact of TB-active agents on replicating and nonreplicating M. tuberculosis bacilli.
PAS and fenamisal were carried over in agar replicating conditions (Table 2). In nonreplicating conditions (Table 2), the impact on including activated charcoal ranged from minor, e.g., 2- to 4-fold, as seen with PAS (Fig. 4a and b), to major, e.g., 128-fold, as seen with the structurally related but mechanistically distinct drug (36) fenamisal (Fig. 4c and d). That is, fenamisal was bacteriostatic at its R-MIC90 and only exerted bactericidal activity at concentrations nearly 2 orders of magnitude greater when charcoal was present in the recovery phase on agar. This suggested that, unless absorbed by charcoal, fenamisal continued to suppress the growth of M. tuberculosis bacilli when it was carried over from the MIC plate to the agar-based outgrowth wells.
FIG 4.

Impact of 0.4% (wt/vol) activated charcoal in 7H11-OADC agar on apparent antimycobacterial activity of certain compounds, likely due to adsorption of compounds carried over to the agar culture from the broth MIC assay. Nonreplicating M. tuberculosis was exposed to para-aminosalicylate (PAS) or fenamisal for 7 days, and aliquots were spotted from the drug-containing MIC microtest plates to microtest plates containing drug-free agar and activated charcoal (CARA) or noncharcoal “normal” agar (NARA). CARA and NARA plates were then incubated 7 to 10 days prior to addition of resazurin and Tween 80. For PAS, there was little difference between growth on CARA (a) and NARA (b) plates. For fenamisal, approximately a 64-fold shift was observed by the addition of activated charcoal (c, d). The CARA data are from the same experiment as in Fig. 7 and are duplicated here for illustrative purposes. Results are means ± SD from 3 replicates in one of two similar experiments.
Comparison of MIC90s and the CARA for TB-active compounds.
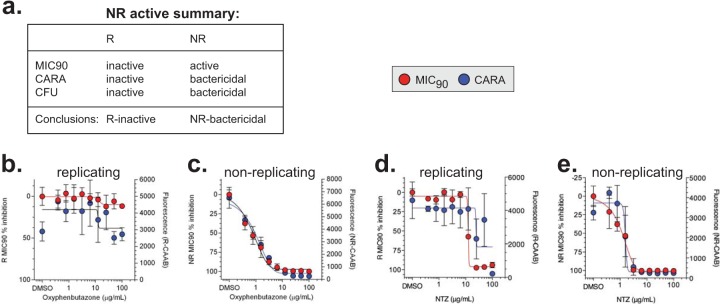
We then tested a compendium of first- and second-line antitubercular drugs and nitazoxanide, an anti-infective used for other indications that has activity against M. tuberculosis in vitro (37, 38) (Fig. 5 to 9). The CARA results distinguished 4 classes of compounds: nonreplicating-bactericidal, replicating-bactericidal, replicating-bacteriostatic, and dual-active, meaning either replicating-bacteriostatic or replicating-bactericidal as well as nonreplicating-bactericidal (Tables 1and 3).
FIG 5.
Nonreplicating-active molecules. (a) An overview of expected results for compounds with predominantly nonreplicating activity. The two examples, oxyphenbutazone (b, c) and nitazoxanide (d, e), were previously demonstrated to have nonreplicating-bactericidal activity using a classical CFU-based assay (6, 37).
TABLE 3.
Summary of CARA data for M. tuberculosis H37Rv exposed to antimycobacterial compounds
| Compound | R activitya |
NR activity(bactericidal)a | Activity prediction based on CARA results | Location of CFU results | |||
|---|---|---|---|---|---|---|---|
| Bactericidal | Bacteriostatic |
||||||
| No PAE | PAE | SW | |||||
| PA-824 | +/− | + | − | − | + | Dual active; equipotent R and NR activity; R-static + weak PAE | Fig. 9 |
| Rifampin | + | n/ab | n/a | n/a | +/−c | Dual active; R-bactericidal with moderate NR activity | Fig. 8 |
| Moxifloxacin | + | n/a | n/a | n/a | +/−c | Dual active; R-bactericidal with weak NR activity | Fig. S3 in the supplemental material |
| Oxyphenbutazone | − | n/a | n/a | n/a | + | NR-bactericidal | Reference 6 |
| Nitazoxanide | +/− | n/a | +/− | +/− | + | NR-bactericidal; weak R activity (likely static) | Reference 37 |
| Isoniazid | + | n/a | n/a | n/a | − | R-bactericidal | Reference 4 |
| Kanamycin | + | n/a | n/a | n/a | −c | R-bactericidal | Fig. S3 in the supplemental material |
| Streptomycin | + | n/a | n/a | n/a | − | R-bactericidal | NTd |
| TMC207 | − | + | − | − | − | R-bacteriostatic | Fig. 9 |
| Ethambutol | − | − | + | +/− | − | R-bacteriostatic + moderate PAE | NT |
| Ethionamide | − | − | + | +/− | − | R-bacteriostatic + moderate PAE | NT |
| Linezolid | − | − | + | + | − | R-bacteriostatic + moderate PAE | Fig. 7 |
| PAS | +/− | − | + | +/−e | − | R-bacteriostatic + strong PAE | Fig. S4 in the supplemental material |
| Fenamisal | +/− | − | + | +/−e | − | R-bacteriostatic + strong PAE | Fig. S4 in the supplemental material |
+, positive; −, negative; +/−, borderline.
n/a, not applicable.
The R-PAE likely impacts NR-CARA results.
NT, not tested.
Minor SW observed when y axis (fluorescence) expanded.
Nonreplicating-active molecules.
The anticipated MIC90, CARA, and CFU results for strict nonreplicating-active molecules are shown in Fig. 5a and 2, example 7. Oxyphenbutazone, a nonsteroidal anti-inflammatory drug, was previously established as selectively bactericidal against nonreplicating M. tuberculosis (6). We tested oxyphenbutazone by MIC and CARA in parallel against replicating M. tuberculosis (Fig. 5b) and nonreplicating M. tuberculosis (Fig. 5c). The NR-CARA data paralleled that of the NR-MIC90 and correctly predicted that oxyphenbutazone was a selectively nonreplicating-active compound. As anticipated from the R-MIC90 data, the R-CARA data indicated that oxyphenbutazone had no discernible replicating-bactericidal activity. Nitazoxanide has weak replicating activity and is bactericidal to M. tuberculosis in a minimal nonreplicating model (7H9, pH 5.0, containing 0.5 mM NaNO2−) (37). The data obtained from the R (Fig. 5d) and NR (Fig. 5e) MIC90 and CARA from M. tuberculosis bacilli treated with nitazoxanide were consistent with those of published results (37).
R-bactericidal molecules.
Strictly replicating-bactericidal molecules display activity in the R-MIC90, R-CARA, and R-CFU assays (Fig. 6a and 2, examples 1 and 2). Drug carryover effects that lead to erroneous NR-MIC90 values for potent replicating actives are mitigated when employing the NR-CARA (Fig. 6a). R- and NR-MIC90 and CARA data for isoniazid (Fig. 6b and c), kanamycin (Fig. 6d and e), and streptomycin (Fig. 6f and g) are consistent with them being replicating-bactericidal compounds. The NR-CARA clearly demonstrates that INH (Fig. 6c) and streptomycin (Fig. 6g) are nonreplicating inactive, which suggests the NR-MIC90 data were a result of compound carryover.
FIG 6.

Replicating-bactericidal molecules. Anticipated results for compounds that are strictly replicating-bactericidal are summarized (a), and experimental examples of INH (b, c), kanamycin (d, e), and streptomycin (f, g) are shown.
Because kanamycin's R/NR MIC90 ratio indicated it was a strict replicating-active compound, we anticipated the NR-CARA results might originate from compound carryover or postantibiotic effect and not from bona fide nonreplicating activity (Fig. 6e). Indeed, detailed analysis of kanamycin's activity against nonreplicating M. tuberculosis by a CFU assay showed a modest decrease of CFU with increased drug concentrations that did not achieve the 2 to 3 log10 kill predicted by the CARA (see Fig. S3b in the supplemental material). Thus, in some cases, the R/NR MIC90 ratio is an important factor in correctly interpreting CARA data.
Replicating-bacteriostatic molecules.
Replicating-bacteriostatic molecules have a profile similar to that of replicating-bactericidal compounds (Fig. 7a and 2, examples 3 and 4), with the following differences: failure to decrease fluorescence in the R-CARA or the presence of a static window in the R-CARA and <99% to 99.9% CFU reduction compared to the starting inoculum (Fig. 7a).
For PAS (Fig. 7b), fenamisal (Fig. 7d), TMC207 (Fig. 7f), ethambutol (Fig. 7h), linezolid (Fig. 7j), and ethionamide (Fig. 7l), R-CARA results did not track with the R-MIC90. The static windows between the R-CARA and R-MIC90 indicated that these compounds were bacteriostatic, with postantibiotic effects, and might only have bactericidal activity at the highest compound concentrations tested. Large static windows were observed for TMC207 (Fig. 7f) and linezolid (Fig. 7j). The results for linezolid confirmed a report that this oxazolidinone is bacteriostatic against replicating M. tuberculosis (39). In contrast, for PAS and fenamisal, the y axis had to be expanded to determine the presence of a static window (Fig. 7b and d, insets). The absence of a more conspicuous static window for PAS and fenamisal was traced to a severe postantibiotic effect (see Fig. S4a and b in the supplemental material).
Turning to the nonreplicating activity of these compounds, the NR-CARA data strongly suggested that neither PAS (Fig. 7c), fenamisal (Fig. 7e), TMC207 (Fig. 7g), ethambutol (Fig. 7i), nor ethionamide (Fig. 7m) had significant nonreplicating activity. PAS, fenamisal, and TMC207 appeared to have nonreplicating activity at concentrations manyfold higher than their R-MIC90. Although the NR-MIC90 indicated that linezolid might kill nonreplicating M. tuberculosis at concentrations far above its R-MIC90 value (Fig. 7k), the NR-CARA result unambiguously predicted that linezolid was inactive against nonreplicating M. tuberculosis (Fig. 7k). CARA predictions for linezolid were confirmed by CFU assays for replicating (Fig. 7n) and nonreplicating (Fig. 7o) M. tuberculosis bacilli (Table 3).
Dual-active molecules.
As noted, dual-active molecules are bactericidal or bacteriostatic against replicating M. tuberculosis and also bactericidal against nonreplicating M. tuberculosis (Fig. 8a and 2, examples 5 and 6). The R-MIC90 to NR-MIC90 ratios of PA-824, moxifloxacin, and rifampin suggested they were dual-active compounds. The CARA data indicated that PA-824 was replicating-bacteriostatic/nonreplicating-bactericidal (Fig. 8b and c), moxifloxacin was replicating-bactericidal/nonreplicating-bactericidal (Fig. 8d and e), and rifampin was replicating-bactericidal/nonreplicating-bactericidal (Fig. 8f and g). CFU assays confirmed the CARA predictions for rifampin (see Fig. 8h and i in the supplemental material), moxifloxacin (see Fig. S3c and d in the supplemental material), and PA-824 (described in more detail in Fig. 9a and b).
Exploring the impact of PA-824 and TMC207 on replicating and nonreplicating M. tuberculosis.
The implication that PA-824 and TMC207 were chiefly bacteriostatic for M. tuberculosis under replicating conditions was unexpected given that other studies using CFU assays reported that PA-824 and TMC207 are bactericidal against M. tuberculosis in vitro and in vivo (40, 41). To test if activated charcoal in the CARA plates might have impacted the conclusion as to whether PA-824 and TMC207 were bacteriostatic or bactericidal, we enumerated CFU on 7H11-OADC plates that lacked or contained 0.4% activated charcoal (Fig. 9a to f). For PA-824 and TMC207, inclusion of activated charcoal significantly increased CFU counts, while inclusion of charcoal did not affect CFU counts for moxifloxacin or kanamycin (see Fig. S3 in the supplemental material). Similar to what has been reported (42), an additional 7 days of TMC207 exposure (14 days total) led to increased killing compared to that at day 7 (Fig. 9e); yet killing remained at <3 log10 when CFU were enumerated on agar plates supplemented with activated charcoal, so the activity can still be classified as bacteriostatic by a strict definition of bactericidal activity. CFU assays enumerated on 7H11-OADC plates supplemented with activated charcoal showed that TMC207 achieved ∼2 log10 kill against M. tuberculosis in the multistress model of nonreplication (Fig. 7g), as predicted by the NR-CARA-MBC≥99 (12.5 μg/ml).
Thus, conducting CFU assays on 7H11-OADC agar plates supplemented or not with 0.4% activated charcoal reinforced the impression that PA-824 and TMC207 were less bactericidal than they are commonly scored, as this scoring reflects continued action of the drugs after the treated M. tuberculosis bacilli are plated. This was confirmed by the following experiment. We treated M. tuberculosis for 7 days with 30 μg/ml of PA-824 or TMC207, and spread undiluted cells on agar plates. We recovered 0 to 2 colonies on 7H11-OADC plates lacking charcoal. In contrast, when the same drug-treated undiluted cells were spread on charcoal-containing 7H11-OADC agar plates, there were too many colonies to count (see Table S1 in the supplemental material). This is a clear illustration of the ability of a carried-over drug to give a false impression of cidality and of activated charcoal to bind a carried-over drug.
Applications of the CARA.
We applied the CARA to monitor dose- and time-dependent killing by two antimycobacterial drugs. The results of a time course testing the action of rifampin and ethambutol against replicating M. smegmatis are shown in Fig. 10. Testing 2 compounds in quadruplicate at 9 drug concentrations plus a DMSO control and at 4 time points (1, 3, 6, and 24 h) used only 4 CARA plates.
FIG 10.

Dose- and time-dependent killing of M. smegmatis revealed by the CARA. Replicating M. smegmatis was exposed to increasing concentrations of rifampin (a to d) or ethambutol (e to h). At indicated time points, 10-μl aliquots were removed and spotted onto CARA plates. Results are means ± SEM from 4 replicates derived from duplicate wells in one of two similar experiments.
Enumerating M. smegmatis in the same experiment by a CFU assay (with experimental triplicates instead of quadruplicates) would have required approximately 20 96-well microplate dilution plates (in each of which one can dilute 4 test conditions, in triplicate columns, from 100 to 10−5) and approximately ∼480 tri-style petri plates containing bacteriologic agar to enumerate surviving cells.
Thus, one CARA plate can replace approximately five 96-well dilution plates and 120 tri-style petri plates used for the CFU assay. These results illustrate the utility of the CARA assay in indicating which drug concentrations and time points will be most suitable for performing a standard CFU assay on a manageable scale.
DISCUSSION
Since the introduction of the CFU assay by Koch in 1883, CFU-based assays have been the gold standard for enumerating those bacilli that survive exposure to various stresses, including antibiotics. Novel approaches to increase CFU assay throughput have been explored (43). Several limitations of the standard CFU assay prompted us to develop the CARA. Among our goals in addressing these needs were to increase throughput, reduce serial dilutions, eliminate laborious counting of colonies, shorten the time to readout, and blunt the impact of test agents that can be carried over into an outgrowth assay (6, 27, 28), either in solution or by adsorption to the bacteria. The CARA facilitates setting up pilot experiments typically considered too laborious and time-consuming for standard CFU assays (Fig. 10). In CFU assays, bacteria that were untreated or treated with various concentrations of test agent, along with any test agent in the suspension, are serially diluted in a test agent-free medium and then plated on growth-supporting agar to allow surviving bacilli to grow as colonies. Plating constitutes another dilution. For example, spreading 10 μl of a bacterial suspension onto 8 ml of solid medium in one section of a tri-petri plate dilutes test agents 800-fold. Thus, after serial dilutions and plating, it is generally assumed that the test agent in solution with the bacterial suspension is diluted to extinction. However, bacterial growth may still be inhibited by test agent carried over into agar (see Table S2 in the supplemental material) or test agent absorbed to bacteria (Fig. 11).
FIG 11.

Activated charcoal may reveal bacterial drug reserves. The diagram shows that when CFU are enumerated on agar plates lacking charcoal, there are three possible scenarios that are indistinguishable: (i) no drug was carried over in the agar plate, (ii) drug was carried over in solution, (iii) or drug was absorbed to bacteria. For most compounds, enumerating CFU on agar plates containing activated charcoal will mitigate carryover effects. Compounds that appear to exert their bactericidal impact when plated on agar plates lacking charcoal and bacteriostatic impact when plated on agar plates containing charcoal may be predictive of long-term potentiation in vivo or a postantibiotic effect in vitro. Mycobacterial cells depict the cytosol (inner layer), membrane/peptidoglycan/arabinogalactan/lipid layer (middle layer), and capsule (outer layer).
In the multistress assay of nonreplication, TMC207 and PA-824 were bactericidal. However, the activity of TMC207 against nonreplicating M. tuberculosis was only found at concentrations far higher than the replicating MIC90. In our studies, the activity of TMC207 and PA-824 against replicating M. tuberculosis was consistent with a bacteriostatic mode of action. This does not rule out that these molecules may be bactericidal at later time points or under conditions that pertain in the human host, where TMC207 and PA-824 have clinical benefit (44–47). Whether fluoroquinolones are bacteriostatic or bactericidal in vitro is dose and time dependent (34, 35), and the distinction does not necessarily relate to treatment outcome (48).
In fact, we speculate that reserves of PA-824 and TMC207 in the cytosol, membrane, lipids, or capsule may contribute to the drugs' efficacy (Fig. 11). The location and extent of drug reserves may be determined by the drug's physicochemical properties. Bacterial drug reserves may contribute to postantibiotic effects between doses or force bacteria to transport their own cache of drug as they migrate to sites in the host in which effective concentrations of the drug might otherwise not be achieved.
Comparing the results of MIC, CARA, and CFU assays illustrated circumstances in which carryover of a test agent into the CFU assay, either in solution or adsorbed to the cells, apparently led to an overestimation of a compound's potency in the given conditions or a misinterpretation of the conditions under which it was effective. To avoid these problems, we recommend inclusion of activated charcoal in agar used to enumerate M. tuberculosis after exposure to antibacterial agents, whether or not the assay is in standard CFU or CARA format.
Concern is not new for carryover of anti-infectives from dilution series into outgrowth assays. The impact of TMC207 when carried over to agar plates inspired the addition of excess bovine serum albumin (BSA) to agar plates to sequester TMC207 for CFU assays (49). Activated charcoal has been used to mitigate drug carryover in vitro and from organ homogenates from M. tuberculosis-infected mice treated with antitubercular drugs (50, 51). Of the antimycobacterial compounds tested, 12 were bound almost completely by activated charcoal within 24 h (Table 2). Streptomycin remained partially free. This may be related to a combination of logP, polar surface area, and rotatable bonds, considering that streptomycin has the lowest logP (−7.65), highest number of H-bond donors and acceptors (14 and 19) and heavy atoms (40), and largest polar surface area (331) (see Table S2 in the supplemental material). Streptomycin and ethambutol had the highest number of rotatable bonds (9) (see Table S2 in the supplemental material).
Activated charcoal may sequester a wider spectrum of compounds than BSA and do so more extensively and thus may have more general utility. Indeed, for 12/12 compounds tested, activated charcoal bound the compound more extensively than BSA by 20 min. Small, hydrophilic drugs such as isoniazid (estimated at <10% protein bound) are predicted to bind BSA with a much lower affinity than TMC207 (estimated at >99% protein bound). Bacteriologic media for the propagation of fastidious microorganisms such as Legionella pneumophila, Campylobacter jejuni, Haemophilus influenzae, and Bordetella pertussis, are often supplemented with activated charcoal to absorb toxic byproducts. In the clinic, activated charcoal is used to absorb chemicals in poisonings and overdoses, including overdoses of rifampin, isoniazid, and PAS (52–55).
Most of the data presented here are consistent with activated charcoal's main effect being to bind carried over compounds and prevent them from exerting an effect on the recovery of bacteria in the outgrowth phase of the assay. However, in some cases, activated charcoal may act differently. For example, activated charcoal may sequester a component in the agar or in the 7H11-OADC medium in which the agar is dissolved, and that component might otherwise synergize with certain test agents to kill M. tuberculosis during the postantibiotic recovery phase of a CFU-based assay. For example, malachite green, a common component in 7H11-based agar, can potentiate the postantibiotic effect of cell-wall-active drugs on mycobacteria (56); perhaps activated charcoal can neutralize this effect. We have observed that M. tuberculosis bacilli form smaller colonies on 7H11-OADC plates containing activated charcoal, which suggests that the activated charcoal may bind essential nutrients, albeit not to an extent that abrogates the ability to form colonies.
A potential confounding factor of using activated charcoal is that it may have a higher affinity for some compounds than others (Table 2). As noted, charcoal poorly bound high concentrations (30 μg/ml) of streptomycin. While ethambutol did not bind charcoal as effectively as other compounds, it was still removed by ∼97% at 20 min. Some bacteriostatic agents, when tested at concentrations at their broth MIC or higher, prevented recovery of bacteria on CARA plates (Fig. 2; see also S6d in the supplemental material). We cannot distinguish between two explanations. Either these drugs may be bacteriostatic over one concentration range and bactericidal at a higher range; or, alternatively, when used at high concentrations, they might exert a prolonged postantibiotic effect that remains static in nature, that is, greatly extend the time for colonies to appear on agar plates without reducing the numbers of viable cells. The latter phenomenon was profound for PAS and fenamisal (see Fig. S4 in the supplemental material), as reflected by the severely decreased colony size on 7H11-OADC agar plates. Because PAEs of some compounds may hinder the predictive power of the CARA, we have proposed specific techniques for data analysis (Table 1 and Fig. 2; see also Fig. S5 and S6 in the supplemental material). It may be possible to adapt the CARA incubations beyond 7 to 10 days on a compound-to-compound basis once a postantibiotic effect is identified; however, this solution would not work for semi-high-throughput analysis in which many compounds are tested head-to-head.
Some molecules classified as replicating-bacteriostatic, such as fenamisal (Fig. 7e), led to a dose-dependent decrease in NR-CARA fluorescence at concentrations significantly above their R-MIC90. Such compounds merit consideration as candidate dual-active molecules. Although moxifloxacin and fenamisal share similar R- and NR- MIC90 and CARA results, fenamisal was noted for significant carryover effects (Fig. 4c and d) and postantibiotic effects (Fig. S4), which led us to suspect fenamisal's dose-dependent fluorescence decrease in the NR-CARA was artifactual. Ultimately, compounds like these with indeterminate activity designations by MIC and CARA will require evaluation by CFU assays.
The CARA is semiquantitative. There is an inverse relationship between the time to half-maximal resazurin fluorescence and starting bacterial numbers (57). By reading fluorescence at a single time point, the CARA has a relatively small dynamic range, such that a well with fluorescence no higher than background only indicates that there has been a ≥2 to 3 log10 reduction in bacterial numbers compared to a well with fluorescence above background. Thus, the CARA may not be appropriate for assays in which the survival differences of interest are <2 log10.
A CARA may be useful for work with other bacterial species, but it will be necessary to tailor the CARA for each. For example, with a detection limit of 2 to 3 log10 CFU, the CARA may fail to accurately distinguish between bactericidal and bacteriostatic actions of drugs against Staphylococcus aureus, as the accepted definition of bacteriostatic for this genus is <3 log10 kill in 24 h. Rapidly dividing organisms like S. aureus presents challenges to the use of the static window, which is dependent on the bacterium's growth rate and the duration of a drug's postantibiotic effect, and may form colonies faster than it takes for some test agents to completely bind to charcoal in CARA plates (Table 2). The factors that we recommend optimizing are: inoculum, choice of bacteriological liquid and solid media, time of drug exposure, time of incubating the CARA plate prior to developing with alamarBlue, and rigorous testing of the assay with compounds that possess well-characterized bacteriostatic or bactericidal activities.
There remain challenges to resolve. Fluorescence bleaching, which can occur in wells from DMSO-treated cells, precludes data normalization. The CARA measures fluorescence of bacterial microcolonies on the agar surface, which can display significant heterogeneity. The large error between replicates gives the CARA poor Z' scores, invalidating the CARA for high-throughput screening, in which one seeks rare active compounds in a collection of mostly inactive compounds. Instead, the CARA's utility is in discerning dose-dependent trends for compounds already identified as active. Potent postantibiotic effects that inhibit the rate of bacterial regrowth on bacteriologic agar medium may negatively impact CARA predictions.
The drive to identify compounds that kill M. tuberculosis rendered nonreplicating in a variety of ways (4–6, 8, 27, 37, 39, 58–62) has been hampered by a paucity of techniques to analyze and progress these molecules post screening. Identifying bona fide nonreplicating- and dual-active compounds is an early step toward prioritizing compounds for efforts that are increasingly resource intensive, such as CFU assays, target identification, and efficacy studies in animal models. The CARA can play a useful role in discerning which compounds should enter this progression.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kyu Rhee, Kristin Burns-Huang, and Madhumitha Nandakumar (Weill Cornell Medical College) for helpful discussions and critical reading of the manuscript.
This work was supported by the TB Drug Accelerator Program of the Bill and Melinda Gates Foundation, the Abby and Howard P. Milstein Program in Translational Medicine, and an NIH TB Research Unit (no. U19 AI111143). The Department of Microbiology and Immunology is supported by the William Randolph Hearst Foundation. S.S.-K. was supported by NIH grant no. K08AI108799.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00803-15.
REFERENCES
- 1.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. 2007. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov 6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 2.Coates A, Hu Y, Bax R, Page C. 2002. The future challenges facing the development of new antimicrobial drugs. Nat Rev Drug Discov 1:895–910. doi: 10.1038/nrd940. [DOI] [PubMed] [Google Scholar]
- 3.Nathan C. 2012. Fresh approaches to anti-infective therapies. Sci Transl Med 4:140sr142. doi: 10.1126/scitranslmed.3003081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bryk R, Gold B, Venugopal A, Singh J, Samy R, Pupek K, Cao H, Popescu C, Gurney M, Hotha S, Cherian J, Rhee K, Ly L, Converse PJ, Ehrt S, Vandal O, Jiang X, Schneider J, Lin G, Nathan C. 2008. Selective killing of nonreplicating mycobacteria. Cell Host Microbe 3:137–145. doi: 10.1016/j.chom.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho SH, Warit S, Wan B, Hwang CH, Pauli GF, Franzblau SG. 2007. Low-oxygen-recovery assay for high-throughput screening of compounds against nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother 51:1380–1385. doi: 10.1128/AAC.00055-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gold B, Pingle M, Brickner SJ, Shah N, Roberts J, Rundell M, Bracken WC, Warrier T, Somersan S, Venugopal A, Darby C, Jiang X, Warren JD, Fernandez J, Ouerfelli O, Nuermberger EL, Cunningham-Bussel A, Rath P, Chidawanyika T, Deng H, Realubit R, Glickman JF, Nathan CF. 2012. Nonsteroidal anti-inflammatory drug sensitizes Mycobacterium tuberculosis to endogenous and exogenous antimicrobials. Proc Natl Acad Sci U S A 109:16004–16011. doi: 10.1073/pnas.1214188109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keren I, Minami S, Rubin E, Lewis K. 2011. Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. mBio 2(3):e00100-11. doi: 10.1128/mBio.00100-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mak PA, Rao SP, Ping Tan M, Lin X, Chyba J, Tay J, Ng SH, Tan BH, Cherian J, Duraiswamy J, Bifani P, Lim V, Lee BH, Ling Ma N, Beer D, Thayalan P, Kuhen K, Chatterjee A, Supek F, Glynne R, Zheng J, Boshoff HI, Barry CE III, Dick T, Pethe K, Camacho LR. 2012. A high-throughput screen to identify inhibitors of ATP homeostasis in non-replicating Mycobacterium tuberculosis. ACS Chem Biol 7:1190–1197. doi: 10.1021/cb2004884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin G, Chidawanyika T, Tsu C, Warrier T, Vaubourgeix J, Blackburn C, Gigstad K, Sintchak M, Dick L, Nathan C. 2013. N,C-capped dipeptides with selectivity for mycobacterial proteasome over human proteasomes: role of S3 and S1 binding pockets. J Am Chem Soc 135:9968–9971. doi: 10.1021/ja400021x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin G, Li D, de Carvalho LP, Deng H, Tao H, Vogt G, Wu K, Schneider J, Chidawanyika T, Warren JD, Li H, Nathan C. 2009. Inhibitors selective for mycobacterial versus human proteasomes. Nature 461:621–626. doi: 10.1038/nature08357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gandotra S, Lebron MB, Ehrt S. 2010. The Mycobacterium tuberculosis proteasome active site threonine is essential for persistence yet dispensable for replication and resistance to nitric oxide. PLoS Pathog 6:e1001040. doi: 10.1371/journal.ppat.1001040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. 2004. Bacterial persistence as a phenotypic switch. Science 305:1622–1625. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- 13.Wakamoto Y, Dhar N, Chait R, Schneider K, Signorino-Gelo F, Leibler S, McKinney JD. 2013. Dynamic persistence of antibiotic-stressed mycobacteria. Science 339:91–95. doi: 10.1126/science.1229858. [DOI] [PubMed] [Google Scholar]
- 14.Orman MA, Brynildsen MP. 2013. Dormancy is not necessary or sufficient for bacterial persistence. Antimicrob Agents Chemother 57:3230–3239. doi: 10.1128/AAC.00243-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mattila JT, Ojo OO, Kepka-Lenhart D, Marino S, Kim JH, Eum SY, Via LE, Barry CE III, Klein E, Kirschner DE, Morris SM Jr, Lin PL, Flynn JL. 2013. Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. J Immunol 191:773–784. doi: 10.4049/jimmunol.1300113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, Dolganov G, Efron B, Butcher PD, Nathan C, Schoolnik GK. 2003. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J Exp Med 198:693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nathan C. 2011. Making space for anti-infective drug discovery. Cell Host Microbe 9:343–348. doi: 10.1016/j.chom.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 18.MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, Nathan CF. 1997. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc Natl Acad Sci U S A 94:5243–5248. doi: 10.1073/pnas.94.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.MacMicking JD, Taylor GA, McKinney JD. 2003. Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science 302:654–659. doi: 10.1126/science.1088063. [DOI] [PubMed] [Google Scholar]
- 20.Vandal OH, Nathan CF, Ehrt S. 2009. Acid resistance in Mycobacterium tuberculosis. J Bacteriol 191:4714–4721. doi: 10.1128/JB.00305-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cunningham-Bussel A, Zhang T, Nathan CF. 2013. Nitrite produced by Mycobacterium tuberculosis in human macrophages in physiologic oxygen impacts bacterial ATP consumption and gene expression. Proc Natl Acad Sci U S A 110:E4256–E4265. doi: 10.1073/pnas.1316894110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Via LE, Lin PL, Ray SM, Carrillo J, Allen SS, Eum SY, Taylor K, Klein E, Manjunatha U, Gonzales J, Lee EG, Park SK, Raleigh JA, Cho SN, McMurray DN, Flynn JL, Barry CE III. 2008. Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and nonhuman primates. Infect Immun 76:2333–2340. doi: 10.1128/IAI.01515-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aly S, Wagner K, Keller C, Malm S, Malzan A, Brandau S, Bange FC, Ehlers S. 2006. Oxygen status of lung granulomas in Mycobacterium tuberculosis-infected mice. J Pathol 210:298–305. doi: 10.1002/path.2055. [DOI] [PubMed] [Google Scholar]
- 24.de Carvalho LP, Fischer SM, Marrero J, Nathan C, Ehrt S, Rhee KY. 2010. Metabolomics of Mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates. Chem Biol 17:1122–1131. doi: 10.1016/j.chembiol.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 25.Munoz-Elias EJ, McKinney JD. 2005. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med 11:638–644. doi: 10.1038/nm1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Warrier T, Martinez-Hoyos M, Marin-Amieva M, Colmenarejo G, Porras E, Alvarez-Pedraglio AI, Fraile-Gabaldon MT, Torres-Gomez PA, Lopez-Quezada L, Gold B, Roberts J, Ling Y, Somersan-Karakaya S, Little D, Cammack N, Nathan CF, Mendoza-Losana A. 17 June 2015. Identification of novel anti-mycobacterial compounds by screening a pharmaceutical small-molecule library against non-replicating Mycobacterium tuberculosis. ACS Infect Dis. doi: 10.1021/acsinfecdis.5b00025. [DOI] [PubMed] [Google Scholar]
- 27.Grant SS, Kawate T, Nag PP, Silvis MR, Gordon K, Stanley SA, Kazyanskaya E, Nietupski R, Golas A, Fitzgerald M, Cho S, Franzblau SG, Hung DT. 2013. Identification of novel inhibitors of nonreplicating Mycobacterium tuberculosis using a carbon starvation model. ACS Chem Biol 8:2224–2234. doi: 10.1021/cb4004817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gold B, Warrier T, Nathan C. 2015. A multi-stress model for high throughput screening against non-replicating Mycobacterium tuberculosis, p 293–315. In Parish T, Roberts D (ed), Mycobacteria protocols, methods in molecular biology, 3rd ed, vol 1285 Springer, New York, NY. [DOI] [PubMed] [Google Scholar]
- 29.Hohman M, Gregory K, Chibale K, Smith PJ, Ekins S, Bunin B. 2009. Novel web-based tools combining chemistry informatics, biology and social networks for drug discovery. Drug Discov Today 14:261–270. doi: 10.1016/j.drudis.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 30.Collins L, Franzblau SG. 1997. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob Agents Chemother 41:1004–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yajko DM, Madej JJ, Lancaster MV, Sanders CA, Cawthon VL, Gee B, Babst A, Hadley WK. 1995. Colorimetric method for determining MICs of antimicrobial agents for Mycobacterium tuberculosis. J Clin Microbiol 33:2324–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barry AL, Craig WA, Nadler H, Reller LB, Sanders CC, Swenson JM. 1999. Methods for determining bactericidal activity of antimicrobial agents: approved guideline, vol 19 National Committee for Clinical Laboratory Standards, Wayne, PA. [Google Scholar]
- 33.Heifets LB, Cynamon MH. 1991. Drug susceptibility in the chemotherapy of mycobacterial infections. CRC Press, Boca Raton, FL. [Google Scholar]
- 34.Chen CR, Malik M, Snyder M, Drlica K. 1996. DNA gyrase and topoisomerase IV on the bacterial chromosome: quinolone-induced DNA cleavage. J Mol Biol 258:627–637. doi: 10.1006/jmbi.1996.0274. [DOI] [PubMed] [Google Scholar]
- 35.Drlica K, Hiasa H, Kerns R, Malik M, Mustaev A, Zhao X. 2009. Quinolones: action and resistance updated. Curr Top Med Chem 9:981–998. doi: 10.2174/156802609789630947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chakraborty S, Gruber T, Barry CE III, Boshoff HI, Rhee KY. 2013. Para-aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science 339:88–91. doi: 10.1126/science.1228980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Carvalho LP, Lin G, Jiang X, Nathan C. 2009. Nitazoxanide kills replicating and nonreplicating Mycobacterium tuberculosis and evades resistance. J Med Chem 52:5789–5792. doi: 10.1021/jm9010719. [DOI] [PubMed] [Google Scholar]
- 38.Shigyo K, Ocheretina O, Merveille YM, Johnson WD, Pape JW, Nathan CF, Fitzgerald DW. 2013. Efficacy of nitazoxanide against clinical isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother 57:2834–2837. doi: 10.1128/AAC.02542-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang M, Sala C, Dhar N, Vocat A, Sambandamurthy VK, Sharma S, Marriner G, Balasubramanian V, Cole ST. 2014. In vitro and in vivo activities of three oxazolidinones against nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother 58:3217–3223. doi: 10.1128/AAC.02410-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andries K, Verhasselt P, Guillemont J, Gohlmann HW, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. 2005. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 41.Stover CK, Warrener P, VanDevanter DR, Sherman DR, Arain TM, Langhorne MH, Anderson SW, Towell JA, Yuan Y, McMurray DN, Kreiswirth BN, Barry CE, Baker WR. 2000. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 405:962–966. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- 42.Koul A, Vranckx L, Dhar N, Gohlmann HW, Ozdemir E, Neefs JM, Schulz M, Lu P, Mortz E, McKinney JD, Andries K, Bald D. 2014. Delayed bactericidal response of Mycobacterium tuberculosis to bedaquiline involves remodelling of bacterial metabolism. Nat Commun 5:3369. doi: 10.1038/ncomms4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaur P, Ghosh A, Krishnamurthy RV, Bhattacharjee DG, Achar V, Datta S, Narayanan S, Anbarasu A, Ramaiah S. 2015. A high-throughput cidality screen for Mycobacterium tuberculosis. PLoS One 10(2):e0117577. doi: 10.1371/journal.pone.0117577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dawson R, Diacon AH, Everitt D, van Niekerk C, Donald PR, Burger DA, Schall R, Spigelman M, Conradie A, Eisenach K, Venter A, Ive P, Page-Shipp L, Variava E, Reither K, Ntinginya NE, Pym A, von Groote-Bidlingmaier F, Mendel CM. 2015. Efficiency and safety of the combination of moxifloxacin, pretomanid (PA-824), and pyrazinamide during the first 8 weeks of antituberculosis treatment: a phase 2b, open-label, partly randomised trial in patients with drug-susceptible or drug-resistant pulmonary tuberculosis. Lancet 385:1738–1747. doi: 10.1016/S0140-6736(14)62002-X. [DOI] [PubMed] [Google Scholar]
- 45.Diacon AH, Dawson R, von Groote-Bidlingmaier F, Symons G, Venter A, Donald PR, van Niekerk C, Everitt D, Winter H, Becker P, Mendel CM, Spigelman MK. 2012. 14-day bactericidal activity of PA-824, bedaquiline, pyrazinamide, and moxifloxacin combinations: a randomised trial. Lancet 380:986–993. doi: 10.1016/S0140-6736(12)61080-0. [DOI] [PubMed] [Google Scholar]
- 46.Diacon AH, Pym A, Grobusch M, Patientia R, Rustomjee R, Page-Shipp L, Pistorius C, Krause R, Bogoshi M, Churchyard G, Venter A, Allen J, Palomino JC, De Marez T, van Heeswijk RP, Lounis N, Meyvisch P, Verbeeck J, Parys W, de Beule K, Andries K, Mc Neeley DF. 2009. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N Engl J Med 360:2397–2405. doi: 10.1056/NEJMoa0808427. [DOI] [PubMed] [Google Scholar]
- 47.Diacon AH, Pym A, Grobusch MP, de los Rios JM, Gotuzzo E, Vasilyeva I, Leimane V, Andries K, Bakare N, De Marez T, Haxaire-Theeuwes M, Lounis N, Meyvisch P, De Paepe E, van Heeswijk RP, Dannemann B, TMC207-C208 Study Group. 2014. Multidrug-resistant tuberculosis and culture conversion with bedaquiline. N Engl J Med 371:723–732. doi: 10.1056/NEJMoa1313865. [DOI] [PubMed] [Google Scholar]
- 48.Pankey GA, Sabath LD. 2004. Clinical relevance of bacteriostatic versus bactericidal mechanisms of action in the treatment of Gram-positive bacterial infections. Clin Infect Dis 38:864–870. doi: 10.1086/381972. [DOI] [PubMed] [Google Scholar]
- 49.Lounis N, Gevers T, Van Den Berg J, Verhaeghe T, van Heeswijk R, Andries K. 2008. Prevention of drug carryover effects in studies assessing antimycobacterial efficacy of TMC207. J Clin Microbiol 46:2212–2215. doi: 10.1128/JCM.00177-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tasneen R, Li SY, Peloquin CA, Taylor D, Williams KN, Andries K, Mdluli KE, Nuermberger EL. 2011. Sterilizing activity of novel TMC207- and PA-824-containing regimens in a murine model of tuberculosis. Antimicrob Agents Chemother 55:5485–5492. doi: 10.1128/AAC.05293-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grosset JH, Tyagi S, Almeida DV, Converse PJ, Li SY, Ammerman NC, Bishai WR, Enarson D, Trebucq A. 2013. Assessment of clofazimine activity in a second-line regimen for tuberculosis in mice. Am J Respir Crit Care Med 188:608–612. doi: 10.1164/rccm.201304-0753OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Neuvonen PJ. 1982. Clinical pharmacokinetics of oral activated charcoal in acute intoxications. Clin Pharmacokinet 7:465–489. doi: 10.2165/00003088-198207060-00001. [DOI] [PubMed] [Google Scholar]
- 53.Olkkola KT. 1985. Effect of charcoal-drug ratio on antidotal efficacy of oral activated charcoal in man. Br J Clin Pharmacol 19:767–773. doi: 10.1111/j.1365-2125.1985.tb02712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Orisakwe OE, Afonne OJ, Agbasi PU, Ilondu NA, Ofoefule SI, Obi E. 2001. Adsorptive capacity of activated charcoal for rifampicin with and without sodium chloride and sodium citrate. Biol Pharm Bull 24:724–726. doi: 10.1248/bpb.24.724. [DOI] [PubMed] [Google Scholar]
- 55.Siefkin AD, Albertson TE, Corbett MG. 1987. Isoniazid overdose: pharmacokinetics and effects of oral charcoal in treatment. Hum Toxicol 6:497–501. doi: 10.1177/096032718700600608. [DOI] [PubMed] [Google Scholar]
- 56.Gelman E, McKinney JD, Dhar N. 2012. Malachite green interferes with postantibiotic recovery of mycobacteria. Antimicrob Agents Chemother 56:3610–3614. doi: 10.1128/AAC.00406-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shiloh MU, Ruan J, Nathan C. 1997. Evaluation of bacterial survival and phagocyte function with a fluorescence-based microplate assay. Infect Immun 65:3193–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Darby CM, Nathan CF. 2010. Killing of non-replicating Mycobacterium tuberculosis by 8-hydroxyquinoline. J Antimicrob Chemother 65:1424–1427. doi: 10.1093/jac/dkq145. [DOI] [PubMed] [Google Scholar]
- 59.Hartkoorn RC, Ryabova OB, Chiarelli LR, Riccardi G, Makarov V, Cole ST. 2014. Mechanism of action of 5-nitrothiophenes against Mycobacterium tuberculosis. Antimicrob Agents Chemother 58:2944–2947. doi: 10.1128/AAC.02693-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang M, Sala C, Hartkoorn RC, Dhar N, Mendoza-Losana A, Cole ST. 2012. Streptomycin-starved Mycobacterium tuberculosis 18b, a drug discovery tool for latent tuberculosis. Antimicrob Agents Chemother 56:5782–5789. doi: 10.1128/AAC.01125-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sala C, Dhar N, Hartkoorn RC, Zhang M, Ha YH, Schneider P, Cole ST. 2010. Simple model for testing drugs against nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother 54:4150–4158. doi: 10.1128/AAC.00821-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang F, Sambandan D, Halder R, Wang J, Batt SM, Weinrick B, Ahmad I, Yang P, Zhang Y, Kim J, Hassani M, Huszar S, Trefzer C, Ma Z, Kaneko T, Mdluli KE, Franzblau S, Chatterjee AK, Johnsson K, Mikusova K, Besra GS, Futterer K, Robbins SH, Barnes SW, Walker JR, Jacobs WR Jr, Schultz PG. 2013. Identification of a small molecule with activity against drug-resistant and persistent tuberculosis. Proc Natl Acad Sci U S A 110:E2510–E2517. doi: 10.1073/pnas.1309171110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.