

Abstract
Background
We have previously characterized 19 ependymal tumors using Giemsa banding and high-resolution comparative genomic hybridization. The aim of this study was to analyze these tumors searching for fusion genes.
Methods
RNA sequencing was performed in 12 samples. Potential fusion transcripts were assessed by seed count and structural chromosomal aberrations. Transcripts of interest were validated using fluorescence in situ hybridization and PCR followed by direct sequencing.
Results
RNA sequencing identified rearrangements of the anaplastic lymphoma kinase gene (ALK) in 2 samples. Both tumors harbored structural aberrations involving the ALK locus 2p23. Tumor 1 had an unbalanced t(2;14)(p23;q22) translocation which led to the fusion gene KTN1-ALK. Tumor 2 had an interstitial del(2)(p16p23) deletion causing the fusion of CCDC88A and ALK. In both samples, the breakpoint of ALK was located between exons 19 and 20. Both patients were infants and both tumors were supratentorial. The tumors were well demarcated from surrounding tissue and had both ependymal and astrocytic features but were diagnosed and treated as ependymomas.
Conclusions
By combining karyotyping and RNA sequencing, we identified the 2 first ever reported ALK rearrangements in CNS tumors. Such rearrangements may represent the hallmark of a new entity of pediatric glioma characterized by both ependymal and astrocytic features. Our findings are of particular importance because crizotinib, a selective ALK inhibitor, has demonstrated effect in patients with lung cancer harboring ALK rearrangements. Thus, ALK emerges as an interesting therapeutic target in patients with ependymal tumors carrying ALK fusions.
Keywords: anaplastic lymphoma kinase, chromosomal aberrations, ependymoma, fusion genes, RNA sequencing
Chromosomal rearrangements resulting in the formation of fusion genes are known to contribute to tumorigenesis in a wide range of human cancers.1 These fusion genes often provide important prognostic and diagnostic information, and their gene products also serve as attractive targets for new, specific therapies. The (9;22)-translocation in chronic myeloid leukemia leading to the fusion of BCR and ABL1 is the most well known example, since the abnormal tyrosine kinase thus generated can be targeted specifically.1
High-throughput RNA sequencing has emerged as an efficient method for the detection of cancer-related fusion genes.2 There are several sophisticated bioinformatic algorithms available for this purpose. However, the computerized software provides long lists of potential fusion transcripts, often numbered in the hundreds, and it can be difficult to distinguish fusions of biological importance from noise. Filtering of false positive results is also a demanding task. One approach to this problem is to combine cytogenetic methods and RNA sequencing. This has previously led to the discovery of several new fusion genes by us as well as other investigators.3–5
Ependymomas are tumors of the CNS that constitute 6%–12% of pediatric intracranial neoplasms6–8; they are also found in adults. The incidence is about 2 cases per million inhabitants per year,9 and 5-year relative survival is about 70%.10,11 Ependymomas are mostly located in relation to the ventricular system of the brain and spinal cord, although supratentorial ependymomas may be located in the brain parenchyma, especially in children.6 Treatment is currently based on surgery and radiation, whereas systemic therapy has not yet proven effective.8,12 Our group has previously investigated 19 ependymomas using Giemsa banding (G-banding) with karyotyping and high-resolution comparative genomic hybridization.13 Four of 19 tumors harbored structural chromosomal rearrangements, 2 of which involved chromosomal band 2p23. The aim of the present study was to analyze these tumors searching for fusion genes.
Materials and Methods
Patients and Tumor Samples
The original cohort of 19 tumor samples from 18 patients has been described.13 RNA was available in 14 of 19 tumors and 12 of these were successfully sequenced. Thus, this study is based on data from 12 ependymoma samples (Table 1). Samples were prospectively collected between January 2005 and December 2012 at the Department of Neurosurgery, Oslo University Hospital, Rikshospitalet. Patient age ranged from 8 months to 75 years at the time of primary surgery. None of the patients had received chemo- or radiotherapy prior to surgery. Both spinal and intracranial tumors were included, and all histopathological diagnoses were reviewed according to the World Health Organization (WHO) 2007 classification system.6
Table 1.
Clinical and pathological details of 12 ependymal tumors analyzed by RNA sequencing
| Case No. | Sex/ Age, y | Location | Histology | ALK FISH | WHO Grade | Primary Tumor/Recurrence | Extent of Resectiona | PFS/OS, y | Current Status | Case No. in Olsen et al, 2014b |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M/0.8 | ST | E | Pos | II or III | Primary | GTR | 6.5c | NED | 11 |
| 2 | F/0.9 | ST | AE/GBM | Pos | IV | Primary | STR | 3.6c | AWD | 15 |
| 3 | F/47 | S | E | NA | II | Primary | GTR | 8.9c | NED | 5 |
| 4 | M/53 | S | E | Neg | II | Primary | GTR | 7.9c | NED | 7 |
| 5 | M/61 | IT | SE | Neg | I | Primary | GTR | 7.5c | NED | 8 |
| 6a | M/38 | ST | AE, giant cell type | Neg | III | Primary | GTR | 1.5 | 9–1 | |
| 6b | AE | NA | III | Recurrent | GTR | 5.3 | AWD | 9–2 | ||
| 7 | M/75 | IT | SE | Neg | I | Primary | GTR | 6.5c | NED | 10 |
| 8 | M/42 | ST | AE | Neg | III | Primary | GTR | 0.8/1.3 | DOD | 12 |
| 9 | F/46 | IT | SE | NA | I | Primary | GTR | 4.9c | NED | 13 |
| 10 | F/46 | IT | E | Neg | II | Primary | GTR | 1.7 | AWD | 14 |
| 11 | F/1 | IT | AE | Neg | III | Primary | STR | 1.2 | AWD | 16 |
Abbreviations:S, spinal; ST, supratentorial; IT, infratentorial; E, ependymoma; AE, anaplastic ependymoma; GBM, glioblastoma multiforme; SE, subependymoma; GTR, gross total resection; STR, subtotal resection; PFS, progression-free survival; OS, overall survival; NED, no evidence of disease; AWD, alive with disease; DOD, dead of disease (January 2015).
aExtent of resection at initial surgery.
bAll cases are referred in Olsen et al, 2014, Tables 1 and 2.
cPatient still alive with no evidence of disease progression (January 2015).
Since the 2 cases described in this paper had marked glial characteristics, analyses of anaplastic lymphoma kinase (ALK) were also performed in a separate series of gliomas (Supplementary Table S1) for comparison. Our 2 indicator patients were both young children. Thus, we selected the youngest glioma patients in our archives (cutoff age was set to 30 in order to get a reasonable number of samples of all WHO grades).
DNA and RNA Extraction, High-throughput Sequencing, and Bioinformatic Analyses
DNA was extracted from frozen tumor material using the Maxwell 16 System (Promega), and DNA quality and concentration were measured and assessed using NanoVue Plus (GE Healthcare Life Sciences). RNA was extracted from frozen tumor material using TRIzol reagent (Life Technologies), and RNA quality was assessed using the Experion Automated Electrophoresis System (Bio-Rad Laboratories). From each sample, 2–4 μg of total RNA was sent for paired-end sequencing at the Norwegian Sequencing Centre (http://www.sequencing.uio.no/, accessed March 9, 2015) using the Illumina HiSeq 2000 platform. An average of 96 million reads was acquired per sample (range, 83–114 million). The Illumina software pipeline was used to process image data into raw sequencing data, and only sequence reads marked as “passed filtering” were used. The regular TruSeq library preparation protocol was used, and the reads obtained had a length of 100 base pairs. The quality of the raw sequence data was assessed using FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/, accessed March 9, 2015). The fusion detection algorithms FusionMap14 (release date 2013-02-01) and FusionFinder15 were used to detect fusion transcripts with Ensembl release 70 (http://jan2013.archive.ensembl.org/Homo_sapiens/Info/Index) as reference. Furthermore, reads were mapped and transcripts quantified using TopHat (2.0.9) and Cufflinks (2.1.1) with the workflow described by Trapnell and colleagues.16 Mapped read counts were generated using htseq-count (http://www-huber.embl.de/users/anders/HTSeq/doc/overview.html, accessed March 9, 2015). Read counts were analyzed with R 3.0.1/Bioconductor and DESeq.17 Unsupervised hierarchical clustering and sample-to-sample distances were calculated from variance-stabilized read counts as described in sections 7.1 and 7.2 of the DESeq manual (http://bioconductor.org/packages/release/bioc/vignettes/DESeq/inst/doc/DESeq.pdf).
PCR and Direct Sequencing
Complementary DNA was synthesized by reverse transcription of extracted total RNA using the iScript Advanced cDNA Synthesis Kit for RT-qPCR (Bio-Rad) according to the manufacturer's protocol. Primers for PCR were designed using Oligo software (http://www.oligo.net/, accessed March 9, 2015) and synthesized at Life Technologies. To validate the presence of the 2 ALK fusion transcripts at the RNA level, 2 different forward primers were used; one for each partner gene: KTN1-3841F1 (GRCh37/hg19, chr14:56142615-56142637; GGT AGC TGG TGA TTT GCA TAA GGC TCA) and CCDC88A-1931F1 (GRCh37/hg19, chr2:55571577-55571603; GGA AGA CCA ACT AGA GGG AAC TCG TGC). One reverse primer, ALK-4351R1 (GRCh37/hg19, chr2:29445414-29445434; TTG GGC ATT CCG GAC ACC TGG), was used. When determining the genomic breakpoint of the CCDC88A/ALK fusion at the DNA level, one forward PCR primer CCDC88A-intr12-F1 (GRCh37/hg19, chr2:55570695-55570720; GCA GTT CTT TAT TTT CCG CAG TGG GA) was used. Two different reverse primers were used: ALKint19-R1 (GRCh37/hg19, chr2:29446386-29446407; GGC GGT ACA CTG CAG GTG GGTG) and ALKint19-R2 (GRCh37/hg19, chr2:29446400-29446422; GGT GGG TGG TCA GCT GCA ACA TG). The 25 µL PCR mix contained ExTaq Premix (TaKaRa), 0.4 µM forward (KTN1-3841F1, CCDC88A-1931F1, or CCDC88A-intr12-F1) and reverse (ALK-4351R1, ALKint19R1, or ALKint19R2) primer and 2 µL cDNA (or 100 ng genomic DNA). Complementary DNA synthesized from qPCR Human Reference Total RNA (Clontech) was used with both primer sets as negative control. PCR was run on a C1000 Thermal Cycler (Bio-Rad) with an initial denaturation at 94°C for 30 s, followed by 34 cycles of 7 s at 98°C and 2 min at 68°C, and a final extension of 5 min at 68°C. Three microliters of the amplified PCR products were stained with GelRed (Biotium) and run on 1% agarose gel electrophoresis. PCR products were then purified using the NucleoSpin Gel and PCR cleanup kit (Macherey-Nagel). Direct Sanger sequencing of the purified DNA was performed (GATC Biotech Light Run Sequencing). To analyze the sequence data, we used the National Center for Biotechnology Information's (NCBI) Basic Local Alignment Search Tool (BLAST; http://www.ncbi.nlm.nih.gov/blast/, accessed March 9, 2015) and the University of California Santa Cruz's BLAST-like alignment tool (http://genome.ucsc.edu/cgi-bin/hgBlat, accessed March 9, 2015).
Fluorescence In situ Hybridization
FISH was performed using the ALK break-apart probe (Cytocell) according to the manufacturer's protocol. In tumor 1 (KTN1-ALK), the probe was hybridized to destained, previously G-banded slides as described elsewhere.18 In the remaining tumors, FISH using the same probe was performed on fresh slides prepared from frozen tumor cell suspension. CytoVision software (Leica Biosystems) was used for image analysis.
Immunohistochemical Staining
Tumors were immunohistochemically stained with anti-ALK (clone D353, Ventana), Olig2 (clone 211F1.1, Cell Marque), and phosphorylated signal transducer and activator of transcription 3 (pSTAT3; Tyr705 M9C6 mouse monoclonal antibody) (Cell Signaling Technology) on Ventana Benchmark XT and OptiView DAB detection system (Ventana), Dako Autostainer, and Dako EnVision FLEX, respectively. Results were assessed according to the manufacturer's guide.
Ethics Statement
The study was approved by the appropriate regional research ethics committee (protocol number S-06046). Written informed consent was obtained from all patients (or their parents). One participant was included postmortem following permission from the Norwegian Directorate of Health.
Results
Sequencing and Fusion Analysis
Material was available for RNA extraction from 14 of the 19 tumors in our original cohort.13 One sample had an insufficient RNA quality indicator for sequencing and another failed the cDNA library preparation. Thus, 12 of 19 tumors were successfully sequenced (Table 1).
Tumor 1, which harbored a der(14)t(2;14)(p23;q22) as its sole aberration (Fig. 1A), was assessed first. When the 934 candidate transcripts from FusionMap were sorted by decreasing seed count, an in-frame chimeric transcript between the kinectin 1 gene (kinesin receptor, KTN1; located at 14q22.3) and the ALK gene (ALK; located at 2p23.2) ranked seventh with a seed count of 27. For tumor 2, which had an interstitial 2p deletion by karyotyping with breakpoints located at 2p16 and 2p23 (Fig. 1B), FusionMap listed 2574 candidate fusion transcripts. When sorted by decreasing seed count, an in-frame gene fusion involving ALK and the coiled-coil domain containing 88A gene (CCDC88A; located at 2p16.1) ranked sixth with a seed count of 25. The 2 fusion genes were also detected by the FusionFinder algorithm. ALK was not found as a potential fusion partner in any of the 10 remaining samples. Because of high seed counts, the known chromosomal aberrations involving 2p23 in both tumors, and the involvement of ALK rearrangements in many malignancies, we decided to investigate these 2 gene fusions further.
Fig. 1.

G-banded karyotypes demonstrating chromosomal rearrangements. Arrowheads indicate chromosomal breakpoints. (A) Tumor 1. Derivative der(14)t(2;14)(p23;q22) chromosome and corresponding normal homologs. (B) Tumor 2. Chromosome 2 carrying an interstitial del(2)(p16p23) deletion and 3 normal homologs in tetraploid karyotype.
PCR and direct sequencing (Fig. 2A and B) verified the presence of the 2 fusion transcripts at the RNA level. The breakpoint in the ALK gene was located between exons 19 and 20 in both tumors. In tumor 1, exons 1–41 of the KTN1 gene (nucleotide 4076, NCBI reference sequence NM_004986.3) were fused to exons 20–29 of ALK. In the shortest PCR fragment from tumor 2 (Fig. 2A, lane 4, white arrow), exons 1–12 of the CCDC88A gene (nucleotide 2175, NCBI reference sequence NM_018084.4) were fused to exons 20–29 of ALK (nucleotide 4125, NCBI reference sequence NM_004304.4).
Fig. 2.

Detection of the gene fusions at the (A, B) RNA and (C) DNA levels. PCR and direct Sanger sequencing verified the presence of 2 fusion transcripts involving the ALK gene. (A) Gel electrophoresis after PCR amplification of cDNA. A 1-kb DNA ladder was used (M). Tumor 1 (KTN1-ALK) is shown in lane 1, tumor 2 (CCDC88A-ALK) in lane 4. White arrows indicate detection of fusion genes. Red arrow marks a longer fragment also containing intronic material. Negative controls are shown in lanes 2 and 5 (water) and 3 and 6 (total human RNA). (B) Direct Sanger sequencing of the amplified PCR products showing the fusion point between ALK and its 2 different partners. (C) Tumor 2. Detection of gene fusion at the DNA level by PCR followed by direct Sanger sequencing. Lane 1: PCR using purified DNA from the longer fragment (red arrow shown in Fig. 2A, lane 4) as template. Lane 2: PCR using genomic DNA as template.
Direct Sanger sequencing of the longer fragment from tumor 2 (Fig. 2A, lane 4, red arrow) revealed material from exon 12 and intron 12 of CCDC88A as well as from exon 20 and part of intron 19 of ALK, but the exact genomic fusion point between the 2 genes could not be determined due to low Sanger sequence quality. To assess the genomic fusion breakpoint at the DNA level, we ran another PCR with primers located closer to the fusion, amplifying purified DNA from the longer gel fragment shown in Fig. 2A (lane 4, red arrow) as well as genomic tumor DNA. Results are shown in Fig. 2C. Direct sequencing of these PCR products revealed the 2p deletion to be located between nucleotides 29446546 and 55570587 (GRCh37/hg19).
The C11orf95-RELA fusion gene previously found in supratentorial pediatric ependymomas19,20 was not detected in any of the 12 samples, of which only our 2 ALK-positive cases were supratentorial pediatric ependymomas, by either fusion algorithm or a search in the raw sequence data directly.
Fluorescence In situ Hybridization
Splitting of ALK due to chromosomal rearrangements was demonstrated in both tumors (Fig. 3). In tumor 1, material from 2p23 was detected in chromosome 14, confirming the unbalanced (2;14) translocation (Fig. 3A). FISH analysis of tumor 2 (Fig. 3B) revealed loss of the 5′ end of ALK, which was to be expected due to the previously known 2p deletion in this tumor. In tumor 2, ALK rearrangement was seen in 175 of 221 (79%) interphase nuclei. Furthermore, interphase FISH was performed in another 11 ependymal tumors from which fixed cell suspension material was available. The analysis was successful in 10 samples (cases 4, 5, 6a, 7, 8, 10, and 11 from this series, as well as cases 2, 6, and 17 described in our previous study),13 but none of these displayed splitting of ALK. FISH results were also negative in all gliomas tested (Supplementary Table S1). Thus, ALK FISH results were negative in 27 of 29 cases tested (93%).
Fig. 3.
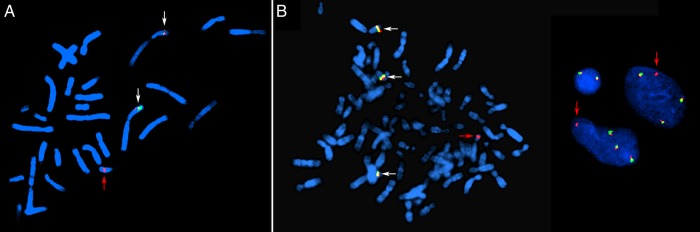
FISH analyses of cases 1 (KTN1-ALK) and 2 (CCDC88A-ALK) using an ALK split-apart probe. Yellow signals (marked with white arrows) indicate nonrearranged ALK. (A) Tumor 1. FISH analysis of destained, previously G-banded metaphase. Two normal ALK signals on chromosome 2. The red arrow indicates a derivative chromosome 14 with genetic material from 2p23. The green signal is lost, indicating loss of the 5′ part of ALK. (B) Tumor 2. Metaphase to the left and interphase nuclei to the right. Four copies of chromosome 2 are present due to a tetraploid karyotype. Three normal ALK signals are marked by white arrows. The red arrow indicates persisting red signal but loss of green signal (ie, loss of exons 1–19 of the ALK gene) due to an interstitial 2p deletion.
Immunohistochemical Staining
In both FISH ALK-positive cases, there was a strong and partly granular cytoplasmic immunohistochemical positivity with a homogeneous distribution when stained for ALK (Fig. 4). The staining pattern was consistent with the presence of the ALK protein. Olig2 staining was negative in both cases. ALK immunostaining was also performed in 8 of the remaining 10 ependymomas as well as in the glioma series (Supplementary Table S1, Supplementary material S6). Although 1 of 8 ependymomas (case 7; 13%) and 6 of 13 gliomas (46%) demonstrated scattered ALK staining, only cases 1 and 2 demonstrated the strong and uniform ALK staining shown in Fig. 4. When considering FISH and immunohistochemistry results in combination, the pathologist's final conclusion was that cases 1 and 2 were the only ALK-positive tumors.
Fig. 4.
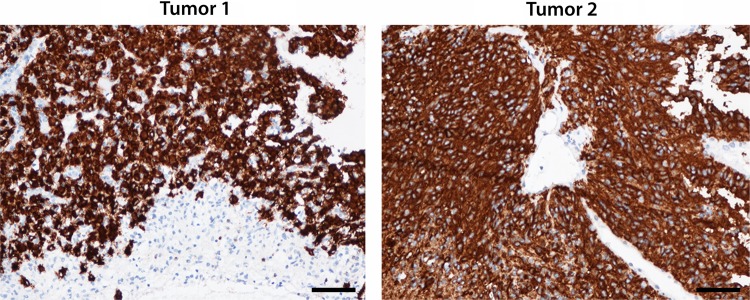
Immunohistochemical ALK staining of formalin-fixed, paraffin-embedded material from tumors 1 and 2. Note the partly granular cytoplasmic pattern and homogeneous distribution. The nuclei and endothelium are ALK negative. Magnification 200×, scale bars 50 µm.
Phosphorylated STAT3 immunostaining was positive in 4 of 13 gliomas (31%) and in 8 of 10 ependymomas (80%). The 2 ALK-rearranged cases demonstrated a strongly positive pSTAT3 staining pattern. Two other tumors had an equally strong pSTAT3 pattern: a pilocytic astrocytoma and an anaplastic astrocytoma of the giant cell type (cases 6a and G1; Table 1 and Supplementary Table S1, respectively).
Clinical and Histopathological Characteristics
The 2 ALK-rearranged tumors had a histologically unusual appearance; however, in both of these cases an ependymoma diagnosis was originally made. There was no relevant family history in either case, and the patients were not related to each other.
Case 1 was of a 9-month-old boy with a diagnosis of frontal cystic, contrast-enhancing tumor following an epileptic seizure (Supplementary material S2). The tumor was completely resected and the patient did not receive adjuvant therapy. There has been no sign of relapse after a follow-up of 6.5 years. Microscopic examination (Fig. 5A and B; Supplementary material S3) revealed an architecturally solid, demarcated tumor consisting of epithelioid glial cells positive for glial fibrillary acidic protein (GFAP) with monotonous round to ovoid nuclei. There were scattered mitoses but no necrosis or microvascular proliferation. Pseudorosettes were not seen, but there were small areas of epithelial membrane antigen (EMA)–positive intracytoplasmic dots. At the time of diagnosis, Professor Bernd W. Scheithauer at the Mayo Clinic was consulted. His pathology report reads as follows: “Intermediate grade glioma with architectural and cytologic features suggestive of ependymoma. The tumor is architecturally solid, demarcated, mitotically active (3 per 10 high power fields) and consists of epitheloid-appearing, GFAP-positive glial cells with monotonous spherical to ovoid nuclei. No EMA staining is noted, either membranous or perinuclear dot-like. Although firm support cannot be histologically provided for a diagnosis of ependymoma, the tumor most closely resembles that lesion. Ultrastructural studies, even on paraffin-embedded tissue, are recommended.” Electron microscopy was thereafter performed on formalin-fixed material, but results were inconclusive. After external consultation, the conclusion was that of an intermediate-grade glioma with architectural and cytological features suggestive of ependymoma. This tumor had the karyotype 46,XY,der(14)t(2;14)(p23;q22)[14]/46,XY[1].13
Fig. 5.

Histopathological features in the 2 tumors harboring ALK rearrangements, both demonstrating ependymal as well as astrocytic differentiation. (A) Tumor 1, hematoxylin-eosin staining. Epithelioid cells with monotonous round to ovoid nuclei. Magnification 400×, scale bar 25 µm. (b) Tumor 1, EMA staining. In small areas intracytoplasmic EMA-positive dots could be seen. Magnification 400×, scale bar 25 µm. (c) Tumor 2, hematoxylin-eosin staining. Perivascular pseudorosette. Magnification 200×, scale bar 50 µm. (d) Tumor 2. GFAP staining highlighted the pseudorosettes. Magnification 400×, scale bar 25 µm.
Case 2 was of an 8-month-old girl who was admitted to hospital because of vomiting and reduced consciousness. She had a diagnosis of large frontoparietal tumor (Supplementary material S2), which was incompletely resected. Widespread leptomeningeal metastatic disease was found. Initially she received chemotherapy according to the Brain Tumor Radiotherapy for Infants and Toddlers with Medulloblastoma (HIT-SKK) 2000 protocol, then later according to the United Kingdom Children's Cancer Study Group CNS 9204 protocol. There has been no disease progression 22 months after discontinuation of chemotherapy. Tumor 2 (Fig. 5C and D; Supplementary material S3) had the microscopic appearance of a well-demarcated cellular glioma with perivascular pseudorosettes, numerous mitoses, pseudopalisading necrosis, and microvascular proliferation. GFAP staining highlighted the pseudorosettes. The tumor was initially diagnosed as an anaplastic glioma with both astrocytic and ependymal features, best compatible with the diagnosis of anaplastic ependymoma. This diagnosis was reviewed by another neuropathologist, who reached the same conclusion. The karyotype was 81-92,XXX,-X,del(2)(p?13p?21).ish cgh dim(2)(p16p23),-10,+22[cp12]/46,XX[4].13
Transcriptome Analysis
FPKM values (fragments per kilobase of transcript per million mapped reads) were calculated by Cufflinks16 (Supplementary material S4). ALK expression was markedly higher in cases 1 and 2. Variance-stabilized read count data are shown in Supplementary material S5, with the 2 ALK-positive cases clustering together in both the Euclidean sample-to-sample distance heatmap (Supplementary material S5A) and the unsupervised hierarchical clustering of the 1000 highest expressed genes (Supplementary material S5B).
Discussion
We combined G-banding cytogenetics with RNA sequencing and identified the first 2 ALK rearrangements ever reported in gliomas. Several ALK fusion proteins of crucial pathogenetic importance have previously been described in various malignancies, including anaplastic large-cell lymphoma, diffuse large B-cell lymphoma, inflammatory myofibroblastic tumors, and non–small cell lung cancer (NSCLC).21–25 The ALK fusion breakpoints in our 2 tumors are similar to those previously reported, with exons 20–29 of ALK comprising the 3′ part of the fusion gene.23,24 Exons 20–29 encode the cytoplasmic part of the ALK protein, including its tyrosine kinase domain,23,24 whose activation promotes cell proliferation and survival.22–24,26 Most tyrosine kinase fusion partners contain coiled-coil or leucine-zipper domains, driving the oligomerization that appears to be necessary for ligand-independent activation of the fusion kinase.23,27 This is also the case in our study. CCDC88A encodes a protein of the girdin family involved in cytoskeletal integrity and cell migration,28 whereas KTN1 encodes a protein located in the endoplasmic reticulum membrane, containing 2 C-terminal leucine-zipper motifs.29
A recent paper20 described a very high frequency of C11orf95-RELA fusion genes in pediatric supratentorial ependymomas. This aberration was not detected by the gene fusion software in any of the 12 ependymomas of our series. Furthermore, none of our raw data (.fastq) files contained any of the 7 different C11orf95-RELA fusion sequences described by Parker et al20 and Pietsch et al.19 There were only 2 pediatric supratentorial tumors in this series and both were ALK positive. It is possible that the C11orf95-RELA fusion and ALK rearrangement are mutually exclusive aberrations.
Several studies indicate that supratentorial ependymomas are a molecularly distinct entity30–32; it has also been suggested that pediatric and adult ependymomas are genetically different.33,34 Pediatric patients have a poorer prognosis than adults,34,35 and supratentorial ependymomas may carry a worse prognosis than infratentorial lesions,35,36 although the latter is debated.37,38 The histopathology of the 2 tumors focused on in this study did not easily fit the established WHO brain tumor entities.6 They were both characterized as gliomas with ependymal features and elements of astrocytic differentiation, one clearly high grade and the other of intermediate grade. Both cases were clinically treated as ependymomas. It is possible that these ALK-positive “hybrid tumors” represent a separate diagnostic entity. This is underlined by the fact that both occurred in infants and were located in the cerebral hemispheres. The possibility that this could constitute a pathogenetic and phenotypic entity might have important diagnostic and therapeutic consequences, not least considering the potential of ALK as an established treatment target.
In light of this possibility, it is interesting to consider the transcriptome-wide analyses. In the sample-to-sample distances plot (Supplementary material S5A), the greatest similarities are found among the 3 subependymomas (cases 5, 7, and 9) as well as between the primary and secondary tumor from one patient (cases 6a and 6b). However, the greatest differences are found between the ALK-positive cases and the low-grade (I or II) tumors (cases 3, 4, 5, 7, 9, and 10). This is also reflected in the unsupervised hierarchical clustering (Supplementary material S5B), where cases 1 and 2 cluster together. Thus, it appears that our ALK-positive tumors bear more resemblance to high-grade than to low-grade ependymomas.
Both tumor specimens in our study had fairly simple karyotypes and harbored only one structural chromosomal aberration. The fusion genes reported here correspond to the previously detected chromosomal rearrangements involving chromosomal band 2p23.13 This is not a common chromosomal breakpoint in ependymomas – in fact, it was not seen in any of the 113 published abnormal ependymoma karyotypes to date.21 However, bearing the limited resolution of chromosomal G-banding in mind, it is possible that cryptic rearrangements affecting ALK may be a recurrent aberration in these tumors. Further molecular studies are therefore needed in order to assess the frequency of ALK rearrangements in ependymomas. They should ideally be accompanied by thorough pathological reassessments in order to investigate whether a pathological-genetic correspondence really exists centered around the ALK theme.
STAT3 is one of the most important downstream targets of oncogenic ALK activity.26 Immunohistochemistry of the phosphorylated STAT3 protein demonstrated strongly positive staining in cases 1 and 2. However, pSTAT3 was positive in 12 of 23 tumors tested (52%; Supplementary Table S1), in both ependymomas and gliomas. Positive pSTAT3 staining in high-grade gliomas39,40 and ependymal tumors41 has been known. This is not surprising, as the STAT3 pathway can be activated by a wide range of mechanisms, including ALK.26,42 Thus, pSTAT3 analysis does not appear to be a good screening method for ALK rearrangements.
ALK immunohistochemistry demonstrated scattered staining both in a subependymoma as well as in several gliomas (Supplementary Table S1, Supplementary material S6). ALK protein is known to be expressed in high-grade gliomas43,44 as well as in normal brain tissue.45 It is important to note that the 2 ALK-rearranged cases described in this paper were the only ones to demonstrate the strong, uniform ALK staining seen in Fig. 4. Furthermore, ALK FISH was negative in all but these 2 cases. Consequently, all tumors with scattered ALK staining were considered negative. Based on these results, we believe that immunohistochemistry and FISH, when held together, represent a good screening method for ALK rearrangement in brain tumors.
Crizotinib is a small-molecule selective tyrosine kinase inhibitor targeting ALK as well as 2 other tyrosine kinases (MET and ROS1). A recent phase III trial investigated crizotinib treatment in the first-line setting. Progression-free survival was significantly longer in the crizotinib group than in the standard chemotherapy group receiving pemetrexed plus platinum (10.9 vs 7.0 mo), with significantly higher objective response rates (74% vs 45%). Crizotinib was also associated with a greater reduction of lung cancer symptoms and greater improvement in quality of life.46
Crizotinib has also demonstrated an effect in the treatment of anaplastic large-cell lymphoma and inflammatory myofibroblastic tumors.47,48 It should be noted that the penetration of crizotinib into the cerebrospinal fluid appears to be very low49 and NSCLC patients receiving crizotinib often develop relapse involving the CNS.50 This might pose a problem for CNS tumors. However, there are several second-generation ALK inhibitors currently under clinical evaluation. For one of these, AP26113, objective responses were detected in 4 of 5 NSCLC patients with brain metastases.50
Surgery and radiotherapy are currently the only well-established treatment modalities for ependymoma patients.12 Systemic therapies have so far been ineffective, although chemotherapy is often used in the pediatric setting. In light of the present findings, ALK emerges as a possible treatment target in some pediatric gliomas, which should in particular be studied for possible ALK involvement. Our data suggest that these 2 tumors represent a new diagnostic entity with the potential of targeted therapy. Whether our 2 tumors represent a new entity has yet to be determined. Further studies are needed to assess the frequency of ALK rearrangements in gliomas, the functional consequences of ALK activation, and the effect of ALK-directed targeted therapy in patients suffering from these neoplasms.
Supplementary Material
Funding
This study was funded by the Norwegian Cancer Society and the Norwegian Children′s Cancer Fund (Barnekreftforeningens forskningsfond – Forsvar mot kreft). The funding source of this study had no role in the development of study design, data collection, or data analysis and interpretation or in the writing of the manuscript. The corresponding and senior author (P.B.) had full access to all data and was responsible for the decision to submit the manuscript for publication.
Supplementary Material
Acknowledgments
The authors thank Lisbeth Haugom, Anne Mette Eng Eibak, and Lene Johannessen for technical assistance. The sequencing service was provided by the Norwegian Sequencing Centre (www.sequencing.uio.no, accessed March 9, 2015), a national technology platform hosted by the University of Oslo/Oslo University Hospital and supported by the Functional Genomics and the Infrastructure programs of the Research Council of Norway and the South-Eastern Regional Health Authorities.
Conflict of interest statement. The authors declare no conflict of interest.
References
- 1.Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;74:233–245. [DOI] [PubMed] [Google Scholar]
- 2.Maher CA, Kumar-Sinha C, Cao X, et al. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;4587234:97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee C-H, Ou W-B, Mariño-Enriquez A, et al. 14-3-3 fusion oncogenes in high-grade endometrial stromal sarcoma. Proc Natl Acad Sci U S A. 2012;1093:929–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tanas MR, Sboner A, Oliveira AM, et al. Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Sci Transl Med. 2011;398:98–82. [DOI] [PubMed] [Google Scholar]
- 5.Panagopoulos I, Thorsen J, Gorunova L, et al. Fusion of the ZC3H7B and BCOR genes in endometrial stromal sarcomas carrying an X;22-translocation. Genes Chromosomes Cancer. 2013;527:610–618. [DOI] [PubMed] [Google Scholar]
- 6.McLendon RE, Schiffer D, Rosenblum MK, et al. Ependymal tumours. In: Louis DN, OhgakiWiestler HOD, Cavenee WK, eds. WHO Classification of Tumours of the Central Nervous System. 4th ed. Lyon: International Agency for Research on Cancer; 2007:69–80. [Google Scholar]
- 7.Dolecek TA, Propp JM, Stroup NE, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005–2009. Neuro Oncol. 2012;14(Suppl 5):v1–v49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilbert MR, Ruda R, Soffietti R. Ependymomas in adults. Curr Neurol Neurosci Rep. 2010;103:240–247. [DOI] [PubMed] [Google Scholar]
- 9.Reni M, Gatta G, Mazza E, et al. Ependymoma. Crit Rev Oncol Hematol. 2007;631:81–89. [DOI] [PubMed] [Google Scholar]
- 10.Crocetti E, Trama A, Stiller C, et al. Epidemiology of glial and non-glial brain tumours in Europe. Eur J Cancer. 2012;4810:1532–1542. [DOI] [PubMed] [Google Scholar]
- 11.Barnholtz-Sloan JS, Sloan AE, Schwartz AG. Relative survival rates and patterns of diagnosis analyzed by time period for individuals with primary malignant brain tumor, 1973–1997. J Neurosurg. 2003;993:458–466. [DOI] [PubMed] [Google Scholar]
- 12.Wright KD, Gajjar A. Current treatment options for pediatric and adult patients with ependymoma. Curr Treat Options Oncol. 2012;134:465–477. [DOI] [PubMed] [Google Scholar]
- 13.Olsen TK, Gorunova L, Meling TR, et al. Genomic characterization of ependymomas reveals 6q loss as the most common aberration. Oncol Rep. 2014;322:483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ge H, Liu K, Juan T, et al. FusionMap: detecting fusion genes from next-generation sequencing data at base-pair resolution. Bioinformatics. 2011;2714:1922–1928. [DOI] [PubMed] [Google Scholar]
- 15.Francis RW, Thompson-Wicking K, Carter KW, et al. Fusionfinder: A software tool to identify expressed gene fusion candidates from RNA-seq data. PLoS One. 2012;76:e39987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anders S, Huber W. DESeq: Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nyquist KB, Thorsen J, Zeller B, et al. Identification of the TAF15-ZNF384 fusion gene in two new cases of acute lymphoblastic leukemia with a t(12;17)(p13;q12). Cancer Genet. 2011;2043:147–152. [DOI] [PubMed] [Google Scholar]
- 19.Pietsch T, Wohlers I, Goschzik T, et al. Supratentorial ependymomas of childhood carry C11orf95-RELA fusions leading to pathological activation of the NF-κB signaling pathway. Acta Neuropathol. 2014;1274:609–611. [DOI] [PubMed] [Google Scholar]
- 20.Parker M, Mohankumar KM, Punchihewa C, et al. C11orf95–RELA fusions drive oncogenic NF-κB signalling in ependymoma. Nature. 2014;5067489:451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitelman F, Johansson B, Mertens F. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. Available at: http://cgap.nci.nih.gov/Chromosomes/Mitelman Accessed January 15, 2015.
- 22.Roskoski R. Anaplastic lymphoma kinase (ALK): structure, oncogenic activation, and pharmacological inhibition. Pharmacol Res. 2013;68:68–94. [DOI] [PubMed] [Google Scholar]
- 23.Shaw AT, Hsu PP, Awad MM, et al. Tyrosine kinase gene rearrangements in epithelial malignancies. Nat Rev Cancer. 2013;1311:772–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mariño-Enríquez A, Dal Cin P. ALK as a paradigm of oncogenic promiscuity: different mechanisms of activation and different fusion partners drive tumors of different lineages. Cancer Genet. 2013;20611:357–373. [DOI] [PubMed] [Google Scholar]
- 25.Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;4487153:561–566. [DOI] [PubMed] [Google Scholar]
- 26.Chiarle R, Voena C, Ambrogio C, et al. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008;81:11–23. [DOI] [PubMed] [Google Scholar]
- 27.Pearson JD, Lee JKH, Bacani JTC, et al. NPM-ALK: The prototypic member of a family of oncogenic fusion tyrosine kinases. J Signal Transduct. 2012;2012:123253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Enomoto A, Ping J, Takahashi M. Girdin, a novel actin-binding protein, and its family of proteins possess versatile functions in the Akt and Wnt signaling pathways. Ann N Y Acad Sci. 2006;1086:169–184. [DOI] [PubMed] [Google Scholar]
- 29.Fütterer A, Kruppa G, Krämer B, et al. Molecular cloning and characterization of human kinectin. Mol Biol Cell. 1995;62:161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Witt H, Mack SC, Ryzhova M, et al. Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell. 2011;202:143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson RA, Wright KD, Poppleton H, et al. Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Nature. 2010;4667306:632–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor MD, Poppleton H, Fuller C, et al. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell. 2005;84:323–335. [DOI] [PubMed] [Google Scholar]
- 33.Kilday JP, Rahman R, Dyer S, et al. Pediatric ependymoma: biological perspectives. Mol Cancer Res. 2009;76:765–786. [DOI] [PubMed] [Google Scholar]
- 34.Korshunov A, Witt H, Hielscher T, et al. Molecular staging of intracranial ependymoma in children and adults. J Clin Oncol. 2010;2819:3182–3190. [DOI] [PubMed] [Google Scholar]
- 35.Rodriguez D, Cheung MC, Housri N, et al. Outcomes of malignant CNS ependymomas: an examination of 2408 cases through the Surveillance, Epidemiology, and End Results (SEER) database (1973–2005). J Surg Res. 2009;1562:340–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Korshunov A, Golanov A, Sycheva R, et al. The histologic grade is a main prognostic factor for patients with intracranial ependymomas treated in the microneurosurgical era: an analysis of 258 patients. Cancer. 2004;1006:1230–1237. [DOI] [PubMed] [Google Scholar]
- 37.McGuire CS, Sainani KL, Fisher PG. Both location and age predict survival in ependymoma: a SEER study. Pediatr Blood Cancer. 2009;521:65–69. [DOI] [PubMed] [Google Scholar]
- 38.Godfraind C, Kaczmarska JM, Kocak M, et al. Distinct disease-risk groups in pediatric supratentorial and posterior fossa ependymomas. Acta Neuropathol. 2012;1242:247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin G-S, Yang L-J, Wang X-F, et al. STAT3 Tyr705 phosphorylation affects clinical outcome in patients with newly diagnosed supratentorial glioblastoma. Med Oncol. 2014;314:924. [DOI] [PubMed] [Google Scholar]
- 40.Mizoguchi M, Betensky RA, Batchelor TT, et al. Activation of STAT3, MAPK, and AKT in malignant astrocytic gliomas: correlation with EGFR status, tumor grade, and survival. J Neuropathol Exp Neurol. 2006;6512:1181–1188. [DOI] [PubMed] [Google Scholar]
- 41.Kong L-Y, Wei J, Haider AS, et al. Therapeutic targets in subependymoma. J Neuroimmunol. 2014;277(1–2):168–175. [DOI] [PubMed] [Google Scholar]
- 42.Yu H, Lee H, Herrmann A, et al. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;1411:736–746. [DOI] [PubMed] [Google Scholar]
- 43.Powers C, Aigner A, Stoica GE, et al. Pleiotrophin signaling through anaplastic lymphoma kinase is rate-limiting for glioblastoma growth. J Biol Chem. 2002;27716:14153–8. [DOI] [PubMed] [Google Scholar]
- 44.Stylianou DC, Auf der Maur A, Kodack DP, et al. Effect of single-chain antibody targeting of the ligand-binding domain in the anaplastic lymphoma kinase receptor. Oncogene. 2009;2837:3296–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu KV, Jong KA, Kim GY, et al. Differential induction of glioblastoma migration and growth by two forms of pleiotrophin. J Biol Chem. 2005;28029:26953–26964. [DOI] [PubMed] [Google Scholar]
- 46.Solomon BJ, Mok T, Kim D-W, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;37123:2167–2177. [DOI] [PubMed] [Google Scholar]
- 47.Gambacorti-Passerini C, Messa C, Pogliani EM. Crizotinib in anaplastic large-cell lymphoma. N Engl J Med. 2011;3648:775–776. [DOI] [PubMed] [Google Scholar]
- 48.Butrynski JE, D'Adamo DR, Hornick JL, et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med. 2010;36318:1727–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Costa DB, Kobayashi S, Pandya SS, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol. 2011;2915:e443–e445. [DOI] [PubMed] [Google Scholar]
- 50.Solomon B, Wilner KD, Shaw AT. Current status of targeted therapy for anaplastic lymphoma kinase-rearranged non-small cell lung cancer. Clin Pharmacol Ther. 2014;951:15–23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.