

Abstract
Background
Ovarian cancer causes more than 15000 deaths per year in the United States. The survival of patients is quite heterogeneous, and accurate prognostic tools would help with the clinical management of these patients.
Methods
We developed and validated two gene expression signatures, the first for predicting survival in advanced-stage, serous ovarian cancer and the second for predicting debulking status. We integrated 13 publicly available datasets totaling 1525 subjects. We trained prediction models using a meta-analysis variation on the compound covariable method, tested models by a “leave-one-dataset-out” procedure, and validated models in additional independent datasets. Selected genes from the debulking signature were validated by immunohistochemistry and quantitative reverse-transcription polymerase chain reaction (qRT-PCR) in two further independent cohorts of 179 and 78 patients, respectively. All statistical tests were two-sided.
Results
The survival signature stratified patients into high- and low-risk groups (hazard ratio = 2.19; 95% confidence interval [CI] = 1.84 to 2.61) statistically significantly better than the TCGA signature (P = .04). POSTN, CXCL14, FAP, NUAK1, PTCH1, and TGFBR2 were validated by qRT-PCR (P < .05) and POSTN, CXCL14, and phosphorylated Smad2/3 were validated by immunohistochemistry (P < .001) as independent predictors of debulking status. The sum of immunohistochemistry intensities for these three proteins provided a tool that classified 92.8% of samples correctly in high- and low-risk groups for suboptimal debulking (area under the curve = 0.89; 95% CI = 0.84 to 0.93).
Conclusions
Our survival signature provides the most accurate and validated prognostic model for early- and advanced-stage high-grade, serous ovarian cancer. The debulking signature accurately predicts the outcome of cytoreductive surgery, potentially allowing for stratification of patients for primary vs secondary cytoreduction.
Ovarian cancer is the most lethal gynecologic malignancy, causing more than 15000 deaths per year in the United States (1). Advanced ovarian cancer (stages III and IV) accounted for the majority of the estimated 22000 new cases of epithelial ovarian cancer in 2012 in the United States (1). Improvements in the treatment of patients with advanced-stage ovarian cancers have extended women’s median survival, but overall survival has not substantially changed. This results from effective upfront treatment of the disease but with subsequent recurrence and the development of drug-resistant disease.
The initial treatment for all patients with advanced-stage ovarian cancer is extensive debulking surgery, with the goal being removal of tumor to less than 1cm. Patients who are optimally debulked (<1cm residual disease) have a substantially improved survival compared with patients who are left with bulky residual disease (>1cm; suboptimally debulked). Suboptimally debulked patients do not benefit from this extensive surgery (2,3). After surgery, all patients undergo combination chemotherapy that is effective in approximately 75% of patients (4). Thus, developing tools to stratify patients according to ability to be cytoreduced or response to primary chemotherapy may help improve patient survival and minimize therapeutic toxicity. It is important to note, however, that most patients with advanced-stage ovarian cancer ultimately suffer recurrence and eventually develop drug-resistant tumors. Thus the development of new and novel therapies for this disease is absolutely critical.
Our laboratory has developed the largest collection of ovarian cancer gene expression data to date (5), allowing us to systematically evaluate a range of previously published prognostic signatures (6–16). We identified the signature developed by The Cancer Genome Atlas (TCGA) consortium as the best available prognostic model (17). The TCGA consortium recently proposed an updated survival signature, trained on twice as many samples and incorporating subtype information and clinicopathologic factors (18). However, it remains unclear whether prognostic accuracy can be improved with additional data.
Here, we use a meta-analytic signature development approach to leverage more than 1500 publicly available, clinically annotated microarray assays of high-grade, primary serous tumors to comprehensively address two objectives for patients with ovarian cancer. These are 1) to develop a prognostic gene signature for overall survival of early- and late-stage patients and 2) to predict suboptimal cytoreductive surgery. We performed extensive signature evaluation demonstrating significant improvement over existing survival signatures. Furthermore, this work establishes the existence of a signature predictive for suboptimal cytoreduction, providing the possibility that unsuccessful surgery can be avoided through a genomic or immunohistochemical test at diagnosis.
Methods
Dataset and Patient Eligibility Criteria
Gene expression data on tumors, with carefully curated clinical annotations, are available in the curatedOvarianData database (5). Criteria for inclusion of studies in this database, the corresponding literature reviews, and the data-processing protocols have been described previously (5). We restricted our meta-analysis to primary, late-stage, high-grade, serous tumors with available overall survival time-to-event data (Table 1). For prediction model training, we required a minimum study sample size of 75. Only datasets published before March 2012 were considered in the training phase. Datasets that did not satisfy these two criteria were set aside as independent validation data for the final model. This led to six training datasets and seven validation datasets. We also tested our final model on the 27 early-stage, high-grade samples from TCGA; these samples contained eight patients with events. Datasets were systematically screened for duplicated samples, as described previously (17).
Table 1.
Datasets used in this study*
| Dataset | Accession ID | Reference | Platform | Number of samples | Stage IV, % | Suboptimal debulked, % | Median survival, mo. | Median follow-up, mo. | Censoring, % | Median age | Time period |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bentink et al. 2012 | E-MTAB-386 | (6) | Illumina HumanRef-8 v2 | 127†,‡ | 15 | 22|| | 42 | 53 | 43 | 66 | 1999–2006 |
| Partheen et al. 2006 | GSE12418 | (19) | Custom | 54† | 0 | 76¶ | Short-term (<3 y) vs long-term survival (>7 y) information only | 60 | 1993–1999 | ||
| Crijns et al. 2009 | GSE13876 | (10) | Operon Human v3 | 157† | — | — | 25 | 72 | 28 | 60 | 1990–2003 |
| Yoshihara et al. 2010 | GSE17260 | (15) | Agilent G4112a | 84†,‡ | 17 | 54|| | 51 | 46 | 55 | — | — |
| Mok et al. 2009 | GSE18520 | (13) | Affymetrix U133 Plus 2.0 | 53† | 0 | — | 25 | 140 | 23 | — | 1990– 2000 |
| Konstantinopoulos et al. 2010 | GSE19829 | (30) | Affymetrix U95 v2 | 42† | — | — | 45 | 50 | 45 | — | — |
| Bonome et al. 2008 | GSE26712 | (8) | Affymetrix U133a | 185† | 19 | 51|| | 46 | 90 | 30 | 63 | 1990– 2003 |
| Gillet et al. 2012 | GSE30009 | (28) | TaqManqRT-PCR 380 | 93† | 18 | 22|| | 41 | 53 | 45 | 61 | 1995–2007 |
| Yoshihara et al. 2012 | GSE32062 | (16) | Agilent G4112a | 91†,‡ | 32 | 69|| | 104 | 56 | 56 | — | 1997–2010 |
| Tothill et al. 2008 | GSE9891 | (63) | Affymetrix U133 Plus 2.0 | 140† | 8 | 37|| | 40 | 40 | 49 | 59 | 1992–2006 |
| Dressman et al. 2007# | — | (29) | Affymetrix U133a | 59† | 15 | 44|| | 42 | 94 | 39 | — | — |
| TCGA 2011 | — | (9) | Affymetrix HT U133a | 413†,‡ | 16 | 72¶ | 41 | 54 | 44 | 59 | 1992–2009 |
| TCGA 2011 early-stage | — | (9) | Affymetrix HT U133a | 27‡,§ | 0 | 26¶ | 70 | 37 | 73 | 62 | 1992–2009 |
* No value is given for datasets for which the corresponding clinical characteristic was not available (columns Stage IV, Subopt. Debulking, Median Age, and Time Period) or for datasets not deposited in the NCBI Gene Expression Omnibus or in the EMBL-EBI ArrayExpress (column Accession ID). TCGA = The Cancer Genome Atlas.
† Only Federation of Gynecology and Obstetrics (FIGO) stage III+IV, high-grade (grade 3), serous histology.
‡ Samples already included in other studies or duplicated within the study were removed (Supplementary Table 1, available online).
§ Only FIGO stage I+II, high-grade, serous subtype.
|| Suboptimal cytoreduction defined as residual tumor mass greater than 1cm.
¶ Suboptimal cytoreduction defined as presence of macroscopic residual tumor.
# Article was retracted because of a misalignment of genomic and survival data; the corrected data were used.
Clinical Endpoints
For the survival signature, the primary endpoint was overall survival from initial diagnosis to death. For the debulking signature, suboptimal debulking was defined as residual tumor mass greater than 1cm, except for datasets (9,19) where we used presence of macroscopic residual tumor.
Statistical Analysis
Overall Strategy.
We trained the overall survival signature on the 6 training datasets (see Figure 1). We then evaluated the performance in three ways: 1) a leave-one-dataset-out validation within the six training datasets (see Figure 2): here each dataset in turn is set aside for cross-study validation, and the model is trained on the remaining five datasets to assess cross-study consistency (Supplementary Figure 1, available online); results of this step are not subject to optimistic bias. At the end of this phase a single model is trained on all six training datasets; 2) independent validation on the seven validation datasets (see Figure 3): none of the samples from the six training datasets are used in this step; and 3) joint visualization of the first two methods to provide a summary view of both leave-one-study-out and independent validation performance (see Figure 4). The third method has no effect on the training. In addition to cross-validation in public datasets (see Supplementary Figure 10, available online), the debulking signature was validated in two independent cohorts by quantitative reverse-transcription polymerase chain reaction (qRT-PCR) and immunohistochemistry (see Figures 6 and 7).
Figure 1.
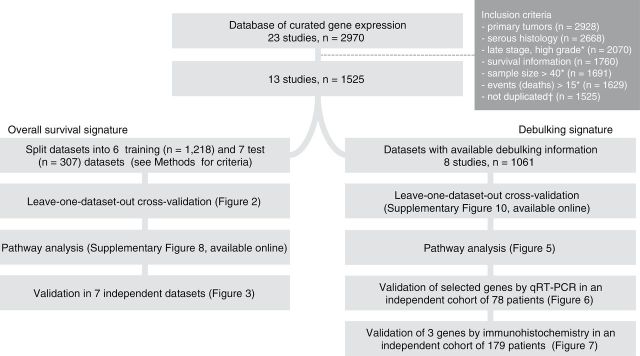
Flowchart of the study. This outlines the steps for training and validating the prognostic models presented in this study. * Including 27 TCGA early stage samples.
†Samples that were included in multiple studies were identified and removed (see Supplementary Table 1, available online). qRT-PCR = quantitative reverse-transcription polymerase chain reaction.
Figure 2.

Leave-one-dataset-out validation of performance of the new gene signature in predicting overall survival in late-stage ovarian cancer. We used our database of the ovarian transcriptome (5) for developing a novel overall survival gene signature by a meta-analytic signature development method. In total, six large datasets passed our training criteria (see Methods). To first test our methodology on the six large datasets we used for training (6,8,9,10,15,63), we applied a leave-one-dataset-out approach (Supplementary Figure 1, available online). Specifically, for each of the six datasets, we trained a prediction model using the remaining five datasets only and then stratified the patients of the dataset not used for training into high- and low-risk groups. P values were calculated with the log-rank test, and cutoffs for patient stratification were the medians of predicted risk scores in the combined training cohorts. All statistical tests were two-sided. TCGA = The Cancer Genome Atlas.
Figure 3.

Validation of the survival signature in independent data (9,13,16,19,28–30). A–E) Risk stratification in five validation microarray datasets of late-stage, high-grade, serous ovarian cancer by a model trained on all six training datasets (Figure 2). The Gillet et al. reverse-transcription polymerase chain reaction validation dataset (28) assayed only 12 of the 200 genes in the signature, which included, however, well-characterized cancer genes such as APC, RB1, and PDGFRB. The remaining four datasets had less than the 75 samples we required for training (13,29,30) or were published after our model was finalized (16). Cutoffs for high and low risk were again determined by calculating the median of the combined patient risk scores in training data. F) Performance in all early-stage, high-grade, serous samples from The Cancer Genome Atlas (TCGA) data. P values were calculated with the log-rank test. All statistical tests were two-sided. G) We further tested the model in a dataset in which survival information was only available as binary outcome (long-term vs short-term survivors) (19). The prediction model here estimated the probability of short term survival and its accuracy is shown with a receiver operating characteristic curve. This curve shows the true- and the false-positive rates for all possible cutoffs of the continuous prediction score. True positives are correctly classified short-term survivors. *Area under the curve (AUC) statistically significantly greater than 0.5 [DeLong test (23)].
Figure 4.

Combined comparison of our novel meta-analysis gene signature with existing prognostic factors and signatures proposed by The Cancer Genome Atlas (TCGA). This figure provides a summary view of both leave-one-study-out cross-validation and independent validation performance shown in Figures 2 and 3. A) Summary of the risk stratifications in Figures 2 and 3 in a single Kaplan–Meier plot [all studies (Table 1), excluding the TCGA cohort so comparison with the TCGA signatures could be made (6,8,10,13,15,16,28–30)]. This plot compares the survival of all patients classified as high risk in this meta-analysis with all low-risk patients. B) Risk stratification based on a fivefold cross-validated multivariable model using the gene signature risk group (high vs low risk) and the categorical covariables tumor stage (III vs IV) and debulking status (optimal vs suboptimal). The smaller sample sizes arise because of missing clinical annotations. C) Risk stratification based on a fivefold cross-validated multivariable model using tumor stage and debulking status only. D) Summary of risk stratifications by the TCGA gene signature (9) over all cohorts excluding TCGA, as in panel A. E) Summary of risk stratifications by the Verhaak et al. survival signature (18), as in panel A. F) Summary of risk stratifications by the multivariable model proposed by Verhaak et al. using their survival signature, continuous TCGA subtype scores, as well as categorical debulking status and tumor stage. P values were calculated with the log-rank test. G) Forest plot providing an alternative visualization of the hazard ratios (HRs) shown in the Kaplan–Meier plots in Figures 2 and 3 and including a comparison with the corresponding hazard ratios of the TCGA and Verhaak et al. models in all datasets separately. Squares show hazard ratio point estimates, with size corresponding to their weighting in the fixed-effects meta-analysis summaries (inverse of squared standard error); horizontal lines show 95% confidence intervals (CIs); and diamonds at the bottom show fixed-effects summaries of the hazard ratios over all shown datasets. Note that this fixed-effects summary corresponds to the hazard ratios shown in the summary Kaplan–Meier plots in panels A, D, and E. All statistical tests were two-sided.
Figure 6.
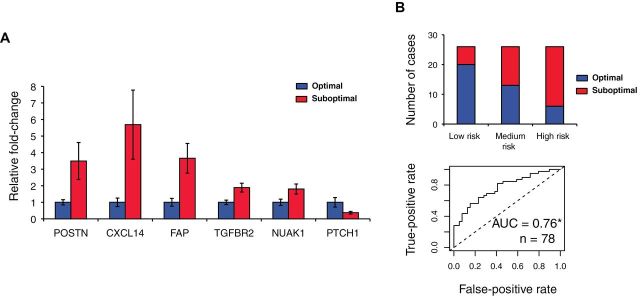
Validation of selected genes associated with debulking status by quantitative reverse-transcription polymerase chain reaction (qRT-PCR) in the Bonome et al. validation data (n = 78). A) Observed fold-changes in suboptimal vs optimal tumors and their standard deviations of the genes with statistically significantly (P < .05, two-sided Student t test) different expression between the two groups. B) The prediction accuracy of a multivariable model in which the qRT-PCR–validated genes were equally weighted. We stratified the samples into groups of high and low risk for suboptimal surgery based on the tertiles of the multivariable risk score: the 33% of patients with highest risk score were classified as high risk, the 33% with lowest risk score were classified as low risk, and all others were classified as medium risk. Between the high- and low-risk groups, 76.9% of samples were classified correctly. The accuracy of the multivariable risk prediction is further shown with a receiver operating characteristic curve. *Area under the curve (AUC) significantly greater than 0.5 [DeLong test (23)].
Figure 7.
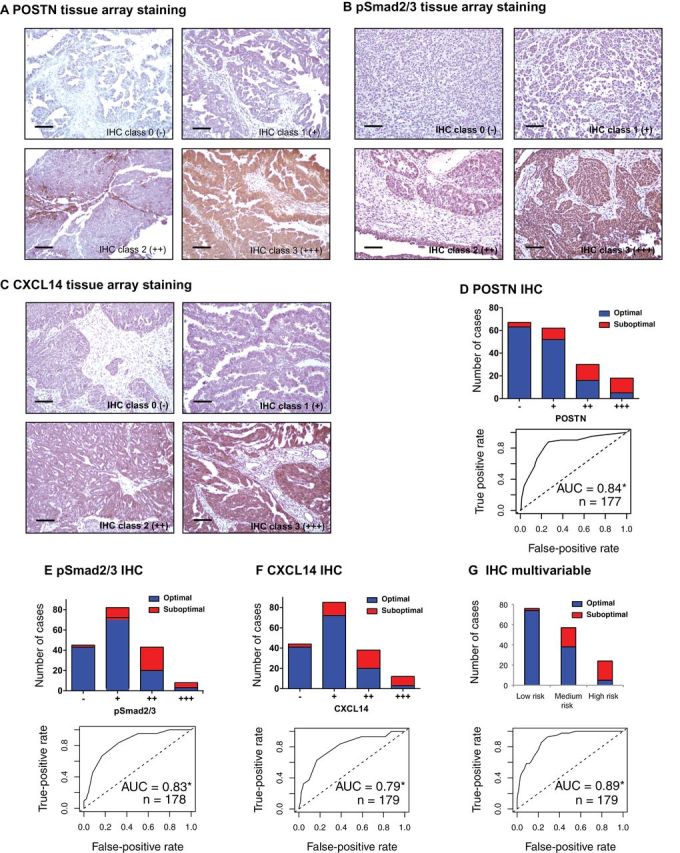
Validation of POSTN, pSmad2/3, and CXCL14 in an independent cohort by immunohistochemistry (IHC). A–C) IHC staining of POSTN, pSmad2/3, and CXCL14. The four IHC classes are categorized by IHC intensity scores as described in the Supplementary Information (available online) and previously (27). Scale bar = 100 μm. D) Histogram visualizing the frequency of optimal and suboptimal tumors stratified by POSTN IHC class (A) in an independent cohort of 177 samples. The true- and false-positive rates of POSTN IHC intensity scores (27) used for classification are further shown with a receiver operating characteristic curve. E and F) Corresponding figures for pSmad2/3 and CXCL14. G) The prediction accuracy of the multivariable model in which the three IHC validated genes were equally weighted (as in Figure 6B). * Area under the curve (AUC) statistically significantly greater than 0.5 [DeLong test (23)].
Model Training.
We aimed to identify robust prognosis genes for which variation in expression was consistently associated with survival. We first calculated the univariable Cox regression coefficients and their standard errors for each gene in each training dataset. We then summarized the Cox coefficients of gene i across training datasets into a single coefficient β i by a fixed effects meta-analysis (20) with coefficients weighted by the inverse of their squared standard errors. We then ranked genes by P value against the null hypothesis that the pooled coefficient is 0. We also compared our results to pooling using a random-effects meta-analysis (21) (Supplementary Figure 2, available online).
We chose a signature size m of 200 genes for all models, a size sufficiently small to be practically useful in a clinical test and sufficiently large to provide robustness in prediction accuracy across technologies. In general, the size of the gene signature had only a modest impact on the prediction accuracy in our algorithm as long as the signature included at least 100 genes (Supplementary Figure 3, available online). The m top-ranked genes were used to calculate a risk score for patient j, r j, using our multistudy variant of the compound covariable score (22). In this approach, the expression of gene i in subject j, x ij, is weighted by the pooled Cox coefficient of gene i (β i) to calculate the risk score rj:
To ensure that gene expression measurements are on the same scale across studies, all gene-specific expression vectors were centered to zero mean and scaled to unit variance within datasets. For binary classification (optimal vs suboptimal debulking), we proceeded analogously, pooling coefficients of a univariable logistic regression model for each gene.
Validation Metrics.
Gene signatures were evaluated by hazard ratio (HR) of dichotomized patient risk scores. Dichotomization cutoffs were the medians of these risk scores in the combined training cohorts. Significance of hazard ratio differences between signatures was estimated by bootstrap. Binary outcome classifiers were evaluated by area under the receiver operating characteristic (ROC) curve (or C-index) and the DeLong test (23). The C-index for censored data was calculated with the survC1 package (24), as previously described (17).
Multivariable Models.
To separately assess the added value of clinical variables, we used multivariable models incorporating the gene signature risk group as a categorical covariable (high vs low risk). These only were evaluated by fivefold cross-validation for a Kaplan–Meier analysis. We further assessed the improvement by a likelihood ratio test.
Details of our implementations of published models (9,18,25) are given in the Supplementary Methods (available online). P values less than .05 were considered significant. All statistical tests were two-sided.
Quantitative RT-PCR
Quantitative RT-PCR was performed as previously described (13) on 20-ng amplified RNA from 39 suboptimally and 39 optimally debulked specimens selected randomly from the Bonome et al. cohort (8) that had not been used in model training. Because tumor stage is associated with debulking status (26), numbers of stage III and IV patients (n = 31 and 8, respectively) in the optimal and suboptimal groups were balanced to disassociate stage and debulking status in the qRT-PCR validation cohort. Primer sets were selected (Supplementary Table 1, available online) for housekeeping genes GAPDH, GUSB, and ACTB and seven genes showing highly differentiated expression levels through the meta-analysis.
Immunohistochemistry
Immunohistochemical staining of POSTN (1.25 μg/mL; BioVendor R&D, Asheville, NC), CXCL14 (ab46010, 2.5 μg/mL; Abcam, Cambridge, UK) and pSmad2/3 (No. 3101, recognizing only phosphorylated Smad2 or Smad3 by transforming growth factor β (TGF-β) receptor, 1:200; Cell Signaling, Danvers, MA) was done on an independent validation tissue microarray consisting of 216 stage III/IV, high-grade, serous ovarian cancers obtained from patients with informed consent at the Massachusetts General Hospital (between 1993 and 2009. Debulking status was available for 179 cancers (n = 136 optimal and 43 suboptimal). Deparaffinized sections were subjected to antigen retrieval (citrate buffer, pH = 6.1, 95 °C for 30 minutes), incubated with each primary antibody at room temperature for 45 minutes, visualized with ImmPRESS Peroxidase Polymer Detection Reagents (Vector Laboratories, Burlingame, CA) and 3,3’-diaminobenzidine, and counterstained with Mayer’s hematoxylin. Intensity scores were calculated as the average difference in staining intensity between the tumor and stroma areas (27).
Results
We applied a meta-analytic signature development approach to publicly available high-grade, advanced-stage, serous ovarian cancer microarray gene expression profiles (Table 1) (5). This included the training and validation of two separate prognostic signatures: the first to identify long- and short-term survivors among patients with early-stage and late-stage, serous ovarian cancer; the second to identify advanced-stage, serous tumors that cannot be optimally debulked to 1cm or less of residual tumor.
Development of an Overall Survival Gene Signature
We created a new gene signature consisting of genes whose expression displayed the most statistically significant association with overall survival across major public datasets (Figure 1). We first used a cross-study validation approach (Supplementary Figure 1, available online) to quantify the extent to which our training approach would generate a signature that is consistent across studies. After applying our leave-one-dataset-out cross-validation to the training datasets, we found patients classified as high risk to consistently have significantly shorter survival than low-risk patients (Figure 2).
We then retrained the signature (Supplementary Table 1, available online) using all six training datasets and performed an external validation in seven additional independent datasets. These included a qRT-PCR dataset (28), three datasets not passing our minimum training sample size of 75 (13,29,30), a dataset that became available after the model was finalized (16), the TCGA early-stage, high-grade samples (9), and a dataset for which survival was annotated with a binary label rather than time to death (19). Our model continued to discriminate short-term from long-term survivors in all of these datasets, as shown by Kaplan–Meier analysis and ROC plots (Figure 3).
Comparison of the Survival Signature With Existing Prognostic Factors and Gene Signatures
We compared Kaplan–Meier stratifications based on our signature to those based on clinical prognostic factors, the original TCGA gene signature (Supplementary Figure 4, available online) (9), which we recently identified as the best performing signature published before July 2012 (6), and a more recent TCGA signature developed by the same investigators (Supplementary Figures 5 and 6, available online) (18). Clinical prognostic factors include optimal debulking (8), age, and tumor stage at diagnosis. Only four datasets provided all three factors; however, seven provided both stage and debulking status (Table 1; Supplementary Table 2, available online). We thus focused on these two factors.
When combining the results of cross-study validation and independent validation into a single summary (Figure 4), patient stratification using our signature was superior to clinical factors, as well as to both of the TCGA gene signatures. Over all cohorts excluding TCGA (n = 1031 patients where direct comparison with the TCGA signatures could be made), patients classified as high risk by our signature had a median survival of 29.6 months (95% confidence interval [CI] = 27.5 to 32.6) compared with 60.1 months (95% CI = 53.2 to 68.0) for the low-risk patients. Our signature (HR = 2.19; 95% CI = 1.84 to 2.61) (Figure 4A) provided an overall increase in hazard ratio of 0.36 (95% CI = −0.04 to 0.81; P = .04) compared with the original TCGA signature (HR = 1.83, 95% CI = 1.54 to 2.17) (Figure 4D). Including stage and debulking status in the prediction model provided only a small improvement over our gene signature alone (P = .02, likelihood ratio test) (Figure 4B). The two signatures proposed by the TCGA consortium performed very similarly compared with each other (HR difference = 0.00; 95% CI = −0.33 to 0.34) (Figure 4, D and E; Supplementary Figure 7, available online). The C-index of our signature improved moderately (0.011; 95% CI = −0.015 to 0.037) to 0.62 compared with the TCGA signature. By simulation, we estimated that an additional hazard ratio improvement of 0.27 would be needed to reach statistical significance on the C-index scale as well (Supplementary Results, available online). Pathway analysis of our signature showed enrichment of TGF-β and PDGF signaling in poor-prognosis patients (Supplementary Figure 8, available online). We finally verified that our meta-analysis training method is superior to single-study training (Supplementary Figure 9, available online).
Development of a Gene Signature for Predicting Suboptimal Debulking Surgery
Using the same meta-analytic signature development approach, we developed a gene signature for predicting unsuccessful debulking surgery (Supplementary Table 1, available online). We expected this signature to differ from the survival signature because the biological basis for optimal surgical removal of tumor tissue is not necessarily the same as that for patient survival. We used all eight late-stage microarray datasets with available debulking information (Table 1), excluding half of the Bonome et al. samples (n = 78) for validation by qRT-PCR.
We first tested our training datasets approach by leave-one-dataset-out validation. Accurate prediction of debulking status proved to be difficult, with an overall area under the curve of 0.59 (95% CI = 0.55 to 0.63) (Supplementary Figure 10, available online). In the same data, the signature published by Berchuck et al. (25) achieved an area under the curve of 0.54 (95% CI = 0.49 to 0.58) (Supplementary Figure 11 and Supplementary Table 3, available online). Expression of the top-ranked hit POSTN alone achieved prediction accuracies very similar to our signature (Supplementary Figure 12, available online). In the 1109 microarray samples with both debulking and survival information, high POSTN levels were prognostic of survival after adjusting for debulking status (P = .04; unadjusted P < .001). The prediction accuracy was robust to the choice of model training parameters (Supplementary Figures 13 and 14, available online).
Analysis of Pathways Contributing to Suboptimal Cytoreductive Surgery
To explore the molecular basis underlying our debulking signature, we analyzed the gene signature to identify biological pathways relevant to suboptimal disease. Application of pathway identification software (Pathway studio 7.1; Ariadne Genomics, Rockville, MD) identified the hyperactivation of TGF-β/Smad signaling (pathway enrichment analysis, P = .004) and the potential activation of RTK/Ras/MAPK/Egr-1, AMPK/Egr-1, and Hedgehog/Gli signaling in suboptimally debulked tumors (Figure 5). The resultant transcriptional network leads to upregulation of genes that support tumor dissemination, which decrease the chance of total surgical removal, reducing the possibility of optimal debulking. Potential molecular events responsible for suboptimal surgical outcome involve migration and invasion (MMP2, PLAU, SERPINE1, TIMP3, POSTN, VCAN, FN1, TGFBI, SPARC, and CYR61) (31–38), angiogenesis (EGR1, SMADs, GLIs, VCAN, POSTN, CNY61, and LOX) (39–45), metastatic colonization (POSTN, VCAN, and LOX) (46–48), and the activation of tumor-associated fibroblasts (ACTA2 and FAP), which play important roles in modulating the tumor microenvironment through the secretion of growth factors and extracellular matrix remodeling to support tumor dissemination through metastasis and angiogenesis (49,50).
Figure 5.

Pathway analysis of the debulking signature. Using the Pathway Studio 7.1 (Ariadne Genomics) software and a novel signature of 200 debulking-associated genes, we identified pathways statistically significantly associated with suboptimal debulking surgery. A gene is labeled in red when it is overexpressed in tumors that were subsequently suboptimally debulked. Conversely, genes overexpressed in tumors with optimal cytoreduction are labeled in blue. Genes with predictive power toward poor prognosis based on the meta-analysis are highlighted with pink borders. Red broken arrows indicate direct stimulatory modification. Green arrows indicate EGR-1–based transcriptional regulations. Orange arrows indicate TGF-β/Smad–based transcriptional regulations. Blue solid arrows indicate other direct regulations. Blue broken arrows indicate other indirect regulations. Purple sticks indicate binding.
Validation of the Debulking Signature by qRT-PCR and Immunohistochemistry in Two Independent Cohorts
Within our debulking signature, we selected seven highly differentially expressed genes with known biological role in ovarian tumorigenesis (six genes enriched in the pathway shown in Figure 5) and validated their expression level by qRT-PCR in an independent cohort of stage III and IV tumors consisting of 78 samples from Bonome et al. (8), which we had excluded from the meta-analysis for this purpose. Of the seven genes tested, six were statistically significantly associated with surgery outcome (POSTN: P = .03; CXCL14: P = .03; FAP: P = .01; NUAK1: P = .03; PTCH1: P = .004; TGFBR2: P = .005; TNFAIP6: P = .95; all Student t test) (Figure 6A). A model using all genes classified 76.9% of all samples correctly, with an area under the curve of 0.76 (95% CI = 0.66 to 0.87) (Figure 6B). Using normalized microarray data for the same set of patients, however, only achieved an area under the curve of 0.65 (95% CI = 0.53 to 0.77) through the same model, possibly because of the higher quantitative accuracy of qRT-PCR over microarray. The area under the curve for POSTN qRT-PCR–measured expression level alone was 0.65 (95% CI = 0.53 to 0.77). Furthermore protein expression of three of these proteins, POSTN, CXCL14 (signature genes), and pSmad2/3 (a surrogate marker of TGF-β pathway activation, a signature pathway), was validated by immunohistochemistry in an independent cohort of 179 patients. This analysis confirmed strong association of their expression with debulking status (Figure 7; Supplementary Table 4, available online). The sum of immunohistochemistry intensities for these three proteins provided a tool that classified 92.8% of samples in the high- and low-risk groups for suboptimal debulking correctly, with an area under the curve of 0.89 (95% CI = 0.84 to 0.93) (Figure 7G).
Discussion
We derived gene expression signatures to predict overall patient survival in early- and late-stage, high-grade, serous ovarian cancer and separately to predict suboptimal debulking surgery. These signatures were developed using the largest gene expression meta-analysis to date for ovarian cancer, incorporating 1525 samples (Figure 1). This analysis triples the sample size of the largest previous study (9). Novel signatures were validated (Figures 2–7) and shown to provide added value compared with known clinical factors, and they consistently outperformed available gene signatures (9,18,25).
Developing outcome predictors for late-stage cancers has been difficult, in part because of methodologic problems. These include: 1) signatures generated on relatively small sample sizes (51–55); 2) a lack of independent validation (53,55); 3) unaudited and unreliable clinical annotation (53,56,57); 4) laboratory-specific biases such as batch effects (57,58); and 5) training performed on nonrepresentative patient cohorts (59). Our comprehensive meta-analysis moves in the direction of addressing these previously missing elements. We trained and validated our models on a large number of carefully curated datasets and used a robust meta-analysis framework that limited the impact of laboratory or cohort-specific biases. Further gains in accuracy will be necessary, and we propose two ways these gains may be made: 1) larger sample size studies and 2) increased standardization and rigor of study design and clinical annotations. Prediction accuracy in validation datasets often shows an increasing trend with training sample size, even up to 1250 samples (Supplementary Figure 9, available online). This continual increase is striking, considering the heterogeneous surgical and medical management used by the different hospitals represented in this meta-analysis. Such heterogeneity highlights the limitations of signatures developed from any single institution and the need for specimens from clinical trials with precisely specified inclusion criteria.
Successful outcome predictors for ovarian cancer are particularly challenging because of the current gap between discrimination ability and clinical utility. Clinical utility is defined on whether the results provide substantial prognostic or predictive information as to alter patient management or behavior in ways that ultimately improve outcomes. It is important to note that the vast majority of advanced-stage ovarian cancer patients initially respond to standard treatment but then suffer recurrence and ultimately die of their disease. As such, we estimate that for a survival signature to be clinically relevant in this patient population, it would have to identify patients with refractory disease (progressive or persistent disease during primary therapy). These patients have a median survival of 9 months. If such a sufficiently accurate signature were identified, one could triage the poor-prognosis patients to phase II trials to attempt to identify active agents against these tumors at the first attempt. The survival signature we generated here, although significantly improved over published signatures, does not meet this standard [for a discussion of the relatively low C-indices of our and published signatures, see (17)]. Although the poor-prognostic patients have a shorter survival, most clearly respond to and benefit from standard combination chemotherapy. Thus, this signature would not alter clinical management of advanced-stage patients. Of interest, although our survival signature was established entirely from late-stage, serous tumors, it demonstrated considerable promise for stratifying early-stage, high-grade, serous tumors. This finding may reflect the underlying biology of recurrent early-stage ovarian cancer because these tumors have gene expression profiles similar to poor-prognosis, advanced-stage cancers. A reliable stratification of early-stage patients could spare low-risk patients unnecessary adjuvant chemotherapy. Unfortunately, our signature is preliminary in this patient group and will require validation in a much larger set of specimens. Our results suggest that meta-analyses like ours can help in achieving adequate sample sizes to establish genomic signatures with clinical utility.
In contrast, the debulking signature will have clinical utility if the 93% accuracy of the immunohistochemistry tool observed in our 179-patient validation cohort is confirmed in prospective validation. Cytoreductive surgery remains an important component of treatment for women with epithelial ovarian cancer (8). Whether this fact is due to 1) the smaller residual tumor mass or 2) an intrinsic biological element of tumors, providing less aggressive and invasive tumors an advantage in surgery, remains unresolved (26). We present the strongest evidence to date for the existence of a biologic basis and a predictive gene signature for debulking ability of ovarian tumors. The immunohistochemistry staining showed high predictive values of our identified biomarkers. It is interesting to note that of the top five misclassified optimal cases (high protein expression for all three biomarkers), two were wrongly annotated and had 1-cm residual disease. This suggests that the signature may be even more accurate than we report, and its limitation is dependent in part on the clinical classification of degree of debulking. The clinical utility in this case results from the identification of those patients who will not benefit from primary debulking surgery, sparing them the toxicity of extensive surgery and the delay until the initiation of chemotherapy. These patients can be triaged to neoadjuvant chemotherapy with interval debulking. An European Organisation for Research and Treatment of Cancer study has recently demonstrated that neoadjuvant therapy with interval debulking is equivalent to primary debulking and adjuvant chemotherapy (60). A genomic tool of this nature will assist surgeons in stratifying patients for these alternative approaches.
The debulking signature also provides biologic insight into ovarian cancer dissemination. TGF-β pathway activation has been documented in ovarian cancer with tumors becoming resistant to the growth inhibitory effects of the ligand (61,62). These data suggest that in a subset of tumors, the TGF-β activated pathway stimulates epithelial-mesenchymal transition, the activation of tumor associated fibroblasts, and other biologic processes that contribute to spread of the tumor with resultant difficulty in surgical debulking. These pathways may also provide therapeutic targets to convert tumors that are not resectable in the neoadjuvant setting to ones that are at interval debulking.
Funding
This work was supported by grants from the National Cancer Institute at the National Institutes of Health (1RC4CA156551-01 and 5R01CA142832 to MB); the Dana-Farber/Harvard Cancer Center NCI 5P30 CA006516-46 to GP and MB); and the National Science Foundation (CAREER DBI-1053486 to CH).
Supplementary Material
The authors are solely responsible for the design of the study; the collection, analysis, and interpretation of the data; the writing of the manuscript; and the decision to submit the manuscript for publication. There are no other conflicts of interest. All authors read and approved the final article.
We thank Benjamin Haibe-Kains for comments on the manuscript.
References
- 1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62(1):10–29. [DOI] [PubMed] [Google Scholar]
- 2. Ibeanu OA, Bristow RE. Predicting the outcome of cytoreductive surgery for advanced ovarian cancer: a review. Int J Gynecol Cancer. 2010;20(Suppl 1):S1–S11. [DOI] [PubMed] [Google Scholar]
- 3. Chang SJ, Bristow RE, Ryu HS. Impact of complete cytoreduction leaving no gross residual disease associated with radical cytoreductive surgical procedures on survival in advanced ovarian cancer. Ann Surg Oncol. 2012;19(13):4059–4067. [DOI] [PubMed] [Google Scholar]
- 4. Barakat R, Berchuck A, Markman M, et al. Principles and Practice of Gynecologic Oncology. 6th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2013. [Google Scholar]
- 5. Ganzfried BF, Riester M, Haibe-Kains B, et al. curatedOvarianData: clinically annotated data for the ovarian cancer transcriptome. Database (Oxford). 10.1093/database/bat013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bentink S, Haibe-Kains B, Risch T, et al. Angiogenic mRNA and microRNA gene expression signature predicts a novel subtype of serous ovarian cancer. PLoS One. 2012;7(2)e30269. 10.1371/journal.pone.0030269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bonome T, Lee J-Y, Park D-C, et al. Expression profiling of serous low malignant potential, low-grade, and high-grade tumors of the ovary. Cancer Res. 2005;65(22):10602–10612. [DOI] [PubMed] [Google Scholar]
- 8. Bonome T, Levine D, Shih J, et al. A gene signature predicting for survival in suboptimally debulked patients with ovarian cancer. Cancer Res. 2008;68(13):5478–5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Crijns A, Fehrmann R, de Jong S, et al. Survival-related profile, pathways, and transcription factors in ovarian cancer. PLoS Med. 2009;6(2)e24. 10.1371/journal.pmed.1000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Denkert C, Budczies J, Darb-Esfahani S, et al. A prognostic gene expression index in ovarian cancer—validation across different independent data sets. J Pathol. 2009;218(2):273–280. [DOI] [PubMed] [Google Scholar]
- 12. Hernandez L, Hsu S, Davidson B, et al. Activation of NF-kappaB signaling by inhibitor of NF-kappaB kinase beta increases aggressiveness of ovarian cancer. Cancer Res. 2010;70(10):4005–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mok SC, Bonome T, Vathipadiekal V, et al. A gene signature predictive for outcome in advanced ovarian cancer identifies a survival factor: microfibril-associated glycoprotein 2. Cancer Cell. 2009;16(6):521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sabatier R, Finetti P, Bonensea J, et al. A seven-gene prognostic model for platinum-treated ovarian carcinomas. Br J Cancer. 2011;105(2):304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yoshihara K, Tajima A, Yahata T, et al. Gene expression profile for predicting survival in advanced-stage serous ovarian cancer across two independent datasets. PLoS One. 2010;5(3)e9615. 10.1371/journal.pone.0009615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yoshihara K, Tsunoda T, Shigemizu D, et al. High-risk ovarian cancer based on 126-gene expression signature is uniquely characterized by downregulation of antigen presentation pathway. Clin Cancer Res. 2012;18(5):1374–1385. [DOI] [PubMed] [Google Scholar]
- 17. Waldron L, Haibe-Kains B, Culhane A, et al. A comparative meta-analysis of prognostic gene signatures for late-stage ovarian cancer. J Natl Cancer Inst. 2014;. [DOI] [PMC free article] [PubMed]
- 18. Verhaak RG, Tamayo P, Yang JY, et al. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J Clin Invest. 2012;123(1):517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Partheen K, Levan K, Osterberg L, et al. Expression analysis of stage III serous ovarian adenocarcinoma distinguishes a sub-group of survivors. Eur J Cancer. 2006;42(16):2846–2854. [DOI] [PubMed] [Google Scholar]
- 20. Hedges LV, Vevea JL. Fixed- and random-effects models in meta-analysis. Psychol Meth. 1998;3(4):486–504. [Google Scholar]
- 21. Viechtbauer W. Bias and efficiency of meta-analytic variance estimators in the random-effects model. J Educ Behav Stat. 2005;30(3):261–293. [Google Scholar]
- 22. Tukey JW. Tightening the clinical trial. Control Clin Trials. 1993;14(4):266–285. [DOI] [PubMed] [Google Scholar]
- 23. DeLong ER, DeLong DM, Clarke-Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44(3):837–845. [PubMed] [Google Scholar]
- 24. Uno H, Cai T, Pencina MJ, et al. On the C-statistics for evaluating overall adequacy of risk prediction procedures with censored survival data. Stat Med. 2011;30(10):1105–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Berchuck A, Iversen ES, Lancaster JM, et al. Prediction of optimal versus suboptimal cytoreduction of advanced-stage serous ovarian cancer with the use of microarrays. Am J Obstet Gynecol. 2004;190(4):910–925. [DOI] [PubMed] [Google Scholar]
- 26. Borley J, Wilhelm-Benartzi C, Brown R, et al. Does tumour biology determine surgical success in the treatment of epithelial ovarian cancer? A systematic literature review. Br J Cancer. 2012;107(7):1069–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Birrer MJ, Johnson ME, Hao K, et al. Whole genome oligonucleotide-based array comparative genomic hybridization analysis identified fibroblast growth factor 1 as a prognostic marker for advanced-stage serous ovarian adenocarcinomas. J Clin Oncol. 2007;25(16):2281–2287. [DOI] [PubMed] [Google Scholar]
- 28. Gillet J-P, Calcagno A, Varma S, et al. Multidrug resistance-linked gene signature predicts overall survival of patients with primary ovarian serous carcinoma. Clin Cancer Res. 2012;18(11):3197–3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dressman H, Berchuck A, Chan G, et al. An integrated genomic-based approach to individualized treatment of patients with advanced-stage ovarian cancer. J Clin Oncol. 2007;25(5):517–525. [DOI] [PubMed] [Google Scholar]
- 30. Konstantinopoulos PA, Spentzos D, Karlan BY, et al. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J Clin Oncol. 2010;28(22):3555–3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen J, Wang M, Xi B, et al. SPARC is a key regulator of proliferation, apoptosis and invasion in human ovarian cancer. PLoS One. 2012;7(8):e42413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25(1):9–34. [DOI] [PubMed] [Google Scholar]
- 33. Mitra AK, Sawada K, Tiwari P, et al. Ligand-independent activation of c-Met by fibronectin and alpha(5)beta(1)-integrin regulates ovarian cancer invasion and metastasis. Oncogene. 2011;30(13):1566–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schmitt M, Harbeck N, Brunner N, et al. Cancer therapy trials employing level-of-evidence-1 disease forecast cancer biomarkers uPA and its inhibitor PAI-1. Expert Rev Mol Diagn. 2011;11(6):617–634. [DOI] [PubMed] [Google Scholar]
- 35. Soikkeli J, Podlasz P, Yin M, et al. Metastatic outgrowth encompasses COL-I, FN1, and POSTN up-regulation and assembly to fibrillar networks regulating cell adhesion, migration, and growth. Am J Pathol. 2010;177(1):387–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tang L, Han X. The urokinase plasminogen activator system in breast cancer invasion and metastasis. Biomed Pharmacother. 2013;67(2):179–182. [DOI] [PubMed] [Google Scholar]
- 37. Ween MP, Oehler MK, Ricciardelli C. Transforming growth factor-beta-induced protein (TGFBI)/(betaig-H3): a matrix protein with dual functions in ovarian cancer. Int J Mol Sci. 2012;13(8):10461–10477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yeung TL, Leung CS, Wong KK, et al. TGF-beta modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res. 2013;73(16):5016–5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Baker AM, Bird D, Welti JC, et al. Lysyl oxidase plays a critical role in endothelial cell stimulation to drive tumor angiogenesis. Cancer Res. 2013;73(2):583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Carpenter RL, Lo HW. Hedgehog pathway and GLI1 isoforms in human cancer. Discov Med. 2012;13(69):105–113. [PMC free article] [PubMed] [Google Scholar]
- 41. Chen Y, Du XY. Functional properties and intracellular signaling of CCN1/Cyr61. J Cell Biochem. 2007;100(6):1337–1345. [DOI] [PubMed] [Google Scholar]
- 42. Khachigian LM. Early growth response-1: blocking angiogenesis by shooting the messenger. Cell Cycle. 2004;3(1):10–11. [PubMed] [Google Scholar]
- 43. Pardali E, ten Dijke P. Transforming growth factor-beta signaling and tumor angiogenesis. Front Biosci. 2009;14:4848–4861. [DOI] [PubMed] [Google Scholar]
- 44. Zheng PS, Wen J, Ang LC, et al. Versican/PG-M G3 domain promotes tumor growth and angiogenesis. FASEB J. 2004;18(6):754–756. [DOI] [PubMed] [Google Scholar]
- 45. Zhu M, Fejzo MS, Anderson L, et al. Periostin promotes ovarian cancer angiogenesis and metastasis. Gynecol Oncol. 2010;119(2):337–344. [DOI] [PubMed] [Google Scholar]
- 46. Erler JT, Bennewith KL, Cox TR, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15(1):35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gao D, Vahdat LT, Wong S, et al. Microenvironmental regulation of epithelial-mesenchymal transitions in cancer. Cancer Res. 2012;72(19):4883–4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Malanchi I, Santamaria-Martinez A, Susanto E, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2012;481(7379):85–89. [DOI] [PubMed] [Google Scholar]
- 49. Ahmed F, Steele JC, Herbert JM, et al. Tumor stroma as a target in cancer. Curr Cancer Drug Targets. 2008;8(6):447–453. [DOI] [PubMed] [Google Scholar]
- 50. Schauer IG, Sood AK, Mok S, et al. Cancer-associated fibroblasts and their putative role in potentiating the initiation and development of epithelial ovarian cancer. Neoplasia. 2011;13(5):393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dobbin KK, Zhao Y, Simon RM. How large a training set is needed to develop a classifier for microarray data? Clin Cancer Res. 2008;14(1):108–114. [DOI] [PubMed] [Google Scholar]
- 52. Koscielny S. Why most gene expression signatures of tumors have not been useful in the clinic. Sci Transl Med. 2010;2(14):14ps2–14ps2. [DOI] [PubMed] [Google Scholar]
- 53. Micheel CM, Nass SJ, Omenn GS. Evolution of Translational Omics Lessons Learned and the Path Forward. Washington, DC: National Academies Press; 2012. [PubMed] [Google Scholar]
- 54. Sabatier R, Finetti P, Cervera N, et al. Gene expression profiling and prediction of clinical outcome in ovarian cancer. Crit Rev Oncol Hematol. 2009;72(2):98–109. [DOI] [PubMed] [Google Scholar]
- 55. Simon R. Development and validation of therapeutically relevant multi-gene biomarker classifiers. J Natl Cancer Inst. 2005;97(12):866–867. [DOI] [PubMed] [Google Scholar]
- 56. Baggerly KA, Coombes KR. Deriving chemosensitivity from cell lines: forensic bioinformatics and reproducible research in high-throughput biology. Ann Appl Stat. 2009;3(4):1309–1334. [Google Scholar]
- 57. Baggerly KA, Coombes KR, Neeley ES. Run batch effects potentially compromise the usefulness of genomic signatures for ovarian cancer. J Clin Oncol. 2008;26(7):1186–1187; author reply 1187–1188. [DOI] [PubMed] [Google Scholar]
- 58. Leek JT, Scharpf RB, Bravo HC, et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet. 2010;11(10):733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Swisher EM, Taniguchi T, Karlan BY. Molecular scores to predict ovarian cancer outcomes: a worthy goal, but not ready for prime time. J Natl Cancer Inst. 2012;104(9):642–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vergote I, Trope CG, Amant F, et al. Neoadjuvant chemotherapy or primary surgery in stage IIIC or IV ovarian cancer. N Engl J Med. 2010;363(10):943–953. [DOI] [PubMed] [Google Scholar]
- 61. Chou JL, Chen LY, Lai HC, et al. TGF-beta: friend or foe? The role of TGF-beta/SMAD signaling in epigenetic silencing of ovarian cancer and its implication in epigenetic therapy. Expert Opin Ther Targets. 2010;14(11):1213–1223. [DOI] [PubMed] [Google Scholar]
- 62. Sunde JS, Donninger H, Wu K, et al. Expression profiling identifies altered expression of genes that contribute to the inhibition of transforming growth factor-beta signaling in ovarian cancer. Cancer Res. 2006;66(17):8404–8412. [DOI] [PubMed] [Google Scholar]
- 63. Tothill RW, Tinker AV, George J, et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res. 2008;14(16):5198–5208. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.