

Summary
Icaritin, can co-target both persistent AR and AR splice variants by arylhydrocarbon-receptor (AhR) mediated proteasomal degradation, inhibit AR transcriptional activity and induce apoptosis in AR-positive prostate cancer (PC) cells.
Abstract
Persistent androgen receptor (AR) signaling is the key driving force behind progression and development of castration-resistant prostate cancer (CRPC). In many patients, AR COOH-terminal truncated splice variants (ARvs) play a critical role in contributing to the resistance against androgen depletion therapy. Unfortunately, clinically used antiandrogens like bicalutamide (BIC) and enzalutamide (MDV), which target the ligand binding domain, have failed to suppress these AR variants. Here, we report for the first time that a natural prenylflavonoid, icaritin (ICT), can co-target both persistent AR and ARvs. ICT was found to inhibit transcription of key AR-regulated genes, such as KLK3 [prostate-specific antigen (PSA)] and ARvs-regulated genes, such as UBE2C and induce apoptosis in AR-positive prostate cancer (PC) cells. Mechanistically, ICT promoted the degradation of both AR and ARvs by binding to arylhydrocarbon-receptor (AhR) to mediate ubiquitin-proteasomal degradation. Therefore, ICT impaired AR transactivation in PC cells. Knockdown of AhR gene restored AR stability and partially prevented ICT-induced growth suppression. In clinically relevant murine models orthotopically implanted with androgen-sensitive and CRPC cells, ICT was able to target AR and ARvs, to inhibit AR signaling and tumor growth with no apparent toxicity. Our results provide a mechanistic framework for the development of ICT, as a novel lead compound for AR-positive PC therapeutics, especially for those bearing AR splice variants.
Introduction
Androgen deprivation therapy (ADT), the mainstay of treatment for recurrent or metastatic prostate cancer (PCa), primarily acts to reduce gonadal androgen synthesis through chemical or surgical castration, and/or disrupts the androgen receptor (AR) signaling axis through antiandrogens, such as bicalutamide (BIC) (1). Despite initial remission with ADT, the development of the castration-resistant prostate cancer (CRPC) becomes inevitable in a majority of the cases (2). Despite castrate levels of circulating testosterone (<1.7 nmol/l) (3), the AR axis is often aberrantly reactivated. Mechanisms critical in the progression of CRPC include intratumoral androgen synthesis (4), AR overexpression and mutations that sensitizes AR to low androgen concentrations or alternative ligands (5,6), changes in the levels of AR transcriptional cofactors (7) and expression of constitutively active AR COOH-terminal truncated variant (ARvs) (8,9).
In 2012, the second-generation AR antagonist MDV (MDV3100) was approved by the United States Food and Drug Administration for use in CRPC following a phase III trial, which demonstrated that MDV was able to prolong median overall survival in men with chemotherapy-refractory CRPC by 4.8 months (10). Despite its success, a recent clinical study indicated that patients with circulating tumors expressing AR-V7, the most abundant ARvs in PCa tissue (11), had a poorer response to MDV compared with those with no detectable AR-V7 (12). Similarly, ARvs were found to be responsible for the resistance to the current ADT, including BIC and MDV (13,14). Therefore, identifying and developing effective inhibitors that can target both AR and ARvs are of paramount importance to improve the clinical management of prostate cancer (PC).
Icaritin (ICT) is a natural prenylflavonoid derived from the genus Epimedium (15). We have previously demonstrated that ICT binds and activates the arylhydrocarbon receptor (AhR) to degrade estrogen receptor alpha in MCF-7 breast cancer cells (16). Importantly, Ohtake et al. (17) have reported that the ligand-activated AhR can promote the proteasomal degradation of sex steroid receptors including estrogen receptor alpha and AR. Therefore, we hypothesized that ICT-mediated proteasomal degradation of AR and its variants via AhR-dependent pathways may be critical to suppress the proliferation of AR-positive PCa cells (including ARvs-positive, if any, in this study).
Here, we show for the first time that ICT inhibited the growth of AR-positive PCa cells in vitro largely via destabilizing AR and ARvs proteins through AhR-mediated proteasomal degradation and subsequently disrupting AR transcriptional activity. Furthermore, intraperitoneal administration of ICT to severe combined immunodeficiency (SCID) mice orthotopically implanted with AR-positive PCa cells corroborated our in vitro findings and demonstrated significant inhibition of PCa growth, and AR signaling without causing toxicity.
Materials and methods
The detailed description for the Materials and methods is available in Supplementary Materials and methods, available at Carcinogenesis Online.
Chemical reagents
ICT (purity 98%) was provided by Shenogen Pharma Group (Beijing, China). BIC, MDV, dihydrotestosterone (DHT), 3-methylcholanthrene (3MC), cycloheximide and MG132 were commercially obtained from Sigma.
Cell lines
The human PCa LNCaP, CWR22Rv1, PC-3, human prostatic epithelial RWPE-1, human breast cancer MCF-7, human cervical cancer HeLa and monkey kidney CV-1 cell lines were obtained from American Type Culture Collection and cultured according to American Type Culture Collection protocol. The cell lines were authenticated by LGC Standards (UK) Cell Line Authentication service. The C4-2 cell line was obtained from ViroMed Laboratories (Minneapolis, MN) and LNCaP-luciferase (LNCaP-luc) cell line was a gift from Dr Patrick Ling, Queensland University of Technology.
Cell Proliferation and Cell-Cycle Assays
Cell viability was determined by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS; Promega) assay as previously described (18). The relative luminescence units were measured at a wavelength of 490nm. Detection and quantitation of apoptotic cell cycle population were studied by flow cytometry as previously described (19).
Cell transfection and Luciferase reporter assay
Stably transfected HeLa-AR-ARE4-Luc cells and LNCaP cells transiently transfected with ARE-Luc reporter gene (20) followed by drug treatments were measured with Luciferase Assay System (Promega). For RNA interference, cells were transfected with ON-TARGET plus SMARTpool four human AhR siRNAs and three human AR exon 7 siRNAs (Thermo Scientific) and one scrambled siRNA (si-Scr) following the manufacturer’s instruction before analysis. CWR22Rv1 cells were transiently transfected with pCDNA3-HA-Ub (Addgene) followed by drug treatments and in vivo ubiquitnation assay. Mammalian two-hybrid assay was conducted in CV1 cells as described previously (21).
Quantitative RT–PCR
Quantitative PCR was carried out using Taqman Gene Expression Assays (Applied Biosystem) or SYBP Green PCR amplification kit (Applied Biosystem). Heatmaps showing relative gene expression normalized to 18S rRNAs were created using TreeView Package version 1.60.
Western blotting
Western blotting of total cell or tumor tissue lysates was performed according to standard protocol. The primary antibodies used for western blotting are as follows: AR (sc-816, Santa Cruz), prostate-specific antigen [PSA (sc-7638, Santa Cruz)], AhR (sc-101104, Santa Cruz), MDM2 (sc-813, Santa Cruz), HDAC1 (sc-7872, Santa Cruz), GAPDH (sc-25778, Santa Cruz), AR-V7 (AG10008, Precision Antibody), UBE2C (A-650, Boston Biochem), poly-ADP-ribose polymerase (PARP) (#9542; Cell Signaling), estrogen receptor alpha (sc-542, Santa Cruz), ERβ (sc-8974, Santa Cruz), RXRα (sc-553, Santa Cruz), progesterone receptor (#8757; Cell Signaling), peroxisome proliferator-activated receptor gamma (PPAR-γ; sc-7273, Santa Cruz), glucocorticoid receptor (#3660; Cell Signaling) and β-actin (Sigma).
AR protein stability assay
The PCa cells were treated with dimethyl sulfoxide (DMSO) (vehicle, Sigma), 30 µmol/l ICT and 30 µmol/l ICT plus 10 µmol/l MG132 (Sigma) (being added 30min earlier than other agents) in the presence of 50 µmol/l translation inhibitor cycloheximide for 0, 1, 3, 6, 12 and 24h, followed by the preparation of whole cell lysates. AR protein was analyzed by western blot assay, quantified by gel-pro analyzer 4.0 and normalized to β-actin.
Co-immunoprecipitation (Co-IP) assay
Androgen-depleted PCa cells were treated with either DMSO (vehicle), 5 μmol/l of 3MC or 30 µmol/l of ICT for 2h. Co-immunoprecipitation (Co-IP) experiments were performed using the Pierce Co-Immunoprecipitation Kit (26149, Pierce) as per manufacturer’s protocol. The co-immunoprecipitate was then eluted and analyzed by the western blot assay along with the 5% input controls. AR antibody (sc-7305, Santa Cruz) were used to pull down and probe for AR (sc-816, Santa Cruz), AhR (sc-5579, Santa Cruz) and Mdm2 (sc-813, Santa Cruz) by western blot. Mouse IgG (sc-2025, Santa Cruz) was used as a negative control for IP.
In vitro ubiquitination
CWR22Rv1 cells were transiently transfected with pCDNA3-HA-Ub (Addgene) by Fugene HD (Roche, USA) for 48h, followed by 24h treatment with DMSO (vehicle), 5 μmol/l 3MC and 30 µmol/l ICT. The cell extracts were harvested and subjected to Co-IP assays. AR antibody (sc-7305, Santa Cruz) was used to pull down the ubiquitinated AR, which was then probed using a HA antibody (sc-805, Santa Cruz) by western blot.
Chromatin immunoprecipitation assay
Androgen-depleted LNCaP and CWR22RV1 cells were treated with either DMSO (vehicle), 30 µmol/l ICT, 30 µmol/l BIC and DHT (10 nmol/l for LNCaP and 1 nmol/l for CWR22RV1) alone or in combinations for 2h. The cells were processed for the chromatin immunoprecipitation assay as described previously (22). Briefly, cells were cross-linked with 1% formaldehyde for 10min at room temperature. The nuclei were isolated and sonicated on ice to break chromatin DNA to an average length of 300–500bp. Soluble chromatin was used in immunoprecipitation with AR antibody (sc-816, Santa Cruz) and IgG (as negative control). The immune complexes were bound using protein A/G agarose beads (Roche Applied Science) at 4°C overnight. After reversing the cross-links, and proteinase K digestion, immunoprecipitated DNA was quantified by quantitative PCR using specific primers.
Murine orthotopic xenograft model
Animal studies were conducted using 6–8 week-old NOD CB17-Prkdcscid/J male mice (Jackson Laboratory, Bar Haror, ME). Orthotopic implantation was performed as previously described (23), where mice were surgically implanted with LNCaP-Luc or CWR22Rv1 cells in the prostate. Mice received intraperitoneal injection of ICT solution at the dose of 0 (vehicle control) and 33mg/kg 5 times per week for 10 weeks (LNCaP-luc) or 5 weeks (CWR22Rv1). For the LNCaP-luc xenograft, tumor growth was measured by an IVIS® imaging system (Xenogen) at 2-week intervals. At the end of the experiment, harvested serum and tissue samples were measured, snap frozen for protein level analysis or paraffin embedded for the immunohistochemistry (IHC) analysis. Pharmacokinetics and toxicity of ICT were performed in tumor-bearing SCID mice and tumor-free SCID mice, respectively. All animal experiments were performed humanely in compliance with guidelines reviewed by the Animal Ethics Committee of the Biological Resource Centre of the Agency for Science, Technology and Research.
Statistical analysis
Statistical analysis was performed using SPSS 13.0. Comparisons between two groups were made using Student’s independent-sample t-test and those with more than two groups, one-way analysis of variance was performed followed by the Tukey tests used for the post-hoc multiple comparisons between individual groups. The statistical significance level was set to be P < 0.05 or P < 0.01.
Results
ICT disrupts the stability of AR and ARvs proteins in human PCa cells
PCa is a heterogeneous disease, which has been shown to acquire genetic and phenotypic variations in response to ADT. We first studied if ICT can alter the stability of AR and ARvs proteins on key models of human PCa namely, LNCaP (androgen-sensitive), CWR22Rv1 (castration-resistant, overexpressing ARvs) and C4-2 (castration-resistant subline of LNCaP) cell lines. Experiments were performed in the presence of 10 nmol/l of DHT to mimic the androgen-sensitive state, or in charcoal stripped serum (CSS) supplemented with 1 nmol/l DHT to mimic the low levels of androgen after castration in patients (3,24), or CSS alone. Strikingly ICT, but not BIC, which targets the ligand binding domain, demonstrated a dose-dependent reduction in the levels of AR (~110kDa) and ARvs (~80kDa), if any, in both androgen-sensitive and castration-resistant PCa cells in the presence or absence of DHT (Figure 1A and Supplementary Figure S1A, available at Carcinogenesis Online). Correspondingly, ICT treatment also resulted in marked dose-dependent reductions in PSA encoded by an AR-regulated KLK3 gene (25), and ubiquitin-conjugating enzyme E2C (UBE2C) encoded by an ARvs-regulated UBE2C gene (14). To assess if the effects of ICT were limited to AR and ARvs, we studied the expression of other nuclear receptors. As expected, ICT induced a reduction of ERα protein content (16). In comparison, peroxisome proliferator-activated receptor gamma, glucocorticoid receptor, progesterone receptor, ERβ and AhR protein stability were not affected (Supplementary Figure S1B, available at Carcinogenesis Online).
Figure. 1.
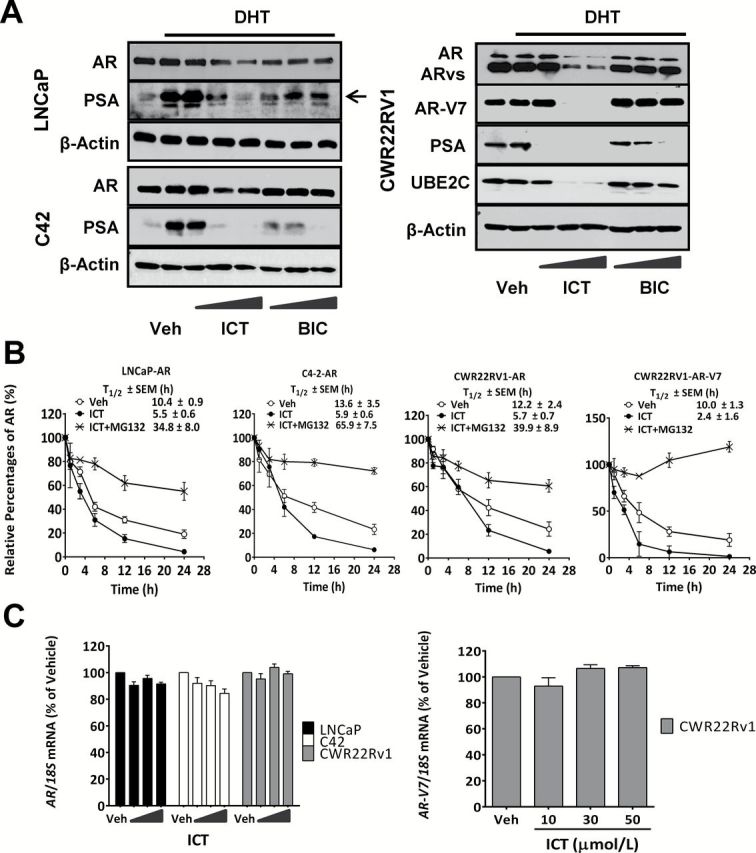
Effect of ICT on the stability of AR and AR splice variant proteins in PCa cells. (A) Androgen-sensitive (LNCaP) and castration-resistant (CWR22Rv1 and C4-2) PCa cells were treated with DMSO (Veh); 10, 30 and 50 µmol/l of ICT and BIC in the presence of DHT (10 nmol/l DHT for LNCaP and 1 nmol/l DHT for CRPC) or in the absence of DHT (CSS) for 24h. AR, AR-V7, UBE2C and PSA (arrows) protein levels were determined by western blotting. β-actin was used as loading control. (B) LNCaP and CRPC cells were incubated with DMSO (Veh), ICT (30 µmol/l) alone or together with MG132 (10 μmol/l) with 50 µmol/l of cycloheximide for indicated durations. AR and AR-V7 proteins were measured by western blotting and quantified by gel-pro analyzer 4.0 and normalized against β-actin. Data shown are mean ± SEM of three independent experiments. The percentage of AR or AR-V7 levels relative to initial level (0h) was plotted over time for estimation of protein half-life (T1/2). (C) PCa cells were treated with DMSO (Veh) and ICT (10, 30 and 50 µmol/l) for 24h. Quantitative RT–PCR (reverse transcription–polymerase chain reaction) analysis of AR and AR-V7 were performed in PCa cells and normalized to 18S rRNA (n = 3, mean ± SEM). mRNA levels of AR and AR-V7 in PCa cells are expressed as a percentage relative to vehicle control. Veh, vehicle.
To further elucidate the mechanistic basis of action of ICT on AR protein stability, a pulse chase experiment was performed. In comparison with vehicle-treated PCa cells, ICT markedly accelerated the rate of AR decay, thereby reducing AR half-lives by more than 50% in all three PCa cells (Figure 1B; Supplementary Figure S2, available at Carcinogenesis Online). Interestingly, ICT-mediated degradation of ARvs was significantly faster than that of full-length AR. Moreover, MG132 inhibited ICT-mediated degradation of AR and ARvs strongly suggesting that proteasomal degradation plays a pivotal role. We then assessed if the changes in AR and ARvs protein levels could be attributed to changes at their transcriptional level with ICT treatment. However, no significant changes in AR and AR-V7 mRNA levels were observed (Figure 1C).
ICT promotes AR and ARvs protein degradation through AhR-mediated ubiquitin proteasome pathway
The ubiquitin-proteasomal pathway has been shown to mediate AR degradation (26,27), with Mdm2, an E3 ubiquitin ligase (28). Thus first, we evaluated if ICT can enhance AR–Mdm2 complex formation using Co-IP. However, we did not observe any significant changes in AR–Mdm2 complex with ICT treatment in LNCaP and CWR22Rv1 cells (Supplementary Figure S3, available at Carcinogenesis Online), suggesting that Mdm2 is unlikely to play an important role in the ICT-mediated AR protein degradation.
It has also been shown that ligand-activated AhR can serve as an E3 ubiquitin ligase, leading to AR degradation (17). Moreover, our group has previously shown that ICT can bind to AhR via a competitive binding assay (16). Thus, we examined if ICT can also modulate the gene expression CYP1A1, a key downstream target of AhR in PCa cells. Both ICT and 3MC (a putative AhR ligand) upregulated the gene expression of CYP1A1, a key downstream target of AhR, in LNCaP and CWR22Rv1 cells (Figure 2A), suggesting that ICT serves as a ligand for AhR in PCa cells. Furthermore, treatment with ICT and 3MC remarkably enhanced the association between full-length AR (possibly ARvs) and AhR proteins in LNCaP and CWR22Rv1 cells (Figure 2B, left and middle panels). To further clarify the association between ARvs and ICT-activated AhR, siRNA targeting AR exon 7 was used to specially abolish the expression of full-length AR in CWR22Rv1 cells (Supplementary Figure S4A, available at Carcinogenesis Online). As expected, ARvs interacted with AhR proteins following ICT treatment (Figure 2B, right panel). These findings indicated that both AR and ARvs degradation is possibly mediated by ICT-activated AhR. To corroborate our findings, the effects of ICT treatment on AR and ARvs (AR-V7) expression were studied following AhR gene silencing (Supplementary Figure S4B, available at Carcinogenesis Online). The dose-dependent destabilization of AR in LNCaP and CWR22Rv1 cells and ARvs (AR-V7) in CWR22Rv1 cells by ICT treatment could be abrogated by AhR gene knockdown (Figure 2C). These findings clearly suggested an essential role for ICT-activated AhR in the degradation of AR and ARvs in PCa cells. Additionally, an in vitro ubiquitination analysis demonstrated that ICT, like 3MC, remarkably enhanced the poly-ubiquitination of AR and ARvs in ICT-treated CWR22Rv1 cells as compared with the vehicle-treated cells (Figure 2D). This finding suggested that the enhanced ubiquitination of AR and ARvs after ICT treatment may be one of the key mechanisms to promote their degradation in PCa cells.
Figure. 2.
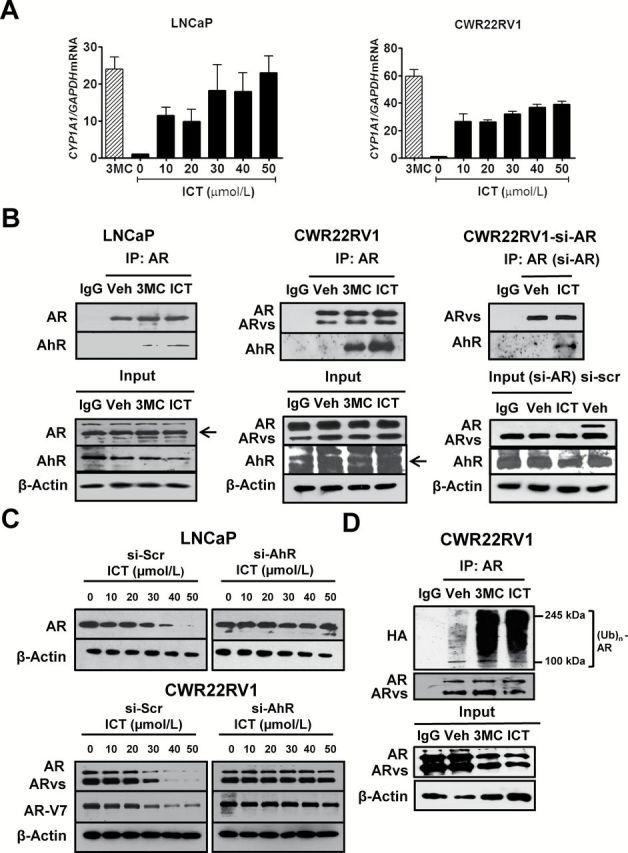
ICT promotes AR and AR splice variant protein degradation through AhR-mediated ubiquitin proteasome pathway. (A) LNCaP and CWR22Rv1 cells were treated with DMSO (Veh), 3MC (5 μmol/l) and ICT for 24h. CYP1A1 mRNA levels were analyzed using quantitative RT–PCR and normalized against 18S rRNA. Data shown are mean ± SEM of three independent experiments. (B) Co-IP of AhR with AR or ARvs. CWR22Rv1 cells were transiently transfected with siRNA targeted to AR exon 7 (si-AR) or si-Scr for 72h followed by drug treatments. Untransfected LNCaP and CWR22Rv1 and transfected CWR22Rv1 cells were treated with either DMSO (Veh), ICT (30 μmol/l) and/or 3MC (5 μmol/l) for 2h. AR- or ARvs-bound proteins were immunoprecipitated from cleared lysates using AR (co-targeting AR full-length and splice variants) or IgG antibody. The presence of AR, AR-V7 and AhR proteins (arrows) were identified using specific antibodies. (C) LNCaP and CWR22Rv1 cells were transiently transfected with siRNA against AhR (si-AhR) or si-Scr for 72h followed by 24h treatment with ICT. Whole cell lysates were then analysed by western blot for AR and AR-V7. β-actin was used as a loading control. (D) Ubiquitination of AR protein. CWR22Rv1 cells were transiently transfected with pCDNA3-HA-Ub for 48h, followed by 24h treatment with either DMSO (Veh), 3MC (5 μmol/l) or ICT (30 μmol/l). Co-IP and immunoblotting were performed, as described for (B), using antibodies against AR and HA tag.
ICT interrupts AR and ARvs signaling pathway via protein degradation
The degradation of AR full-length and splice variants with ICT treatment might also lead to the interruption of the androgen/AR signaling pathway in human PCa cells. In addition to the global protein degradation (Figure 1A and Supplementary Figure S1A, available at Carcinogenesis Online), the subcellular fractionation study also confirmed that ICT treatments markedly lowered nuclear AR and ARvs protein levels in LNCaP and CWR22Rv1 cells (Supplementary Figure S5, available at Carcinogenesis Online).
We next investigated if ICT can interfere with the binding of AR to the androgen response element (ARE) of AR-regulated genes. As expected, 24h treatment with ICT demonstrated suppressive effects on DHT-stimulated ARE luciferase reporter gene activity in both HeLa cells stably transfected with AR and ARE-Luc reporter genes and LNCaP cells transiently transfected with ARE-Luc reporter genes (Figure 3A). Consistently, chromatin immunoprecipitation experiments also showed that ICT treatments disrupted DHT-mediated AR recruitment to the enhancers of AR-regulated genes, KLK3, KLK2, TMPRSS2 and FKBP5 (Figure 3B and Supplementary Figure S6, available at Carcinogenesis Online) in LNCaP and CWR22Rv1 cells.
Figure. 3.
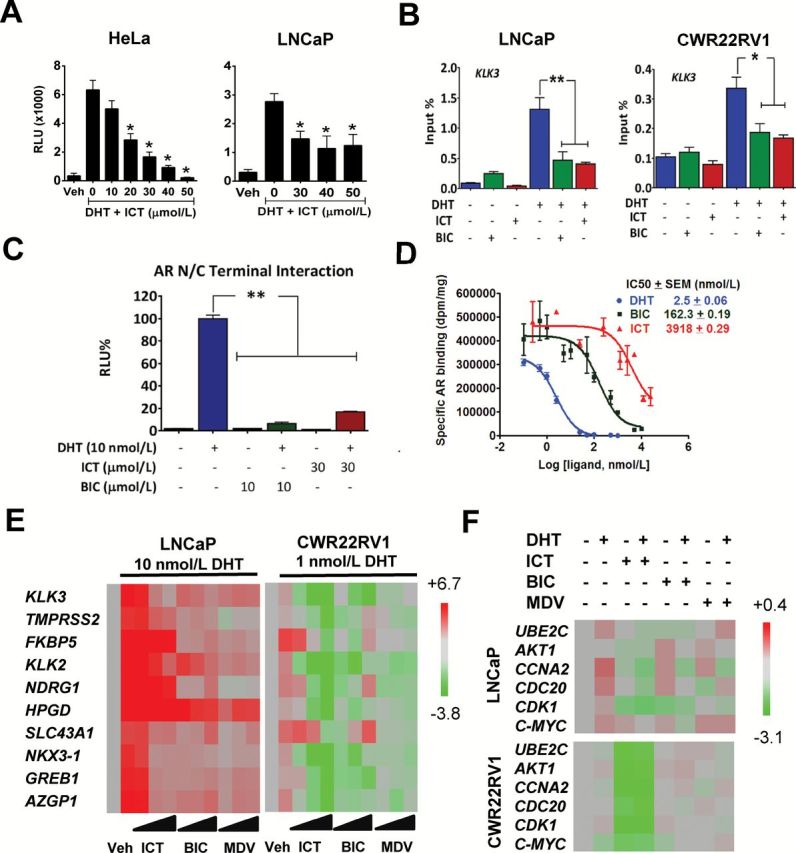
Effect of ICT on AR- or ARvs-signaling pathway. (A) AR transcriptional activity on consensus ARE was assessed in HeLa and LNCaP cells transfected with AR and ARE-Luc reporter genes. Luciferase activity was measured after exposure to either DMSO (Veh) or ICT with or without 10 nmol/l of DHT for 24h. (B) Chromatin immunoprecipitation of AR binding to the ARE of KLK3 gene. LNCaP and CWR22Rv1 cells were treated with 30 μmol/l of ICT or 10 μmol/l BIC alone or in combinations in the presence or absence of DHT for 2h. DNA fragments bound to AR were immunoprecipitated and analysed by the quantitative PCR. (C) AR NH2- and COOH-terminal (N–C) interactions were measured by a mammalian two-hybrid assay in CV-1 cells following exposure to indicated compounds for 24h. (D) Representative competition binding curves showing inhibition of 3H-DHT equilibrium binding to AR with increasing doses of unlabeled DHT, BIC and ICT in HeLa cells stably expressing AR. The IC50 values were determined using a one-site model using Graphpad Prism. (E) Full-length AR-regulated gene expression in LNCaP and CWR22Rv1 cells was assessed by quantitative RT–PCR following exposure to DMSO (Veh), 10, 30 and 50 µmol/l of ICT, BIC or MDV with or without DHT for 24h. (F) ARvs-regulated gene expression in LNCaP and CWR22Rv1 was assessed by quantitative RT–PCR following exposure to DMSO (Veh), 30 µmol/l of ICT, 10 µmol/l of BIC or MDV with or without 1 nmol/l DHT for 24h. Heatmaps show relative gene expression normalized to 18S rRNA. All data shown are mean ± SEM of at least three independent experiments. *P < 0.05; **P < 0.01.
To further elucidate the effects of ICT on AR transcriptional activity (29), we examined AR amino- and carboxyl-terminal (N–C) interaction following ICT treatment, via a mammalian two hybrid assay in CV1 cells. ICT displayed a substantial disruption of the DHT-stimulated AR N–C interaction 24h post-treatment, albeit weaker than BIC (Figure 3C). This led us to question if ICT may impair AR N–C interaction by directly binding to AR protein. To that end, a whole cell binding assay using HeLa cells stably transfected with AR was used to study the binding affinities. ICT competitively displaced [H3]-DHT with an IC50 of 3.9 µmol/l, a binding affinity which was approximately 24-fold weaker than that of BIC (0.16 µmol/l, Figure 3D), suggesting that ICT is less likely to regulate AR transcriptional activity via direct AR binding.
ICT exerted an inferior suppression than MDV on the transcript level of known AR-regulated genes including KLK3 and TMPRSS2 in ARvs-null LNCaP and C4-2 cells (Figure 3E and Supplementary Figure S7, available at Carcinogenesis Online), especially in a high concentration of DHT (10 nmol/l). Interestingly, ICT robustly antagonized DHT-mediated expression of those genes to a greater extent than MDV in ARvs-expressing CWR22Rv1 cells, possibly because that ARvs can modulate some AR-regulated genes (9). Thus, we next investigated the effects of ICT on ARvs-mediated transcription in CWR22Rv1 cells compared with LNCaP cells. As expected, ICT did not elicit any effects on the expression of the ARvs-regulated genes such as UBE2C in LNCaP cells (Figure 3F). However ICT, unlike BIC and MDV, significantly reduced the expression of the ARvs-regulated genes in CWR22Rv1 cells. Taken together, these findings clearly demonstrate that ICT can markedly suppress AR and ARvs transcriptional activity possibly through protein degradation, especially in ARvs-expressing human PCa cells.
ICT can inhibit growth and induce apoptosis in human AR-positive PCa cells via AhR-mediated AR destabilization
Given that ICT can destabilize AR and disrupt AR transcriptional signaling, we next investigated if ICT can suppress the growth of AR-positive PCa cells. ICT had no effect on the growth of the non-tumorigenic human prostate epithelial cells (RWPE-1) in the presence of DHT, indicating minimal adverse effect of ICT on the normal prostate epithelial cells (Figure 4A). However, in both androgen-rich and CSS conditions, in comparison with BIC and MDV, ICT (≥ 30 µmol/l) markedly reduced cell proliferation by around 50% in LNCaP cells, and by more than 70% in C4-2 and CWR22Rv1 cells (Figure 4A). In contrast, under androgen-replete and androgen-deplete milieu, both BIC and MDV slightly or barely suppressed CWR22Rv1 cell growth, which was consistent with the reported resistance to BIC or MDV in ARvs-expressing CWR22Rv1 cells (13).
Figure. 4.
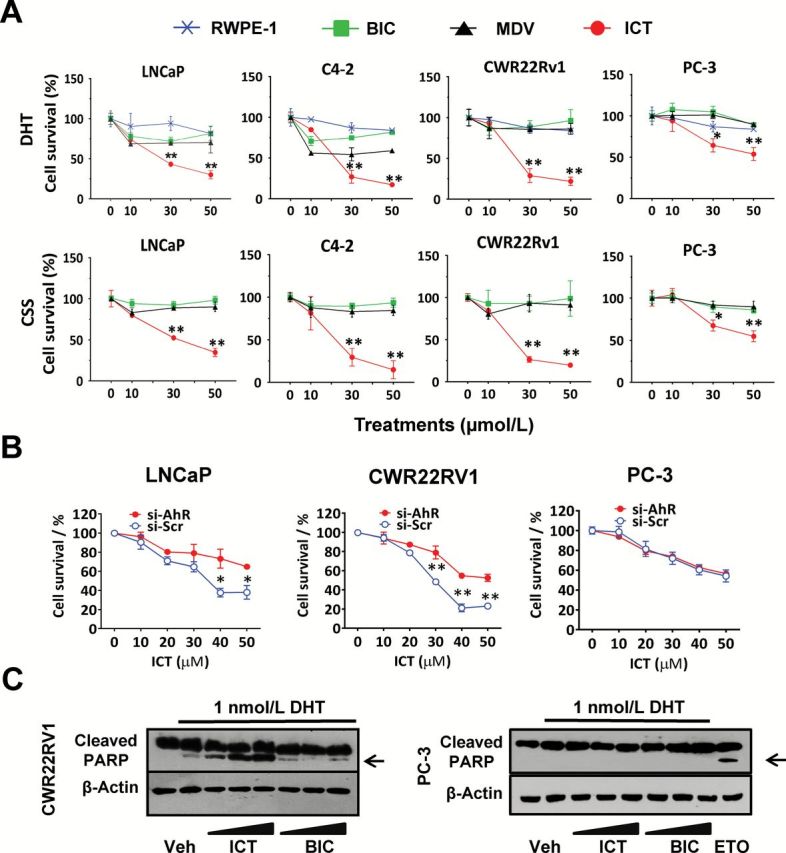
ICT inhibits growth and induces apoptosis in human AR-positive PCa cells largely via AhR-mediated AR destabilization. (A) PCa cells were treated with DMSO (Veh); 10, 30 and 50 µmol/l of ICT; BIC or MDV, in the presence of DHT (10 nmol/l DHT for LNCaP and 1 nmol/l DHT for CWR22Rv1, C4-2 and PC-3) or the absence of DHT (CSS) for 48h. Normal prostate epithelial cells (RWPE-1) were exposed to ICT in the presence of DHT for 48h. Cell survival was assessed by the MTS cell proliferation assay and expressed as a percentage of DMSO (Veh) or Veh plus DHT. All data shown are mean ± SEM of at least three independent experiments. *P < 0.05, **P < 0.01, ICT-treated compared to BIC- and MDV-treated PCa cells. (B) LNCaP, CWR22Rv1 and PC-3 cells were transiently transfected with siRNA against AhR (si-AhR) or si-Scr for 72h followed by 24h treatment with ICT. Cell survival was measured, as described in (A). The cell survival rates were expressed as percentage of si-AhR-treated and si-Scr-treated control (without ICT) for the cells treated with 10–50 µmol/l ICT. Data shown are mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01. (C) CWR22Rv1 and PC-3 cells from above treatments were analysed by western blot for PARP cleavage fragments (arrows). Etoposide (ETO), 100 µmol/l, served as a positive control in treated PC-3 cells. β-actin was used as a loading control.
Interestingly despite being weaker than BIC and MDV in suppressing AR transcriptional activity (Figure 3E and Supplementary Figure S7, available at Carcinogenesis Online), ICT demonstrated a more potent growth inhibitory effect than the two drugs in AR-positive LNCaP and C4-2 cells (Figure 4A). Therefore, the antiproliferative effects of ICT on AR-positive PCa cells shown in this study may not exclude the contributions of AR-independent cell death pathways. Indeed, in both androgen-rich and CSS conditions, 30 µmol/l of ICT diminished cell proliferation by around 35% in AR-null PC-3 cells (Figure 4A), which is in agreement with previous findings (19). Notably, AhR gene silencing rescued LNCaP and CWR22Rv1 cells from ICT-mediated cell death by 20% to 30% (relative to cells treated with a si-Scr sequence) while exerting negligible effects on AR-null PC-3 cells (Figure 4B). This finding strongly suggests that ICT-mediated cell death is largely related to AhR-dependent AR degradation.
Treatment of AR-positive PCa cells with 30 and 50 µmol/l of ICT, noticeably induced PARP cleavage, the apoptosis indicator, whereas BIC in PCa cells and ICT in PC-3 cells did not (Figure 4C and Supplementary Figure S8A, available at Carcinogenesis Online), which was corroborated by previous published report that showed ICT causes cell cycle arrest with no apparent apoptosis in PC-3 cells (19). Consistently, flow cytometric analysis demonstrated a dose-dependent increase in the hypodiploid sub-G1 phase indicative of DNA fragmentation, and possibly apoptosis following ICT treatment in both LNCaP and C4-2 but not in PC-3 cells (Supplementary Figure S8B, available at Carcinogenesis Online). These results imply that the ICT-mediated suppression of AR-positive PCa cells also goes through an AR-dependent apoptosis pathway compared with AR-null PCa cells.
Put together, our data indicate that ICT-mediated inhibition of AR-positive PCa cell proliferation is mediated, at least in part, via AhR-dependent ubiquitin-proteasomal degradation of AR and/or ARvs.
ICT inhibits growth of PC tumors in mice
We next investigated the effects of ICT in a physiologically relevant animal model (30). Androgen-sensitive LNCaP-luciferase and castration-resistant CWR22Rv1 cells were orthotopically implanted into the prostates of SCID mice. The progression of LNCaP tumor growth was studied in the presence or absence of ICT. Bioluminescent imaging (BLI) indicates that intraperitoneal administration of 33mg/kg ICT strongly suppressed the growth of LNCaP tumors from week 4 of treatment relative to controls (P < 0.05; Figure 5A). After 10 weeks of treatment, there was a significant increase of tumor BLI signal by 4-fold in the control group compared with the ICT group.
Figure. 5.
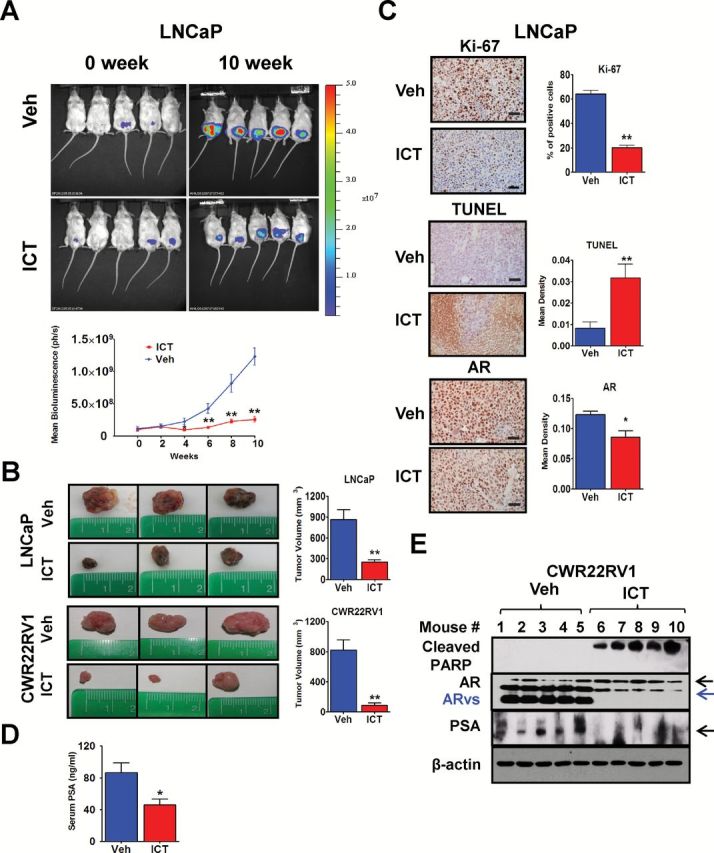
In vivo antitumor effect of ICT is associated with AR and/or ARvs protein content and signaling. (A) BLI in vivo of five representative male mice harboring LNCaP-luc tumors at each group after 0 and 10 weeks treatment (top panel). Mice (n = 12 per group) were treated with intraperitoneal injection of vehicle control or ICT at 33mg/kg 5 times per week for 10 weeks. Change in tumor volume was measured bi-weekly as the bioluminescence in photons /second (ph/s) (bottom panel). (B) Representative images of the excised tumor from three mice-bearing LNCaP or CWR22Rv1 xenografts in vehicle- and ICT-treated groups. Tumor volumes are plotted in the right panel (n = 12 for LNCaP and n = 10 for CWR22Rv1, mean ± SEM). (C) Representative IHC staining of Ki-67, TUNEL and AR proteins in primary LNCaP tumors from vehicle- or ICT-treated mice after 10 weeks of treatment. Scale bars are 50 µm. Percentage of Ki-67 positive cells and densities of TUNEL and AR staining were quantified using Image-Pro Plus software, each with 5 randomly chosen ×400 fields (n = 8, mean ± SEM). (D) Serum PSA levels were analysed by ELISA in SCID mice-bearing LNCaP tumors after 10 weeks of vehicle or 33mg/kg ICT treatment (n = 12, mean ± SEM). (E) Representative primary CWR22Rv1 tumors from five vehicle- or ICT-treated mice after 5 weeks of treatment were western blotted for cleaved PARP, AR, AR-V7, PSA and β-actin. *P < 0.05, **P < 0.01. TUNEL, Terminal Deoxynucleotide Transferase dUTP Nick End Labeling.
Consistently after 10 weeks of ICT administration, LNCaP tumors harvested from mice of the ICT group were significantly smaller than those from control mice (250 versus 861mm3, P < 0.01; Figure 5B). This result was also unequivocally observed in the CWR22Rv1 xenograft mouse model. Furthermore, there was a strong correlation between tumor volumes and endpoint BLI signal in LNCaP tumor-bearing mice (R 2 = 0.732; Supplementary Figure S9, available at Carcinogenesis Online). There was a relief of tumor burden by ICT treatment, reduced weight loss in two PCa xenograft mouse models (P < 0.05; Supplementary Figure S10, available at Carcinogenesis Online), and improved survival rate of LNCaP tumor-bearing mice at the point of sacrifice (100% in ICT group versus 58% in vehicle group).
IHC staining on excised LNCaP tumors revealed that ICT significantly suppressed proliferation of LNCaP xenografts compared with vehicle control (Ki-67 positive cells: 20.3% versus 64.3%, P < 0.01; Figure 5C). Consistently, ICT treatment significantly increased the mean density of Terminal Deoxynucleotide Transferase dUTP Nick End Labeling staining compared with vehicle control (0.032 versus 0.008, P < 0.01; Figure 5C), indicative of increased DNA fragmentation, hence apoptosis in these sections. The western blot analysis demonstrated that ICT induced more PARP cleavage compared with vehicle control in CWR22Rv1 xenograft tissues (Figure 5E). Overall, these findings indicated that ICT can apoptosis dependently reduce tumor growth in both androgen-sensitive and castration-resistant models of PCa in vivo.
In vivo antitumor effect of ICT is partially associated with AR and ARvs protein content and signaling
ELISA assay showed that serum PSA levels in mice-bearing LNCaP xenografts treated with ICT were lower compared with those administered vehicle (45.7 versus 86.5ng/ml, P < 0.05; Figure 5D). IHC indicate that AR protein content was lower in LNCaP tumors exposed to ICT (mean density: 0.086 for ICT versus 0.123 for vehicle, P < 0.05; Figure 5C). ICT also suppressed AR, AR-V7 and PSA protein in castration-resistant CWR22Rv1 tumors (Figure 5E). These results further reiterate our in vitro findings that the growth inhibitory effects of ICT on PCa tumor growth could be partially mediated by targeting the AR and/or ARvs.
ICT has favorable pharmacokinetics and safety profiles
Due to the rapid conversion to the glucuronidated and/or sulfated metabolites in vivo (31), ICT exhibits high systemic clearance, and short serum half-life in mice (Figure 6A). Maximal ICT concentrations in serum were lower than the effective concentrations (≥ 30 µM) associated with antiproliferative effects in our in-vitro studies. Thus, we investigated if ICT could achieve higher concentrations in the tumor tissues, necessary to drive the observed in-vivo responses. Comparative analysis of the ICT concentrations in serum and tumor tissues 6–9h after final dosing revealed that the tissue-to-plasma partition coefficient of ICT was 89 (ICT concentration: 41.8±19.7 µM in tumor, n = 6 versus 471±175nM in serum, n = 4). This result clearly demonstrates a preferential enrichment of ICT in the prostate tumor tissues and explains the significant inhibition in LNCaP cell growth in vivo despite the low serum levels.
Figure. 6.

ICT has favorable pharmacokinetics and safety profiles. (A) The serum concentrations of ICT in SCID mice-bearing LNCaP tumors were quantified after intraperitoneal administration of ICT at doses of 33mg/kg. The mean serum concentrations of ICT (nmol/l) are expressed as mean ± SD (n = 2). The lower dotted line indicates the lower limit of quantitation (20 nmol/l) of ICT in the mouse serum. The upper dotted line indicates 30 µmol/l ICT, the effective concentrations associated with anti-proliferative effects. The terminal serum half-life (T 1/2, z), the serum clearance (CL) and the tissue-to-plasma partition coefficient of ICT were assessed using non-compartmental analysis with WinNonLin 6.2.1. (B) Mean body weight-time profiles were computed following treatment of intact SCID mice with intraperitoneally injected physiological saline (n = 5) or 33mg/kg ICT (n = 10) 5 times per week for 10 weeks. Bars represents mean ± SEM. (C) The figures show hematoxylin and eosin staining of representative sections (×200) of heart, liver, spleen, lung and kidney harvested at the end of the experiment from mice receiving intraperitoneal delivery of vehicle control (top) or 33mg/kg ICT (bottom). Scale bars represent 100 µm in all micrographs.
After 10 weeks of treatment, histopathology, clinical chemistry, hematological evaluation and body weight revealed minimal toxicological effects in healthy tumor-free SCID mice treated with ICT formulation over 10 weeks (Figure 6B and C; Supplementary Table S1, available at Carcinogenesis Online).
Discussion
Our data provide evidence that ICT exerted potent antiproliferative and proapoptotic effects on androgen-sensitive and castration-resistant PCa models both in vitro and in vivo. ICT effectively suppressed AR- and/or ARvs-regulated gene transcription through acceleration of the AhR-mediated proteasomal degradation of proteins in AR-positive PCa cells. More importantly, ICT demonstrated a potent inhibition of ARvs-regulated genes or proliferation in the CWR22Rv1 cells compared with both BIC and MDV (Figure 3F and 4A) highlighting its immense therapeutic potential in ARvs overexpressing PCs which often show the MDV resistance.
At present, there are four documented small molecule inhibitors, namely, EPI-001, ASC-J9, mahanine and niclosamide, which have also demonstrated this unique capacity to target ARvs (24,32–35). EPI-001 and its analogs target the AR N-terminal domain by covalently binding to it. Similar to ICT, ASC-J9 and mahanine accelerate the proteasomal degradation of both AR and ARvs, albeit through different mechanisms. ASC-J9 augments AR degradation by enhancing the association of AR–Mdm2 complex (24), whereas as a ligand, ICT works uniquely via activating AhR (Figure 2B). Noticeably, AhR gene silencing could partially restore tumor growth (Figure 4B) suggesting that the proteasomal degradation of AR and ARvs via ICT-activated AhR pathway plays a vital role in the growth inhibition of AR-positive PCa cells.
In this study, the antiproliferative effects of ICT on AR-positive PCa cells also included AR-independent cell death pathways (Figure 4A and B). Interestingly, the ICT-mediated suppression of the growth of PC-3 cells were independent of apoptosis pathway (Figure 4C and Supplementary Figure S8B, available at Carcinogenesis Online), which was also observed in a previous study (19). In contrast, ICT induced an apoptosis-dependent cell death in the AR-positive PCa cells (Figure 4C and Supplementary Figure S8A and B, available at Carcinogenesis Online). These findings suggest a fundamental difference in the mechanisms of ICT-induced antiproliferation between AR positive and negative PCa cells. Similarly, Liao et al. (36) showed that siRNA-induced AR silencing led to apoptotic death in AR-positive PCa cells. In fact, accumulating evidences indicate that there are multiple mechanisms by which AR protects cells from apoptosis, including but not limited to p21, p53 and mitogen-activated protein kinases pathways (37–39). Elucidating which pathway is responsible for apoptotic cell death induced by ICT-stimulated AR proteasomal degradation warrants further study.
The ability of ICT to target different type of PCa cells is important as PCa is composed of a mixture of cells (basal, intermediate and luminal) (40). The current ADT has been shown to diminish the majority of CK5−/CK8+ luminal epithelial cells (e.g. LNCaP and CWR22Rv1), while boosting or un-changing CK5+/CK8+ basal epithelial and intermediate cells (e.g. PC-3) (40). As such, cellular heterogeneity of PCa offers a critical explanation for why current ADT would eventually fail. Thus, the ability of ICT to inhibit a wider variety of PCa cells confers ICT a significant advantage over currently available ADT.
While our findings underscore the potential of specific AhR ligands such as ICT in targeting AR, this therapeutic benefit cannot be arbitrarily conferred to other AhR agonists. For example, the putative AhR agonist 2, 3, 7,8-tetrachlorodibenzo-p-dioxin has been shown to suppress PCa proliferation in vitro (41,42). However, in a population study on Vietnam warfare veterans exposed to TCDD-contaminated Agent Orange, an increased rate of PC incidence and malignancy was observed (43). In this light, it is noteworthy to mention that our 10-week intervention study demonstrated that ICT was well-tolerated in healthy animals (Figure 6B and C, and Supplementary Table S1, available at Carcinogenesis Online). More importantly, a phase II clinical study on ICT for advanced hepatocellular carcinoma [NCT01972672] is underway following the successful completion of phase I study. This study showed that ICT has satisfactory safety and tolerance on subjects and high bioavailability after subjects orally received 600mg of ICT.
In conclusion, we have shown mechanistic evidence to demonstrate that ICT can effectively promote the proteasomal degradation of both AR and ARvs, effectively target the AR transcriptional regulatory system, and consequently inhibit AR-positive PC growth. In addition, ICT has demonstrated a satisfactory long-term safety profiles. Together, this work provides the platform and preclinical evidence for the development of ICT, alone or in combination with other PCa drugs, to target key pathways of prostate tumorigenesis.
Supplementary material
Supplementary Table 1, Figures 1–10 and Supplementary Materials and methods can be found at http://carcin.oxfordjournals.org/
Funding
Singapore National Medical Research Council(R-174-000-137-275 to E.-L.Y.); the Jay and Betty Van Andel Foundation80801 to (H.E.X.); National Institutes of Health(5R01DK071662-08 to H.E.X.).
Supplementary Material
Acknowledgements
We are grateful to Professor C.S. Chang from University of Rochester and Dr Lei Li for their valuable suggestions in the experimental designs.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- 3MC
3-methylcholanthrene
- ADT
androgen deprivation therapy
- AhR
arylhydrocarbon receptor
- AR
androgen receptor
- ARE
androgen response element
- ARvs
AR COOH-terminal truncated variants
- BIC
bicalutamide
- BLI
bioluminescent imaging
- Co-IP
co-immunoprecipitation
- CSS
charcoal stripped serum
- CRPC
castration-resistant prostate cancer
- DHT
dihydrotestosterone
- DMSO
dimethyl sulfoxide
- ERα
estrogen receptor alpha
- ICT
icaritin
- IHC
immunohistochemistry
- MDV
enzalutamide
- PARP
poly-ADP-ribose polymerase
- PCa
metastatic prostate cancer
- PSA
prostate-specific antigen
- RLUs
relative luminescence units
- RT–PCR
reverse transcription–polymerase chain reaction
- SEM
standard error of the mean
- SCID
severe combined immunodeficiency
- si-Scr
scrambled siRNA
References
- 1. Huggins C., et al. (1972) Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA. Cancer J. Clin., 22, 232–240. [DOI] [PubMed] [Google Scholar]
- 2. Harris W.P., et al. (2009) Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat. Clin. Pract. Urol., 6, 76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dason S., et al. (2013) Defining a new testosterone threshold for medical castration: Results from a prospective cohort series. Can. Urol. Assoc. J., 7, E263–E267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Montgomery R.B., et al. (2008) Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res., 68, 4447–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen C.D., et al. (2004) Molecular determinants of resistance to antiandrogen therapy. Nat. Med., 10, 33–39. [DOI] [PubMed] [Google Scholar]
- 6. Taplin M.E., et al. (1995) Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N. Engl. J. Med., 332, 1393–1398. [DOI] [PubMed] [Google Scholar]
- 7. Xu J., et al. (2009) Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat. Rev. Cancer, 9, 615–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dehm S.M., et al. (2008) Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res., 68, 5469–5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hu R., et al. (2009) Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res., 69, 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Scher H.I., et al. AFFIRM Investigators. (2012) Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med., 367, 1187–1197. [DOI] [PubMed] [Google Scholar]
- 11. Dehm S.M., et al. (2011) Alternatively spliced androgen receptor variants. Endocr. Relat. Cancer, 18, R183–R196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Antonarakis E.S., et al. (2014) AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med., 371, 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li Y., et al. (2013) Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res., 73, 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu R., et al. (2012) Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res., 72, 3457–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. (2010) Chinese materia medica and prepared slices of Chinese crude Drugs. In Wang Z.Tet al. (eds.), Pharmacopoeia of the People’s Republic of China. vol. 1 China Medical Science Press, Beijing, China, pp. 169–170. [Google Scholar]
- 16. Tiong C.T., et al. (2012) A novel prenylflavone restricts breast cancer cell growth through AhR-mediated destabilization of ERα protein. Carcinogenesis, 33, 1089–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ohtake F., et al. (2007) Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature, 446, 562–566. [DOI] [PubMed] [Google Scholar]
- 18. Clegg N.J., et al. (2012) ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res., 72, 1494–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang X., et al. (2007) A novel anticancer agent, icaritin, induced cell growth inhibition, G1 arrest and mitochondrial transmembrane potential drop in human prostate carcinoma PC-3 cells. Eur. J. Pharmacol., 564, 26–36. [DOI] [PubMed] [Google Scholar]
- 20. Wong S.P., et al. (2007) Ultrasensitive cell-based bioassay for the measurement of global estrogenic activity of flavonoid mixtures revealing additive, restrictive, and enhanced actions in binary and higher order combinations. Assay Drug Dev. Technol., 5, 355–362. [DOI] [PubMed] [Google Scholar]
- 21. Loy C.J., et al. (2003) Filamin-A fragment localizes to the nucleus to regulate androgen receptor and coactivator functions. Proc. Natl. Acad. Sci. U. S. A., 100, 4562–4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chng K.R., et al. (2012) A transcriptional repressor co-regulatory network governing androgen response in prostate cancers. EMBO J., 31, 2810–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stephenson R.A., et al. (1992) Metastatic model for human prostate cancer using orthotopic implantation in nude mice. J. Natl. Cancer Inst., 84, 951–957. [DOI] [PubMed] [Google Scholar]
- 24. Lai K.P., et al. (2013) New therapeutic approach to suppress castration-resistant prostate cancer using ASC-J9 via targeting androgen receptor in selective prostate cells. Am. J. Pathol., 182, 460–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tran C., et al. (2009) Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science, 324, 787–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jaworski T. (2006) Degradation and beyond: control of androgen receptor activity by the proteasome system. Cell. Mol. Biol. Lett., 11, 109–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sheflin L., et al. (2000) Inhibiting proteasomes in human HepG2 and LNCaP cells increases endogenous androgen receptor levels. Biochem. Biophys. Res. Commun., 276, 144–150. [DOI] [PubMed] [Google Scholar]
- 28. Lin H.K., et al. (2002) Phosphorylation-dependent ubiquitylation and degradation of androgen receptor by Akt require Mdm2 E3 ligase. EMBO J., 21, 4037–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hsu C.L., et al. (2005) Androgen receptor (AR) NH2- and COOH-terminal interactions result in the differential influences on the AR-mediated transactivation and cell growth. Mol. Endocrinol., 19, 350–361. [DOI] [PubMed] [Google Scholar]
- 30. Sato N., et al. (1997) A metastatic and androgen-sensitive human prostate cancer model using intraprostatic inoculation of LNCaP cells in SCID mice. Cancer Res., 57, 1584–1589. [PubMed] [Google Scholar]
- 31. Wong S.P., et al. (2009) Pharmacokinetics of prenylflavonoids and correlations with the dynamics of estrogen action in sera following ingestion of a standardized Epimedium extract. J. Pharm. Biomed. Anal., 50, 216–223. [DOI] [PubMed] [Google Scholar]
- 32. Andersen R.J., et al. (2010) Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell, 17, 535–546. [DOI] [PubMed] [Google Scholar]
- 33. Amin K.S., et al. (2014) A naturally derived small molecule disrupts ligand-dependent and ligand-independent androgen receptor signaling in human prostate cancer cells. Mol. Cancer Ther., 13, 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Myung J.K., et al. (2013) An androgen receptor N-terminal domain antagonist for treating prostate cancer. J. Clin. Invest., 123, 2948–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu C., et al. (2014) Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin. Cancer Res., 20, 3198–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liao X., et al. (2005) Small-interfering RNA-induced androgen receptor silencing leads to apoptotic cell death in prostate cancer. Mol. Cancer Ther., 4, 505–515. [DOI] [PubMed] [Google Scholar]
- 37. Lu S., et al. (1999) Androgen regulation of the cyclin-dependent kinase inhibitor p21 gene through an androgen response element in the proximal promoter. Mol. Endocrinol., 13, 376–384. [DOI] [PubMed] [Google Scholar]
- 38. Rokhlin O.W., et al. (2005) Androgen regulates apoptosis induced by TNFR family ligands via multiple signaling pathways in LNCaP. Oncogene, 24, 6773–6784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nguyen T.V., et al. (2005) Androgens activate mitogen-activated protein kinase signaling: role in neuroprotection. J. Neurochem., 94, 1639–1651. [DOI] [PubMed] [Google Scholar]
- 40. Wen S., et al. (2014) Androgen receptor (AR) positive vs negative roles in prostate cancer cell deaths including apoptosis, anoikis, entosis, necrosis and autophagic cell death. Cancer Treat. Rev., 40, 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Morrow D., et al. (2004) Aryl hydrocarbon receptor-mediated inhibition of LNCaP prostate cancer cell growth and hormone-induced transactivation. J. Steroid Biochem. Mol. Biol., 88, 27–36. [DOI] [PubMed] [Google Scholar]
- 42. Jana N.R., et al. (1999) Cross-talk between 2,3,7,8-tetrachlorodibenzo-p-dioxin and testosterone signal transduction pathways in LNCaP prostate cancer cells. Biochem. Biophys. Res. Commun., 256, 462–468. [DOI] [PubMed] [Google Scholar]
- 43. Chamie K., et al. (2008) Agent Orange exposure, Vietnam War veterans, and the risk of prostate cancer. Cancer, 113, 2464–2470. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.