

Abstract
The regulation of surface levels of protein is critical for proper cell function and influences properties including cell adhesion, ion channel contributions to current flux, and the sensitivity of surface receptors to ligands. Here we demonstrate a two-color labeling system in live cells using a single fluorogen activating peptide (FAP) based fusion tag, which enables the rapid and simultaneous quantification of surface and internal proteins. In the nervous system, BK channels can regulate neural excitability and neurotransmitter release, and the surface trafficking of BK channels can be modulated by signaling cascades and assembly with accessory proteins. Using this labeling approach, we examine the dynamics of BK channel surface expression in HEK293 cells. Surface pools of the pore-forming BKα subunit were stable, exhibiting a plasma membrane half-life of >10 h. Long-term activation of adenylyl cyclase by forskolin reduced BKα surface levels by 30%, an effect that could not be attributed to increased bulk endocytosis of plasma membrane proteins. This labeling approach is compatible with microscopic imaging and flow cytometry, providing a solid platform for examining protein trafficking in living cells.
Introduction
Protein trafficking is tightly regulated in all cells and mediates important functions such as receptor signaling, cell–cell contacts, cell adhesion, nutrient uptake, and membrane excitability. For excitable cells, channel distribution at the plasma membrane (PM) can strongly influence membrane potential and stimulus–response coupling.1 Analysis of protein surface expression is limited by cumbersome techniques, including biotinylation of surface proteins, immunofluorescence using ectofacial epitopes, and the use of pH-dependent fluorophores. Similarly, real-time dynamics of surface protein trafficking have been difficult to visualize due to the time scales required for these experiments. Biotinylation is useful to measure population surface protein levels, but cannot label intracellular stores. While immunofluorescence can be employed to quantify surface fraction of a protein, labeling in this manner employs subsequent permeabilization and staining steps, requiring cell fixation. Although pH-dependent fluorophores such as pHluorin2 are useful for imaging surface proteins in live cells, whole cell quantification is obscured by fluorescence from neutral intracellular compartments such as endoplasmic reticulum (ER),3 and detection of protein contained within acidic compartments requires alkaline unmasking steps. Surface levels of protein are influenced by a number of cellular mechanisms including changes in gene expression, protein synthesis, trafficking, and degradation. We sought to create a method that enables quantitation of surface and internal protein levels that could be used to characterize these dynamic processes.
The large conductance, voltage- and calcium-activated potassium (BK) channel, (KCNMA1/Maxi-K/Slo1), requires both depolarization and increases in intracellular Ca2+ for channel opening.4,5 BK channels regulate membrane potential and excitability in multiple cell types, notably in neurons and vascular smooth muscle.6−13 The diversity of BK channel properties in different tissues is driven by extensive alternative splicing of the pore-forming α subunit, which then exhibits varied degrees of surface localization and voltage gating.9,12,14−17 In addition, the tetrameric BKα complex associates with various tissue and cell-type specific β subunits that control channel currents and subcellular localization.6,7,9,16,18−21
Post-translational modifications to the α subunit, including phosphorylation and palmitoylation, can exert robust and rapid changes in channel function, effects that are isoform dependent.22,23 For example, phosphorylation of BKα by protein kinase A (PKA) promotes BK channel opening for some splice isoforms but not others.23−27 Multiple kinases, including PKA, cyclic GMP-dependent protein kinase (PKG), and protein kinase C (PKC), can regulate BK channel currents.26,28−30 Although the biophysical effects of BK channel phosphorylation have been extensively investigated, the regulation of BK channel trafficking to the PM has been relatively ignored. Whole-cell BK channel currents are determined in part by the PM localization of the channel,15,31,32 a point that is especially relevant to pathological alterations in BK channel function in epilepsy,6,11,33 hypertension, and bladder dysfunction.8,28 While modulation of channel opening can induce a rapid and reversible change in cell excitability, alterations in BK channel trafficking and localization could underlie long-lasting changes in cell activity, especially relevant to the development and progression of disease states.6,11,20 Our motivation to determine if intracellular signaling modifies BKα trafficking to increase or decrease surface expression calls for a new quantitative technique.
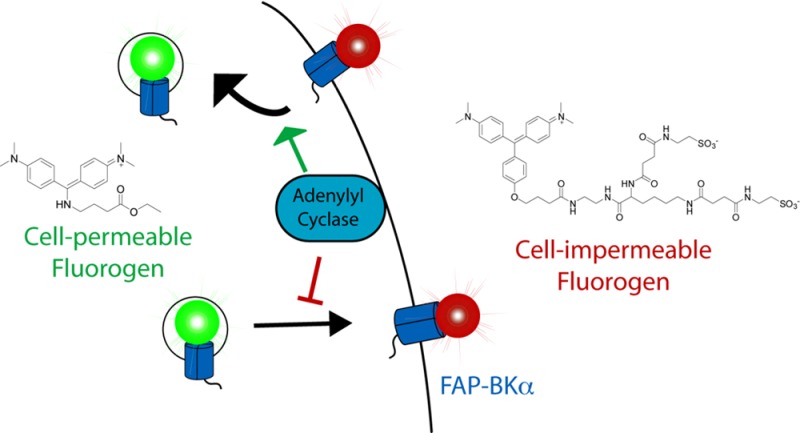
Here we demonstrate a novel method for the rapid and simultaneous detection of both surface and internal protein in living cells using fluorogen-activating peptides (FAPs). Using the previously established dL5** FAP,34 we generated an N-terminal, FAP-BKα fusion construct, where the FAP is localized on the extracellular portion of the channel. We synthesized a novel, cell-permeable, rapidly activated, and highly fluorogenic dye (4-{[Bis(4-dimethylamino-phenyl)-methylene]-amino}-butyric acid ethyl ester; MHN-ester) that produces an activated fluorescence spectra similar to GFP when bound by FAP. In combination with a malachite green (MG)-based fluorogen emitting in the far-red spectral range,34,35 this enables simultaneous detection of two subpopulations of FAP depending on bound dye. When MHN-ester is used together with a cell-impermeable MG, extra- and intracellular stores of the FAP-BKα channel can be distinguished, effectively producing a system for green-inside red-outside (GIRO) labeling in living cells. This labeling qualitatively recapitulates surface and internal labeling by immunofluorescence methods, but is performed in live cells to monitor real-time changes in protein distribution, is highly quantitative (since each FAP binds a single dye molecule), and takes minutes rather than hours. We used GIRO labeling to reveal the dynamics of BKα PM residency. We found that the rate of surface turnover of BKα expressed in HEK293 cells is slow, and that chronic adenylyl cyclase (AC) activation can preferentially reduce the surface levels of BKα. These results suggest a cellular mechanism by which intracellular signaling cascades can alter the abundance of surface BK channels, a route for modifying neural excitability.
Results and Discussion
The FAP system is uniquely suited to studying trafficking; fluorescent signals are generated based on the specific interaction of the dye-binding peptide and fluorogen dyes.34,35 Chemical modifications to the dye can limit its access to the FAP, producing spatially constrained fluorescence activation, such as at the cell surface.36−38 Because the chromophore is formed by the fluorogen dye, generation of alternative dyes which are bound by the same FAP enables the control of fluorescent colors based on which dyes are used.39 By combining this color-selection with control of cell permeability, we aimed to develop a method to label surface and internal protein pools with simultaneous detection using a pair of fluorogen dyes, one designed to be cell-impermeable, thus only labeling cell-surface FAP, and a second, spectrally distinct, cell-permeable dye that would be bound at the remaining intracellular sites (Figure 1A).
Figure 1.
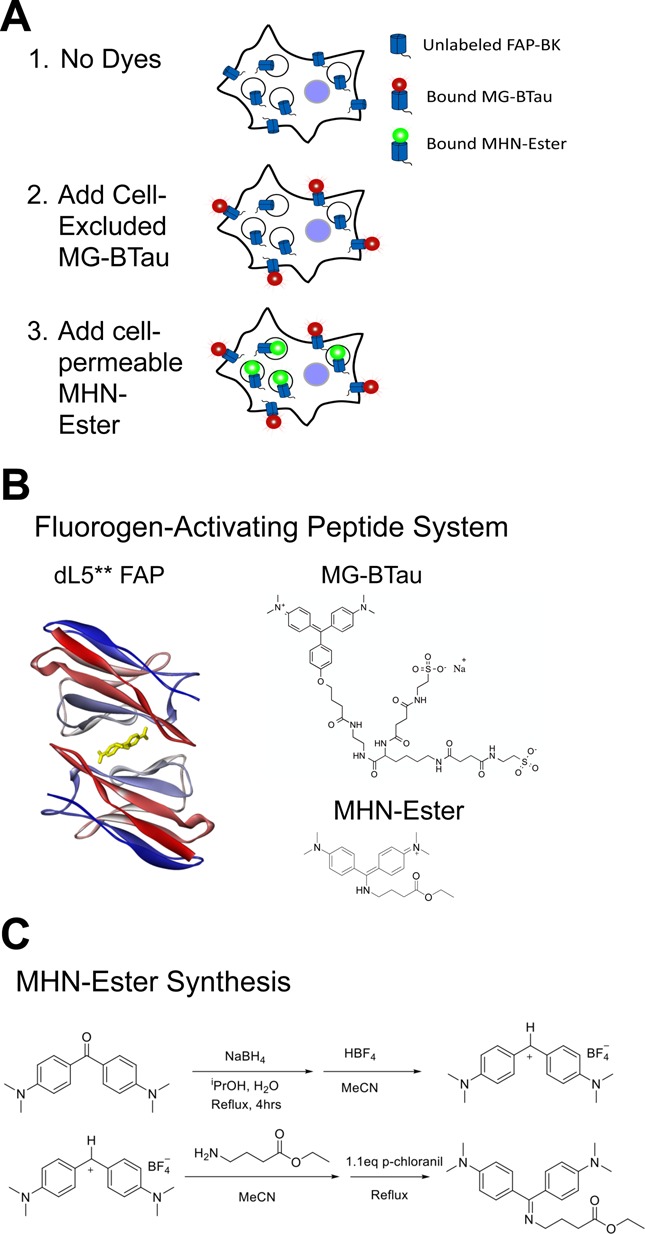
Design of green-inside red-outside (GIRO) labeling (A) schematized paradigm for dye additions. MG-BTau is added first to label surface-exposed FAP; MHN-Ester is then added to label all remaining sites to produce a surface-red internal-green fluorescent signal pattern. (B) Schematic of dL5** FAP, cell excluded MG-BTau, and cell permeable MHN-Ester. Structure shown is dimer of L5** FAP,33 dL5** is formed by addition of a G4S peptide linker. (C) Condensed schematic of MHN-Ester synthesis (see Supporting Information for detailed synthesis).
We have previously characterized a cell-excluded MG-derivative (2-({4-[(2,5-dioxopyrrolidin-1-yl)oxy]-4-oxobutanoyl}amino)ethanesulfonate; MG-BTau) to label surface-exposed FAP.38 For a spectrally distinct counterpart, we developed the novel cell-permeable dye, MHN-Ester (Figure 1B,C). Both dyes are nonfluorescent in solution, but produce spectrally distinct absorption and emission when bound to the FAP (Table 1 and Figure S.1A), allowing for simultaneous detection of different protein populations. In all cases, the FAP used was dL5**, a tandem dimer of L5 originally developed as a tight binder and activator of MG;34,35 this results in a complete high-affinity (Kd = 18 pM) dye-binding module expressed as a single fusion peptide. MHN-Ester was designed to exploit the binding properties of dL5** identified from the L5+MG crystal structure.34 MHN-Ester is prepared from a four-step synthesis (Figure 1C). Purified FAP tightly binds MHN-Ester in solution, with a measured dissociation constant of 42.5 ± 5.7 pM (Table 1 and Figure S.1B). The quantum yield for the activated fluorogen complex was determined to be 0.30 (Table 1 and Figure S.1C). Cell-permeable ester dyes showed proper intracellular accessibility at concentrations below 1 μM, while sulfonated analogs were cell-excluded (Figure S.2).
Table 1. Properties of dL5** FAP Fluorogen Activationa.
| λmax | λex | λem | εmax | Φ | εB/εFb | ΦB/ΦF | ARc | KD | |
|---|---|---|---|---|---|---|---|---|---|
| MHN-Ester | 422 | (422) | (488) | 5.9 × 104 | 4.2 × 10–4 | 12 | 710 | 8520 | 42.5 |
| MHN-Ester/dL5** | 456 | 456 | 532 | 6.4 × 104 | 0.3 | ||||
| MG-BTau | 606 | (606) | (636) | 9.1 × 104 | 9.5 × 10–5 | 3.5 | 2010 | 7035 | 18 |
| MG-BTau/dL5** | 633 | 633 | 668 | 1.1 × 105 | 0.19 |
Units: λ, nm; ε, M–1 cm–1; KD, pM.
εB and εF: Extinction coefficients of free and bound dye at microscopy excitation wavelength (488 and 640 nm for MHN-ester and MG-BTau respectively).
Activation Ratio: (εB/εF) × (ΦB/ΦF).
Design and Validation of FAP-BKα
FAP was fused to the extracellular N-terminus of the ZERO isoform of BKα, allowing FAP access to the extracellular environment (Figure 2A). An HA tag was included at the very N-terminus of the FAP for immunohistochemical verification, since there are no antibodies against either the FAP or extracellular BKα epitopes. HEK293 cells stably expressing FAP-BKα were established. Sequential dye addition provided optimal quantitative labeling of surface and internal proteins. MG-BTau (300 nM) was added first in order to saturate surface-exposed FAP. After a 5 min incubation, MHN-Ester (300 nM) was added to occupy all unbound sites. With its low Kd and slow off-rate,38 MG-BTau labeling is effectively irreversible over these short experimental time scales. We identified no measurable displacement of complexed MG-BTau by MHN-Ester either microscopically or in suspension measured by flow cytometry (Figure S.3), even after washing. These features of the fluorogen+FAP complexes enabled the design of a surface and internal dual-sensor.
Figure 2.
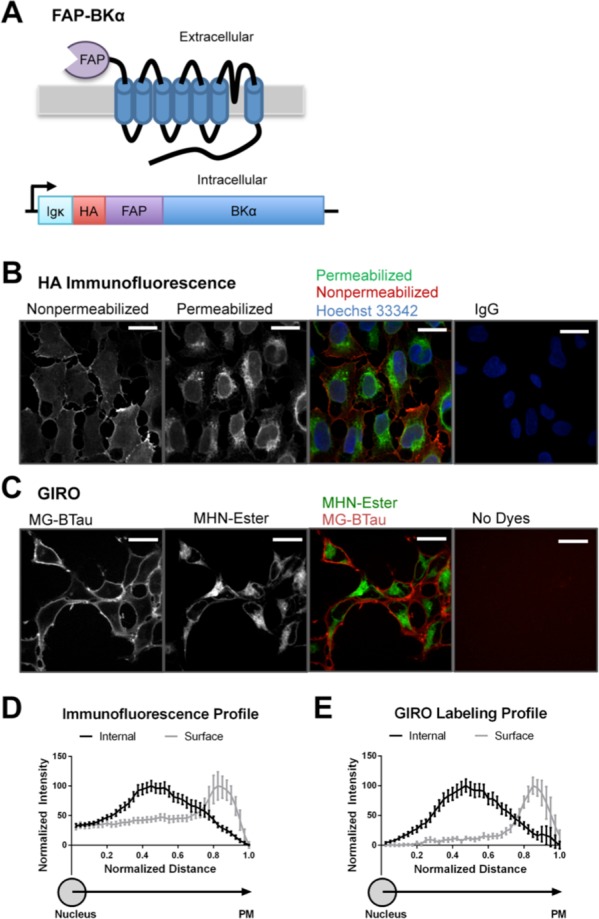
GIRO labeling recapitulates immunofluorescence methods. (A) Schematic of FAP-BKα construct showing protein topology and DNA configuration. An HA tag is included at the very N-terminus of FAP-BKα. (B) Immunofluorescence against HA without (red) and with (green) permeabilization to label surface and internal protein, respectively. (C) Live cells labeled with MG-BTau (red) and MHN-Ester (green) to label surface and internal protein in FAP-BKα expressing live cells. (D) Normalized profile plot for fixed cells quantified from nucleus to cell periphery (distance 0.0 corresponds to nuclear center; 1.0 is just beyond PM). Peak intensities are at 0.42 for internal and 0.81 for surface labeling. (E) Cumulative profile plots for dye-labeled live cells. Peak intensities are at 0.44 for internal and 0.82 for surface labeling. Profile plot distance bins are average pixel values. n = 25 cells from 3 experiments for each condition. Scale bars = 40 μm.
A clear advantage of the FAP system is that unbound dye remains nonfluorescent in solution, allowing for the simple addition of dyes to the cellular media without any need for fixation or washout, and enabling live-cell imaging.35 This can be compared to the traditional method of using immunofluorescence against an ectofacial epitope in nonpermeabilized cells. Immunofluorescence using an anti-BKα antibody showed similar localization for FAP-BKα and untagged BKα transfected into HEK-293 cells (Figure S.4), suggesting that the addition of the FAP tag does not measurably disrupt BKα trafficking. In stably transfected FAP-BKα cells, HA-immunofluorescence without permeabilization resulted in a strong PM signal with a distinct restriction to the cell surface (Figure 2B). Subsequent staining following permeabilization revealed clear internal localization. This labeling process took approximately 8 h to perform. In contrast, sequential labeling with MG-BTau and MHN-Ester can be complete in as little as 7 min and similarly showed visible segregation of surface and internal signals (Figure 2C). Profile plots were drawn from the center of the nucleus to the outside edge of the PM along the longest axis. Because cells are not uniform in size or shape, and fixed cells tend to be wider due to the mounting process, distance was normalized (Figure 2D,E). We found that surface and internal labeling by immunofluorescence or GIRO yielded similar distributions for surface and internal channels. Interestingly, GIRO labeling exhibited lower intracellular background compared to immunostaining as shown by the magnitude of signal to the left of the peaks; this is potentially a result of flattening of the cells and a limited degree of membrane permeability conferred by fixation and coverslip mounting. Thus, MG-BTau successfully labels the cell-surface FAP while MHN-Ester labels internal protein; both channels can be quantified simultaneously with clear spectral discrimination. GIRO labeling recapitulates established antibody methods with the distinct advantages of being more rapid with lower background and applicable to live cells.
Dye Properties and Activation in Flow Cytometry
The GIRO labeling system functions as a “smart-probe” distinguishing and reporting protein localization simply based on color in live cells. In this way, we can quantify levels of surface protein, internal protein, and use the ratio of surface to internal signal to generate a measure of relative surface expression (RSE). Flow cytometry presents an advantageous application of this system, as it is significantly higher throughput and more unbiased than region-of-interest selection or cell segmentation in microscopy. We confirmed specific binding using whole-cell fluorescence by flow cytometry. By measuring MHN-Ester fluorescence intensity in cells through a range of concentrations from 5 pM to 600 nM, we found a labeling Keff of 7.62 (±0.82) nM (Figure S.5A), with saturating levels below 300 nM. We opted to use 300 nM MHN-Ester in experiments since this saturates signal without unnecessary contribution to nonspecific background (Figure S.5B). Next, in order to optimize our labeling paradigm for flow cytometry, we characterized the rates of dye activation and saturation using flow-cytometric timecourse measurements. MHN-Ester binding saturates rapidly, reaching a plateau in less than 2 min (Figure 3A). Application of MG-BTau prior to MHN-Ester addition causes a roughly 6% decrease in maximum MHN-ester intensity (Figure 3B) demonstrating that the MG-BTau labeled population reduces available sites for MHN-ester binding and that the majority of FAP-BKα is internal. The tetrameric BKα subunit contains 4 FAP molecules each of which can independently bind only one molecule of dye. This consistent labeling stoichiometry enables reliable quantitation not possible with standard immunofluorescence, which can vary in the number of bound antibodies and fluorophores per antibody.
Figure 3.

MHN-Ester addition to FAP-BKα expressing cells shows rapid fluorescence activation. (A) MHN-Ester activation rate was measured by flow cytometry. Representative smoothed curves show rapid activation and signal saturation within 2 min of dye addition. A small decrease in MHN-Ester signal as a result of MG-BTau precomplexing is evident. (B) Quantification of end point MHN-Ester signal with and without precomplexing of MG-BTau, showing a small but statistically significant decrease in saturation level signal (3 experiments for each condition, ** p ≤ 0.01, Student’s t test with Welch’s correction).
Disruption of Global Trafficking Pathways Alters Relative Surface Expression
To validate the response of the two-color labeling system, cellular trafficking pathways were pharmacologically suppressed to observe changes in surface and internal labeling in FAP-BKα expressing cells. This approach validates the sensitivity of this labeling approach to subtle changes in protein distribution, and establishes the lifetime of BKα at the cell surface, which has not previously been characterized. These experiments were carried out in stably transfected HEK293 cells, due to their lack of endogenous BKα or β subunits, which can alter trafficking.14,15,18−20,40 To confirm that changes in trafficking can be monitored using GIRO labeling, forward trafficking was blocked with the ER-export inhibitor brefeldin A (BFA; 5 μM), allowing measurement of the removal of channels from the PM over time. Representative flow cytometry histograms from BFA-treated, FAP-BKα-expressing cells show a large fluorescence reduction from surface MG-BTau emission, with a comparatively meager reduction in the MHN-Ester signal after 18 h BFA treatment (Figure 4A). Quantification of median surface MG-BTau signal shows a continuous decrease over time from 1 to 18 h (Figure 4B), with significant effects appearing at 3 h. In the same cells, internal signal from MHN-Ester labeling showed a minimal change. A statistically significant reduction in internal labeling is observable after 18 h, possibly due to diversion of BKα to degradation pathways with this extended blockade of ER export. By taking the relative ratio of median surface and internal signal, a significant 22.3% reduction in RSE was observed after 6 h. At 18 h, RSE was reduced by 71.6% compared to vehicle-treated control cells (Figure 4C). We conclude that FAP-BKα turnover in HEK293 cells is slow, with a PM residency half-time in the tens of hours.
Figure 4.

Two-color labeling tracks changes in relative surface expression. (A) Representative fluorescence intensity histograms for Brefeldin A (BFA) treated cells over 18 h. (B) Median fluorescence values for labeled samples for surface (MG-BTau) and internal (MHN-Ester). The normalized relative surface expression (MG-Btau/MHN-Ester) is shown as a measure of surface residence. (C) RSE declines to 77.7% after 6 h, and to 38.4% after 18 h. Error bars are 95% confidence intervals, n = 9–12 replicates per condition from 4 experiments for all conditions except for 18 h, for which n = 9 from 3 experiments. (D) Representative fluorescence intensity histograms for dynasore treated cells over 6 h. (E) Quantification of surface and internal labeling in dynasore treated cells. Median fluorescence values were averaged across samples. (F) RSE of dynasore treatment shows a steady increase to 127.0% after 6 h (n = 9 biological replicates from 3 experiments). Error bars are 95% confidence intervals, one-way ANOVA with Dunnett’s multiple comparison test. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
We next analyzed delivery to the PM by blocking endocytosis (and hence removal from the PM) with the dynamin 1/2 inhibitor dynasore. Dynasore (50 μM) was applied for 1, 3, and 6 h. Representative histograms of fluorescence intensity (Figure 4D) at 6 h show an increase in surface MG-BTau fluorescence but not in MHN-ester fluorescence (Figure 4E). We observed a steady and significant increase in RSE over time; with a significant 27.0% increase after 6 h (Figure 4F). This increase in surface levels induced by dynasore appears to be in equilibrium with the measured decrease in BFA treated cells. Taken together, BKα delivery and removal from the cell surface in these stable cells appears to be in equilibrium, and GIRO labeling has sufficient precision to detect these changes.
Forskolin (Fsk) Reduces BKα Surface Expression
In the brain, AC acts as a signaling nexus for neuromodulators, critical in effecting synaptic plasticity by activation of downstream signaling targets. One such target, PKA, is well-characterized in regulating the biophysical properties of BK channels.23,25−27,29,30,41 Phosphorylation-dependent modification of channel opening properties provides short- to medium-term regulation of BK currents, readily reversed by phosphatases. Longer-term regulation of cellular BK channel currents could involve reduction or enhancement of channel density at the PM, and changes in overall expression level of the channel. Given PKA’s established interactions with the BKα C-terminal domain and its known involvement in trafficking of receptors and other ion channels,42−44 we decided to examine the effect of AC activation on surface expression of FAP-BKα by overnight treatment with 25 μM fsk.
Examination of FAP-BKα expressing cells qualitatively by microscopy (Figure 5A) and quantitatively by flow cytometry (Figure 5B) showed a substantial and significant decrease in cell surface labeling accompanied by a meager reduction in internal labeling after fsk treatment. Indeed, quantification of median fluorescence in flow cytometry (Figure 5C–E) showed a 38% decrease in surface labeling compared to vehicle-treated control samples. Internal labeling of FAP-BKα was also significantly decreased, but this decrease was to a lesser extent than surface labeling (15.3% decrease). This led to a significant 31% decrease in RSE. For comparison, a constitutively expressed FAP construct targeted to the PM (FAP-TM, Figure 5A) was expressed in HEK293 cells. FAP-TM contains a transmembrane domain derived from platelet-derived growth factor receptor 1 to confer surface expression; it contains no PKA phosphorylation sites or characterized trafficking signals. In contrast to FAP-BKα, FAP-TM showed no change in localization response to fsk (Figure 5B–E, Figure S.6). These data demonstrate that overnight application of fsk preferentially reduces surface levels of BKα. We have not yet determined whether the observed effect is due to alterations in endocytosis, degradation, or ER export. The effect of fsk apears specific to FAP-BKα, and overnight incubation is not altering global trafficking pathways reported by FAP-TM.
Figure 5.

Overnight forskolin application reduces surface level of FAP-BKα. (A) Representative microscope images of FAP-BKα stable cells treated with 25 μM forskolin or 0.1% DMSO overnight. FAP-TM expression is shown below; note the high surface expression without substantial internal labeling. Scale bars are 40 μm. (B) Representative flow cytometry histograms of FAP-BKα and FAP-TM surface and internal labeling with overnight fsk treatment. Nonexpressing cells run with dyes were used as a background control. Note the relative lack of MHN-Ester labeling in FAP-TM cells compared to FAP-BKα. (C) Quantification of median surface fluorescence for FAP-BKα and FAP-TM with fsk treatment (FAP-BKα = 62.9% of control, FAP-TM = 96.6% of control). (D) Median internal fluorescence for FAP-BKα and FAP-TM with fsk treatment (FAP-BKα = 89.2% of control, FAP-TM = 98.2% of control). (E) RSE for FAP-BKα and FAP-TM (FAP-BKα = 70.4% of control, FAP-TM = 97.9% of control). Samples were normalized to DMSO intensity for each experiment. n = 19 biological replicates from 7 experiments for FAP-BKα. n = 9 biological replicates from 3 experiments for FAP-TM. RSE was determined by (median MG-BTau)/(median MHN-Ester) for each sample. Error bars are ±95% confidence intervals. Significance was determined by Student’s t test with Welch’s correction. Significant p-values are shown.
Conclusions
In this report, we have demonstrated GIRO labeling, a two-color, compartment selective FAP-based approach that generates distinct signals from surface and internal proteins in live cells for simultaneous detection. GIRO reliably and quantitatively measures changes in surface and internal levels of target proteins with distinct advantages compared to immunofluorescence including speed, capacity for high throughput, use in live cells, and fixed binding stoichiometry. The availability of cell-permeable and impermeable variants of MHN and MG dyes (Figure S.3) allow a variety of pulse-chase experiments to distinguish endocytic, exocytic, and internal resident cargoes.45 The FAP is genetically encoded analogously to other fusion tags or proteins, but the modular nature of the FAP–dye interaction also enables the use of physiological sensors by changing the applied dye.46
We identified a PM half-life for BKα in the tens of hours, demonstrating that GIRO labeling can be applied to quantitate changes in protein surface expression. The intrinsic stability of BKα at the cell surface is likely to vary among splicing isoforms and be influenced by the incorporation of certain β subunits which contain their own endocytic and trafficking signals.18−21 This imaging platform can be used with fluorescence microscopy to identify cellular locations where BKα surface residence is high, or where BKα is retained inside the cell, even in positions juxtaposed to the membrane which would be optically indistinguishable from PM. HEK293 cells are nonpolarized and not known to express BKα or β subunits, and while BKα is sufficient to assemble into a functional tetrameric channel, it is unknown whether β-subunit lacking BK channels are commonly found in cells. Future studies aim to address the roles of β subunits and alternative splicing in modifying BK channel trafficking, and their roles in BK channel localization to the cell surface and in PM microdomains.
Methods
Synthesis of Dyes
MG-BTau was prepared as described previously.38 Synthesis of MHN-Ester is schematized in Figure 1D and described in detail, along with spectroscopic and biochemical properties and methods in Supporting Information.
DNA Constructs
Murine BKα ZERO (MDAL start and EMVYR end, accession# NM_010610.2) was cloned previously.20 FAP-BKα was generated by addition of FAP to the N-terminus. A murine Igκ signal sequence was added to the N-terminus to ensure proper membrane orientation, addition of such a signal sequence has previously been shown to not adversely affect BKα topology.47 FAP-BKα was subcloned into pcDNA3.1 using EcoRI and BamHI sites for generation of stable cells (see Supporting Information). FAP-TM was generated by the addition of a PDGFR1 transmembrane domain to dL5** in a pBabe backbone.
Cell Lines and Culture
HEK293 cells were maintained in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% fetal bovine serum. Stable HEK293 cells expressing FAP-BKα were generated by transfection of PacI linearized pcDNA3.1 encoding FAP-BKα. Cells were selected for 1 week with 1 μg/mL G418. Selected cells were subjected to two rounds of fluorescence-activated cell sorting (FACS) after labeling with cell-permeant MG-Ester. Single cells were sorted into wells of a 96-well plate to generate clonal lines. Clones were identified by MG-ester fluorescence, and two clones with different expression levels were selected for use in experiments; these clones had distinct baseline GIRO profiles (Figure S.7A). Polyclonal cells stably expressing FAP-TM were generated by transfection of FAP-TM and selection with 2 μg/mL puromycin. The dynamin 1/2 inhibitor dynasore, the ER-Golgi trafficking inhibitor brefeldin A, and adenylyl cyclase activator forskolin were acquired from Cayman Chemical Corp (Ann Arbor, Michigan). Dynasore was prepared as a 50 mM stock in DMSO and aliquotted. Brefeldin A was prepared as a 5 mM stock in DMSO, Forskolin was prepared as 50 mM stock in DMSO. Cells were deprived of serum for 2 h before dynasore treatment. Cells were treated with inhibitors for the indicated times (1–18 h), with equivalent volumes of DMSO as vehicle control.
Immunofluorescence
Antibodies against the HA epitope were acquired from Abcam (clone HA.C5, www.abcam.com, Cambridge, MA), Anti-BKα (clone L6/60) monoclonal antibodies were acquired from NeuroMab (http://neuromab.ucdavis.edu/). Anti-HA was used for surface/total labeling of FAP-BKα due to the ectofacial HA epitope. Cells were seeded on 25 mm coverslips (Corning). Cells were fixed in 4% paraformaldehyde (Electron Microscopy Sciences, www.emsdiasum.com) for 10 min and washed twice in PBS. Cells were permeabilized as needed with 0.5% Triton-X for 5 min; blocking was done by adding PBS containing 10% fetal bovine serum for 20 min. Anti-HA antibody was applied at a dilution of 1:1000 for 1 h at room temperature. Anti-BKα antibody was applied at a dilution of 1:250–1:500 for 4 h at room temperature. After primary antibody incubation, coverslips were washed three times with PBS. Alexa 568 or Alexa 488 conjugated anti-mouse secondary antibody was applied at a 1:500 dilution for 1 h at room temperature. For surface and total labeling against HA, this process was done once with permeabilization omitted, then repeated with permeabilization and a different color secondary. Coverslips were mounted onto slides using a homemade poly(vinyl alcohol)-based mounting media. HA slides were imaged on a Nikon spinning disk confocal microscope (Andor Technologies) using a 40× Nikon Plan Fluor objective (NA 1.30) with oil immersion, BKα immunostained slides were imaged on a Zeiss LSM 510 using a Zeiss Plan-Neofluar 40× objective (NA 1.30).
Live Cell Imaging
Live cells were seeded in 35 mm glass-bottom dishes (MatTek corporation). Prior to imaging, media was switched to Fluorobrite DMEM (Life Technologies). Images were acquired prior to dye addition to assess cellular autofluorescence. MG-BTau was added directly to cell media to a final concentration of 300 nM. After 5 min, MHN-Ester was added to the cellular media to a final concentration of 300 nM. Cells were imaged using a Nikon spinning disk confocal microscope (Andor Technology) using a Zeiss Plan Fluor objective (NA 1.30) with oil immersion.
Quantification of Localization
Profile plots were drawn from the center of the nucleus to the PM of fixed and live cells along the longest axis in order to include perinuclear and endoplasmic reticulum staining (n = 25 cells from 3 experiments for each condition). Nuclear center was identified in fixed cells by Hoechst staining and in live cells by BKα nuclear exclusion. Pixel values were measured using ImageJ (NIH). Profile plots were processed using a custom Python script in which distance was normalized by aggregating mean pixel values into 40 distance bins.
Flow Cytometry and RSE Measurement
Cells were grown and treated with drugs in 12-well plates (Greiner). Adherent cells were labeled at room temperature for 5 min by addition of MG-BTau directly to cell media to a concentration of 300 nM. After the 5 min incubation, all media was aspirated and cells were moved to ice and detached using cold PBS containing 4 mM EDTA. Single-cell suspensions were generated by vigorous pipetting and cells were moved to round-bottom 96-well plates (Greiner) for flow cytometry. MHN-Ester was added to all wells to a final concentration of 300 nM 5 min prior to flow cytometry initiation. Live cells were analyzed using an Accuri C6 cell analyzer with Intellicyte plate sampler attachment. Populations for analysis were selected by forward and side scatter; MG-BTau fluorescence (FL4-A channel, 640 nm excitation, 675/30 nm emission filter) and MHN-Ester fluorescence (FL1-A channel, 488 nm excitation, 525/15 nm emission filter) values were collected and analyzed using FlowJo (FlowJo LLC). RSE was determined by (Median MG-BTau)/(Median MHN-Ester). Due to potential variability in instrument sensitivity from day to day as well as differences expression between clones, all experiments were normalized to contemporaneous vehicle controls. Data from clones were thus normalized and pooled, as both clones responded similarly to drug treatments (Figure S7.B). This particular protocol was chosen for ease of labeling surface proteins without removing drug treatments; however, similar results were obtained if cells were detached and suspended, followed by sequential addition of dyes prior to analysis without any washing steps. This may not hold true for more rapidly recycling proteins.
Measurement of Dye Activation Rate in Cells
Dye activation was measured using flow cytometry. Cells were prepared by detachment with cold PBS containing 8 mM EDTA and moved to 1.5 mL microcentrifuge tubes. Flow cytometry was initiated at a flow rate of 40 μL/min. After 1 min of run, a 10× concentrate of the requisite dye was added during continuous sampling to produce the 300 μM final concentration. Events were discretized into 0.1-s time bins, each bin containing 100–200 cells. Mean fluorescence intensities per time bin were quantified using Python 3.2. Nearest neighbor smoothing was performed using GraphPad Prism (20 neighbors on each side to obtain a 4 s moving average, second-order polynomial fit) to generate plots of mean cell fluorescence vs time.
Acknowledgments
We thank Christopher Szent-Gyorgyi for help in illustrating the FAP-Fluorogen complex. This work was supported in part by the National Institutes of Health (R21MH100612: M.P.B., C.P.P., A.L.B.; P30DK072506: J.H.) and the Shurl and Kay Curci Foundation (Y.W., J.H., M.P.B.).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconjchem.5b00409.
Detailed experimental procedures, MHN dye synthesis and photophysical properties, BKα construct details and localization, and dye dissociation data (PDF)
The authors declare the following competing financial interest(s): MPB is a founder of Sharp Edge Labs, a company that is utilizing the fluorogen activating peptide commercially.
Supplementary Material
References
- Shah M. M.; Hammond R. S.; Hoffman D. A. (2010) Dendritic ion channel trafficking and plasticity. Trends Neurosci. 33, 307–316 10.1016/j.tins.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenböck G.; de Angelis D.; Rothman J. E. (1998) Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394, 192. 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Paroutis P. (2004) The pH of the Secretory Pathway: Measurement, Determinants, and Regulation. News Physiol. Sci. 19, 207–215 10.1152/physiol.00005.2004. [DOI] [PubMed] [Google Scholar]
- Toro L.; Wallner M.; Meera P.; Tanaka Y. (1998) Maxi-KCa, a Unique Member of the Voltage-Gated K Channel Superfamily. Physiology 13, 112–117. [DOI] [PubMed] [Google Scholar]
- Vergara C.; Latorre R.; Marrion N. V.; Adelman J. P. (1998) Calcium-activated potassium channels. Curr. Opin. Neurobiol. 8, 321–329 10.1016/S0959-4388(98)80056-1. [DOI] [PubMed] [Google Scholar]
- Brenner R.; Chen Q. H.; Vilaythong A.; Toney G. M.; Noebels J. L.; Aldrich R. W. (2005) BK channel beta4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nat. Neurosci. 8, 1752–1759 10.1038/nn1573. [DOI] [PubMed] [Google Scholar]
- Jaffe D. B.; Wang B.; Brenner R. (2011) Shaping of action potentials by type I and type II large-conductance Ca2+-activated K+ channels. Neuroscience 192, 205–218 10.1016/j.neuroscience.2011.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph B. K.; Thakali K. M.; Moore C. L.; Rhee S. W. (2013) Ion channel remodeling in vascular smooth muscle during hypertension: Implications for novel therapeutic approaches. Pharmacol. Res. 70, 126–138 10.1016/j.phrs.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orio P.; Rojas P.; Ferreira G.; Latorre R. (2002) New disguises for an old channel: MaxiK channel beta-subunits. Physiology 17, 156–161 10.1152/nips.01387.2002. [DOI] [PubMed] [Google Scholar]
- Sachse G.; Faulhaber J.; Seniuk A.; Ehmke H.; Pongs O. (2014) Smooth muscle BK channel activity influences blood pressure independent of vascular tone in mice. J. Physiol. 592, 2563–74 10.1113/jphysiol.2014.272880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shruti S.; Clem R. L.; Barth A. L. (2008) A seizure-induced gain-of-function in BK channels is associated with elevated firing activity in neocortical pyramidal neurons. Neurobiol. Dis. 30, 323–330 10.1016/j.nbd.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng-Crank J.; Foster C. D.; Krause J. D.; Mertz R.; Godinot N.; DiChiara T. J.; Reinhart P. H. (1994) Cloning, expression, and distribution of functionally distinct Ca(2+)-activated K+ channel isoforms from human brain. Neuron 13, 1315–1330 10.1016/0896-6273(94)90418-9. [DOI] [PubMed] [Google Scholar]
- Wang B.; Rothberg B. S.; Brenner R. (2009) Mechanism of increased BK channel activation from a channel mutation that causes epilepsy. J. Gen. Physiol. 133, 283–294 10.1085/jgp.200810141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu Y.-H.; Alvarez-Baron C.; Kim E.; Dryer S. (2010) Dominant-negative regulation of cell surface expression by a pentapeptide motif at the extreme COOH terminus of an Slo1 calcium-activated potassium channel splice variant. Mol. Pharmacol. 77, 497–507 10.1124/mol.109.061929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E. Y.; Ridgway L. D.; Zou S.; Chiu Y.-H.; Dryer S. E. (2007) Alternatively spliced C-terminal domains regulate the surface expression of large conductance calcium-activated potassium channels. Neuroscience 146, 1652–1661 10.1016/j.neuroscience.2007.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen A. N.; Wulf H.; Hay-Schmidt A.; Jansen-Olesen I.; Olesen J.; Klaerke D. A. (2009) Differential expression of BK channel isoforms and beta-subunits in rat neuro-vascular tissues. Biochim. Biophys. Acta, Biomembr. 1788, 380–389 10.1016/j.bbamem.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Fodor A. A.; Aldrich R. W. (2009) Convergent Evolution of Alternative Splices at Domain Boundaries of the BK Channel. Annu. Rev. Physiol. 71, 19–36 10.1146/annurev.physiol.010908.163124. [DOI] [PubMed] [Google Scholar]
- Toro B.; Cox N.; Wilson R. J.; Garrido-Sanabria E.; Stefani E.; Toro L.; Zarei M. M. (2006) KCNMB1 regulates surface expression of a voltage and Ca2+-activated K+ channel via endocytic trafficking signals. Neuroscience 142, 661–669 10.1016/j.neuroscience.2006.06.061. [DOI] [PubMed] [Google Scholar]
- Zarei M. M.; Song M.; Wilson R. J.; Cox N.; Colom L. V.; Knaus H. G.; Stefani E.; Toro L. (2007) Endocytic trafficking signals in KCNMB2 regulate surface expression of a large conductance voltage and Ca2+-activated K+ channel. Neuroscience 147, 80–89 10.1016/j.neuroscience.2007.04.019. [DOI] [PubMed] [Google Scholar]
- Shruti S.; Urban-Ciecko J.; Fitzpatrick J. A.; Brenner R.; Bruchez M. P.; Barth A. L. (2012) The Brain-Specific Beta4 Subunit Downregulates BK Channel Cell Surface Expression. PLoS One 7, e33429–e33429 10.1371/journal.pone.0033429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox N.; Toro B.; Pacheco-Otalora L. F.; Garrido-Sanabria E. R.; Zarei M. M. (2014) An endoplasmic reticulum trafficking signal regulates surface expression of β4 subunit of a voltage- and Ca(2+)-activated K(+) channel. Brain Res. 1553, 12–23 10.1016/j.brainres.2014.01.028. [DOI] [PubMed] [Google Scholar]
- Jeffries O.; Tian L.; McClafferty H.; Shipston M. J. (2012) An electrostatic switch controls palmitoylation of the large conductance voltage- and calcium-activated potassium (BK) channel. J. Biol. Chem. 287, 1468–77 10.1074/jbc.M111.224840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L.; Jeffries O.; McClafferty H.; Molyvdas A.; Rowe I. C.; Saleem F.; Chen L.; Greaves J.; Chamberlain L. H.; Knaus H.-G.; et al. (2008) Palmitoylation gates phosphorylation-dependent regulation of BK potassium channels. Proc. Natl. Acad. Sci. U. S. A. 105, 21006–21011 10.1073/pnas.0806700106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart P. H.; Chung S.; Martin B. L.; Brautigan D. L.; Levitan I. B. (1991) Modulation of calcium-activated potassium channels from rat brain by protein kinase A and phosphatase 2A. J. Neurosci. 11, 1627–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L.; Duncan R. R.; Hammond M. S. L.; Coghill L. S.; Wen H.; Rusinova R.; Clark A. G.; Levitan I. B.; Shipston M. J. (2001) Alternative Splicing Switches Potassium Channel Sensitivity to Protein Phosphorylation. J. Biol. Chem. 276, 7717–7720 10.1074/jbc.C000741200. [DOI] [PubMed] [Google Scholar]
- Zhou X.-B.; Wulfsen I.; Utku E.; Sausbier U.; Sausbier M.; Wieland T.; Ruth P.; Korth M. (2010) Dual role of protein kinase C on BK channel regulation. Proc. Natl. Acad. Sci. U. S. A. 107, 8005–10 10.1073/pnas.0912029107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G.; Shi J.; Yang L.; Cao L.; Park S. M.; Cui J.; Marx S. O. (2004) Assembly of a Ca2+-dependent BK channel signaling complex by binding to beta2 adrenergic receptor. EMBO J. 23, 2196–2205 10.1038/sj.emboj.7600228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyle B. D., and Braun A. P.. The regulation of BK channel activity by pre- and post-translational modifications. Front. Physiol. 2014, 5, DOI: 10.3389/fphys.2014.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert R.; Nelson M. T. (2001) Protein kinases: tuners of the BKCa channel in smooth muscle. Trends Pharmacol. Sci. 22, 505–512 10.1016/S0165-6147(00)01775-2. [DOI] [PubMed] [Google Scholar]
- Velàzquez-Marrero C.; Seale G. E.; Treistman S. N.; Martin G. E. (2014) BK Channel β4 Subunit Influences Sensitivity and Tolerance to Alcohol by Altering its Response to Kinases. J. Biol. Chem. 289, 29261–29272 10.1074/jbc.M114.604306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipston M. J. (2011) Ion channel regulation by protein palmitoylation. J. Biol. Chem. 286, 8709–8716 10.1074/jbc.R110.210005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C.; Cavolo S. L.; Levitan E. S. (2012) Delayed endosome-dependent CamKII and p38 kinase signaling in cardiomyocytes destabilizes Kv4.3 mRNA. J. Mol. Cell. Cardiol. 52, 971–977 10.1016/j.yjmcc.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Ye J.; Zou X.; Xu Z.; Feng Y.; Zou X.; Chen Z.; Li Y.; Cang Y. (2014) CRL4A(CRBN) E3 ubiquitin ligase restricts BK channel activity and prevents epileptogenesis. Nat. Commun. 5, 3924. 10.1038/ncomms4924. [DOI] [PubMed] [Google Scholar]
- Szent-Gyorgyi C.; Stanfield R. L.; Andreko S.; Dempsey A.; Ahmed M.; Capek S.; Waggoner A. S.; Wilson I. A.; Bruchez M. P. (2013) Malachite Green Mediates Homodimerization of Antibody VL Domains to Form a Fluorescent Ternary Complex with Singular Symmetric Interfaces. J. Mol. Biol. 425, 4595. 10.1016/j.jmb.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szent-Gyorgyi C.; Schmidt B. A.; Creeger Y.; Fisher G. W.; Zakel K. L.; Adler S.; Fitzpatrick J. A. J.; Woolford C. A.; Yan Q.; Vasilev K. V.; Berget P. B.; et al. (2008) Fluorogen-activating single-chain antibodies for imaging cell surface proteins. Nat. Biotechnol. 26, 235–240 10.1038/nbt1368. [DOI] [PubMed] [Google Scholar]
- Fisher G. W.; Adler S. A.; Fuhrman M. H.; Waggoner A. S.; Bruchez M. P.; Jarvik J. W. (2010) Detection and Quantification of 2AR Internalization in Living Cells Using FAP-Based Biosensor Technology. J. Biomol. Screening 15, 703–709 10.1177/1087057110370892. [DOI] [PubMed] [Google Scholar]
- Holleran J.; Brown D.; Fuhrman M. H.; Adler S. A.; Fisher G. W.; Jarvik J. W. (2010) Fluorogen-activating proteins as biosensors of cell-surface proteins in living cells. Cytometry, Part A 77A, 776–782 10.1002/cyto.a.20925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q.; Schmidt B. F.; Perkins L. A.; Naganbabu M.; Saurabh S.; Andreko S. K.; Bruchez M. P. (2015) Near-instant surface-selective fluorogenic protein quantification using sulfonated triarylmethane dyes and fluorogen activating proteins. Org. Biomol. Chem. 13, 2078–2086 10.1039/C4OB02309A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo E.; Wienbar S.; Snyder A. C.; Vasilev K. V.; Armitage B. A.; Jarvik J. W. (2014) A Single-Chain-Variable-Fragment Fluorescence Biosensor Activates Fluorogens from Dissimilar Chemical Families. Protein Pept. Lett. 21, 1289–1294. [PubMed] [Google Scholar]
- Zarei M. M.; Zhu N.; Alioua A.; Eghbali M.; Stefani E.; Toro L. (2001) A novel MaxiK splice variant exhibits dominant-negative properties for surface expression. J. Biol. Chem. 276, 16232–9 10.1074/jbc.M008852200. [DOI] [PubMed] [Google Scholar]
- Nara M.; Dhulipala P. D.; Wang Y.-X.; Kotlikoff M. I. (1998) Reconstitution of β-Adrenergic Modulation of Large Conductance, Calcium-activated Potassium (Maxi-K) Channels in Xenopus Oocytes. J. Biol. Chem. 273, 14920–14924 10.1074/jbc.273.24.14920. [DOI] [PubMed] [Google Scholar]
- Diering G. H.; Gustina A. S.; Huganir R. L. (2014) PKA-GluA1 Coupling via AKAP5 Controls AMPA Receptor Phosphorylation and Cell-Surface Targeting during Bidirectional Homeostatic Plasticity. Neuron 84, 790–805 10.1016/j.neuron.2014.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Pena y Valenzuela I.; Pires-Oliveira M.; Akaaboune M. (2013) PKC and PKA Regulate AChR Dynamics at the Neuromuscular Junction of Living Mice. PLoS ONE 8, e81311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vistein R.; Puthenveedu M. A. (2013) Reprogramming of G protein-coupled receptor recycling and signaling by a kinase switch. Proc. Natl. Acad. Sci. U. S. A. 110, 15289–15294 10.1073/pnas.1306340110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher G. W.; Fuhrman M. H.; Adler S. A.; Szent-Gyorgyi C.; Waggoner A. S.; Jarvik J. W. (2014) Self-Checking Cell-Based Assays for GPCR Desensitization and Resensitization. J. Biomol. Screening 19, 1220. 10.1177/1087057114534299. [DOI] [PubMed] [Google Scholar]
- Grover A.; Schmidt B. F.; Salter R. D.; Watkins S. C.; Waggoner A. S.; Bruchez M. P. (2012) Genetically Encoded pH Sensor for Tracking Surface Proteins through Endocytosis. Angew. Chem., Int. Ed. 51, 4838–4842 10.1002/anie.201108107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallner M.; Meera P.; Toro L. (1996) Determinant for beta-subunit regulation in high-conductance voltage-activated and Ca(2+)-sensitive K+ channels: an additional transmembrane region at the N terminus. Proc. Natl. Acad. Sci. U. S. A. 93, 14922–14927 10.1073/pnas.93.25.14922. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.