

Abstract
Lactobacilli are used widely in food, feed, and health applications. The taxonomy of the genus Lactobacillus, however, is confounded by the apparent lack of physiological markers for phylogenetic groups of lactobacilli and the unclear relationships between the diverse phylogenetic groups. This study used the core and pan-genomes of 174 type strains of Lactobacillus and Pediococcus to establish phylogenetic relationships and to identify metabolic properties differentiating phylogenetic groups. The core genome phylogenetic tree separated homofermentative lactobacilli and pediococci from heterofermentative lactobacilli. Aldolase and phosphofructokinase were generally present in homofermentative but not in heterofermentative lactobacilli; a two-domain alcohol dehydrogenase and mannitol dehydrogenase were present in most heterofermentative lactobacilli but absent in most homofermentative organisms. Other genes were predominantly present in homofermentative lactobacilli (pyruvate formate lyase) or heterofermentative lactobacilli (lactaldehyde dehydrogenase and glycerol dehydratase). Cluster analysis of the phylogenomic tree and the average nucleotide identity grouped the genus Lactobacillus sensu lato into 24 phylogenetic groups, including pediococci, with stable intra- and intergroup relationships. Individual groups may be differentiated by characteristic metabolic properties. The link between phylogeny and physiology that is proposed in this study facilitates future studies on the ecology, physiology, and industrial applications of lactobacilli.
INTRODUCTION
Lactobacilli are significant members of animal and human microbiota and of the plant phyllosphere. Owing to their stable association with humans as well as raw material for food production, lactobacilli also occur in many or most food fermentations. Lactobacilli are at the interface of aerobic and anaerobic life. Many lactobacilli retain the conditional capacity for respiration (1), but their ecology and physiology are mainly related to the fermentative conversion of sugars to organic acids (2, 3). Lactobacilli employ the Embden-Meyerhof pathway (glycolysis) and/or the phosphoketolase pathway for conversion of hexoses (2). These pathways have a low energetic efficiency; lactobacilli compensate for this disadvantage by rapid depletion of carbon sources and by accumulation of organic acids to inhibit competitors. The evolution and ecology of lactobacilli are shaped by niche adaptation and reduction of genome size (4). Many species, e.g., Lactobacillus plantarum, maintain a genetic diversity that enables occupation of diverse ecological niches (5). Other species are highly specialized and reduce their genomes more extensively. Examples include the insect-associated Lactobacillus fructivorans (6); Lactobacillus iners, a species specialized for the female human urogenital tract (7); and Lactobacillus reuteri, a host-specific intestinal symbiont of vertebrate animals (8). Lactobacillus delbrueckii has undergone a very recent reduction of genome size to adapt to dairy fermentations (9).
Lactobacillus spp. have been used for food production since the onset of agriculture (10) and contribute to the fermentation of a majority of fermented foods (11, 12). Their role as symbiotic members of human and animal microbiota and their long history of use in food constitute an exceptional safety record (13). Their industrial applications related to food, feed, and human and animal health make organisms in the genus Lactobacillus one of the economically most important bacterial taxa. However, the taxonomy of this genus is confounded, impeding the correlation of phylogenetic relationships with physiological properties or ecotype (14). The exceptional size and diversity of the genus Lactobacillus are one of the reasons for the uncertain taxonomy of the genus (14). The genus comprises close to 200 species, and the range of the DNA G+C content exceeds the typical range of a bacterial genus by more than 2-fold (15). Prior to the use of DNA-based taxonomic markers, lactic acid bacteria, including lactobacilli, were characterized on the basis of their growth requirements and on the basis of their carbohydrate metabolism as a central element of their physiology and ecology (14, 16). The genus Lactobacillus is an exception among lactic acid bacteria, however, as it comprises species that employ homolactic metabolism (glucose metabolism via glycolysis) as well as species that employ heterolactic metabolism (glucose metabolism via the phosphoketolase pathway) (12, 17). The most recent comprehensive revision of the taxonomy of the genus (14) employed 16S rRNA sequence data to establish 18 phylogenetic groups. However, this classification does not relate to the phenotype and does not reflect relationships between phylogenetic groups (14, 18–20). This lack impedes research aiming to understand the ecology of lactobacilli in human and animal microbiota and to improve their applications in food, green biotechnology, and pharmacology. This study therefore aimed to use a genome-wide approach to study the ecology, phylogeny, and physiology of lactobacilli. Bioinformatic analyses were carried out with genome sequence data of 174 type strains of Lactobacillus spp. or Pediococcus spp.
MATERIALS AND METHODS
Data collection and preparation.
All genome sequence data for type strains of the genera Lactobacillus and Pediococcus (see Table S1 in the supplemental material) that were available on 1 March 2015 were retrieved from GenBank (http://www.ncbi.nlm.nih.gov). Of these genomes, 68 had complete or draft assemblies, and the remaining 106 whole-genome shotgun sequences were downloaded as read data in SRA format. Genome sequence data encompassed all of the 18 groups of Lactobacillus (14), all Pediococcus spp. (21), and 174 of the 201 validly described species in the genus Lactobacillus sensu lato (see Table S1). We also downloaded the read data of Eggerthia catenaformis DSM 20559. SRA data were converted into fastq format with the SRA Toolkit (http://www.ncbi.nlm.nih.gov/Traces/sra/?view=software). The quality of the processed reads was assessed using the FastQC tool (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc), and low-quality reads were filtered by Quake (22). For each strain, the reads were assembled by the ABySS 1.5.2 software (23) with different k-mer values, and the best assemblies with the largest contig N50 were chosen for further analysis. The resulting draft genome sequences were validated by comparing the 16S rRNA gene sequences to those published in GenBank using BLASTn (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Only if the query sequence was identical to the previously deposited target sequence of the same type strain was the assembled draft genome sequence used for further analysis.
The assignment of the fermentation type of lactobacilli was based on information provided by Hammes and Hertel (19) and Pot et al. (14). The species description was consulted when these sources were inconsistent (24–30). The assignment of the fermentation type of pediococci was based on the work of Franz et al. (21) and the description of the species (31–35).
Nucleotide sequence analysis and annotation.
The average nucleotide sequence identity (ANI) between a pair of genomes was calculated by use of JSpecies software (http://imedea.uib-csic.es/jspecies/) with the BLAST algorithm (36). Clustered regularly interspaced short palindromic repeats (CRISPRs) and spacers were predicted by CRT (CRISPR recognition tool) (37). To avoid the differences caused by different annotation tools, each genome sequence was reannotated using the local annotation tool Prokka (38).
Gene clustering and function assignment.
Protein sequences were extracted from the annotation of each strain. Protein sequences with more than 50 amino acids were combined and searched using BLAST (39) with an all-against-all style and an E value cutoff of 10−10. These protein sequences were then clustered into families using the MCL algorithm with an inflation value of 2 (40). As most of the genome sequences were draft sequences, some protein coding sequences (CDS) may be interrupted by sequence gaps, which can cause fragments of the same protein clustered in the same family. To reduce the occurrence of fragmented protein sequences, we filtered the clustering results as follows. For each family, if there were multiple members from the same genome, we compared each of these sequences to the remaining proteins. Those belonging to different regions of the normal sequence were considered; only the longest sequences among them were maintained in that family. Core genes were defined as those present in all of the genomes that were analyzed.
For each family, one of the sequences was chosen as representative and searched against the local CDD (http://www.ncbi.nlm.nih.gov/cdd), COG (http://www.ncbi.nlm.nih.gov/COG), and Pfam (http://pfam.xfam.org) databases. For each family, the top 3 target functions were assigned by an in-house Perl script.
Phylogenomic analysis.
Protein sequences of each of the 172 single-copy core gene families were aligned by MUSCLE (41); each alignment was trimmed by TrimAl (42). The 172 single-copy core genes are listed in Table S2 in the supplemental material. Alignments were concatenated by an in-house script. A maximum-likelihood core protein phylogenetic tree was constructed by PhyML (43) using the best model (LG+I+G+F) predicted by ProtTest (44). Bootstrap support values were calculated from 1,000 replicates and drawn with iTOL (45).
Gene gain and loss.
Gene gain and loss over the evolutionary history of the genera Lactobacillus and Pediococcus were analyzed using COUNT software (46). COUNT uses phylogenetic birth-and-death models in a stochastic probability inference. Rate models were analyzed and optimized using the gain-loss-duplication model with the Poisson distribution. For optimization, 100 rounds were set to use the convergence criteria with a likelihood threshold of 0.1. The family history was determined by the Dollo parsimony assumption.
Identification of the presence or absence of metabolic genes.
Protein sequences were obtained from the UniProt database (http://www.uniprot.org/). Query sequences and the corresponding references are listed in Table S3 in the supplemental material. Homologous sequences were extracted from each of the analyzed genomes by sequence similarity using BLASTp with an E value of 10−10. Results were validated by searching for the same conserved domains of queries using HMMER or RPSblast; proteins are listed as present if BLASTp and domain searches were all positive. All results were checked manually to verify >75% coverage and to identify proteins that were interrupted by sequence gaps. Genes coding for bacteriocins or bacteriolysins were obtained as described previously (47). The heat map depicting the presence or absence of metabolic genes was drawn with iTOL (45).
RESULTS
A phylogenomic tree of the genera Lactobacillus and Pediococcus is shown in Fig. 1. All major nodes in the tree have high bootstrap support, demonstrating that intergroup relationships are reflected accurately. The phylogenomic tree clearly separates heterofermentative lactobacilli and homofermentative lactobacilli. Organisms in the Lactobacillus rossiae, Lactobacillus vaccinostercus, L. reuteri, Lactobacillus collinoides, Lactobacillus brevis, Lactobacillus kunkeei, L. fructivorans, and Lactobacillus buchneri groups contain only heterofermentative organisms and cluster together in the phylogenetic tree (Fig. 1). Lactobacillus coleohominis, a species in the L. reuteri group, is the only species in this cluster that was designated a homofermentative organism based on the lack of gas formation from glucose (25). Moreover, the phylogenetic tree places the genus Pediococcus as an integral, not a peripheral, part of the genus Lactobacillus. We will subsequently use the term “Lactobacillus sensu lato,” encompassing Lactobacillus spp. and Pediococcus spp.
FIG 1.

Phylogenomic analysis and basic features of 174 Lactobacillus and Pediococcus type strains based on the concatenated protein sequences of single-copy core genes. Eggerthia catenaformis was used as an outlier for the phylogenetic analysis. The maximum likelihood tree was inferred by PhyML using the best model (LG+I+G+F) predicted by ProtTest. Bootstrap support values were calculated from 1,000 replicates, and only values above 70% are shown in the figure. Members of the same group are indicated by the same color for branches, and the type strain of each group is printed in bold. Names of homofermentative species are printed in red; names of heterofermentative species are printed in blue. The color gradient in red represents the GC content of each genome sequence; higher GC contents are indicated by darker shading. The solid circles in brown represent genome sizes of these type strains; the area of the circle correlates with the genome size. The solid circles in dark purple represent the number of CRISPR spacers (clustered regularly interspaced short palindromic repeats) in each genome; the area of the circles correlates with the number of spacers. The assignment of species to phylogenetic groups shown here confirms previous assignments of species to phylogenetic groups (14) with the following exceptions or additions: L. kunkeei and Lactobacillus ozensis are now grouped together in a new L. kunkeei group; Lactobacillus selangorensis (previously in the L. perolens group) forms a separate group adjacent to L. sakei; L. amylophilus (previously in the L. delbrueckii group) forms a separate group adjacent to the L. delbrueckii group; Lactobacillus algidus (previously L. salivarius) forms a separate group adjacent to the L. salivarius group. Lactobacillus concavus was assigned to a new group with the reclassified L. dextrinicus (93). L. mellis and L. mellifer (66) form a separate group.
The assignment of species in Lactobacillus sensu lato to phylogenetic groups (14) was reassessed with two criteria, the cluster analysis of the phylogenomic tree and the average nucleotide identity (ANI). All assignments to groups have 100% bootstrap support (Fig. 1) and confirm most previous assignments of species to phylogenetic groups (14). However, relative to the last taxonomic update on the genus Lactobacillus, the assignment of species to phylogenetic groups was changed for some peripheral species, which increases the number of phylogenetic groups to 24 (Fig. 1). The intragenus ANI of Pediococcus spp. is greater than 68.1% (see Table S4 in the supplemental material); intragroup ANIs in the genus Lactobacillus are generally higher than 67.5%, and intergroup ANIs are generally lower than 67.5% (Fig. 2; see also Table S4). An intragroup ANI of below 67.5% was obtained for L. fructivorans to Lactobacillus florum (67.1%; see Table S4), for members of the Lactobacillus salivarius group, and for L. delbrueckii to Lactobacillus equicursoris. The latter two species have an ANI of 64 to 68% to other species in the same group (see Table S4) and have a noticeably higher GC content (Fig. 1). Other species in the group, however, exhibit a high intragroup ANI, and L. delbrueckii does not form a cluster that is separate from other species in the group. Of note, Lactobacillus amylotrophicus shares 99% ANI with Lactobacillus amylophilus (Fig. 1; see also Table 4). L. amylotrophicus is thus a later synonym of L. amylophilus. With the same justification, Lactobacillus parakefiri should be considered a later synonym of Lactobacillus kefiri.
FIG 2.
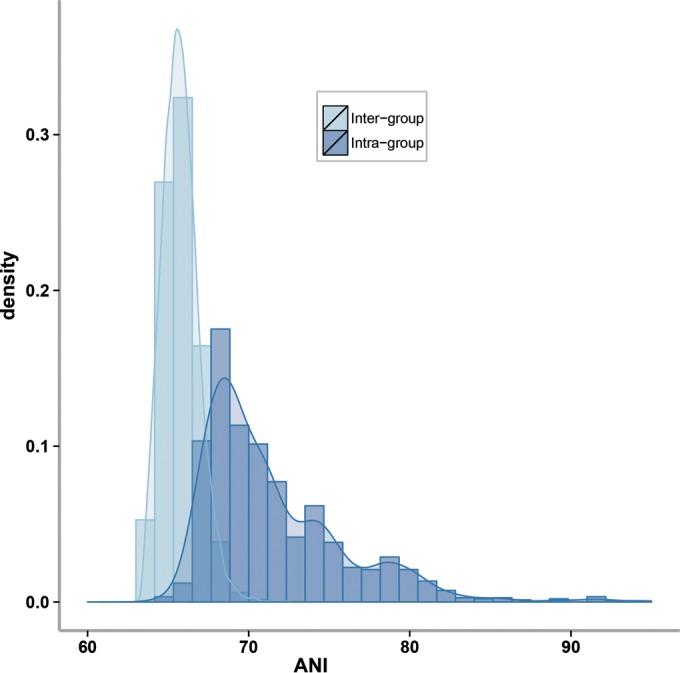
ANI distribution based on all the analyzed genome sequences of the genera Lactobacillus and Pediococcus. Dark blue represents pairwise ANI values calculated between strains in the same group, and light blue represents those between strains from different groups.
The GC content was not a suitable criterion for the demarcation of groups; however, with the exceptions of the L. delbrueckii (discussed below) and L. reuteri groups, the intragroup variation of the GC content is less than 10% (Fig. 1; see also Table S1 in the supplemental material). The genome size exhibits substantial intragroup variation (Fig. 1; see also Table S1); significant intraspecies variation is also observed. For example, the genome size ranges from 1.9 to 2.4 Mbp in L. reuteri (12 genomes), from 2.3 to 2.9 Mbp in L. brevis (13 genomes), and from 2.9 to 3.5 Mbp in L. plantarum (32 genomes) (references 5 and 48 and data not shown). The number of CRISPR spacers in the bacterial genomes was unrelated to the size of the genome or the phylogenetic position of the species (Fig. 1), indicating that the role of phages in the ecology of the organisms is species or strain specific.
Gain and loss of genes during the evolution of Lactobacillus sensu lato.
The phylogenetic birth-and-death model imposed on the phylogenomic tree was used to reconstruct the gene content dynamics during the evolution of lactobacilli (Fig. 3A). All groups of Lactobacillus sensu lato underwent genome reduction. Gene loss was a major force in all phylogenetic groups driving the distinct genome sizes of different species. Groups with smaller genomes correspond to greater gene loss. For example, the Lactobacillus floricola group lost the most genes and has the smallest genomes, while the L. delbrueckii group lost the fewest genes and has the largest genome among the groups.
FIG 3.

Gene gain and loss of the genera Lactobacillus and Pediococcus. (A) Gene gain and loss by phylogenetic groups of the genus Lactobacillus and the genus Pediococcus. The analysis was based on the phylogenomic analysis mentioned in Fig. 1 and the clustered results of protein sequences from all the genomes analyzed by the software COUNT. Numbers in black are predicted gene family numbers for ancestral nodes, with numbers of gains (blue) and losses (red) in parentheses. (B) Functions of genes lost in all heterofermentative lactobacilli but not in homofermentative lactobacilli.
Gene families that were lost by all heterofermentative species were compared to gene families lost by all homofermentative species (Fig. 3B). More than one-third (34%) of the gene families lost by heterofermentative lactobacilli relate to carbohydrate transport and metabolism. The most characteristic feature is the loss of phosphotransferase systems (PTS), which account for one-third of the gene loss in the gene family carbohydrate transport and metabolism. Accordingly, heterofermentative species harbor significantly fewer PTS than do homofermentative species (P < 10−4 [see Fig. S1 in the supplemental material]). Analysis of the functions of genes lost by each group revealed no differences in the proportions of each COG category (see Fig. S2).
Metabolic potential of the lactobacilli: matching phylogeny to ecology and physiology. (i) Genes related to carbohydrate metabolism.
To determine whether the phylogenetic separation of lactobacilli into clusters of homofermentative and heterofermentative organisms is reflected by their metabolic potential, we analyzed the presence of key genes for carbohydrate metabolism in genomes of lactobacilli (Fig. 4). Phosphofructokinase and fructose-bisphosphate aldolase represent the Embden-Meyerhof pathway. Phosphofructokinase catalyzes the dedicated and irreversible phosphorylation of fructose-6-phosphate; the gene coding for this enzyme is present in all homofermentative lactobacilli and absent in all heterofermentative lactobacilli (Fig. 4). Aldolase catalyzes the reversible conversion of fructose-1,6-bisphosphate to glyceraldehyde-3-phosphate and dihydroxyacetone-phosphate. The gene coding for this enzyme was identified in all but two homofermentative lactobacilli; its occasional presence in heterofermentative lactobacilli matches prior reports (49) and may reflect the role of aldolase in gluconeogenesis. The two isoforms of pyruvate formate lyase were exclusively present in homofermentative lactobacilli (Fig. 4) (12). Pyruvate formate lyase was frequently present in the L. plantarum, L. salivarius, Lactobacillus coryniformis, Lactobacillus sakei, Lactobacillus casei, Lactobacillus perolens, and Lactobacillus dextrinicus groups and found only occasionally in other groups of homofermentative lactobacilli.
FIG 4.

Metabolic potential of all the strains indicated by the presence/absence of key enzyme genes for different pathways. The topology of the phylogenetic tree to the left is identical to the tree shown in Fig. 1. Different colors of the heat map represent different gene sets: gray for lactic metabolism, blue for acid resistance, purple for oligosaccharide metabolism, green for peptidases, and red for bacteriocins. Darker colors represent multiple gene copies. Query sequences and Pfam domains used for the bioinformatics analysis are shown in Table S3 in the supplemental material. Genes are abbreviated as follows: for central carbon metabolism, ADH, alcohol dehydrogenase; ADL, aldolase; CydABCD, cytochrome d ubiquinol oxidase; LactAldDH, lactaldehyde dehydrogenase; MDH, mannitol dehydrogenase; PDH, pyruvate dehydrogenase; PFK, phosphofructokinase; PFL, pyruvate formate lyase; PKT, phosphoketolase; PduCDE, glycerol dehydratase; TAK, transketolase; TAL, transaldolase; acid resistance, ADI, arginine deiminase; AgDI, agmatine deiminase; GAD, glutamate decarboxylase; GLS, glutaminase; Hdc, histidine decarboxylase; Nhac, Na+/H+ antiporter; TyrDC, tyrosine decarboxylase; OrnDC, ornithine decarboxylase; F1Fo, F1Fo-ATPase; for oligosaccharide and organic acid metabolism, AmyX, intracellular amylopullulanase; MalEFG and MsmK, four-component ATP-binding cassette (ABC) transport system, importing maltodextrins into the cytosol; MalL and MalN, intracellular amylopullulanase; DexB hydrolyzes isomaltooligosaccharides, panose, and dextran but not maltose or sucrose; MalH, phospho-α-glucosidase; MalP, maltose phosphorylase; GlgP, glycogen phosphorylase; beta-phosphoglucomutase; MsmEFGK, four-component ATP-binding cassette (ABC) transport system that imports fructooligosaccharides into the cytosol; BfrA, (phospho)-fructofuranosidase; SacA, beta-fructofuranosidase; ScrP, sucrose phosphorylase; Pts1BCA, PTS, oligosucrose-specific EIIBCA component; SacK1, fructokinase; LevS, levansucrase; GtfA, sucrose phosphorylase; LacZ, β-galactosidase; MelA, α-galactosidase; LacM and LacL, β-galactosidase subunits; LacEF, lactose phosphotransferase system; LacG, phospho-β-galactosidase; CitCDEFG, citrate lyase, converting citrate to acetate and oxaloacetate; OadABCD, oxaloacetate metabolism; for proteases, peptidases, and bacteriocin production, PrtH, PrtH2, PrtH3, and PrtH4, cell envelope-associated proteinase; PrtM and PrtM2, protease maturation protein precursor; PepE, PepE2, PepF, PepO, PepO2, and PepO3, endopeptidase; Gcp, o-sialoglycoprotein transmembrane endopeptidase; PepN, aminopeptidase; PepX, X-prolyl dipeptidyl aminopeptidase; PepI and PepPN, proline iminopeptidase; PepQ and PepQ2, Xaa-Pro dipeptidase; PepD, PepD2, PepD3, and PepD4 dipeptidase; PepV, carnosinase; PepT and PepT2, tripeptide aminopeptidase; YsdC, peptidase M42 family; Map, methionine aminopeptidase; Pcp, pyrrolidone carboxyl peptidase; SprT, SprT family metallopeptidase; Hyp_prt2, peptidase M16 family; PepM1, membrane alanine aminopeptidase; PepCE, peptidase CE.
Phosphoketolase was invariably present, reflecting the role of this enzyme in anabolic reactions in addition to its role in the phosphoketolase pathway. Two enzymes discriminated for heterofermentative lactobacilli, mannitol dehydrogenase and a two-domain enzyme combining acetyl coenzyme A (acetyl-CoA) and alcohol dehydrogenase domains. Mannitol dehydrogenase reduces fructose to mannitol; this activity is found in most heterofermentative lactobacilli but not in homofermentative organisms (50, 51). Accordingly, mdh was present in all heterofermentative lactobacilli except the L. vaccinostercus and L. collinoides groups but in only 5 homofermentative lactobacilli. The two-domain aldehyde/alcohol dehydrogenase encoded by aad catalyzes conversion of acetyl-CoA to ethanol (52). Homologues of aad were invariably present in genomes of heterofermentative lactobacilli; its occasional presence in homofermentative lactobacilli was linked to pyruvate formate lyase (Fig. 4), which generates acetyl-CoA (12).
Lactate and diol catabolism was probed with gene sequences of lactaldehyde dehydrogenase, the key enzyme for lactate-to-1,2-propanediol conversion (53), and glycerol/diol dehydratase (PduCDE), the key enzyme for conversion of 1,2-propanediol to propionate and propanol (54). Lactaldehyde dehydrogenase was frequently found in the L. buchneri group, in keeping with the physiological description of this conversion in this group (12), but absent in all but three homofermentative species. The diol dehydratase was frequently present in the L. reuteri, L. collinoides, L. brevis, and L. buchneri groups, matching known physiological properties (12). The enzyme was also occasionally present in homofermentative lactobacilli, including L. coryniformis, for which the conversion was described previously (55).
Genes coding for the respiratory cytochromes CytABCD were present in most lactobacilli, irrespective of their phylogenetic position. Transaldolase and transketolase were present throughout, and genes coding for these enzymes usually occur together (Fig. 4). Transaldolase and transketolase are the key enzymes for the homofermentative conversion of pentoses to pyruvate, which was first described in an isolate later classified as Lactobacillus vini (3).
(ii) Genes related to acid resistance, oligosaccharide metabolism, and the proteolytic system.
To identify whether cluster-specific differences extend to other metabolic pathways, genes related to acid resistance, oligosaccharide metabolism, and the proteolytic system were identified (Fig. 4). Query sequences for acid resistance mechanisms focused on metabolic genes related to pH homeostasis (56, 57). The housekeeping FoF1-ATPase was identified in all genomes (Fig. 4). Remarkably, ornithine decarboxylase was the most frequent amino acid decarboxylase. It was present in most homofermentative lactobacilli, with the exceptions of Pediococcus species and the L. sakei and L. plantarum groups, but only sporadically found in heterofermentative lactobacilli (Fig. 4). The occasional presence of ornithine decarboxylase in heterofermentative lactobacilli has been attributed to horizontal gene transfer (58, 59). The arginine deiminase pathway was present in 74 genomes and matched in most cases data related to NH3 production from arginine that were provided in the species description (14, 19). The occurrence of the agmatine deiminase pathway in the L. collinoides, L. brevis, and L. buchneri groups also matches the biochemical characterization of the pathway (60) and was typically associated with the arginine deiminase pathway (Fig. 4). Genes coding for glutaminase, urease, and other amino acid decarboxylases were detected only sporadically and not linked to specific groups (Fig. 4); their presence in lactobacilli is known to be strain specific rather than species specific (12). NhaC, a Na+/H+ antiporter, was absent in the L. floricola, L. amylophilus, L. dextrinicus, L. perolens, and L. casei groups but present in virtually all other genomes. NhaC mediates proton import in exchange of Na+ extrusion in the alkaliphile Bacillus firmus (61) but has not been biochemically characterized in lactobacilli.
Identification of genes coding for metabolism of oligo- and polysaccharides (62, 63) revealed that the L. delbrueckii and L. plantarum groups are the metabolically most versatile groups, matching physiological data for individual members of these groups (5, 64). The exceptionally restricted carbohydrate fermentation pattern in L. delbrueckii was attributed to recent gene decay (9). The insect-associated Lactobacillus mellis group (65) also has a restricted pattern of genes related to oligosaccharide metabolism. Generally, heterofermentative lactobacilli harbor fewer genes coding for oligosaccharide utilization; among these, the L. reuteri group is the metabolically most versatile group. The L. fructivorans group metabolizes only α-glucosides and sucrose. The “nothing but maltose and sucrose” diet of Lactobacillus sanfranciscensis (66) thus likely pertains to other members of the group.
Most lactobacilli harbor genes coding for maltodextrins, isomaltooligosaccharides, and sucrose; genes coding for metabolism of α- and β-galactosides are less frequent (Fig. 4). AmyX is an amylopullulanase which is structurally and functionally related to MalL/MalN but carries an export signal to allow hydrolysis of extracellular starch. Its gene is present in only a few strains and, remarkably, absent in 3 of the 5 species for which starch utilization is eponymous (Fig. 4). Several genes are generally absent in heterofermentative lactobacilli; these include genes for the ABC transporter MalEFG, the isomaltase DexB, and the lactose PTS LacEF. The lack of lacEF in heterofermentative lactobacilli is matched by a higher frequency of the proton symport transporter lacS (Fig. 4). Sak1 is associated with the sucrose-PTS PTS1BCA in most homofermentative lactobacilli but in only a few heterofermentative lactobacilli, reflecting the loss of PTS in heterofermentative lactobacilli (see above) and the dual role of this enzyme as a phosphofructosidase and invertase (62). Sucrose phosphorylase is found mainly in the L. delbrueckii, L. reuteri, and L. buchneri groups. Genes citCDEFG, coding for citrate lyase, are broadly distributed; the genes oadABCD, coding for oxaloacetate decarboxylase, however, are virtually exclusive to the L. sakei and L. casei groups, reflecting the preference of lactobacilli for citrate conversion to succinate (12).
Lactobacilli metabolize proteins by extracellular proteases, followed by peptide transport and intracellular hydrolysis of peptides (67–69). Lactobacilli harbor a broad array of strain-specific genes coding for intracellular peptidases (Fig. 4) (69). Extracellular protease activity is required for growth in milk but not for growth in other substrates (68, 70, 71). Accordingly, the gene coding for the extracellular protease PrtH was frequently found in dairy isolates of the L. delbrueckii, L. casei, L. salivarius, and L. buchneri groups but was rarely present in other lactobacilli. The role of the proteolytic system as niche-specific enzymes (72) rather than taxonomic markers is also reflected in the array of genes coding for peptidases in dairy-associated strains. The pattern of the distribution of yadC, a putative aminopeptidase with homology to PepA of Lactococcus lactis (73), matches that of prtH (Fig. 4). Genes coding for the pyrrolidone carboxyl peptidase Pcp (74) were most frequent in the L. delbrueckii and L. casei groups; the alanine aminopeptidase PepM1 was exclusive to the L. delbrueckii group (Fig. 4).
Bacteriocins and bacteriolysins of lactic acid bacteria are used in food preservation but also provide insights into microbial ecology (75–77). Genome-wide screening identified peptides with homology to the class II bacteriocins carnocin, helveticin, pediocin, and plantaricin and the bacteriolysin enterolysin (Fig. 4). Genes coding for bacteriocins and bacteriolysins were present most frequently in the L. delbrueckii, L. plantarum, and L. buchneri groups (Fig. 4). Lantibiotic production has been described for a few lactobacilli (76), but genes coding for synthesis for lantibiotics were absent in type strains.
DISCUSSION
Current data on the phylogeny of lactobacilli are in apparent disagreement with their phenotype and ecotype (14, 18). In this study, we argue that a revised metabolic classification of lactobacilli combined with phylogenomic analysis allows the distinction of two major clusters and 24 groups which exhibit metabolic properties that are consistent with their ecology and phylogenetic position.
The classification into “obligate homofermentative,” “facultative heterofermentative,” and “obligate heterofermentative” lactobacilli differentiated organisms depending on the use of two alternative pathways of carbohydrate metabolism, the Embden-Meyerhof pathway and the phosphoketolase pathway (3, 14, 16). This classification, however, does not account for the third metabolic pathway for carbohydrate metabolism that is present in lactic acid bacteria, the pentose phosphate pathway (12, 78). Moreover, pentose fermentation, and hence, the differentiation between “obligate homofermentative” and “facultative heterofermentative” lactobacilli, depends on the strain-specific presence of single genes and thus is not suitable for use as a taxonomic marker. The differentiation between homofermentative and heterofermentative lactobacilli, however, reflects major physiological and ecological differences between species (2, 12). We show that the terms homofermentative and heterofermentative in regard to lactobacilli also differentiate between two major phylogenetic clusters of lactobacilli (Fig. 1 and 4).
Of the type strains included in this study, two heterofermentative lactobacilli were described as “facultative heterofermentative” based on the lack of gas formation from glucose: Lactobacillus spicheri (24) and Lactobacillus coleohominis (25). Many heterofermentative lactobacilli, however, grow poorly with glucose as the sole carbon source (12), making gas production from glucose an unreliable criterion. Both species show the typical heterofermentative pattern of metabolic genes, i.e., the absence of aldolase and phosphofructokinase and the presence of aad2 and mdh. Metabolite analysis confirmed that L. spicheri is a heterofermentative organism (79).
Multilocus sequence analysis (MLSA) clarified the disputed taxonomy of Bacillus cereus sensu lato and the genera Escherichia and Shigella (80–82). Extending MLSA to all proteins in the core genome typically confirms clusters obtained by MLSA but provides a more powerful tool to elucidate phylogenetic relationships and evolutionary history (83). MLSA established the intraspecies relationships of several Lactobacillus spp. (8, 84, 85) but has not been used to establish interspecies or intergroup relationships of lactobacilli. The phylogenomic tree provides high (>70%) bootstrap support for all nodes defining the relationship between groups and the two major clusters formed by homo- and heterofermentative lactobacilli (Fig. 1).
An ANI of 95 to 96%, corresponding to 98.65% 16S rRNA gene sequence similarity, is widely accepted as a threshold for species demarcation (36, 86). A threshold for demarcation of bacterial genera or families, however, has to our knowledge not been proposed. We determined the ANI threshold for demarcation of groups in the genus Lactobacillus sensu lato by assessing the intragroup ANI of homogenous groups, including the genus Pediococcus. The ANI threshold of 67.5 to 68% confirms the phylogenetic groups described by Pot et al. (14) and groups bacterial species that are related with respect to their ecology and metabolic potential. Together with the phylogenomic analysis, it can guide the assignment of new species to phylogenetic groups in Lactobacillus sensu lato. The L. salivarius and L. delbrueckii groups are most heterogenous with respect to ANI and physiological properties (Fig. 1 and 4; see also Table S4 in the supplemental material), but the cluster analysis justifies maintaining these diverse groups. The diversity of phylogenetic groups in the genus Lactobacillus may justify their formal recognition as genera, as exemplified by the genus Pediococcus. Previous authors, however, maintained the genus designation Lactobacillus for all groups (14, 19, 20). We follow this suggestion but propose the term Lactobacillus sensu lato to include Lactobacillus spp. and Pediococcus spp.
This study used a genome-wide screening for multiple metabolic genes to identify physiological differences between clusters or groups. Our multipronged approach eliminated truncated genes and identified proteins with sequence gaps in the original sequence data. Remaining false-positive or negative results may originate from silent genes or from sequence gaps. The absence of aldolase, a key enzyme of glycolysis, in two homofermentative lactobacilli is a case in point. To ensure the validity of conclusions, we used multiple genes for identification of differences between groups. Moreover, our results were validated by comparison with the substantial body of literature providing a detailed metabolic characterization of lactobacilli (for reviews, see references 1–3, 12, 50, 56, 57, and 62–64). A clear distinction between heterofermentative and homofermentative lactobacilli is provided by genes coding for phosphofructokinase and aldolase (homofermentative lactobacilli) as well as the two-domain acetaldehyde/alcohol dehydrogenase and mannitol dehydrogenase (heterofermentative lactobacilli) (Fig. 4).
Several other key enzymes are exclusive to one of the two clusters but are not found in all hetero- or homofermentative organisms. Lactate catabolism to 1,2-propanediol is virtually exclusive to heterofermentative lactobacilli; pyruvate formate lyase is exclusive to homofermentative lactobacilli; 1,2-propanediol or glycerol catabolism is most frequent in heterofermentative organisms (Fig. 4). The fermentation type apparently also relates to the preference of several other metabolic pathways (12). Carbohydrate utilization in lactobacilli is strain or species specific; the rapid decay of genes coding for carbohydrate metabolism and the acquisition of genes by horizontal gene transfer account for substantial intraspecies or intragroup variability of sugar fermentation (5, 9). Nevertheless, the homo- and heterofermentative lactobacilli exhibited characteristic differences with respect to their carbohydrate utilization; the loss of PTS by heterofermentative lactobacilli is the most significant feature. The loss of PTS also has repercussions for carbon catabolite repression and the preferential use of carbohydrates (12).
The phylogenetic position and metabolic potential of lactobacilli are linked to their ecotype. Niche adaptation typically corresponds to small genome size and gene decay (9, 66). Remarkably, organisms that are associated with vertebrate animals, which particularly include species in the L. reuteri and L. delbrueckii groups, maintain the capacity to metabolize a broad spectrum of carbohydrates while insect-associated organisms in the L. mellis and L. fructivorans group exhibit a highly restricted carbohydrate metabolism. Lactate and diol metabolism in the L. buchneri and L. reuteri groups are indicative of trophic relationships with other lactic acid bacteria, yeasts producing glycerol, or intestinal microbiota that produce 1,2-propanediol from fucose (87). Indeed, diol metabolism in L. reuteri is found in human-lineage strains that colonize the large intestine but not in rodent- or swine-lineage strains that colonize the upper intestine (8, 12). Acid resistance also demonstrates the importance of trophic relationships. Ornithine decarboxylase and the agmatine deiminase pathway use microbial metabolites as the substrates (Fig. 4). Although bacteriocin production as a means of bacterial warfare or communication is a strain-specific trait, bacteriocin production also clusters in specific groups, the L. delbrueckii and L. buchneri groups. However, the ecological context of this distribution remains unclear. With the possible exception of L. delbrueckii, food-fermenting lactobacilli do not cluster separately from species that originate from animal or plant microbiota (Fig. 1), reflecting the plant or animal origin of food-fermenting lactobacilli (88–90). Remarkably, stable associations of organisms that are typical for animal microbiota are also found in food fermentations. Examples include the association of L. fructivorans and L. plantarum group organisms in sourdough and Drosophila melanogaster (6, 91) and the association of L. reuteri and L. delbrueckii group organisms in cereal fermentations and in the forestomach of rodents (88, 92).
In conclusion, a combined phylogenomic and metabolic analysis of lactobacilli established that physiological properties of lactobacilli are congruent with their ecotype and phylogenetic position. The phylogenetic and physiological distinction between hetero- and homofermentative lactobacilli proposed here reestablishes the use of carbohydrate fermentation as a tool for the taxonomic and ecological characterization of lactic acid bacteria. We believe that the link between phylogeny and physiology that is proposed in this study may be a cornerstone for future studies on the ecology, physiology, and industrial applications of lactobacilli.
Supplementary Material
ACKNOWLEDGMENTS
Michael Gänzle acknowledges funding from the National Science and Engineering Council and the Canada Research Chairs Program. Ming Sun acknowledges support from the National High Technology Research and Development Program (863) of China (2011AA10A203) and China 948 Program of Ministry of Agriculture (2011-G25).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02116-15.
REFERENCES
- 1.Pedersen MB, Gaudu P, Lechardeur D, Petit MA, Gruss A. 2012. Aerobic respiration metabolism in lactic acid bacteria and uses in biotechnology. Annu Rev Food Sci Technol 3:37–58. doi: 10.1146/annurev-food-022811-101255. [DOI] [PubMed] [Google Scholar]
- 2.Axelsson L. 2003. Lactic acid bacteria: classification and physiology, p 1–66. In Salminen S, von Wright A, Ouwehand A (ed), Lactic acid bacteria: microbiological and functional aspects, 3rd ed Marcel Dekker, New York, NY. [Google Scholar]
- 3.Kandler O. 1983. Carbohydrate metabolism in lactic acid bacteria. Antonie Van Leeuwenhoek 49:209–224. doi: 10.1007/BF00399499. [DOI] [PubMed] [Google Scholar]
- 4.Makarova K, Slesarev A, Wolf Y, Sorokin A, Mirkin B, Koonin E, Pavlov A, Pavlova N, Karamychev V, Polouchine N, Shakhova V, Grigoriev I, Lou Y, Rohksar D, Lucas S, Huang K, Goodstein DM, Hawkins T, Plengvidhya V, Welker D, Hughes J, Goh Y, Benson A, Baldwin K, Lee JH, Diaz-Muniz I, Dosti B, Smeianov V, Wechter W, Barabote R, Lorca G, Altermann E, Barrangou R, Ganesan B, Xie Y, Rawsthorne H, Tamir D, Parker C, Breidt F, Broadbent J, Hutkins R, O'Sullivan D, Steele J, Unlu G, Saier M, Klaenhammer T, Richardson P, Kozyavkin S, Weimer B, Mills D. 2006. Comparative genomics of the lactic acid bacteria. Proc Natl Acad Sci U S A 103:15611–15616. doi: 10.1073/pnas.0607117103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siezen RJ, van Hylckama Vlieg JE. 2011. Genomic diversity and versatility of Lactobacillus plantarum, a natural metabolic engineer. Microb Cell Fact 10(Suppl 1):S3. doi: 10.1186/1475-2859-10-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wong CN, Ng P, Douglas AE. 2011. Low-diversity bacterial community in the gut of the fruitfly Drosophila melanogaster. Environ Microbiol 13:1889–1900. doi: 10.1111/j.1462-2920.2011.02511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mendes-Soares H, Suzuki H, Hickey RJ, Forney LJ. 2014. Comparative functional genomics of Lactobacillus spp. reveals possible mechanisms for specialization of vaginal lactobacilli to their environment. J Bacteriol 196:1458–1470. doi: 10.1128/JB.01439-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oh PL, Benson AK, Peterson DA, Patil PB, Moriyama EN, Roos S, Walter J. 2010. Diversification of the gut symbiont Lactobacillus reuteri as a result of host-driven evolution. ISME J 4:377–387. doi: 10.1038/ismej.2009.123. [DOI] [PubMed] [Google Scholar]
- 9.van de Guchte M, Penaud S, Grimaldi C, Barbe V, Bryson K, Nicolas P, Robert C, Oztas S, Mangenot S, Couloux A, Loux V, Dervyn R, Bossy R, Bolotin A, Batto JM, Walunas T, Gibrat JF, Bessieres P, Weissenbach J, Ehrlich SD, Maguin E. 2006. The complete genome sequence of Lactobacillus bulgaricus reveals extensive and ongoing reductive evolution. Proc Natl Acad Sci U S A 103:9274–9279. doi: 10.1073/pnas.0603024103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayden BCN, Shanse J. 2013. What was brewing in the Natufian? An archaeological assessment of brewing technology in the Epipaleolithic. J Archaeol Method Theory 20:102–150. [Google Scholar]
- 11.Bourdichon F, Casaregola S, Farrokh C, Frisvad JC, Gerds ML, Hammes WP, Harnett J, Huys G, Laulund S, Ouwehand A, Powell IB, Prajapati JB, Seto Y, Ter Schure E, Van Boven A, Vankerckhoven V, Zgoda A, Tuijtelaars S, Hansen EB. 2012. Food fermentations: microorganisms with technological beneficial use. Int J Food Microbiol 154:87–97. doi: 10.1016/j.ijfoodmicro.2011.12.030. [DOI] [PubMed] [Google Scholar]
- 12.Gänzle MG. 2015. Lactic metabolism revisited: metabolism of lactic acid bacteria in food fermentations and food spoilage. Curr Opin Food Sci 2:106–117. doi: 10.1016/j.cofs.2015.03.001. [DOI] [Google Scholar]
- 13.Bernardeau M, Guguen M, Vernoux JP. 2006. Beneficial lactobacilli in food and feed: long-term use, biodiversity and proposals for specific and realistic safety assessments. FEMS Microbiol Rev 30:487–513. doi: 10.1111/j.1574-6976.2006.00020.x. [DOI] [PubMed] [Google Scholar]
- 14.Pot B, Felis GE, De Bruyne K, Tsakalidou E, Papadimitriou K, Leisner J, Vandamme P. 2014. The genus Lactobacillus, p 249–353. In Holzapfel WP, Wood BJB (ed), Lactic acid bacteria: biodiversity and taxonomy. John Wiley & Sons, Inc, Hoboken, NJ. [Google Scholar]
- 15.Schleifer KH, Stackebrandt E. 1983. Molecular systematics of prokaryotes. Annu Rev Microbiol 37:143–187. doi: 10.1146/annurev.mi.37.100183.001043. [DOI] [PubMed] [Google Scholar]
- 16.Orla-Jensen S. 1919. The lactic acid bacteria. Host and Son, Copenhagen, Denmark. [Google Scholar]
- 17.Holzapfel WH, Wood BJB (ed). 2014. Lactic acid bacteria: biodiversity and taxonomy. John Wiley & Sons, Inc, Hoboken, NJ. [Google Scholar]
- 18.Hammes WP, Vogel RF. 1995. The genus Lactobacillus, p 19–54. In Wood BJB, Holzapfel WP (ed), The genera of lactic acid bacteria, vol 2 Springer, Heidelberg, Germany. [Google Scholar]
- 19.Hammes WP, Hertel C. 2006. The genera Lactobacillus and Carnobacterium. Prokaryotes 4:320–403. [Google Scholar]
- 20.Salvetti E, Torriani S, Felis GE. 2012. The genus Lactobacillus: a taxonomic update. Probiotics Antimicrob Proteins 4:217–226. doi: 10.1007/s12602-012-9117-8. [DOI] [PubMed] [Google Scholar]
- 21.Franz CMAP, Endo A, Abriouel H, Van Reenen CA, Gálvez A, Dicks LMT. 2014. The genus Pediococcus, p 359–376. In Holzapfel WP, Wood BJB (ed), Lactic acid bacteria: biodiversity and taxonomy. John Wiley & Sons, Inc, Hoboken, NJ. [Google Scholar]
- 22.Kelley DR, Schatz MC, Salzberg SL. 2010. Quake: quality-aware detection and correction of sequencing errors. Genome Biol 11:R116. doi: 10.1186/gb-2010-11-11-r116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. 2009. ABySS: a parallel assembler for short read sequence data. Genome Res 19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meroth CB, Hammes WP, Hertel C. 2004. Characterisation of the microbiota of rice sourdoughs and description of Lactobacillus spicheri sp. nov. Syst Appl Microbiol 27:151–159. doi: 10.1078/072320204322881763. [DOI] [PubMed] [Google Scholar]
- 25.Nikolaitchouk N, Wacher C, Falsen E, Andersch B, Collins MD, Lawson PA. 2001. Lactobacillus coleohominis sp. nov., isolated from human sources. Int J Syst Evol Microbiol 51:2081–2085. doi: 10.1099/00207713-51-6-2081. [DOI] [PubMed] [Google Scholar]
- 26.Ehrmann MA, Brandt M, Stolz P, Vogel RF, Korakli M. 2007. Lactobacillus secaliphilus sp. nov., isolated from type II sourdough fermentation. Int J Syst Evol Microbiol 57:745–750. doi: 10.1099/ijs.0.64700-0. [DOI] [PubMed] [Google Scholar]
- 27.Endo A, Okada S. 2007. Lactobacillus farraginis sp. nov. and Lactobacillus parafarraginis sp. nov., heterofermentative lactobacilli isolated from a compost of distilled shochu residue. Int J Syst Evol Microbiol 57:708–712. doi: 10.1099/ijs.0.64618-0. [DOI] [PubMed] [Google Scholar]
- 28.Hiraga K, Ueno Y, Sukontasing S, Tanasupawat S, Oda K. 2008. Lactobacillus senmaizukei sp. nov., isolated from Japanese pickle. Int J Syst Evol Microbiol 58:1625–1629. doi: 10.1099/ijs.0.65677-0. [DOI] [PubMed] [Google Scholar]
- 29.Ehrmann MA, Preissler P, Danne M, Vogel RF. 2010. Lactobacillus paucivorans sp. nov., isolated from a brewery environment. Int J Syst Evol Microbiol 60:2353–2357. doi: 10.1099/ijs.0.018077-0. [DOI] [PubMed] [Google Scholar]
- 30.Liang ZQ, Srinivasan S, Kim YJ, Kim HB, Wang HT, Yang DC. 2011. Lactobacillus kimchicus sp. nov., a beta-glucosidase-producing bacterium isolated from kimchi. Int J Syst Evol Microbiol 61:894–897. doi: 10.1099/ijs.0.017418-0. [DOI] [PubMed] [Google Scholar]
- 31.Zhang B, Tong H, Dong X. 2005. Pediococcus cellicola sp. nov., a novel lactic acid coccus isolated from a distilled-spirit-fermenting cellar. Int J Syst Evol Microbiol 55:2167–2170. doi: 10.1099/ijs.0.63778-0. [DOI] [PubMed] [Google Scholar]
- 32.Liu L, Zhang B, Tong H, Dong X. 2006. Pediococcus ethanolidurans sp. nov., isolated from the walls of a distilled-spirit-fermenting cellar. Int J Syst Evol Microbiol 56:2405–2408. doi: 10.1099/ijs.0.64407-0. [DOI] [PubMed] [Google Scholar]
- 33.Franz CM, Vancanneyt M, Vandemeulebroecke K, De Wachter M, Cleenwerck I, Hoste B, Schillinger U, Holzapfel WH, Swings J. 2006. Pediococcus stilesii sp. nov, isolated from maize grains. Int J Syst Evol Microbiol 56:329–333. doi: 10.1099/ijs.0.63944-0. [DOI] [PubMed] [Google Scholar]
- 34.De Bruyne K, Franz CM, Vancanneyt M, Schillinger U, Mozzi F, de Valdez GF, De Vuyst L, Vandamme P. 2008. Pediococcus argentinicus sp. nov. from Argentinean fermented wheat flour and identification of Pediococcus species by pheS, rpoA and atpA sequence analysis. Int J Syst Evol Microbiol 58:2909–2916. doi: 10.1099/ijs.0.65833-0. [DOI] [PubMed] [Google Scholar]
- 35.Doi K, Nishizaki Y, Fujino Y, Ohshima T, Ohmomo S, Ogata S. 2009. Pediococcus lolii sp. nov., isolated from ryegrass silage. Int J Syst Evol Microbiol 59:1007–1010. doi: 10.1099/ijs.0.005793-0. [DOI] [PubMed] [Google Scholar]
- 36.Richter M, Rossello-Mora R. 2009. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A 106:19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bland C, Ramsey TL, Sabree F, Lowe M, Brown K, Kyrpides NC, Hugenholtz P. 2007. CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 8:209. doi: 10.1186/1471-2105-8-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 39.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li L, Stoeckert CJ Jr, Roos DS. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. 2009. TrimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 44.Darriba D, Taboada GL, Doallo R, Posada D. 2011. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27:1164–1165. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Letunic I, Bork P. 2011. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res 39:W475–W478. doi: 10.1093/nar/gkr201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Csuros M. 2010. Count: evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics 26:1910–1912. doi: 10.1093/bioinformatics/btq315. [DOI] [PubMed] [Google Scholar]
- 47.Zheng J, Gänzle MG, Lin XB, Ruan L, Sun M. 2015. Diversity and dynamics of bacteriocins from human microbiome. Environ Microbiol 17:2133–2143. doi: 10.1111/1462-2920.12662. [DOI] [PubMed] [Google Scholar]
- 48.Spinler JK, Sontakke A, Hollister EB, Venable SF, Oh PL, Balderas MA, Saulnier DM, Mistretta TA, Devaraj S, Walter J, Versalovic J, Highlander SK. 2014. From prediction to function using evolutionary genomics: human-specific ecotypes of Lactobacillus reuteri have diverse probiotic functions. Genome Biol Evol 6:1772–1789. doi: 10.1093/gbe/evu137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arskold E, Svensson M, Grage H, Roos S, Radstrom P, van Niel EW. 2007. Environmental influences on exopolysaccharide formation in Lactobacillus reuteri ATCC 55730. Int J Food Microbiol 116:159–167. doi: 10.1016/j.ijfoodmicro.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 50.von Weymarn N, Hujanen M, Leisola M. 2002. Production of d-mannitol by heterofermentative lactic acid bacteria. Process Biochem 37:1207–1213. doi: 10.1016/S0032-9592(01)00339-9. [DOI] [Google Scholar]
- 51.Korakli M, Vogel RF. 2003. Purification and characterisation of mannitol dehydrogenase from Lactobacillus sanfranciscensis. FEMS Microbiol Lett 220:281–286. doi: 10.1016/S0378-1097(03)00129-0. [DOI] [PubMed] [Google Scholar]
- 52.Sillers R, Al-Hinai MA, Papoutsakis ET. 2009. Aldehyde-alcohol dehydrogenase and/or thiolase overexpression coupled with CoA transferase downregulation lead to higher alcohol titers and selectivity in Clostridium acetobutylicum fermentations. Biotechnol Bioeng 102:38–49. doi: 10.1002/bit.22058. [DOI] [PubMed] [Google Scholar]
- 53.Oude Elferink SJ, Krooneman J, Gottschal JC, Spoelstra SF, Faber F, Driehuis F. 2001. Anaerobic conversion of lactic acid to acetic acid and 1,2-propanediol by Lactobacillus buchneri. Appl Environ Microbiol 67:125–132. doi: 10.1128/AEM.67.1.125-132.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dishisha T, Pereyra LP, Pyo SH, Britton RA, Hatti-Kaul R. 2014. Flux analysis of the Lactobacillus reuteri propanediol-utilization pathway for production of 3-hydroxypropionaldehyde, 3-hydroxypropionic acid and 1,3-propanediol from glycerol. Microb Cell Fact 13:76. doi: 10.1186/1475-2859-13-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martin R, Olivares M, Marin ML, Xaus J, Fernandez L, Rodriguez JM. 2005. Characterization of a reuterin-producing Lactobacillus coryniformis strain isolated from a goat's milk cheese. Int J Food Microbiol 104:267–277. doi: 10.1016/j.ijfoodmicro.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 56.De Angelis M, Gobbetti M. 2004. Environmental stress responses in Lactobacillus: a review. Proteomics 4:106–122. doi: 10.1002/pmic.200300497. [DOI] [PubMed] [Google Scholar]
- 57.Teixeira JS, Seeras A, Sanchez-Maldonado AF, Zhang C, Su MS, Gänzle MG. 2014. Glutamine, glutamate, and arginine-based acid resistance in Lactobacillus reuteri. Food Microbiol 42:172–180. doi: 10.1016/j.fm.2014.03.015. [DOI] [PubMed] [Google Scholar]
- 58.Romano A, Trip H, Lolkema JS, Lucas PM. 2013. Three-component lysine/ornithine decarboxylation system in Lactobacillus saerimneri 30a. J Bacteriol 195:1249–1254. doi: 10.1128/JB.02070-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Romano A, Ladero V, Alvarez MA, Lucas PM. 2014. Putrescine production via the ornithine decarboxylation pathway improves the acid stress survival of Lactobacillus brevis and is part of a horizontally transferred acid resistance locus. Int J Food Microbiol 175:14–19. doi: 10.1016/j.ijfoodmicro.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 60.Lucas PM, Blancato VS, Claisse O, Magni C, Lolkema JS, Lonvaud-Funel A. 2007. Agmatine deiminase pathway genes in Lactobacillus brevis are linked to the tyrosine decarboxylation operon in a putative acid resistance locus. Microbiology 153:2221–2230. doi: 10.1099/mic.0.2007/006320-0. [DOI] [PubMed] [Google Scholar]
- 61.Ivey DM, Guffanti AA, Bossewitch JS, Padan E. 1991. Molecular cloning and sequencing of a gene from alkaliphilic Bacillus firmus OF4 that functionally complements an Escherichia coli strain carrying a deletion in the nhaA Na+/H+ antiporter gene. J Biol Chem 266:23483–24489. [PubMed] [Google Scholar]
- 62.Gänzle MG, Follador R. 2012. Metabolism of oligosaccharides and starch in lactobacilli: a review. Front Microbiol 3:340. doi: 10.3389/fmicb.2012.00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goh YJ, Klaenhammer TR. 2015. Genetic mechanisms of prebiotic oligosaccharide metabolism in probiotic microbes. Annu Rev Food Sci Technol 6:137–156. doi: 10.1146/annurev-food-022814-015706. [DOI] [PubMed] [Google Scholar]
- 64.Barrangou R, Azcarate-Peril MA, Duong T, Conners SB, Kelly RM, Klaenhammer TR. 2006. Global analysis of carbohydrate utilization by Lactobacillus acidophilus using cDNA microarrays. Proc Natl Acad Sci U S A 103:3816–3821. doi: 10.1073/pnas.0511287103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Olofsson TC, Alsterfjord M, Nilson B, Butler E, Vasquez A. 2014. Lactobacillus apinorum sp. nov., Lactobacillus mellifer sp. nov., Lactobacillus mellis sp. nov., Lactobacillus melliventris sp. nov., Lactobacillus kimbladii sp. nov., Lactobacillus helsingborgensis sp. nov. and Lactobacillus kullabergensis sp. nov., isolated from the honey stomach of the honeybee Apis mellifera. Int J Syst Evol Microbiol 64:3109–3119. doi: 10.1099/ijs.0.059600-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vogel RF, Pavlovic M, Ehrmann MA, Wiezer A, Liesegang H, Offschanka S, Voget S, Angelov A, Böcker G, Liebl W. 2011. Genomic analysis reveals Lactobacillus sanfranciscensis as stable element in traditional sourdoughs. Microb Cell Fact 10(Suppl 1):S6. doi: 10.1186/1475-2859-10-S1-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Savijoki K, Ingmer H, Varmanen P. 2006. Proteolytic systems of lactic acid bacteria. Appl Microbiol Biotechnol 71:394–406. doi: 10.1007/s00253-006-0427-1. [DOI] [PubMed] [Google Scholar]
- 68.Griffiths MW, Tellez AM. 2013. Lactobacillus helveticus: the proteolytic system. Front Microbiol 4:30. doi: 10.3389/fmicb.2013.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gänzle MG, Loponen J, Gobbetti M. 2008. Proteolysis in sourdough fermentations: mechanisms and potential for improved bread quality. Trend Food Sci Technol 19:513–521. doi: 10.1016/j.tifs.2008.04.002. [DOI] [Google Scholar]
- 70.Vermeulen N, Pavlovic M, Ehrmann MA, Gänzle MG, Vogel RF. 2005. Functional characterization of the proteolytic system of Lactobacillus sanfranciscensis DSM 20451T during growth in sourdough. Appl Environ Microbiol 71:6260–6266. doi: 10.1128/AEM.71.10.6260-6266.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu E, Zheng H, Hao P, Konno T, Yu Y, Kume H, Oda M, Ji ZS. 2012. A model of proteolysis and amino acid biosynthesis for Lactobacillus delbrueckii subsp. bulgaricus in whey. Curr Microbiol 65:742–751. doi: 10.1007/s00284-012-0214-4. [DOI] [PubMed] [Google Scholar]
- 72.O'Sullivan O, O'Callaghan J, Sangrador-Vegas A, McAuliffe O, Slattery L, Kaleta P, Callanan M, Fitzgerald GF, Ross RP, Beresford T. 2009. Comparative genomics of lactic acid bacteria reveals a niche-specific gene set. BMC Microbiol 9:50. doi: 10.1186/1471-2180-9-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.l'Anson KJA, Movahedi S, Griffin HG, Gasson MJ, Mulholland F. 1995. A non-essential glutamyl aminopeptidase is required for optimal growth of Lactococcus lactis MG1363 in milk. Microbiology 141:2873–2881. doi: 10.1099/13500872-141-11-2873. [DOI] [PubMed] [Google Scholar]
- 74.Cleuziat P, Awade A, Robert-Baudouy J. 1992. Molecular characterization of pcp, the structural gene encoding the pyrrolidone carboxylyl peptidase from Streptococcus pyogenes. Mol Microbiol 6:2051–2063. doi: 10.1111/j.1365-2958.1992.tb01378.x. [DOI] [PubMed] [Google Scholar]
- 75.Nes IF, Holo H. 2000. Class II antimicrobial peptides from lactic acid bacteria. Biopolymers 55:50–61. doi:. [DOI] [PubMed] [Google Scholar]
- 76.Twomey D, Ross RP, Ryan M, Meaney B, Hill C. 2002. Lantibiotics produced by lactic acid bacteria: structure, function and applications. Antonie Van Leeuwenhoek 82:165–185. doi: 10.1023/A:1020660321724. [DOI] [PubMed] [Google Scholar]
- 77.Cotter PD, Ross RP, Hill C. 2013. Bacteriocins—a viable alternative to antibiotics? Nat Rev Microbiol 11:95–105. doi: 10.1038/nrmicro2937. [DOI] [PubMed] [Google Scholar]
- 78.Tanaka K, Komiyama A, Sonomoto K, Ishizaki A, Hall SJ, Stanbury PF. 2002. Two different pathways for d-xylose metabolism and the effect of xylose concentration on the yield coefficient of l-lactate in mixed-acid fermentation by the lactic acid bacterium Lactococcus lactis IO-1. Appl Microbiol Biotechnol 60:160–167. doi: 10.1007/s00253-002-1078-5. [DOI] [PubMed] [Google Scholar]
- 79.Ianniello RG, Zheng J, Zotta T, Ricciardi A, Gänzle MG. 20 May 2015. Biochemical analysis of respiratory metabolism in the heterofermentative Lactobacillus spicheri and Lactobacillus reuteri. Appl Microbiol doi: 10.1111/jam.12853. [DOI] [PubMed] [Google Scholar]
- 80.Hoffmaster AR, Ravel J, Rasko DA, Chapman GD, Chute MD, Marston CK, De BK, Sacchi CT, Fitzgerald C, Mayer LW, Maiden MC, Priest FG, Barker M, Jiang L, Cer RZ, Rilstone J, Peterson SN, Weyant RS, Galloway DR, Read TD, Popovic T, Fraser CM. 2004. Identification of anthrax toxin genes in a Bacillus cereus associated with an illness resembling inhalation anthrax. Proc Natl Acad Sci U S A 101:8449–8454. doi: 10.1073/pnas.0402414101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jaureguy F, Landraud L, Passet V, Diancourt L, Frapy E, Guigon G, Carbonnelle E, Lortholary O, Clermont O, Denamur E, Picard B, Nassif X, Brisse S. 2008. Phylogenetic and genomic diversity of human bacteremic Escherichia coli strains. BMC Genomics 9:560. doi: 10.1186/1471-2164-9-560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cardazzo B, Negrisolo E, Carraro L, Alberghini L, Patarnello T, Giaccone V. 2008. Multiple-locus sequence typing and analysis of toxin genes in Bacillus cereus food-borne isolates. Appl Environ Microbiol 74:850–860. doi: 10.1128/AEM.01495-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Touchon M, Hoede C, Tenaillon O, Barbe V, Baeriswyl S, Bidet P, Bingen E, Bonacorsi S, Bouchier C, Bouvet O, Calteau A, Chiapello H, Clermont O, Cruveiller S, Danchin A, Diard M, Dossat C, Karoui ME, Frapy E, Garry L, Ghigo JM, Gilles AM, Johnson J, Le Bouguenec C, Lescat M, Mangenot S, Martinez-Jehanne V, Matic I, Nassif X, Oztas S, Petit MA, Pichon C, Rouy Z, Ruf CS, Schneider D, Tourret J, Vacherie B, Vallenet D, Medigue C, Rocha EP, Denamur E. 2009. Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet 5:e1000344. doi: 10.1371/journal.pgen.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Raftis EJ, Salvetti E, Torriani S, Felis GE, O'Toole PW. 2011. Genomic diversity of Lactobacillus salivarius. Appl Environ Microbiol 77:954–965. doi: 10.1128/AEM.01687-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nyquist OL, McLeod A, Brede DA, Snipen L, Aakra A, Nes IF. 2011. Comparative genomics of Lactobacillus sakei with emphasis on strains from meat. Mol Genet Genomics 285:297–311. doi: 10.1007/s00438-011-0608-1. [DOI] [PubMed] [Google Scholar]
- 86.Kim M, Oh HS, Park SC, Chun J. 2014. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol 64:346–351. doi: 10.1099/ijs.0.059774-0. [DOI] [PubMed] [Google Scholar]
- 87.Reichardt N, Duncan SH, Young P, Belenguer A, McWilliam Leitch C, Scott KP, Flint HJ, Louis P. 2014. Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. ISME J 8:1323–1335. doi: 10.1038/ismej.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vogel RF, Knorr R, Müller MR, Steudel U, Gänzle MG, Ehrmann MA. 1999. Non-dairy lactic fermentations: the cereal world. Antonie Van Leeuwenhoek 76:403–411. doi: 10.1023/A:1002089515177. [DOI] [PubMed] [Google Scholar]
- 89.Walter J. 2008. Ecological role of lactobacilli in the gastrointestinal tract: implications for fundamental and biomedical research. Appl Environ Microbiol 74:4985–4996. doi: 10.1128/AEM.00753-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Su MSW, Oh PL, Walter J, Gänzle MG. 2012. Intestinal origin of sourdough Lactobacillus reuteri isolates as revealed by phylogenetic, genetic, and physiological analysis. Appl Environ Microbiol 78:6777–6780. doi: 10.1128/AEM.01678-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lattanzi A, Minervini F, Di Cagno R, Diviccaro A, Antonielli L, Cardinali G, Cappelle S, De Angelis M, Gobbetti M. 2013. The lactic acid bacteria and yeast microbiota of eighteen sourdoughs used for the manufacture of traditional Italian sweet leavened baked goods. Int J Food Microbiol 163:71–79. doi: 10.1016/j.ijfoodmicro.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 92.Schwab C, Tveit AT, Schleper C, Urich T. 2014. Gene expression of lactobacilli in murine forestomach biofilms. Microb Biotechnol 7:347–359. doi: 10.1111/1751-7915.12126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Haakensen M, Dobson CM, Hill JE, Ziola B. 2009. Reclassification of Pediococcus dextrinicus (Coster and White 1964) back 1978 (Approved Lists 1980) as Lactobacillus dextrinicus comb. nov., and emended description of the genus Lactobacillus. Int J Syst Evol Microbiol 59:615–621. doi: 10.1099/ijs.0.65779-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.