

Abstract
Saliva of blood-sucking arthropods contains a complex cocktail of pharmacologically active compounds that assists feeding by counteracting their hosts’ hemostatic and inflammatory reactions. Panstrongylus megistus (Burmeister) is an important vector of Chagas disease in South America, but despite its importance there is only one salivary protein sequence publicly deposited in GenBank. In the present work, we used Illumina technology to disclose and publicly deposit 3,703 coding sequences obtained from the assembly of >70 million reads. These sequences should assist proteomic experiments aimed at identifying pharmacologically active proteins and immunological markers of vector exposure. A supplemental file of the transcriptome and deducted protein sequences can be obtained from http://exon.niaid.nih.gov/transcriptome/P_megistus/Pmeg-web.xlsx.
Keywords: Chagas disease, vector biology, salivary gland, transcriptome, medical entomology
While attempting to feed, hematophagous animals have to deal with their host’s inflammatory and hemostatic responses against tissue injury and blood loss, which include the phenomena of platelet aggregation, vasoconstriction, and blood coagulation. Against these challenging obstacles, blood-sucking arthropods have evolved a complex salivary potion that antagonizes these responses (Ribeiro and Arca 2009, Ribeiro et al. 2010). The diversity of the salivary potion in blood-sucking arthropods is large because the blood-feeding mode evolved independently many times, even within related organisms, such as the flies, and also because salivary coding genes appear to be evolving quickly, perhaps in response to the relentless host immune pressure (Ribeiro and Arca 2009). Indeed, evidence of strong positive selection effects has been found for salivary protein coding genes of mosquitoes (Arca et al. 2014). The combination of convergent evolution and positive selection thus create a diverse landscape of salivary protein families in blood-sucking animals. From the practical side, salivary proteins of such animals may have interesting pharmacological properties (Champagne 2005, Francischetti 2010, Chmelar et al. 2012), may be vaccine targets to prevent transmission of vector-borne diseases (Valenzuela et al. 2001b, Gomes et al. 2008), and also may be used as unique immunological markers of vector exposure, when their sequence is known (Valenzuela 2002, Ribeiro and Francischetti 2003, Chmelar et al. 2012).
Among the Hemiptera, the blood-sucking mode has evolved independently at least two times within the Cimicomorpha, namely within the family Cimicidae (bed bugs) and within the subfamily Triatominae (kissing bugs) of the family Reduviidae (Beaty and Marquartd 1996). The subfamily Triatominae contains 140 described species within 15 genera and five tribes, a number of which are known vectors of Chagas disease (Schofield and Galvao 2009). Sialotranscriptomes of species from the Rhodnius, Dipetalogaster, and Triatoma genera have been analyzed, and hundreds of derived coding sequences (CDS) have been deposited to GenBank (Ribeiro et al. 2004, 2012a; Santos et al. 2007; Assumpcao et al. 2008, 2011, 2012). A single salivary transcriptome from Panstrongylus megistus (Burmeister) reported the expansion of the lipocalin family of proteins, but no protein sequences have been publicly deposited (Bussacos et al. 2011). Indeed there is only one publicly available salivary protein sequence from the genus Panstrongylus deposited in GenBank, coding for a serine protease (Meiser et al. 2010). P. megistus recently became the most important Chagas disease vector in Brazil since successful control of Triatoma infestans (Marcilla et al. 2002, Cavassin et al. 2014).
More recently, the RNAseq revolution and associated bioinformatic tools (Miller et al. 2010) allowed for cheap and massive transcriptome sequencing and coding sequence discovery, which has already been used to increase the knowledge of insect and tick sialomes (Chagas et al. 2013; Ribeiro et al. 2013; Schwarz et al. 2013, 2014). Here we used Illumina sequencing to assemble and disclose thousands of CDS derived from a sialotranscriptome from P. megistus, 4,357 of which have been deposited to GenBank. It is expected that these sequences will help proteomic studies attempting to identify interesting salivary pharmacological activities from this insect as well as for designing specific immunological markers of P. megistus exposure.
Materials and Methods
Ethics Statement
All experimental exposures of animals to triatomines were carried out in the Czech Republic in accordance with the Animal Protection Law of the Czech Republic (§17, Act Number 246/1992 Sb) and with the approval of the Academy of Science of the Czech Republic (protocol approval number 172/2010), which complies with the regulations of the European Directive 2010/63/EU on the protection of animals used for scientific purposes in Europe.
Insects
P. megistus originated from Minas Gerais, Brazil (obtained from J. Jurberg, Departamento de Entomologia, Instituto Oswaldo Cruz, Rio de Janeiro, Brazil) and was kept in the laboratory for 17 yrs. The colony was maintained at an air temperature of 28 ± 1°C, a relative humidity of 60–70%, and on a photoperiod of 12:12 (L:D) h. For colony maintenance, the bugs were regularly fed on guinea pigs or rabbits. Two-week-old, starved fifth-instar nymphs were used for this study. Salivary glands (SG) were dissected from two insects every other day after one full bloodmeal as a fifth-instar nymph for 28 d.
RNA Extraction, Library Preparation, and Sequencing
RNA preparation, library construction, and sequencing were performed essentially as described previously (Ribeiro et al. 2013), and will be repeated here with small modifications for reference: SG were stored in RNAlater at 4°C for 48 h before being transferred to −70°C until RNA extraction. SG RNA was extracted and isolated using the Micro FastTrack mRNA isolation kit (Invitrogen, Grand Island, NY) per manufacturer’s instructions. The integrity of the total RNA was checked on a Bioanalyser (Agilent Technologies, Santa Clara, CA). mRNA library construction and sequencing were done by the NIH Intramural Sequencing Center. The SG library was constructed using the TruSeq RNA sample prep kit, v. 2 (Illumina Inc., San Diego, CA). The resulting cDNA was fragmented using a Covaris E210 (Covaris, Woburn, MA). Library amplification was performed using eight cycles to minimize the risk of overamplification. Sequencing was performed on a HiSeq 2000 (Illumina) with v. 3 flow cells and sequencing reagents. One lane of the HiSeq machine was used for this and four other libraries, distinguished by bar coding. Libraries from the tick Hyalloma annatolicum excavatum, the horse fly Tabanus bromius, and the bat Diphila ecaudata were co-sequenced with Panstrongylus, and we found that some cross-contamination of sequences occurred between the libraries (see below). A total of 71,755,660 sequences of 101 nucleotides in length were obtained. A paired-end protocol was used.
Bioinformatic Tools Used
Raw data were processed using RTA 1.12.4.2 and CASAVA 1.8.2. Reads were trimmed of low quality regions (<10), and only those with an average quality of 20 or more were used, comprising a total of 70,612,224 high-quality reads. These were assembled with the ABySS software (Genome Sciences Centre, Vancouver, BC, Canada; Birol et al. 2009, Simpson et al. 2009) using various kmer (k) values (every tenth from 21 to 91). Because the ABySS assembler tends to miss highly expressed transcripts (Zhao et al. 2011), the SOAPdenovo-Trans assembler (Luo et al. 2012) was also used, again with odd kmers from 21–91. The resulting assemblies were joined by an iterative BLAST and cap3 assembler (Karim et al. 2011). Sequence contamination between bar-coded libraries were identified and removed when their sequence identities were over 98% but their abundance of reads were >10 fold between libraries. CDS were extracted using an automated pipeline based on similarities to known proteins or by obtaining CDS containing a signal peptide (Nielsen et al. 1999). CDS and their protein sequences were mapped into a hyperlinked Excel spreadsheet (presented as Supp File 1 [online only]). Signal peptide, transmembrane domains, furin cleavage sites, and mucin-type glycosylation were determined with software from the Center for Biological Sequence Analysis (Technical University of Denmark, Lyngby, Denmark; Sonnhammer et al. 1998, Nielsen et al. 1999, Duckert et al. 2004, Julenius et al. 2005). Reads were mapped into the contigs using blastn (Altschul et al. 1997) with a word size of 25, masking homonucleotide decamers and allowing mapping to up to three different CDS if the BLAST results had the same score values. Mapping of the reads was also included in the Excel spreadsheet. RPKM values (Trapnell et al. 2012) for each coding sequence were also mapped to the spreadsheet. To compare relative expression of transcripts, we use the “expression index” defined as the number of reads mapped to a particular CDS divided by the largest found number of reads mapped to a single CDS, which in the case of this transcriptome was a value of 6,015,741 mapped to a single lipocalin coding sequence. Automated annotation of proteins was based on a vocabulary of nearly 290 words found in matches to various databases, including Swissprot, Gene Ontology, KOG, Pfam, and SMART, Refseq-invertebrates, and a subset of the GenBank sequences containing Hemiptera[organism] protein sequences. Further manual annotation was done as required. Detailed bioinformatics analysis of our pipeline can be found in our previous publication (Karim et al. 2011). For determination of synonymous and nonsynonymous sites within CDS, the tool BWA aln (Li and Durbin 2010) was used to map the reads to the CDS, producing SAI files that were joined by BWA sampe module, converted to BAM format, and sorted. The sequence alignment and map tools (samtools) package (Li et al. 2009) was used to do the mpileup of the reads (samtools mpileup), and the binary call format tools (bcftools) program from the same package was used to make the final vcf file containing the single-nucleotide polymorphic (SNP) sites, which were only taken if the site coverage was at least 100 (–D100), the quality was 13 or better, and the SNP frequency was 5 or higher (default). Determination of whether the SNPs lead to a synonymous or nonsynonymous codon change was achieved by a program written in Visual Basic by J.M.C.R., the results of which are mapped into the Excel spreadsheet and color visualized in hyperlinked rtf files within Supp File 1 (online only). Sequence alignments were done with the ClustalX software package (Thompson et al. 1997). Phylogenetic analysis and statistical neighbor-joining bootstrap tests (1,000 iterations) of the phylogenies were done with the Mega package (Kumar et al. 2004).
Data Access
The raw reads were deposited on the Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI) under bioproject ID PRJNA249079 and run SRR1304639. This Transcriptome Shotgun Assembly project has been deposited at DDBJ/EMBL/GenBank under the accession GBGD00000000. The version described in this paper is the first version, GBGD01000000. Hyperlinked excel spreadsheets containing the CDS and their annotation are available at http://exon.niaid.nih.gov/transcriptome/P_megistus/Pmeg-web.xlsx (hyperlinked excel spreadsheet, 12 MB) and http://exon.niaid.nih.gov/transcriptome/P_megistus/Pmeg-SA.zip (Standalone excel with all local links, 306 MB).
Results and Discussion
Overview of the Sialotranscriptome of P. megistus
Following assembly of 70,612,224 reads, a total of 53,228 sequences were obtained (Supp Table 1 [online only]), from which we extracted 5,488 CDS. These CDS mapped 46,592,833 reads, or 66% of the total reads. Their average length was 1,247 nucleotides (nt) with 2,619 CDS being larger than 1,000 nt. These CDS were classified generally into five classes, namely “secreted” (S), “housekeeping” (H), “unknown” (U), “transposable elements” (TE), and “viral” (V; Table 1). The S class had 633 assigned CDS, which mapped 71% of all reads, in accordance with the secretory nature of the organ. The H class produced 4,414 CDS, mapping 28% of the reads. TE’s accounted for 3.5% of the CDS and 0.56% of the reads, a somewhat typical finding when comparing with other sialotranscriptomes. Seven putative viral transcripts were also found, mapping 0.12% of the reads. These include a transcript (Pm-2405) coding for a putative replicase from the Euprosterna elaeasa virus that has 50% amino acid similarity over 700 amino acids, and expressed with an RPKM = 94. This virus belongs to the Tetraviridae family, a lepidopteran-restricted family (Zeddam et al. 2010). Three transcripts matched the Picornavirus Sacbrood virus polyprotein, a virus infecting bees (Ghosh et al. 1999). PmegSigP-513 matches Melanoplus sanguinipes entomopoxvirus, a virus described from grasshoppers (Afonso et al. 1999). Finally, 243 CDS were not able to be classified, representing 0.41% of the reads.
Table 1.
General classification of the CDS derived from the sialotranscriptome of P. megistus
| Class | Number of CDS | Percent of CDS | Number of reads mapped | Percent of reads |
|---|---|---|---|---|
| Secreted | 633 | 11.53 | 33,199,075 | 71.25 |
| Housekeeping | 4,414 | 80.43 | 12,887,452 | 27.66 |
| Unknown | 243 | 4.43 | 191,448 | 0.41 |
| Transposable elements | 191 | 3.48 | 260,081 | 0.56 |
| Viral | 7 | 0.13 | 54,777 | 0.12 |
| Total | 5,488 | 100 | 46,592,833 | 100 |
The housekeeping CDS were further classified by their function (Table 2), not surprisingly showing the category “protein synthesis” to be the most expressed and accruing 10% of the reads of the H class. Further classification of the S class reveals, as previously indicated (Bussacos et al. 2011), that lipocalins abound in the P. megistus sialotranscriptome, as it does in all other triatomine sialome studies (Ribeiro et al. 2012b; Table 3). Indeed we found 87 transcripts coding for lipocalins that accrued 87% of all reads of the S class. This is equivalent to stating that about 87% of the mRNA mass associated with the S class code for lipocalins. Indeed a single lipocalin CDS (Pm-27201) accrued over 6 million reads, which is 8.5% of the total number of sequenced reads and accordingly has an expression index of 1 (see methods). Other classes of transcripts that are relatively well expressed are for the enzymes apyrase (1% of the transcriptional mass of the S class), inositol phosphatase (1.7%), serine proteases (1.1%), Kazal-domain containing peptides (2.1%), antigen-5 family (0.9%), hemolysin and trialysin family (1.3%), and some deorphanized protein families (1.2%), which were previously identified in triatomine transcriptomes but only at a single species level and not having similarities to other known proteins.
Table 2.
Classification of the CDS of the housekeeping class derived from the sialotranscriptome of P. megistus
| Class | Number of CDS | Percent of CDS | Number of reads mapped | Percent of reads |
|---|---|---|---|---|
| Protein synthesis machinery | 237 | 5.37 | 1,383,467 | 10.73 |
| Cytoskeletal proteins | 181 | 4.10 | 1,197,908 | 9.30 |
| Lipid metabolism | 192 | 4.35 | 1,111,559 | 8.63 |
| Unknown conserved | 669 | 15.16 | 1,092,340 | 8.48 |
| Transcription machinery | 430 | 9.74 | 1,006,864 | 7.81 |
| Signal transduction | 622 | 14.09 | 970,747 | 7.53 |
| Protein modification | 201 | 4.55 | 886,377 | 6.88 |
| Oxidant metabolism/Detoxification | 42 | 0.95 | 792,334 | 6.15 |
| Protein export | 271 | 6.14 | 595,017 | 4.62 |
| Transporters and channels | 226 | 5.12 | 530,812 | 4.12 |
| Proteasome machinery | 186 | 4.21 | 518,726 | 4.03 |
| Energy metabolism | 129 | 2.92 | 437,013 | 3.39 |
| Extracellular matrix | 117 | 2.65 | 367,581 | 2.85 |
| Carbohydrate metabolism | 129 | 2.92 | 328,566 | 2.55 |
| Nuclear regulation | 223 | 5.05 | 322,378 | 2.50 |
| Intermediary metabolism | 46 | 1.04 | 310,341 | 2.41 |
| Unknown conserved membrane protein | 148 | 3.35 | 209,169 | 1.62 |
| Amino acid metabolism | 62 | 1.40 | 166,980 | 1.30 |
| Transcription factor | 95 | 2.15 | 157,537 | 1.22 |
| Detoxification | 48 | 1.09 | 151,032 | 1.17 |
| Nucleotide metabolism | 76 | 1.72 | 147,693 | 1.15 |
| Immunity | 45 | 1.02 | 123,071 | 0.95 |
| Nuclear export | 30 | 0.68 | 52,541 | 0.41 |
| Storage | 9 | 0.20 | 27,399 | 0.21 |
| Total | 4,414 | 100 | 12,887,452 | 100 |
Table 3.
Classification of the CDS of the secretory class derived from the sialotranscriptome of P. megistus
| Class | Number of CDS | Percent of CDS | Number of reads mapped | Percent of reads |
|---|---|---|---|---|
| Enzymes | ||||
| Apyrase | 6 | 0.95 | 344,772 | 1.038 |
| Ectonucleotide pyrophosphatase | 1 | 0.16 | 502 | 0.002 |
| Inositol phosphatase | 7 | 1.11 | 567,399 | 1.709 |
| Endonuclease | 2 | 0.32 | 3,301 | 0.010 |
| Esterase | 3 | 0.47 | 7,327 | 0.022 |
| Serine proteases | 11 | 1.74 | 370,825 | 1.117 |
| Metalloproteases | 2 | 0.32 | 229,703 | 0.692 |
| Other peptidases | 18 | 2.84 | 49,102 | 0.148 |
| Lipases | 11 | 1.74 | 58,834 | 0.177 |
| Protease inhibitor domains | ||||
| Kazal-domain peptides | 13 | 2.05 | 689,500 | 2.077 |
| Serpin | 4 | 0.63 | 16,122 | 0.049 |
| Tyropin and SPARC domain | 1 | 0.16 | 269 | 0.001 |
| Pacifastin domains | 1 | 0.16 | 3,980 | 0.012 |
| VIT domain | 1 | 0.16 | 390 | 0.001 |
| Small molecule binding domains | ||||
| Lipocalins | 87 | 13.74 | 28,919,147 | 87.108 |
| Nitrophorin-like protein | 2 | 0.32 | 18,162 | 0.055 |
| Classical lipocalins | 2 | 0.32 | 892 | 0.003 |
| Odorant binding protein family | 7 | 1.11 | 2,922 | 0.009 |
| Odorant binding protein family II | 7 | 1.11 | 28,714 | 0.086 |
| Yellow/Major royal jelly family | 1 | 0.16 | 389 | 0.001 |
| Phosphatidyl-ethanolamine-binding protein | 1 | 0.16 | 5,655 | 0.017 |
| Mys-3 family | 8 | 1.26 | 69,649 | 0.210 |
| Antigen 5 family | 6 | 0.95 | 302,608 | 0.911 |
| Pheromone-binding protein | 5 | 0.79 | 2,244 | 0.007 |
| Immune-related products | ||||
| Antimicrobial peptides | 9 | 1.42 | 7,031 | 0.021 |
| Histidine-rich peptide | 2 | 0.32 | 332 | 0.001 |
| GGY-rich peptide | 1 | 0.16 | 3,441 | 0.010 |
| C type lectin | 5 | 0.79 | 2,379 | 0.007 |
| Other lectins | 2 | 0.32 | 10,478 | 0.032 |
| Mucins | 1 | 0.16 | 5,561 | 0.017 |
| Conserved insect secreted proteins | 135 | 21.33 | 288,191 | 0.868 |
| Triatomine hemolysin and trialysin family | 4 | 0.63 | 426,640 | 1.285 |
| Deorphanized triatomine proteins | ||||
| 33.5 kDa triatomine family | 2 | 0.32 | 23,880 | 0.072 |
| Other deorphanized proteins | 11 | 1.74 | 409,693 | 1.234 |
| Orphan proteins | ||||
| Family 74–4 | 4 | 0.63 | 1,742 | 0.005 |
| Family 159–2 | 2 | 0.32 | 2,600 | 0.008 |
| Other putative secreted peptides | 248 | 39.18 | 324,699 | 0.978 |
| Total | 633 | 100 | 33,199,075 | 100 |
Analysis of the Secretory Complement of the P. megistus Sialotranscriptome
Enzymes
Apyrases (enzymes that hydrolyze ADP and ATP) of the 5′-nucleotidase family, ectonucleotide pyrophosphatase, inositol phosphatases, endonucleases, esterases, serine proteases, metalloproteases, other peptidases, and lipases were identified as putative secreted enzymes in the sialotranscriptome of P. megistus (Supp File 1 [online only]). Several of these may have a housekeeping role, having functions in cellular organelles such as lysosomes or the endoplasmic reticulum. However, indication of high relative expression may point these enzymes as truly secreted in saliva.
Six genes coding for apyrases have relatively high expression indexes (EI > 0.01), with RPKM varying from 210–1,790 and were assembled by a minimum of 17,000 reads each. Apyrase activity is ubiquitous in the saliva of blood-sucking arthropods (Ribeiro and Francischetti 2003) and an example of the convergent evolution scenario shaping the sialome of these animals, where Triatoma and mosquito enzymes were shown to be from members of the 5′-nucleotidase family (Champagne et al. 1995, Faudry et al. 2004), and those from bed bugs and sand flies to be derived from a novel enzyme family (Valenzuela et al. 1998, 2001a). Phylogenetic analysis of five of the apyrase CDS that appear full length shows they are most certainly products of different genes, where they cluster with homologs of other Triatoma species, indicative of gene duplication events (Supp Fig. 1 [online only]).
Inositol phosphatases are well expressed in triatomine and bed bug sialotranscriptomes (Ribeiro et al. 2012b), although their function is still unknown, unless they are delivered to the cytoplasm of their host cells, perhaps via exosomes. Eight transcripts were found coding for this class of enzymes; six appear to be full length. Phylogenetic analysis indicate that these derive from distinct genes, four of which are well expressed and are represented in Clade I of Supp Fig. 2 (online only), where they cluster with other enzymes previously described from triatomine and bed bug sialotranscriptomes. Two additional enzymes represented in Clade III are less well expressed and cluster with pea aphid enzymes, possibly being of housekeeping significance. PmegSigP-24706 and -26167 are well expressed, with EI = 0.037 and 0.027, having been assembled with >160,000 reads each.
Several transcripts coding for serine proteases were disclosed in this transcriptome. Three transcripts coded for truncated versions of gi| 295315341, a fibrinolytic salivary serine protease of P. megistus (Meiser et al. 2010). However, six full length CDS were obtained, including PmegSigP-24641 that best matches a secreted salivary trypsin of T. infestans, being assembled from >70,000 reads and having an EI = 0.01. Two transcripts coding for distantly related metalloproteases were also found, both relatively well expressed, and matching homologs found in previous T. matogrossensis sialotranscriptome (Assumpcao et al. 2012). The function of these metalloproteases is unknown but could have fibrinolytic function as occurs with tick metalloproteases (Francischetti et al. 2003).
Protease Inhibitor Domains
The sialotranscriptome of P. megistus revealed 13 CDS with single Kazal domains, four serpins, and one transcript each with a Tyropin (cysteine protease inhibitor domain), a pacisfastin and another with a vWA_interalpha trypsin inhibitor domain. The Kazal family of peptides includes protease inhibitors found in the gut of triatomines, usually with two Kazal domains (Campos et al. 2002, 2004; Lovato et al. 2006) as well as the horse fly salivary vasodilator named vasotab (Takac et al. 2006). Several of these are well expressed with EI > 0.01. Supp File 3 (online only) displays the phylogenetic relationship of 12 Kazal-domain containing peptides from P. megistus derived from the alignments with related Hemiptera-derived peptides, highlighting the diversity and expansion of this family in triatomines.
Small Molecule Binding Domains
Saliva of blood-sucking animals have proteins that have high affinity to agonists of inflammation such as biogenic amines and eicosanoids, named generically as kratagonists, from the Greek kratos = seize (Ribeiro and Arca 2009). Among the protein families recruited for this function and found in the sialotranscriptome of P. megistus are the lipocalins, odorant-binding proteins (OBP), similar to the modified OBP named D7 family of the Culicomorpha, and members of the Yellow protein family, shown in sand flies to be kratagonists of serotonin (Xu et al. 2011). A phosphatidyl-ethanolamine-binding protein was also found, which is truncated. Additionally, members of the juvenile hormone-binding protein family were found.
Among the 87 CDS coding for lipocalins, 67 have a length larger than 150 amino acids and appear full or near full length. Two additional lipocalins have their best matches to Rhodnius nitrophorins, and two additional CDS are most similar to traditional housekeeping lipocalins, such as apolipoprotein D. Pm-27201, PmegSigP-27197, PmegSigP-24561, PmegSigP-25319, and PmegSigP-24819 are the most expressed, having EIs of 1, 0.91, 0.39, 0.3, and 0.28, summing up 636,422 reads altogether.
Antigen 5 Family
This is a ubiquitous family consistently found in the sialome of blood-sucking arthropods. Recently a member of this family was shown to have antiplatelet and superoxide dismutase activity (Assumpcao et al. 2013). Six transcripts matches members of this family, one of which, Pm-24252, is well expressed with an EI = 0.04.
Immunity related
Transcripts related to antimicrobial peptides and lectins are commonly found in blood-feeding sialomes. Here we report CDS related to lysozyme, diptericin, attacin, and defensins, as well as histidine-rich peptides and glycine-tyrosine rich peptides related to worm antimicrobials (Ribeiro et al. 2012b).
Triatomine-Specific Families
Eight members of the MYS-3 family of proteins, first described in R. prolixus, were found in the sialotranscriptome of P. megistus. This is a very divergent family with unknown function. Members of the trialysin and triatomine hemolysin family were also found, two of which being well expressed.
Deorphanized Triatomine Proteins
Thirteen proteins previously found as unique in triatomine sialomes were here deorphanized, including several that are relatively well expressed such as PmegSigP-24618, PmegSigP-24094, PmegSigP-25075, and PmegSigP-25814 that have EIs larger than 0.01.
Other Putative Secreted Proteins
Conserved proteins belonging to uncharacterized families amounted to 135 CDS, only four of which have EIs of 0.01 or larger. Additionally we have found 258 CDS coding for putative secreted peptides that have no significant matches to known proteins, only three of which have EIs on the order of 0.01.
Analysis of Putative Housekeeping Proteins That Might be Relevant to the Salivary Function Based on Their Transcript Abundance
An arylsulfatase B CDS displaying a signal peptide indicative of secretion was relatively well expressed, the 565 amino acid protein being assembled from 35,718 reads and having an EI of 0.01. To the extent this enzyme is secreted in saliva and not lysosomal, it could affect sulfated polysaccharides in the host skin. Six transcripts coding for members of the cytochrome P450 family were found, all with EI larger than 0.01, Pm-24484 being assembled from >279,000 reads and having an EI = 0.05. These enzymes could be involved in the production of bioactive eicosanoids, such as prostaglandin E2, which is known to be secreted in tick saliva (Dickinson et al. 1976, Higgs et al. 1976), but not found so far in other blood-sucking arthropods. Paradoxically, within the lipid metabolism class, CDS for a putative 15-hydroxyprostaglandin dehydrogenase is highly expressed, with an EI = 0.02. This enzyme is linked to the catabolism of prostaglandins (Tai et al. 2006). A similar finding of a highly transcribed pair of a P450 enzyme and 15-hydroxyprostaglandin dehydrogenase was observed in the sialotranscriptome of T. rubida (Ribeiro et al. 2012a) where it was proposed that the dehydrogenase could be modulating the production of salivary prostaglandins.
Polymorphism Analysis
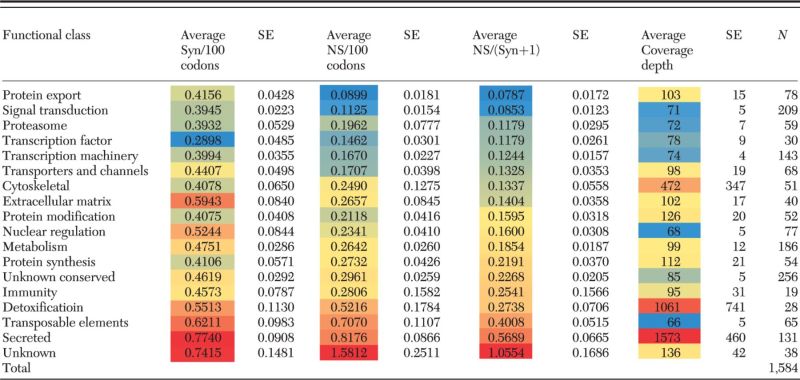
A total of 1,584 CDS were found to have polymorphisms and a minimum base coverage of 30 (RPKM = 7) following read mapping as described in the methods section (Supp File 1 [online only]). Transposable elements, the secreted and the unknown classes have the highest synonymous and nonsynonymous rates (Table 4), as well as the highest rates of nonsynonymous to synonymous mutations, further supporting the faster rate of evolution of salivary proteins from blood-sucking arthropods.
Table 4.
P. megistus polymorphism detected on a set of 1,584 CDS of 18 functional classes
 |
Conclusions
Sialomes of blood-sucking arthropods are objects of study due to their vast array of pharmacologically active components, their use as epidemiological markers of vector exposure and, in some cases, for their role in pathogen transmission and the possibility to target salivary proteins as vaccines to prevent vector-borne diseases. Presently, we have described and publicly deposited 3,703 protein and coding sequences to GenBank that should assist future proteomic work attempting to identify P. megistus pharmacologically active peptides as well as unique P. megistus epidemiological markers of vector exposure. These sequences should also help future P. megistus genome annotation by determining transcript exon–intron boundaries. Novel virus sequences were also identified.
Supplementary Data
Supplementary data are available at Journal of Medical Entomology online.
Acknowledgments
We are thankful to Dr. John Andersen for critical review of the manuscript. This work was partially supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, USA, (project Z01 AI000810-16: Vector-Borne Diseases: Biology Of Vector Host Relationship) and by a grant from the Grant Agency of the Czech Republic (Grant P302/11/P798), the Ministry of Education, Youth and Sports of the Czech Republic (KONTAKT II grant LH12002), and the Academy of Sciences of the Czech Republic (grant Z60220518).
References Cited
- Afonso C. L., Tulman E. R., Lu Z., Oma E., Kutish G. F., Rock D. L. 1999. The genome of Melanoplus sanguinipes entomopoxvirus. J. Virol. 73: 533–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F., Madden T. L., Schaffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arca B., Struchiner C. J., Pham V. M., Sferra G., Lombardo F., Pombi M., Ribeiro J. M. 2014. Positive selection drives accelerated evolution of mosquito salivary genes associated with blood-feeding. Insect Mol. Biol. 23: 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assumpcao T. C., Francischetti I. M., Andersen J. F., Schwarz A., Santana J. M., Ribeiro J. M. 2008. An insight into the sialome of the blood-sucking bug Triatoma infestans, a vector of Chagas' disease. Insect Biochem. Mol. Biol. 38: 213–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assumpcao T. C., Eaton D. P., Pham V. M., Francischetti I. M., Aoki V., Hans-Filho G., Rivitti E. A., Valenzuela J. G., Diaz L. A., Ribeiro J. M. 2012. An insight into the sialotranscriptome of Triatoma matogrossensis, a kissing bug associated with fogo selvagem in South America. Am. J. Trop. Med. Hyg. 86: 1005–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assumpcao T. C., Ma D., Schwarz A., Reiter K., Santana J. M., Andersen J. F., Ribeiro J. M., Nardone G., Yu L. L., Francischetti I. M. 2013. Salivary Antigen-5/CAP family members are Cu2+-dependent antioxidant enzymes that scavenge O2− and inhibit collagen-induced platelet aggregation and neutrophil oxidative burst. J. Biol. Chem. 288: 14341–14361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assumpcao T. C., Charneau S., Santiago P. B., Francischetti I. M., Meng Z., Araujo C. N., Pham V. M., Queiroz R. M., de Castro C. N., Ricart C. A., et al. 2011. Insight into the salivary transcriptome and proteome of Dipetalogaster maxima. J. Proteome Res. 10: 669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaty B. J., Marquartd W. C. 1996. Biology of disease vectors. University Press of Colorado, CO. [Google Scholar]
- Birol I., Jackman S. D., Nielsen C. B., Qian J. Q., Varhol R., Stazyk G., Morin R. D., Zhao Y., Hirst M., Schein J. E., et al. 2009. De novo transcriptome assembly with ABySS. Bioinformatics 25: 2872–2877. [DOI] [PubMed] [Google Scholar]
- Bussacos A. C., Nakayasu E. S., Hecht M. M., Assumpcao T. C., Parente J. A., Soares C. M., Santana J. M., Almeida I. C., Teixeira A. R. 2011. Redundancy of proteins in the salivary glands of Panstrongylus megistus secures prolonged procurement for blood meals. J. Proteomics 74: 1693–1700. [DOI] [PubMed] [Google Scholar]
- Campos I. T., Tanaka-Azevedo A. M., Tanaka A. S. 2004. Identification and characterization of a novel factor XIIa inhibitor in the hematophagous insect, Triatoma infestans (Hemiptera: Reduviidae). FEBS Lett. 577: 512–516. [DOI] [PubMed] [Google Scholar]
- Campos I. T., Amino R., Sampaio C. A., Auerswald E. A., Friedrich T., Lemaire H. G., Schenkman S., Tanaka A. S. 2002. Infestin, a thrombin inhibitor presents in Triatoma infestans midgut, a Chagas' disease vector: Gene cloning, expression and characterization of the inhibitor. Insect Biochem. Mol. Biol. 32: 991–997. [DOI] [PubMed] [Google Scholar]
- Cavassin F. B., Kuehn C. C., Kopp R. L., Thomaz-Soccol V., Da Rosa J. A., Luz E., Mas-Coma S., Bargues M. D. 2014. Genetic variability and geographical diversity of the main Chagas' disease vector Panstrongylus megistus (Hemiptera: Triatominae) in Brazil based on ribosomal DNA intergenic sequences. J. Med. Entomol. 51: 616–628. [DOI] [PubMed] [Google Scholar]
- Chagas A. C., Calvo E., Rios-Velasquez C. M., Pessoa F. A., Medeiros J. F., Ribeiro J. M. 2013. A deep insight into the sialotranscriptome of the mosquito, Psorophora albipes. BMC Genomics 14: 875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champagne D. E. 2005. Antihemostatic molecules from saliva of blood-feeding arthropods. Pathophysiol. Haemost. Thromb. 34: 221–227. [DOI] [PubMed] [Google Scholar]
- Champagne D. E., Smartt C. T., Ribeiro J. M., James A. A. 1995. The salivary gland-specific apyrase of the mosquito Aedes aegypti is a member of the 5'-nucleotidase family. Proc. Natl. Acad. Sci. USA 92: 694–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmelar J., Calvo E., Pedra J. H., Francischetti I. M., Kotsyfakis M. 2012. Tick salivary secretion as a source of antihemostatics. J. Proteomics 75: 3842–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson R. G., O'Hagan J. E., Shotz M., Binnington K. C., Hegarty M. P. 1976. Prostaglandin in the saliva of the cattle tick Boophilus microplus. Aust. J. Exp. Biol. Med. Sci. 54: 475–486. [DOI] [PubMed] [Google Scholar]
- Duckert P., Brunak S., Blom N. 2004. Prediction of proprotein convertase cleavage sites. Protein Eng. Des. Sel. 17: 107–112. [DOI] [PubMed] [Google Scholar]
- Faudry E., Lozzi S. P., Santana J. M., D'Souza-Ault M., Kieffer S., Felix C. R., Ricart C. A., Sousa M. V., Vernet T., Teixeira A. R. 2004. Triatoma infestans apyrases belong to the 5'-nucleotidase family. J. Biol. Chem. 279: 19607–19613. [DOI] [PubMed] [Google Scholar]
- Francischetti I. M. 2010. Platelet aggregation inhibitors from hematophagous animals. Toxicon 56: 1130–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francischetti I. M., Mather T. N., Ribeiro J. M. 2003. Cloning of a salivary gland metalloprotease and characterization of gelatinase and fibrin(ogen)lytic activities in the saliva of the Lyme disease tick vector Ixodes scapularis. Biochem. Biophys. Res. Commun. 305: 869–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh R. C., Ball B. V., Willcocks M. M., Carter M. J. 1999. The nucleotide sequence of sacbrood virus of the honey bee: An insect picorna-like virus. J. Gen. Virol. 80: 1541–1549. [DOI] [PubMed] [Google Scholar]
- Gomes R., Teixeira C., Teixeira M. J., Oliveira F., Menezes M. J., Silva C., de Oliveira C. I., Miranda J. C., Elnaiem D. E., Kamhawi S., et al. 2008. Immunity to a salivary protein of a sand fly vector protects against the fatal outcome of visceral leishmaniasis in a hamster model. Proc. Natl. Acad. Sci. USA 105: 7845–7850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs G. A., Vane J. R., Hart R. J., Porter C., Wilson R. G. 1976. Prostaglandins in the saliva of the cattle tick, Boophilus microplus(Canestrini) (Acarina, Ixodidae). Bull. Entomol. Res. 66: 665–670. [Google Scholar]
- Julenius K., Molgaard A., Gupta R., Brunak S. 2005. Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology 15: 153–164. [DOI] [PubMed] [Google Scholar]
- Karim S., Singh P., Ribeiro J. M. 2011. A deep insight into the sialotranscriptome of the Gulf Coast tick, Amblyomma maculatum. PLOS ONE 6: e28525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Tamura K., Nei M. 2004. MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief. Bioinform. 5: 150–163. [DOI] [PubMed] [Google Scholar]
- Li H., Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26: 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., and S. Genome Project Data Processing. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovato D. V., Nicolau de Campos I. T., Amino R., Tanaka A. S. 2006. The full-length cDNA of anticoagulant protein infestin revealed a novel releasable Kazal domain, a neutrophil elastase inhibitor lacking anticoagulant activity. Biochimie 88: 673–681. [DOI] [PubMed] [Google Scholar]
- Luo R., Liu B., Xie Y., Li Z., Huang W., Yuan J., He G., Chen Y., Pan Q., Liu Y., et al. 2012. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 1: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcilla A., Bargues M. D., Abad-Franch F., Panzera F., Carcavallo R. U., Noireau F., Galvao C., Jurberg J., Miles M. A., Dujardin J. P., et al. 2002. Nuclear rDNA ITS-2 sequences reveal polyphyly of Panstrongylus species (Hemiptera: Reduviidae: Triatominae), vectors of Trypanosoma cruzi. Infect. Genet. Evol. 1: 225–235. [DOI] [PubMed] [Google Scholar]
- Meiser C. K., Piechura H., Meyer H. E., Warscheid B., Schaub G. A., Balczun C. 2010. A salivary serine protease of the haematophagous reduviid Panstrongylus megistus: Sequence characterization, expression pattern and characterization of proteolytic activity. Insect Mol. Biol. 19: 409–421. [DOI] [PubMed] [Google Scholar]
- Miller J. R., Koren S., Sutton G. 2010. Assembly algorithms for next-generation sequencing data. Genomics 95: 315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen H., Brunak S., von Heijne G. 1999. Machine learning approaches for the prediction of signal peptides and other protein sorting signals. Protein Eng. 12: 3–9. [DOI] [PubMed] [Google Scholar]
- Ribeiro J. M., Francischetti I. M. 2003. Role of arthropod saliva in blood feeding: Sialome and post-sialome perspectives. Ann. Rev. Entomol. 48: 73–88. [DOI] [PubMed] [Google Scholar]
- Ribeiro J. M., Mans B. J., Arca B. 2010. An insight into the sialome of blood-feeding Nematocera. Insect Biochem. Mol. Biol. 40: 767–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro J. M., Assumpcao T. C., Pham V. M., Francischetti I. M., Reisenman C. E. 2012a. An insight into the sialotranscriptome of Triatoma rubida (Hemiptera: Heteroptera). J. Med. Entomol. 49: 563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro J. M., Chagas A. C., Pham V. M., Lounibos L. P., Calvo E. 2013. An insight into the sialome of the frog biting fly, Corethrella appendiculata. Insect Biochem. Mol. Biol. 1748: 191–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro J. M., Andersen J., Silva-Neto M. A., Pham V. M., Garfield M. K., Valenzuela J. G. 2004. Exploring the sialome of the blood-sucking bug Rhodnius prolixus. Insect Biochem. Mol. Biol. 34: 61–79. [DOI] [PubMed] [Google Scholar]
- Ribeiro J.M.C., Arca B. 2009. From sialomes to the sialoverse: An insight into the salivary potion of blood feeding insects. Adv. Insect Physiol. 37: 59–118. [Google Scholar]
- Ribeiro J.M.C., Assumpcao T.C.F., Francischetti I. M. B. 2012b. An insight into the sialomes of bloodsucking Heteroptera. Psyche 2012: 16 p. [Google Scholar]
- Santos A., Ribeiro J. M., Lehane M. J., Gontijo N. F., Veloso A. B., Sant' Anna M. R., Nascimento Araujo R., Grisard E. C., Pereira M. H. 2007. The sialotranscriptome of the blood-sucking bug Triatoma brasiliensis (Hemiptera, Triatominae). Insect Biochem. Mol. Biol. 37: 702–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield C. J., Galvao C. 2009. Classification, evolution, and species groups within the Triatominae. Acta Trop. 110: 88–100. [DOI] [PubMed] [Google Scholar]
- Schwarz A., von Reumont B. M., Erhart J., Chagas A. C., Ribeiro J. M., Kotsyfakis M. 2013. De novo Ixodes ricinus salivary gland transcriptome analysis using two next-generation sequencing methodologies. FASEB J. 27: 4745–4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz A., Medrano-Mercado N., Schaub G. A., Struchiner C. J., Bargues M. D., Levy M. Z., Ribeiro J. M. 2014. An updated insight into the sialotranscriptome of Triatoma infestans: Developmental stage and geographic variations. PLoS Negl. Trop. Dis. 8: e3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson J. T., Wong K., Jackman S. D., Schein J. E., Jones S. J., Birol I. 2009. ABySS: A parallel assembler for short read sequence data. Genome Res. 19:1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnhammer E. L., von Heijne G., Krogh A. 1998. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 6: 175–182. [PubMed] [Google Scholar]
- Tai H. H., Cho H., Tong M., Ding Y. 2006. NAD+-linked 15-hydroxyprostaglandin dehydrogenase: Structure and biological functions. Current pharmaceutical design 12: 955–962. [DOI] [PubMed] [Google Scholar]
- Takac P., Nunn M. A., Meszaros J., Pechanova O., Vrbjar N., Vlasakova P., Kozanek M., Kazimirova M., Hart G., Nuttall P. A., et al. 2006. Vasotab, a vasoactive peptide from horse fly Hybomitra bimaculata (Diptera, Tabanidae) salivary glands. J. Exp. Biol. 209: 343–352. [DOI] [PubMed] [Google Scholar]
- Thompson J. D., Gibson T. J., Plewniak F., Jeanmougin F., Higgins D. G. 1997. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25: 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D. R., Pimentel H., Salzberg S. L., Rinn J. L., Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Prot. 7: 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela J. G. 2002. High-throughput approaches to study salivary proteins and genes from vectors of diseaseInsect Biochem. Mol. Biol. 32: 1199–1209. [DOI] [PubMed] [Google Scholar]
- Valenzuela J. G., Charlab R., Galperin M. Y., Ribeiro J. M. 1998. Purification, cloning, and expression of an apyrase from the bed bug Cimex lectularius. A new type of nucleotide-binding enzyme. J. Biol. Chem. 273: 30583–30590. [DOI] [PubMed] [Google Scholar]
- Valenzuela J. G., Belkaid Y., Rowton E., Ribeiro J. M. 2001a. The salivary apyrase of the blood-sucking sand fly Phlebotomus papatasi belongs to the novel Cimex family of apyrases. J. Exp. Biol. 204: 229–237. [DOI] [PubMed] [Google Scholar]
- Valenzuela J. G., Belkaid Y., Garfield M. K., Mendez S., Kamhawi S., Rowton E. D., Sacks D. L., Ribeiro J. M. 2001b. Toward a defined anti-Leishmania vaccine targeting vector antigens: Characterization of a protective salivary protein. J. Exp. Med. 194: 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Oliveira F., Chang B. W., Collin N., Gomes R., Teixeira C., Reynoso D., My Pham V., Elnaiem D. E., Kamhawi S., et al. 2011. Structure and function of a “yellow” protein from saliva of the sand fly Lutzomyia longipalpis that confers protective immunity against Leishmania major infection. J. Biol. Chem. 286: 32383–32393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeddam J. L., Gordon K. H., Lauber C., Alves C. A., Luke B. T., Hanzlik T. N., Ward V. K., Gorbalenya A. E. 2010. Euprosterna elaeasa virus genome sequence and evolution of the Tetraviridae family: Emergence of bipartite genomes and conservation of the VPg signal with the dsRNA Birnaviridae family. Virology 397: 145–154. [DOI] [PubMed] [Google Scholar]
- Zhao Q. Y., Wang Y., Kong Y. M., Luo D., Li X., Hao P. 2011. Optimizing de novo transcriptome assembly from short-read RNA-Seq data: A comparative study. BMC Bioinform. 12: S2. [DOI] [PMC free article] [PubMed] [Google Scholar]