

Abstract
A better understanding about the mechanisms involved in the pathogenesis of type 2 diabetes mellitus (T2D) showed that inflammatory cytokines such as tumour necrosis factor (TNF) and interleukin (IL)-1β play a pivotal role, mirroring data largely reported in rheumatoid arthritis (RA). IL-1β is produced mainly by monocytes (MO), and hyperglycaemia may be able to modulate, in the cytoplasm of these cells, the assembly of a nucleotide-binding domain and leucine-rich repeat containing family pyrin (NLRP3)-inflammosome, a cytosolic multi-protein platform where the inactive pro-IL-1β is cleaved into active form, via caspase-1 activity. In this paper, we evaluated the production of IL-1 β and TNF, in peripheral blood MO of patients affected by RA or T2D or both diseases, in order to understand if an alteration of the glucose metabolism may influence their proinflammatory status. Our data showed, after 24 h of incubation with different glucose concentrations, a significantly increased production of IL-1β and TNF in all evaluated groups when compared with healthy controls. However, a significant increase of IL-1β secretion by T2D/RA was observed when compared with other groups. The analysis of relative mRNA expression confirmed these data. After 24 h of incubation with different concentrations of glucose, our results showed a significant increase in NLRP3 expression. In this work, an increased production of IL-1β by MO obtained from patients affected by both RA and T2D via NLRP3-inflammasome activation may suggest a potential IL-1β targeted therapy in these patients.
Keywords: IL-1β, NLRP3-inflammasome, rheumatoid arthritis, type 2 diabetes mellitus
Introduction
Rheumatoid arthritis (RA) is a chronic and disabling disease characterized by persistent synovitis and systemic inflammation, in which over-expression of tumour necrosis factor (TNF) and interleukin (IL)-1β drive both synovial inflammation and joint destruction 1. It is well known that RA is associated with an increased mortality rate due to the increased risk of cardiovascular (CV) events when compared with the general population 2–5, and related to the chronic inflammatory process, which is able to modulate an accelerated atherosclerosis and CV disease (CVD) 6. Both atherosclerosis and RA share common inflammatory pathways, mediated mainly by TNF and IL-1β 7–9. In fact, increased levels of TNF and IL-1β may promote endothelial dysfunction, structural vessel abnormalities, oxidative stress and changes in lipid levels, thus favouring the appearance of traditional CV risk factors such as hypertension, insulin resistance and type 2 diabetes (T2D) 2,10,11. It is well known that patients affected by T2D show a higher CV morbidity and mortality when compared with non-diabetic subjects 12. In addition, the diabetic vascular alterations may be responsible for both diffuse coronary artery disease and stroke, which are increased significantly in T2D patients, and the risk for the occurrence of a CV event in these patients, without a previous history of CVD, mirrors the CV risk observed in patients with a history of previous myocardial infarction 13,14. On this basis, we understand why patients affected by both RA and T2D displayed the highest risk for CVD and related mortality 15,16, as suggested by a prospective cohort study, conducted in the setting of the CORRONA registry (Comparative Effectiveness of Biologic and Oral Agents for the Treatment of Rheumatoid Arthritis), aimed at investigating the association between CVD and RA showing that both T2D and RA may contribute to increasing the severity of CVD and in predicting the risk of fatal CV events 17. At present, a growing body of evidence shows the positive association between RA and T2D: different databases, from clinical studies and from national registers, confirmed that RA patients displayed a double risk of developing T2D when compared with the general population 18–20, and epidemiological data strongly support the association between RA and insulin resistance 21,22.
In recent years, many papers have supported the role of inflammatory mechanisms in the pathogenesis of T2D 23 and recently, as already described for type 1 diabetes mellitus (T1D), the pathogenetic role of insulitis in T2D has been proposed 23–26. Although the precise aetiology of the insulitis in both types of diabetes remains to be understood fully, some specific differences have been described. At present we know that the T1D insulitis is driven by an autoimmune process, whereas in T2D it is linked to an autoinflammatory process, involving the IL-1β pathway 23,24.
Although different cell types may produce IL-1β, this cytokine is produced mainly by monocytes which, as shown by preclinical and clinical studies, may be involved in the pathogenesis of both these diseases 27,28. Different pathological stimuli are able to modulate the assembly of a cytosolic platform, the inflammasome, composed of at least five different protein components integral to this complex, including nucleotide-binding domain and leucine-rich repeat containing family pyrin 3(NLRP3), caspase recruitment domain containing protein 8 (CARD8), pyrin, apoptosis-associated speck-like protein containing CARD (ASC) and pro-caspase-1, which is involved mainly in host defence against microbes and where the inactive pro-IL-1β is cleaved into a small, mature, active form 29,30. NLRP3-inflammasome is also activated by host-derived metabolites, such as monosodium urate crystals, cholesterol crystals and free fatty acids, and involved in the development of some inflammatory diseases such as gout, atherosclerosis and T2D 23–30.
Of interest, it must be pointed out that recently, in RA patients, a significantly higher gene expression of ASC, NLRP3 and CASP1, associated with an increase of caspase-1 and IL-18 levels, was observed, suggesting that this up-regulation of NLRP3-inflammasome-related transcripts may reflect an increased inflammasome activity 31, and supports the hypothesis that autoinflammatory pathways may play a pathogenetic role in both diseases.
In this study, we evaluated the production of IL-1β, TNF, and NLRP3 expression in peripheral blood monocytes (MO) of patients affected by both RA and T2D, after exposure to different glucose concentrations, to assess the influence of altered glucose metabolism on their proinflammatory status.
Patients and methods
Patients
Between January 2010 and December 2014 we enrolled 10 RA patients, 10 T2D patients and 10 patients affected by both diseases (T2D/RA patients), according to standard criteria 32,33, and five healthy controls (HC). Demographic and clinical features were summarized in Table1. T2D patients were eligible if the disease duration was more than 3 months but less than 1 year, the glycated haemoglobin value between >7·0 and < 9·0% at screening time and no changes in diabetes medications within the last 3 months. RA patients were eligible if their disease duration was diagnosed from more than 3 months and receiving only symptomatic therapy [non-steroidal anti-inflammatory drugs (NSAIDs)]. At the time of enrolment no patient showed signs and/or symptoms of early atherosclerosis or CVD, as assessed by flow-mediated dilation of the brachial artery and by intimamedia thickness, transthoracic Doppler and tissue Doppler echocardiography, aortic root echocardiography and pulse-wave velocity and colour Doppler ultrasounds of the neck vessels. Furthermore, no patient showed clinical and laboratory parameters compatible with the diagnoses of metabolic syndrome, essential hypertension, hypercholesterolaemia or hypertriglyceridaemia. Finally, no patient had smoking and alcohol habits.
Table 1.
Demographic and clinical features.
| HC | T2D | RA | T2D/RA | |
|---|---|---|---|---|
| Patients | 5 | 10 | 10 | 10 |
| Female (male) | 3 (2) | 5 (5) | 6 (4) | 6 (4) |
| Age (years), median (range) | 48 (40; 50) | 46 (40; 62) | 44 (39; 58) | 45 (41; 58) |
| Disease duration (years), median (range) | 0·6 (0·5; 1) | 5·1 (1·2; 6·3) | T2D 0·6 (0·5; 1) | |
| RA 5·2 (2·7; 7·2) | ||||
| Fasting blood glucose (mg/dl) | 80 (70; 90) | 126 (100; 164) | 90 (70; 105) | 129 (105; 156) |
| Glycated haemoglobin (%) | 5·2 (4·8; 5·3) | 7·4 (7·1; 7·8) | 5·4 (4·9; 5·8) | 7·5 ( 7·2; 7·8) |
| Erythrocytes sedimentation rate (mm/h) | 4 (0; 8) | 8 (4; 12) | 18 (10; 25) | 16 (8; 30) |
| C reactive protein (mg/l) | 1·2 (0·1; 3·4) | 4·4 (1·8; 6·2) | 4·2 (2·8; 5·2) | 4·4 (1·8; 6·2) |
| Metformin (dosage, mg/daily) median (range) | None | 1500 (1000; 2000) | None | 1500 (1000; 2000) |
HC = healthy control; T2D = type 2 diabetes mellitus; RA = rheumatoid arthritis.
An independent ethics committee approved the study and all subjects provided written informed consent before participation; the protocol was according to the Declaration of Helsinki.
Monocyte preparations
Human peripheral blood mononuclear cells (PBMCs) were isolated from heparinized blood using Ficoll-Hypaque gradients (Histopaque-1077; Sigma, St Louis, MO, USA). MO were isolated from PBMCs either by positive selection using magnetic CD14 MicroBeads (human; Miltenyi Biotech, Bergisch Gladbach, Germany), according to the manufacturer’s instructions. Following isolation, MO from our patients and controls were washed three times in phosphate-buffered saline, suspended in RPMI-1640 medium (Gibco/BRL, Grand Island, NY, USA) containing 10% heat-inactivated autoserum, 1 × 105 IU/1 penicillin (Gibco/BRL), 100 mg/1 streptomycin (Gibco/BRL) and 1000 p-mol/1 L-glutamine (Gibco/BRL) and seeded into autoserum-coated culture plates (Nunclon; Nunc, Roskilde, Denmark). Aliquots of 1 × 106 MO were suspended in 1 ml of complete medium seeded into 24-well culture dishes (Falcon 3047; Becton Dickinson, Parsippany, NJ, USA) and incubated with 11 mmol/1 glucose (11G) and 33 mmol/1 glucose (33G) for 24 h in a humidified 5% CO2 incubator at 37°C. The choice of 11G was suggested by the observation that, in humans, glucose is degraded continuously and formed in order to maintain a stable concentration of 5 mmol/1 in the bloodstream. However, in our cell cultures, where glucose is metabolized but not replaced, cells incubated with a concentration of 5 mmol/1 glucose for 24 h metabolized this molecule strongly, lowering the concentration to less than 2 mmol/1. For this reason, we chose a concentration of 11G, as in our experience a glucose concentration of 11G does not lead to glucose deficiency after 24 h of incubation. In high glucose studies, cells were exposed to a high glucose concentration of 33G. Many previous studies have reported that glucose concentrations as high as 50 mmol/1 have been found in the blood of patients with uncontrolled diabetes 34,35. It is true that blood glucose levels in patients are not likely to remain as high as 33G for 24 h. However, tissue damage in diabetic patients occurs over many years of countless hyperglycaemic episodes. Thus, the glucose concentration of 33G used in this cell culture study does not seem unreasonable, as observed in previous work 35. Lipopolysaccharide (2 ng/ml) was used to stimulate the secretion of cytokines in cell culture. Media were collected and stored after 24 h of incubation for the IL-1β and TNF using a specific enzyme-linked immunosorbent assay (ELISA).
Measurement of IL-1β and TNF levels by ELISA
The amounts of IL-1β and TNF released in the supernatants were determined by using specific Quantikine Human Immunoassay ELISA kits (all by R&D Systems, Minneapolis, MN, USA), according to the manufacturer’s protocol. Our results were expressed as median (range). For circulating levels of IL-1β and TNF, sera were collected from patients and HC and tested by the same ELISA assays used for MO culture.
qRT–PCR analysis
RNA were extracted from collected MO using Trizol (Sigma, St Louis, MO, USA) and reverse-transcribed into complementary DNA (cDNA) with the ThermoScript reverse transcription–PCR system (Invitrogen, Carlsbad, CA, USA). Results were analysed after 40 cycles of amplification using the ABI 7500 Fast Real Time PCR System. IL-1β, TNF and NLRP3 gene expression were assessed by commercial TaqMan gene expression assay (Hs01555410_m1, Hs01113624_g1 and Hs00918082_m1, respectively). All gene expression data were normalized to those for glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
Western blot
Human MO were washed three times with cold Ca2+-free and Mg-free phosphate-buffered saline (PBS) and lysed by NP-40 lysis buffer (Boston Biosciences, Ashland, MA, USA) containing 150 µM phenylmethanesulphonylfluoride (PMSF) (Fluka Biochemika-Sigma Aldrich, St Louis, MO, USA) and 1% protease inhibitor cocktail [dissolved in dimethylsulphoxide (DMSO), Sigma Aldrich]. Lysates were centrifuged and protein concentrations in supernatants were determined using the bicinchoninic acid (BCA) assay (Pierce, Rockford, IL, USA). Equal amounts of protein were added to the sodium dodecyl sulphide (SDS) sample buffer containing 2-mercaptoethanol (without boiling), and electrophoresed on SDS–polyacrylamide gels.
Gels were blotted on nitrocellulose membrane (Invitrogen). Blots were blocked overnight in TTBS (TBS-buffer containing 5% milk powder and 0.05% Tween-20). Blots were incubated with primary antibodies [room temperature (RT), 1 h, TTBS], washed three times with TTBS and then probed with secondary horseradish peroxidase (HRP)-linked antibodies (RT, 1 h, TTBS; GE Healthcare, Piscataway, NJ, USA). After repeated washes, blots were developed by chemiluminescence using the Lumigen DS detection kit (GE Healthcare). The primary antibodies used were: NLRP3 (R&D Systems). All the signals were quantified by normalizing to the tubulin signal (Abcam, Cambridge, MA, USA). Immunoreactive bands were quantified with densitometry using ImageJ software (NIH, Bethesda, MD, USA).
Statistical analysis
GraphPad Prism version 5·0 software was used for statistical analyses. Results are expressed as median (range). Due to the non-parametric distribution of our data the Mann–Whitney U-test was used as appropriate for analyses. Despite the small numbers of patients enrolled into each studied group, the strong a priori evidence of an increased NLPR3 activation associated with both RA and T2D 24,29,31 allowed us to not apply Bonferroni’s correction in our statistical analyses. Statistical significance was expressed by a P-value < 0·05.
Results
Increased secretion of IL-1β after high concentration glucose stimulation in supernatants of cultured cells
Our data showed that, after 24 h of incubation with 11G, a significant increase of IL-1β levels was shown when T2D, RA and T2D/RA patients were compared with HC [HC: 4·5 (4·1; 5·8) pg/ml; T2D: 48·0 (20·1; 63·6) pg/ml; RA: 55·0 (34·5; 105·2); T2D/RA: 54·5 (32·5; 102·4) pg/ml; P < 0·01 for each comparison]. Furthermore, no significant differences in IL-1β levels were observed among patient groups. On the contrary, after 24 h of incubation with 33G a significant increase in IL-1β levels were observed in the supernatants of T2D/RA patients when compared with HC and among other evaluated groups [HC: 135·3 (108·1; 256·8) pg/ml; T2D: 208·2 (123·1; 300·6) pg/ml; RA: 201·0 (168·5; 208·2) pg/ml; T2D/RA 352·5 (257·5; 711·9) pg/ml; P < 0.01 for each comparison]. No significant differences were observed when we compared RA patients and T2D patients with HC.
Our results showed that, after 24 h of incubation with 11G, a significant increase in TNF levels was shown when T2D, RA and T2D/RA patients were compared with HC [HC: 21·4 (20·2; 23·6) pg/ml; T2D: 356·1 (234·1; 695·3) pg/ml; RA: 437·5 (356·5; 970·2) pg/ml; T2D/RA: 496·2 (426·7; 770·5) pg/ml in T2D/RA patients; P < 0.01 for each comparison].No significant differences in TNF levels were observed among patient groups. Our data showed that, after 24 h of incubation with 33G, a significant increase in TNF levels was shown when T2D, RA and T2D/RA patients were compared with HC [HC: 135·4 (108·2; 256·3) pg/ml; T2D: 733·5 (500·2; 1008·1) pg/ml; RA: 778·5 (570·1; 1007·0) pg/ml; T2D/RA: 800·4 (437·1; 1130·6) pg/ml; P < 0.01 for each comparison]. No significant differences in TNF levels were observed among patient groups. These results are summarized in Fig. 1.
Figure 1.
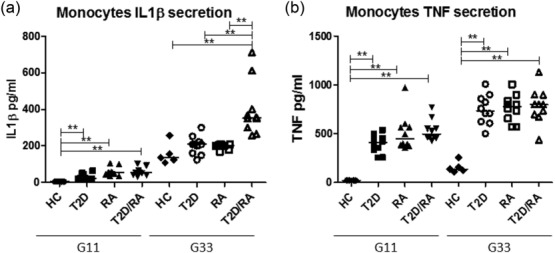
Increased secretion of interleukin (IL)-1β (a) and tumour necrosis factor (TNF) (b) after glucose stimulation in supernatants of cultured cells. Monocytes were incubated with 11 mmol/1 glucose (11G) and 33 mmol/1 glucose (33G) for 24 h. Our data showed that, after 24 h of incubation with 11G, a significant increase of IL-1β and TNF levels were shown when type 2 diabetes (T2D), rheumatoid arthritis (RA) and T2D/RA patients were compared with healthy controls (HC) (P < 0·01 for each comparison). After 24 h of incubation with 33G, a significant increase in IL-1β levels were observed in the supernatants of T2D/RA patients when compared with all evaluated groups (P < 0·01 for each comparison) (**P < 0·01).
Increased expression of IL-1β after high concentration glucose stimulation
Our data showed that, after 24 h of incubation with 11G, a significant increase in relative IL-1β mRNA expression was reported when T2D, RA, and T2D/RA patients were compared with HC [HC: 1·5 (1·0; 2·0); T2D: 5·2 (4·5; 6·9); RA: 4·8 (3·2; 6·2); T2D/RA 5·7 (4·5; 6·9) patients; P < 0·01 for each comparison]. No significant differences in relative IL-1β mRNA expression were observed among patient groups. However, after 24 h of incubation with 33G a significant increase in relative IL-1β mRNA expression was observed in the supernatants of T2D/RA patients when compared with HC and among other evaluated groups [HC: 3·2 (2·0; 5·7); T2D: 14·5 (12·0; 18·0); RA: 15·8 (14·2; 19·4); T2D/RA: 24·3 (20·0; 27·2); P < 0·001 for each comparison]. Significant differences were observed when we compared both RA patients and T2D patients with HC (P < 0.01 for each comparison).
Our results showed that, after 24 h of incubation with 11G, a significant increase of relative TNF mRNA expression was shown when T2D, RA and T2D/RA patients were compared with HC [HC: 1·2 (1·0; 2·2); T2D: 5·7 (4·5; 6·8); RA: 6·2 (4·8; 9·0); T2D/RA: 5·8 (4·5; 7·6); P < 0·001 for each comparison]. Our work showed that, after 24 h of incubation with 33G, a significant increase of relative TNF mRNA expression was shown when T2D, RA and T2D/RA patients were compared with HC [HC: 6·2 (6·0; 7·2); T2D: 14·7 (14·1; 18·2);RA: 15·8 (14·0; 18·0); T2D/RA: 15·5 (14·8; 18·6); P < 0·01 for each comparison]. No significant differences in TNF levels were observed among patient groups. All these data are reported in Fig. 2.
Figure 2.
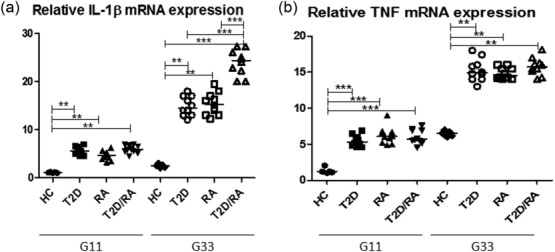
Increased relative expression of interleukin (IL)-1β (a) and tumour necrosis factor (TNF) (b) after glucose stimulation in cultured cells. Monocytes were incubated with 11 mmol/1 glucose (11G) and 33 mmol/1 glucose (33G) for 24 h. Our data showed that, after 24 h of incubation with 11G, a significant increase of relative IL-1β and TNF mRNA expressions were reported when type 2 diabetes (T2D), rheumatoid arthritis (RA) and T2D/RA patients were compared with healthy controls (HC) (P < 0·001 for each comparison). However, after 24 h of incubation with 33G a significant increase in relative IL-1β mRNA expression was observed in the supernatants of T2D/RA patients when compared with HC and among other evaluated groups (P < 0·01 for each comparison) (***P < 0·001).
Increased expression of NLRP3-inflammosome in T2D/RA patients
After 24 h of incubation to 11G our results showed a significant increase in relative NLRP3 mRNA expression of T2D/RA patients when compared with other groups [HC: 1·2 (1·0; 2·0); T2D: 5·1 (4·5; 7·9); RA: 3·6 (3·2; 5·9); T2D/RA: 7·5 (6·9; 10·9); P < 0·001 for each comparison]. In addition, we reported a significantly higher relative NLRP3 mRNA expression when compared T2D and RA patients with HC (P < 0·01 for each comparison). A significantly higher relative NLRP3 mRNA expression in T2D patients with respect to RA patients was shown (P < 0·01) at the same glucose concentration. After 24 h of incubation with 33G our results showed a significant increase in relative NLRP3 mRNA expression when we compared T2D/RA patients to other groups [HC: 4·2 (2·0; 5·0); T2D: 16·4 (15·4; 18·8); RA: 13·8 (10·0; 18·0); T2D/RA: 22·8 (20·9; 27·0); P < 0·001 for each comparison]. We described a significantly higher relative NLRP3 mRNA expression when we compared T2D and RA patients with HC (P < 0·01 for each comparison). A significantly higher relative NLRP3 mRNA expression in T2D patients in respect to RA patients was shown (P < 0·01) at the same glucose concentration. These data were confirmed by Western blot analyses. This analysis mirrored the results obtained by evaluation of NLRP3 mRNA expression; Fig. 3 shows these findings.
Figure 3.
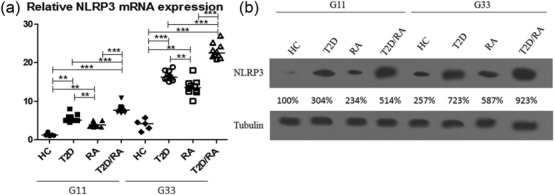
Increased relative expression of nucleotide-binding domain and leucine-rich repeat containing family pyrin (NLRP3) (a) after glucose stimulation and Western blot analyses (b). Monocytes were incubated with 11 mmol/1 glucose (G11), 33 mmol/1 glucose (G33). The expression levels of healthy controls (HC) incubated with 11 mmol/1 glucose (G11) were set to 100%, and the results were normalized to this value. Tubulin was measured as a loading control for normalization. After 24 h of incubation to 11G our results showed a significant increase in relative NLRP3 mRNA expression in type 2 diabetes/rheumatoid arthritis (T2D/RA) patients when compared with other groups (P < 0·001 for each comparison). After 24 h of incubation to 33G the results showed a significant increase in relative NLRP3 mRNA expression when we compared T2D/RA patients to other groups (P < 0·001 for each comparison). This analysis mirrored the results obtained by evaluation of NLRP3 mRNA expression by Western blot (**P < 0·01; ***P < 0·001, respectively).
No differences in circulating levels of IL-1β and TNF were observed
When we analysed the circulating levels of IL-1β among enrolled patients we did not find significant differences [HC: 1·5 (1·1; 1·8) pg/ml; T2D: 1·1 (1·2; 1·8) pg/ml; RA: 5·9 (1·5; 2·8) pg/ml; T2D/RA: 1,9 (1·5;2·3) pg/ml]. Similarly, the analysis of the circulating levels of TNF among enrolled patients did not show significant differences [HC: 2·9 (0·2; 3·6) pg/ml; T2D: 2·8 (1·2; 3·9) pg/ml; RA: 2·7 (0·5; 4·1) pg/ml; T2D/RA: 3·1 (0·4; 4·6) pg/ml].
Discussion
In recent years, the involvement of innate immunity has been suggested in the pathogenesis of T2D 23–30, and several studies have described elevated circulating levels of acute-phase proteins, cytokines and chemokines in patients with T2D, and elevated levels of IL-1β, IL-6 and C-reactive protein have been suggested as predictive factors of T2D development 36–39. In addition, an immune cell infiltration surrounding the pancreatic islets in human T2D, characterized largely by MO, may be observed 39. On this basis, a better knowledge of the metabolically driven inflammation observed in T2D may open future perspectives, identifying new potential biomarkers and/or new therapeutic targets 23–30. In this regard, several papers have suggested, both in experimental models and in humans, the usefulness of cytokine antagonism therapies in T2D, mirroring what has been suggested for RA and other inflammatory arthritis 23,24.
In this paper, we show that increased concentrations of glucose strongly influence the proinflammatory status of MO derived from patients affected by RA and concomitant T2D, suggesting that the altered glucose metabolism, increasing the production of inflammatory cytokines, may affect the clinical picture of RA and influences the response to targeted therapies 1,2,5,8–10.
After incubation of MO with different glucose concentrations, we observed an increased production of IL-1β in any studied subset correlating directly with glucose concentrations, the highest levels observed in patients affected by both diseases. It is well known that hyperglycaemia levels may up-regulate the expression of different transcription factors of many proinflammatory cytokine genes 40–42. Among them, the transcription factor nuclear factor (NF)-κB may mediate the increased IL-1β secretion from MO exposed to hyperglycaemic conditions 43,44. Analyses of IL-1β mRNA confirmed these results, showing a significant increase of its expression in T2D/RA patients when compared with other groups.
It is well known that, during T2D, the polarity of islet-infiltrating MO is shifted towards the activated proinflammatory M1-type, releasing higher concentrations of IL-1β 45. Experimental models of T2D showed that during euglycaemic conditions MO exhibit a M2-type phenotype, promoting wound healing and modulating immune responses. Conversely, under the influence of hyperglycaemia, these M2-type MO shift to a M1-type phenotype, starting the production of inflammatory cytokines such as IL-1β, and leading to a fibrotic degeneration associated with amyloid deposits of Langerhans islets 27,45. In this setting, the higher levels of IL-1β have been shown to drive the progressive failure of insulin production and apoptosis of β cells 46,47. Furthermore, glucose-induced IL-1β production by MO involved up-regulation of the NLRP3-inflammasome 48. Higher concentrations of glucose may induce the dissociation of thioredoxin-interacting protein (TXNIP) from thioredoxin, under the influence of reactive oxygen species, allowing the binding of TXNIP to the NLRP3-inflammasome. This event leads to the activation of caspase-1 and subsequent release of mature IL-1β from inactive pro-IL-1β 49. Interestingly, confirming the results concerning IL-1β production, a significantly higher expression of NLRP3-inflammasome was observed after incubation with different glucose concentrations in T2D/RA patients when compared with the other groups. Regarding the role of NLRP3-inflammasome in glucose and insulin homeostasis, several preclinical studies showed that the genetic deletion of NLRP3 in high-fat diet-fed mice improved glucose tolerance and enhanced insulin sensitivity 50,51. These data were confirmed in NLRP3−/− knock-out animals, where the IL-1β levels in the adipose tissue decreased significantly; this reduction is associated with lower levels of caspase-1 activity 51. Furthermore, inhibition of NLRP3-inflammasome is associated with improved insulin signalling in adipose tissue, liver and skeletal muscles, which are considered insulin-sensitive tissue, and with increased insulin secretion from β islets 50–53.
Our data also showed increased production of TNF from MO obtained from T2D/RA, T2D and RA patients after exposure to different glucose concentrations when compared to HC, although no significant difference was observed among the studied subsets. The available literature reports that the expression of TNF in adipose tissue was increased in multiple rodent obesity models, and this cytokine may decrease insulin signalling in insulin-sensitive tissues 54–56. Furthermore, in these experimental models, weight loss may induce an improvement in insulin sensitivity that has been associated with a reduction in TNF expression 54–57. According to these experimental findings, studies using TNF−/− knock-out mice, or alternatively neutralizing TNF activity by specific antibodies, showed an improvement in glycaemia levels, suggesting TNF as potential therapeutic target for T2D 54–57. Despite the experimental evidence supporting a role for TNF in regulating insulin production and function, the translation from basic researches to the clinical setting by using TNF inhibitors in the treatment of human diabetes showed disappointing results 58–60. Although further studies are needed to understand more clearly the failure of TNF inhibition in human diabetes, it must be pointed out that the intracellular TNF concentration is responsible for insulin resistance via paracrine effects, and this intracellular cytokine cannot be blocked by TNF inhibitors 61.
In contrast, several published studies suggest the possible therapeutic role of IL-1 antagonism in T2D 62,63. In this context, 70 T2D patients, assigned randomly to receive 100 mg of anakinra, an IL-1 receptor antagonist licensed for RA or placebo, showed a significant decrease in the glycated haemoglobin level and an increase of C-peptide secretion after 13 weeks of treatment 64. The follow-up of these patients showed that this improvement was still detectable 39 weeks after discontinuation of anakinra 65. Furthermore, the results of a 1-year prospective observational study enrolling 470 RA patients treated with anakinra showed that RA patients with concomitant T2D displayed a better response to this drug 66. These data suggest that T2D co-morbidity in RA patients predicts those patients with a better response to IL-1 inhibition. In addition, different papers suggest that IL-1 blockade may be helpful in patients with T2D and inflammatory rheumatic diseases 67,68. On this basis, an open, randomized, controlled, double-armed, multi-centre study, whose primary end-point is the efficacy of anakinra in controlling signs and symptoms of T2D in RA patients and T2D co-morbidity, is ongoing (TRACK study, NCT02236481, www.clinicaltrial.gov).
It is well known that both T2D and RA are independent factors increasing the risk of CV events in affected patients and these data are related to the chronic inflammatory activity observed in both diseases, mediated by the over-production of inflammatory cytokines 16,17,23,24,28. In addition, a large body of preclinical data showed that IL-1β is involved strongly in the progression of atherosclerosis 69,70. During the inflammatory process occurring in RA, it has been shown that TNF may contribute to endothelial dysfunction 71. However, conflicting results have been published concerning the possibility that an anti-TNF therapy might restore normal endothelial function 72,73. Furthermore, it has been shown recently that the decrease of inflammatory markers, after anti-TNF treatment, were not associated with an improvement in vascular function 74,75. On the contrary, different works suggest that the IL-1 inhibition may be associated with a greater improvement of endothelial function in RA patients 76,77. Heart failure is a common evolution of atherosclerosis, and several studies have shown the safety and the usefulness of anti-IL-1 treatment in this condition, a clinical setting in which anti-TNF treatment is contraindicated 78–81. It is well known that the incidence of impaired cardiac function is increased strongly both in RA and T2D, leading to a poor prognosis 82,83. In this context, our experimental data, showing a strong up-regulation of IL-1β MO derived from patients with both RA and T2D, may support the hypothesis that blocking IL-1β may be considered the best therapeutic strategy to treat these patients, due to the up-regulation of this cytokine in these patients and to the safety shown during chronic heart failure. On this basis, the scientific community is still waiting for the results of the CANTOS (Canakinumab Anti-inflammatory Thrombosis Outcome Study) trial, a study planned to test the hypothesis that the inhibition of IL-1β by Canakinumab, a human IL-1β blocking monoclonal antibody, might reduce CV events in T2D 84,85.
IL-1 blocking agents are generally considered safe drugs. Although the occurrence of upper-airway infections may increase in patients receiving anti-IL-1 therapy, as observed with the other biological agents, the lack of opportunistic and tuberculosis infections confirms their safety profile 86,87.
In conclusion, although further studies are needed to understand fully the complex interplay between inflammation and metabolic disorders and consequent therapeutic implications, our study, showing an increased production of IL-1β by MO obtained from patients affected by both RA and T2D via NLRP3-inflammasome activation, supports the potential role of IL-1β-targeted therapy in these patients.
Acknowledgments
The authors thank Mrs Federica Sensini for her technical assistance.
Author contributions
P.R.: study conception and design, data interpretation, literature search, figure creation, writing, paper revision and acceptance; P.C.: study conception and design, data interpretation, literature search, figure creation, writing, paper revision and acceptance; P.D.B.: data collection, data interpretation, literature search, paper revision and acceptance; V.L.: data collection, data interpretation, literature search, paper revision and acceptance; O.B.: data collection, literature search, paper revision and acceptance; F.C.A.: data collection, data interpretation, literature search, paper revision and acceptance; F.C.I.: data collection, literature search, paper revision and acceptance; S.A.: data collection, literature search, paper revision and acceptance; G.T.: data collection, data interpretation, literature search, paper revision and acceptance; R.G.: study design, data interpretation, writing, paper revision and acceptance. All authors gave final approval for submitting the manuscript for review and agree to be accountable for all aspects of the work.
Disclosure
All the authors have no conflicts of interest to disclose.
References
- McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205–19. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- Choy E. Cardiovascular risk in rheumatoid arthritis: recent advances in the understanding of the pivotal role of inflammation, risk predictors and the impact of treatment. Rheumatology. 2014;53:2143–54. doi: 10.1093/rheumatology/keu224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviña-Zubieta JA, Choi HK, Sadatsafavi M, et al. Risk of cardiovascular mortality in patients with rheumatoid arthritis: a meta-analysis of observational studies. Arthritis Rheum. 2008;59:1690–7. doi: 10.1002/art.24092. [DOI] [PubMed] [Google Scholar]
- Avina-Zubieta JA, Thomas J, Sadatsafavi M, et al. Risk of incident cardiovascular events in patients with rheumatoid arthritis: a meta-analysis of observational studies. Ann Rheum Dis. 2012;71:1524–9. doi: 10.1136/annrheumdis-2011-200726. [DOI] [PubMed] [Google Scholar]
- Peters MJ, Symmons DP, McCarey D, et al. EULAR evidence-based recommendations for cardiovascular risk management in patients with rheumatoid arthritis and other forms of inflammatory arthritis. Ann Rheum Dis. 2010;69:325–31. doi: 10.1136/ard.2009.113696. [DOI] [PubMed] [Google Scholar]
- Libby P. Role of inflammation in atherosclerosis associated with rheumatoid arthritis. Am J Med. 2008;121:S21–31. doi: 10.1016/j.amjmed.2008.06.014. [DOI] [PubMed] [Google Scholar]
- Rho YH, Chung CP, Oeser A, et al. Inflammatory mediators and premature coronary atherosclerosis in rheumatoid arthritis. Arthritis Rheum. 2009;61:1580–5. doi: 10.1002/art.25009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waehre T, Yndestad A, Smith C, et al. Increased expression of interleukin-1 in coronary artery disease with downregulatory effects of HMG-CoA reductase inhibitors. Circulation. 2004;109:1966–72. doi: 10.1161/01.CIR.0000125700.33637.B1. [DOI] [PubMed] [Google Scholar]
- Miller AM, McInnes IB. Cytokines as therapeutic targets to reduce cardiovascular risk in chronic inflammation. Curr Pharm Des. 2011;17:1–8. doi: 10.2174/138161211795049796. [DOI] [PubMed] [Google Scholar]
- Ku IA, Imboden JB, Hsue PY, et al. Rheumatoid arthritis: model of systemic inflammation driving atherosclerosis. Circ J. 2009;73:977–85. doi: 10.1253/circj.cj-09-0274. [DOI] [PubMed] [Google Scholar]
- del Rincon ID, Williams K, Stern MP, et al. High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional cardiac risk factors. Arthritis Rheum. 2001;44:2737–45. doi: 10.1002/1529-0131(200112)44:12<2737::AID-ART460>3.0.CO;2-%23. [DOI] [PubMed] [Google Scholar]
- Martín-Timón I, Sevillano-Collantes C, Segura-Galindo A, et al. Type 2 diabetes and cardiovascular disease: have all risk factors the same strength? World J Diabetes. 2014;5:444–70. doi: 10.4239/wjd.v5.i4.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner SM, Lehto S, Rönnemaa T, et al. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–34. doi: 10.1056/NEJM199807233390404. [DOI] [PubMed] [Google Scholar]
- Evans JM, Wang J, Morris AD. Comparison of cardiovascular risk between patients with type 2 diabetes and those who had had a myocardial infarction: cross sectional and cohort studies. BMJ. 2002;324:939–42. doi: 10.1136/bmj.324.7343.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon DH, Karlson EW, Rimm EB, et al. Cardiovascular morbidity and mortality in women diagnosed with rheumatoid arthritis. Circulation. 2003;107:1303–7. doi: 10.1161/01.cir.0000054612.26458.b2. [DOI] [PubMed] [Google Scholar]
- Gabriel SE. Cardiovascular morbidity and mortality in rheumatoid arthritis. Am J Med. 2008;121:S9–14. doi: 10.1016/j.amjmed.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon DH, Kremer J, Curtis JR, et al. Explaining the cardiovascular risk associated with rheumatoid arthritis: traditional risk factors versus markers of rheumatoid arthritis severity. Ann Rheum Dis. 2010;69:1920–5. doi: 10.1136/ard.2009.122226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer JF, Gourraud PA, Cantagrel A, et al. Traditional cardiovascular risk factors in rheumatoid arthritis: a meta-analysis. Joint Bone Spine. 2011;78:179–183. doi: 10.1016/j.jbspin.2010.07.016. [DOI] [PubMed] [Google Scholar]
- Han C, Robinson DW, Jr, Hackett MV, et al. Cardiovascular disease and risk factors in patients with rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis. J Rheumatol. 2006;33:2167–72. [PubMed] [Google Scholar]
- Solomon DH, Love TJ, Canning C, et al. Risk of diabetes among patients with rheumatoid arthritis, psoriatic arthritis and psoriasis. Ann Rheum Dis. 2010;69:2114–7. doi: 10.1136/ard.2009.125476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasko MC, Kay J, Hsia EC, et al. Diabetes mellitus and insulin resistance in patients with rheumatoid arthritis: risk reduction in a chronic inflammatory disease. Arthritis Care Res (Hoboken) 2011;63:512–21. doi: 10.1002/acr.20414. [DOI] [PubMed] [Google Scholar]
- Liao KP, Solomon DH. Traditional cardiovascular risk factors, inflammation and cardiovascular risk in rheumatoid arthritis. Rheumatology (Oxf) 2013;52:45–52. doi: 10.1093/rheumatology/kes243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 11:98–107. doi: 10.1038/nri2925. 201; [DOI] [PubMed] [Google Scholar]
- Grant RW, Dixit VD. Mechanisms of disease: inflammasome activation and the development of type 2 diabetes. Front Immunol. 2013;4:50. doi: 10.3389/fimmu.2013.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauner H, Lüthje P, Grünler J, et al. Markers of innate immune activity in patients with type 1 and type 2 diabetes mellitus and the effect of the anti-oxidant coenzyme Q10 on inflammatory activity. Clin Exp Immunol. 2014;177:478–82. doi: 10.1111/cei.12316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haseda F, Imagawa A, Murase-Mishiba Y, et al. CD4+CD45RA– FoxP3high activated regulatory T cells are functionally impaired and related to residual insulin-secreting capacity in patients with type 1 diabetes. Clin Exp Immunol. 2013;173:207–16. doi: 10.1111/cei.12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi K, Manabe I. Macrophages and islet inflammation in type 2 diabetes. Diabetes Obes Metab. 2013;3:152–8. doi: 10.1111/dom.12168. [DOI] [PubMed] [Google Scholar]
- Skeldon AM, Faraj M, Saleh M. Caspases and inflammasomes in metabolic inflammation. Immunol Cell Biol. 2014;92:304–13. doi: 10.1038/icb.2014.5. [DOI] [PubMed] [Google Scholar]
- Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science. 2010;327:296–300. doi: 10.1126/science.1184003. [DOI] [PubMed] [Google Scholar]
- Akira S, Misawa T, Satoh T, et al. Macrophages control innate inflammation. Diabetes Obes Metab. 2013;15:10–8. doi: 10.1111/dom.12151. [DOI] [PubMed] [Google Scholar]
- Mathews RJ, Robinson JI, Battellino M, et al. Evidence of NLRP3-inflammasome activation in rheumatoid arthritis (RA); genetic variants within the NLRP3-inflammasome complex in relation to susceptibility to RA and response to anti-TNF treatment. Ann Rheum Dis. 2014;73:1202–10. doi: 10.1136/annrheumdis-2013-203276. [DOI] [PubMed] [Google Scholar]
- Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid arthritis classification criteria: an American College of heumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62:2569–81. doi: 10.1002/art.27584. [DOI] [PubMed] [Google Scholar]
- American Diabetes Association. Standards of medical care in diabetes-2010. Diabetes Care. 2010;33:S11–61. doi: 10.2337/dc10-S011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candiloros H, Muller S, Zeghari N, et al. Decreased erythrocyte membrane fluidity in poorly controlled IDDM. Influence of ketone bodies. Diabetes Care. 1995;18:549–51. doi: 10.2337/diacare.18.4.549. [DOI] [PubMed] [Google Scholar]
- Manna P, Jain SK. L-cysteine and hydrogen sulfide increase PIP3 and AMPK/PPARγ expression and decrease ROS and vascular inflammation markers in high glucose treated human U937 monocytes. J Cell Biochem. 2013;114:2334–45. doi: 10.1002/jcb.24578. [DOI] [PubMed] [Google Scholar]
- Spranger J. Kroke A, Möhlig M, et al. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European prospective investigation into cancer and nutrition (EPIC) – Potsdam study. Diabetes. 2003;52:812–7. doi: 10.2337/diabetes.52.3.812. [DOI] [PubMed] [Google Scholar]
- Herder C, Brunner EJ, Rathmann W, et al. Elevated levels of the anti-inflammatory interleukin-1 receptor antagonist precede the onset of type 2 diabetes: the Whitehall II study. Diabetes Care. 2009;32:421–3. doi: 10.2337/dc08-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AD, Manson JE, Rifai N, et al. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA. 2001;286:327–34. doi: 10.1001/jama.286.3.327. [DOI] [PubMed] [Google Scholar]
- Ehses JA, Perren A, Eppler E, et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes. 2007;56:2356–70. doi: 10.2337/db06-1650. [DOI] [PubMed] [Google Scholar]
- Shanmugam N, Reddy MA, Guha M, et al. High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes. 2003;52:1256–64. doi: 10.2337/diabetes.52.5.1256. [DOI] [PubMed] [Google Scholar]
- Guha M, Bai W, Nadler JL, Natarajan R. Molecular mechanisms of tumor necrosis factor alpha gene expression in monocytic cells via hyperglycemia-induced oxidant stress-dependent and -independent pathways. J Biol Chem. 2000;275:17728–39. doi: 10.1074/jbc.275.23.17728. [DOI] [PubMed] [Google Scholar]
- Dasu MR, Devaraj S, Jialal I. High glucose induces IL-1beta expression in human monocytes: mechanistic insights. Am J Physiol Endocrinol Metab. 2007;293:E337–46. doi: 10.1152/ajpendo.00718.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann MA, Schiekofer S, Kanitz M, et al. Insufficient glycemic control increases NF-κB binding activity in peripheral blood mononuclear cells isolated from patients with type 1 diabetes. Diabetes Care. 1998;21:1310–6. doi: 10.2337/diacare.21.8.1310. [DOI] [PubMed] [Google Scholar]
- Aljada A, Friedman J, Ghanim H, et al. Glucose ingestion induces an increase in intranuclear NF-κB, a fall in cellular inhibitor κB, and an increase in TNF-alpha mRNA by mononuclear cells in healthy human subjects. Metabolism. 2006;55:1177–85. doi: 10.1016/j.metabol.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Eguchi K, Manabe I, Oishi-Tanaka Y, et al. Saturated fatty acid and TLR signaling link β cell dysfunction and islet inflammation. Cell. Metab. 2012;5:518–33. doi: 10.1016/j.cmet.2012.01.023. [DOI] [PubMed] [Google Scholar]
- Maedler K, Sergeev P, Ris F, et al. Glucose-induced β-cell production of interleukin-1β contributes to glucotoxicity in human pancreatic islets. J. Clin. Invest. 2002;110:851–60. doi: 10.1172/JCI15318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann DM, Maedler K, Franklin I, et al. The Fas pathway is involved in pancreatic β cell secretory function. Proc Natl Acad Sci USA. 2007;104:2861–6. doi: 10.1073/pnas.0611487104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HM, Kim JJ, Kim HJ, et al. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62:194–204. doi: 10.2337/db12-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Tardivel A, Thorens B, et al. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nature Immunol. 2010;11:136–40. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- Stienstra R, van Diepen JA, Tack CJ, et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci USA. 2011;108:15324–9. doi: 10.1073/pnas.1100255108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandanmagsar B, Youm YH, Ravussin A, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–88. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youm YH, Adijiang A, Vandanmagsar B, et al. Elimination of the NLRP3-ASC inflammasome protects against chronic obesity-induced pancreatic damage. Endocrinology. 2011;152:4039–45. doi: 10.1210/en.2011-1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Gris D, Lei Y, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12:408–15. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Murray DL, Choy LN, et al. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci USA. 1994;91:4854–8. doi: 10.1073/pnas.91.11.4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Budavari A, Murray D, et al. Reduced tyrosine kinase activity of the insulin receptor in obesity-diabetes. Central role of tumor necrosis factor-alpha. J Clin Invest. 1994;94:1543–9. doi: 10.1172/JCI117495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Arner P, Caro JF, et al. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. 1995;95:2409–15. doi: 10.1172/JCI117936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Spiegelman BM. Tumor necrosis factor alpha: a key component of the obesity-diabetes link. Diabetes. 1994;43:1271–8. doi: 10.2337/diab.43.11.1271. [DOI] [PubMed] [Google Scholar]
- Ofei F, Hurel S, Newkirk J, et al. Effects of an engineered human anti-TNF-alpha antibody (CDP571) on insulin sensitivity and glycemic control in patients with NIDDM. Diabetes. 1996;45:881–5. doi: 10.2337/diab.45.7.881. [DOI] [PubMed] [Google Scholar]
- Paquot N, Castillo MJ, Lefèbvre PJ, et al. No increased insulin sensitivity after a single intravenous administration of a recombinant human tumor necrosis factor receptor: Fc fusion protein in obese insulin-resistant patients. J Clin Endocrinol Metab. 2000;85:1316–9. doi: 10.1210/jcem.85.3.6417. [DOI] [PubMed] [Google Scholar]
- Di Rocco P, Manco M, Rosa G, et al. Lowered tumor necrosis factor receptors, but not increased insulin sensitivity, with infliximab. Obes Res. 2004;12:734–9. doi: 10.1038/oby.2004.86. [DOI] [PubMed] [Google Scholar]
- Bernstein LE, Berry J, Kim S, et al. Effects of etanercept in patients with the metabolic syndrome. Arch Intern Med. 2006;166:902–8. doi: 10.1001/archinte.166.8.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauter NS, Schulthess FT, Galasso R, et al. The antiinflammatory cytokine interleukin-1 receptor antagonist protects from high-fat diet-induced hyperglycemia. Endocrinology. 2008;149:2208–18. doi: 10.1210/en.2007-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Asseldonk EJ, Stienstra R, Koenen TB, et al. Treatment with Anakinra improves disposition index but not insulin sensitivity in nondiabetic subjects with the metabolic syndrome: a randomized, double-blind, placebo-controlled study. J Clin Endocrinol Metab. 2011;96:2119–26. doi: 10.1210/jc.2010-2992. [DOI] [PubMed] [Google Scholar]
- Larsen CM, Faulenbach M, Vaag A, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356:1517–26. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]
- Larsen CM, Faulenbach M, Vaag A, et al. Sustained effects of interleukin-1 receptor antagonist treatment in type 2 diabetes. Diabetes Care. 2009;32:1663–8. doi: 10.2337/dc09-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missler-Karger B, Leu M, M. A. Raboisson MA, B. Pilstrom B. Disease severity, no steroid use and type II diabetes predict response to anakinra (kineret®) in patients with rheumatoid arthritis. [FRI0219] Ann Rheum Dis. 2013;72(Suppl3):447. [Google Scholar]
- Vitale A, Cantarini L, Rigante D, et al. Anakinra treatment in patients with gout and type 2 diabetes. Clin Rheumatol. 2015;34:981–4. doi: 10.1007/s10067-014-2601-7. [DOI] [PubMed] [Google Scholar]
- Ruscitti P, Cipriani P, Cantarini L, et al. Efficacy of inhibition of IL-1 in patients with rheumatoid arthritis and type 2 diabetes mellitus: two case reports and review of the literature. J Med Case Rep. 2015;9:123. doi: 10.1186/s13256-015-0603-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo S, Palacios E, Romacho T, et al. The interleukin-1 receptor antagonist anakinra improves endothelial dysfunction in streptozotocin-induced diabetic rats. Cardiovasc Diabetol. 2014;13:158. doi: 10.1186/s12933-014-0158-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isoda K, Akita K, Isobe S, et al. Interleukin-1 receptor antagonist originating from bone marrow-derived cells and non-bone marrow-derived cells helps to suppress arterial inflammation and reduce neointimal formation after injury. J Atheroscler Thromb. 2014;21:1208–18. doi: 10.5551/jat.25668. [DOI] [PubMed] [Google Scholar]
- Hansel S, Lassig G, Pistrosch F, et al. Endothelial dysfunction in young patients with long-term rheumatoid arthritis and low disease activity. Atherosclerosis. 2003;170:177–80. doi: 10.1016/s0021-9150(03)00281-8. [DOI] [PubMed] [Google Scholar]
- Sandoo A, Kitas GD, Carroll D, et al. The role of inflammation and cardiovascular disease risk on microvascular and macrovascular endothelial function in patients with rheumatoid arthritis: a cross-sectional and longitudinal study. Arthritis Res Ther. 2012;14:R117. doi: 10.1186/ar3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Juanatey C, Testa A, Garcia-Castelo A, et al. Active but transient improvement of endothelial function in rheumatoid arthritis patients undergoing long-term treatment with anti-tumor necrosis factor alpha antibody. Arthritis Rheum. 2004;51:447–50. doi: 10.1002/art.20407. [DOI] [PubMed] [Google Scholar]
- Bosello S, Santoliquido A, Zoli A, et al. blockade induces a reversible but transient effect on endothelial dysfunction in patients with long-standing severe rheumatoid arthritis. Clin Rheumatol. 2008;27:833–39. doi: 10.1007/s10067-007-0803-y. TNFalpha. [DOI] [PubMed] [Google Scholar]
- Foster W, Carruthers D, Lip GY, et al. Inflammation and microvascular and macrovascular endothelial dysfunction in rheumatoid arthritis: effect of treatment. J Rheumatol. 2010;37:711–16. doi: 10.3899/jrheum.090699. [DOI] [PubMed] [Google Scholar]
- Ikonomidis I, Lekakis JP, Nikolaou M, et al. Inhibition of interleukin-1 by anakinra improves vascular and left ventricular function in patients with rheumatoid arthritis. Circulation. 2008;117:2662–9. doi: 10.1161/CIRCULATIONAHA.107.731877. [DOI] [PubMed] [Google Scholar]
- Ikonomidis I, Tzortzis S, Lekakis J, et al. Lowering interleukin-1 activity with anakinra improves myocardial deformation in rheumatoid arthritis. Heart. 2009;95:1502–7. doi: 10.1136/hrt.2009.168971. [DOI] [PubMed] [Google Scholar]
- Ikonomidis I, Tzortzis S, Andreadou I, et al. Increased benefit of interleukin-1 inhibition on vascular function, myocardial deformation, and twisting in patients with coronary artery disease and coexisting rheumatoid arthritis. Circ Cardiovasc Imaging. 2014;7:619–28. doi: 10.1161/CIRCIMAGING.113.001193. [DOI] [PubMed] [Google Scholar]
- Van Tassell BW, Raleigh JM, Abbate A. Targeting interleukin-1 in heart failure and inflammatory heart disease. Curr Heart Fail Rep. 2015;12:33–41. doi: 10.1007/s11897-014-0231-7. [DOI] [PubMed] [Google Scholar]
- Mann DL, McMurray JJ, Packer M, et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL) Circulation. 2004;109:1594–602. doi: 10.1161/01.CIR.0000124490.27666.B2. [DOI] [PubMed] [Google Scholar]
- Chung ES, Packer M, Lo KH, et al. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003;107:3133–40. doi: 10.1161/01.CIR.0000077913.60364.D2. [DOI] [PubMed] [Google Scholar]
- Dei Cas A, Spigoni V, Ridolfi V, et al. Diabetes and chronic heart failure: from diabetic cardiomyopathy to therapeutic approach. Endocr Metab Immune Disord Drug Targets. 2013;13:38–50. doi: 10.2174/1871530311313010006. [DOI] [PubMed] [Google Scholar]
- Wright K, Crowson CS, Gabriel SE. Cardiovascular comorbidity in rheumatic diseases: a focus on heart failure. Heart Fail Clin. 2014;10:339–52. doi: 10.1016/j.hfc.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Thuren T, Zalewski A, et al. Interleukin-1β inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) Am Heart J. 2011;162:597–605. doi: 10.1016/j.ahj.2011.06.012. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Howard CP, Walter V, et al. Effects of interleukin-1β inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: a phase IIb randomized, placebo-controlled trial. Circulation. 2012;126:2739–48. doi: 10.1161/CIRCULATIONAHA.112.122556. [DOI] [PubMed] [Google Scholar]
- Cantarini L, Lopalco G, Caso F, et al. Effectiveness and tuberculosis-related safety profile of interleukin-1 blocking agents in the management of Behçet’s disease. Autoimmun Rev. 2015;14:1–9. doi: 10.1016/j.autrev.2014.08.008. [DOI] [PubMed] [Google Scholar]
- Rossi-Semerano L, Fautrel B, Wendling D, et al. Tolerance and efficacy of off-label anti-interleukin-1 treatments in France: a nationwide survey. Orphanet J Rare Dis. 2015;10:19. doi: 10.1186/s13023-015-0228-7. [DOI] [PMC free article] [PubMed] [Google Scholar]