

Abstract
Mutations that affect calcium homeostasis (Ca2+) in rod photoreceptors are linked to retinal degeneration and visual disorders such as retinitis pigmentosa and congenital stationary night blindness (CSNB). It is thought that the concentration of Ca2+ in rod outer segments is controlled by a dynamic balance between influx via cGMP-gated (CNG) channels and extrusion via Na+/Ca2+, K+ exchangers (NCKX1). The extrusion-driven lowering of rod [Ca2+]i following light exposure controls their light adaptation and response termination. Mutant NCKX1 has been linked to autosomal-recessive stationary night blindness. However, whether NCKX1 contributes to light adaptation has not been directly tested and the mechanisms by which human NCKX1 mutations cause night blindness are not understood. Here, we report that the deletion of NCKX1 in mice results in malformed outer segment disks, suppressed expression and function of rod CNG channels and a subsequent 100-fold reduction in rod responses, while preserving normal cone responses. The compensating loss of CNG channel function in the absence of NCKX1-mediated Ca2+ extrusion may prevent toxic Ca2+ buildup and provides an explanation for the stationary nature of the associated disorder in humans. Surprisingly, the lack of NCKX1 did not compromise rod background light adaptation, suggesting additional Ca2+-extruding mechanisms exist in these cells.
Introduction
Calcium (Ca2+) signaling controls a wide range of biological functions. In photoreceptors, Ca2+ is tightly regulated and abnormal Ca2+ homeostasis has adverse effects on their function and survival. For example, mutations of phosphodiesterase (1) or the guanylyl cyclase-activating protein GCAP1 (2) that produce abnormally high Ca2+ levels result in retinal degeneration. Similarly, the abnormally low Ca2+ level produced by prolonged light exposure (3), constitutive activation of the phototransduction cascade by mutant rhodopsin (4), vitamin A deprivation, or defective visual cycle (5–8) and delayed shutoff of phototransduction owing to mutant rhodopsin kinase or arrestin (9,10) has also been proposed to regulate the life and death of photoreceptors (11). Finally, mutations that reduce Ca2+ influx through the cGMP-gated (CNG) transduction channels such as deletion of guanylyl cyclase (12,13), mutations (14) or deletion of CNG channel subunits in rods (15) or cones (16) (channelopathies) also lead to photoreceptor degeneration. Common among all these conditions is the resulting prolonged exposure to abnormal Ca2+ levels in photoreceptors (17,18).
In darkness, Ca2+ flow into the rod outer segment through open CNG channels is matched by its extrusion, believed to be mediated exclusively by the Na+/Ca2+, K+ exchanger NCKX1 [for recent review, see (19)]. NCKX1 removes Ca2+ by exchanging four Na+ for one Ca2+ and one K+ ions. Light stimulation activates the phototransduction cascade, causing closure of CNG channels that reduces Ca2+ influx. The continuing extrusion of Ca2+, presumably by NCKX1, leads to decreased [Ca2+]i (20–22). This decrease orchestrates a feedback modulation of phototransduction gain and response termination kinetics to extend the operating range of photoreceptors in a process commonly known as light adaptation (23–25). Therefore, it is reasonable to assume that NCKX1 plays a critical role in light adaptation. Additionally, as disrupting Ca2+ homeostasis is implicated in photoreceptor cell death (11,26,27), NCKX1 is likely to play a role in supporting rod survival as well. Indeed, mutant human NCKX1 has been linked to autosomal-recessive night blindness [CSNB (28)]. However, it is unknown how the function of the rods is affected by the lack of Ca2+ extrusion via NCKX1. In addition, the stationary nature of the disease is surprising considering the expected dramatic change in Ca2+ homeostasis in these rods.
Although the biochemical properties of NCKX1 have been extensively studied in vitro, its role in vivo is poorly understood. Surprisingly, deletion of the presumed cone exchanger NCXK2 in mice appeared to have no effect on cone photoreceptor function (29) raising the possibility of NCKX-independent mechanisms for Ca2+ extrusion in photoreceptors. To address the role of NCKX1 in regulating the mammalian rod Ca2+ homeostasis, cell survival and phototransduction, and to understand the mechanisms by which mutant NCKX1 causes night blindness in humans, we generated NCKX1-deficient (Nckx1−/−) mice. We then analyzed their vision, retina morphology and photoreceptor survival, as well as their rod phototransduction and light adaptation.
Results
Deletion of NCKX1 in mouse rods
To remove NCKX1, most of the second exon of the NCKX1-coding gene Slc24a1 was deleted and replaced by neomycin cassette (Fig. 1A). To confirm the deletion of NCKX1 from the rod photoreceptors, we performed western blot analysis on whole retinal extracts from WT controls and Nckx1−/− mice. We first used several commercially available antibodies. However, these proved not to be useful as they showed similar nonspecific bands in both WT and Nckx1−/− retinal extracts, and the antibody against bovine NCKX1 (30) did not cross-react with the murine protein. We therefore developed a monoclonal antibody (NCKX1 8H6) from a mouse immunized with a GST-fusion protein containing amino acid residues 700–760 of mouse NCKX1. The 8H6 antibody strongly reacted with a band at 230 kDa and two lower bands of various intensities in the WT samples (Fig. 1B). This pattern is identical to that seen in bovine retinal extracts using the bovine-specific NCKX1 antibody PMe1B3, with the full-length protein migrating at 230 kDa and the lower-molecular-weight bands being proteolytic fragments (30). As expected, these bands were not present in the Nckx1−/− samples, confirming the successful removal of NCKX1 expression. To evaluate the distribution of NCKX1 in mouse retina, we performed immunohistochemistry on WT and Nckx1−/− retinal sections. The 8H6 antibody showed specific labeling in the outer segment layer of the photoreceptors in WT mouse retinas (Fig. 1C). At higher magnification, the fluorescent signal appeared to outline the rod outer segment, a staining pattern consistent with its localization to the plasma membrane (30). No fluorescent signal was detected in the outer segments of Nckx1−/− mouse retina (Fig. 1C; green fluorescence signal at the outer plexiform layer is due to nonspecific staining of mouse vessels by the secondary antibody), although the outer segment layer was present as indicated by the robust rhodopsin staining (red). Together, these results demonstrate the successful deletion of NCKX1 and reveal that its absence does not prevent rod outer segment formation.
Figure 1.
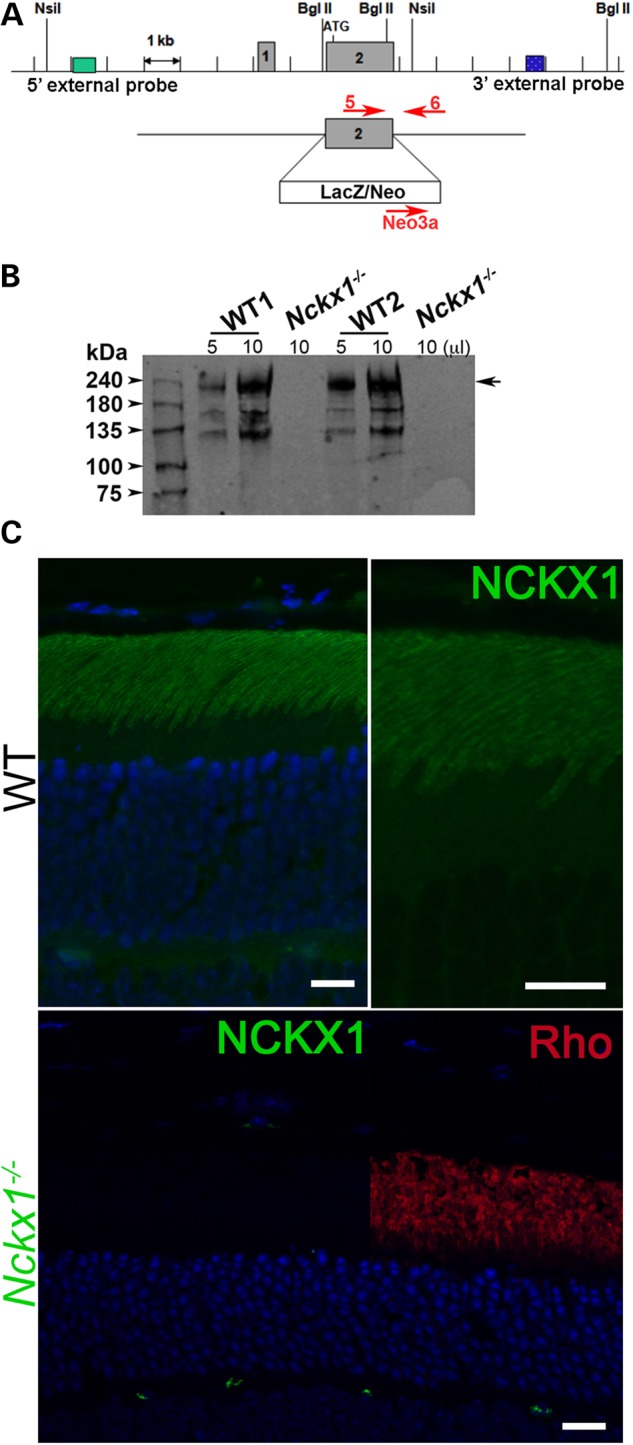
Generation of Nckx1−/− mice. (A) The second exon of the Slc24a1 gene encoding the Nckx1 protein was replaced with a LacZ/Neo cassette by homologous recombination in mouse embryonic stem cells. The positions of the DNA oligos used for genotyping are shown. (B) Western blot of whole retinal homogenates from two independent 1-month-old WT control and Nckx1−/− mice. (C) Localization of the NCKX1 protein (green) and rhodopsin (red) in WT and Nckx1−/− retinal sections. Nuclei were visualized by DAPI staining (blue). Scale bar = 25 μm.
NCKX1 deletion causes slow retinal degeneration and disrupts disk morphogenesis
We next evaluated the effect of NCKX1 knockout on retinal morphology. Figure 2A–D shows representative retinal sections from Nckx1−/− mice at ages 1, 6 and 12 months and from a WT mouse at 12 months (N ≥ 3). At 1 month of age (Fig. 2A), Nckx1−/− retina appeared similar to that of age-matched WT mouse (not shown). As the Nckx1−/− mice aged to 6 months, slight thinning of the outer segment and outer nuclear layer was observed (Fig. 2B). Progressive thinning of these retinal layers continued to 1 year of age (Fig. 2C). For comparison, retinal sections from a 1-year-old WT mouse are shown (Fig. 2D). To see whether these degenerative changes are associated with an injury response, retinal sections were stained with an antibody against glial fibrillary acid protein (GFAP), whose expression is often induced in Müller cells as a consequence of retinal degeneration (31). Nckx1−/− retinal section from 1-month-old mice showed GFAP immunoreactivity extending from the neurofilament layer toward the outer nuclear layer (Fig. 2E), a stereotypical pattern observed in degenerating retinas (31). Interestingly, GFAP staining attenuated as the mice aged despite ongoing degeneration, such that at 1 year of age, GFAP reactivity was largely confined to the neurofilament layer, similar to age-matched WT retina (Figs 2F–H). We then performed transmission electron microscopy (TEM) to detect ultrastructural changes. The Nckx1−/− retinal outer nuclear layer at 3 months of age was only slightly thinner compared with age-matched controls (compare Fig. 2I with K). However, TEM images of outer segments showed many of them to be disrupted, with their discs occasionally arranged longitudinally rather than horizontally (Fig. 2J, arrows), a phenotype reminiscent of Cngβ1−/− rods (32). In contrast, the discs of WT rods appeared uniform in size and were regularly stacked and arranged perpendicular to the longitudinal axis of the outer segment (Fig. 2H). Together, these results demonstrate that the deletion of NCKX1 in rods disrupts the morphology of their outer segments and results in slow progressive retinal degeneration.
Figure 2.

Age-dependent retinal degeneration in Nckx1−/− mice. Light microscope images of semithin retinal sections prepared from Nckx1−/− mice at the following ages: 1 month (A), 6 months (B) and 12 months (C) and WT control at 12 months (D). Immunofluorescence of glial fibrillary protein (GFAP, green) and nuclei stained with DAPI (blue) of frozen retinal sections prepared from Nckx1−/− mice at 1 month (E), 6 months (F) and 12 months (G) and WT control at 12 months (H). Light (I) and electron (J) micrograph from Nckx1−/− at 3 months of age, and light (K) and electron (L) micrographs from age-matched WT control. onl: outer nuclear layer; inl, inner nuclear layer; gcl, ganglion cell layer. Scale bar = 20 μm for light and 2 μm for electron microscope sections.
NCKX1 deletion leads to reduced expression of CNG channels
To examine how the absence of NCKX1 affects expression of other phototransduction proteins, we performed western blot analysis of whole retinal extracts from age-matched WT and Nckx1−/− mice aged 1 to 2 months (Fig. 3A). Expression levels were decreased for the majority of the proteins, perhaps owing to ongoing rod cell death. The lowered expression observed for the subunits of the CNG channels was particularly striking, by as much as 50% and exceeding the loss of rods at this age. The rod CNGβ1 protein contains a GARP domain at its amino-terminus. An antibody against the GARP epitope [Garp 4B1 (33)] detects both CNGβ1 as well as GARP2, a soluble protein arising from alternative splicing of the same gene (34,35). Interestingly, expression of the GARP2 was also downregulated and the protein appeared degraded (Fig. 3A, left panel). To further examine the relationship between NCKX1 and the CNG channel, localization of these proteins was investigated in frozen retinal sections prepared from Nckx1−/− and Cngβ1−/− mice (Fig. 3B). Consistent with previous reports (36,37), WT rod outer segments showed strong labeling of CNGβ1 whereas CNGβ1 immunofluorescence appeared dimmer in the Nckx1−/− retina but expression was still restricted to the rod outer segment layer [visualized by rhodopsin immunoreactivity (red) at the right]. To see whether the absence of CNG channels in turn affected NCKX1 localization, we also performed NCKX1 immunofluorescence labeling of Cngβ1−/− retinas (Fig. 3B, lower panel). The Cngβ1−/− retina showed normal NCKX1 localization to the outer segments. However, some NCKX1 labeling appeared punctate, a pattern characteristic of aggregated proteins. Together, these results show a marked downregulation of CNG channels in rods lacking NCKX1 as well as an altered staining pattern of NCKX1 in rods lacking CNGβ1.
Figure 3.
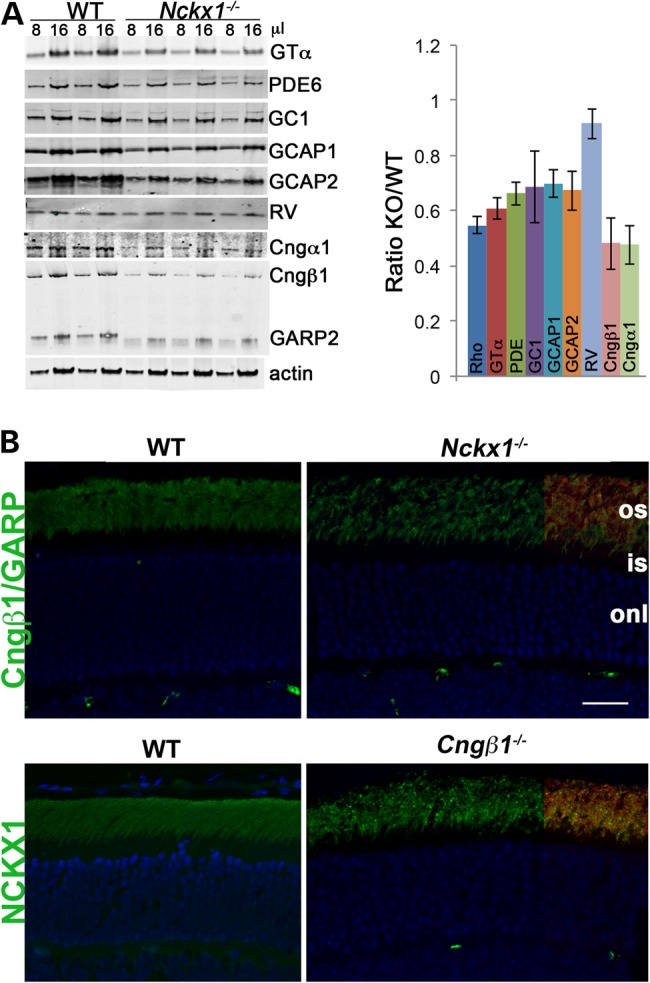
Altered expression of phototransduction proteins in Nckx1−/− retinas. (A) Representative western blots of the indicated phototransduction proteins in whole retinal homogenates prepared from WT control and Nckx1−/− mice at 1–2 months of age. Two different amounts (8 and 16 μl) were loaded per indicated genotype. Rhodopsin levels were quantified by the difference in absorption at 500 nm in dark and fully bleached solubilized retinas and the molar extinction coefficient of rhodopsin (n = 3) as described in Materials and Methods. In each experiment, protein levels were normalized to actin and then presented as a ratio of Nckx1−/− to WT (bar graph, n ≥ 7, mean ± SD). (B) Immunofluorescence localization of Cngβ1 and NCKX1 (green) and rhodopsin (red) in retinal sections from WT and the indicated knockout mice. Rhodopsin signal was shown only at the right side of the sections to demonstrate the outer segment layer. Nuclei were visualized by DAPI stain (blue). Scale bar = 25 μm.
Rod-mediated vision is desensitized and slow in NCKX1-deficient mice
Although the removal of NCKX1 led to slowly progressing rod degeneration, the retinas of 1- to 3-month-old Nckx1−/− mice appeared largely normal, suggesting that mutant rods might still contribute to mouse vision. To test that, we assessed the sensitivity of rod-driven dim light vision by OptoMotory (CerebralMechanics, Inc.) experiments in 2-month-old mice. This behavioral assay is based on an optokinetic tracking reflex when the animal is presented a moving grating, which can be adjusted for different spatial frequency or contrast. We determined the lowest contrast of moving gratings that the mice could resolve at different background light levels starting from the dimmest background. Results, illustrated in Figure 4A, demonstrate that Nckx1−/− mice attain the best contrast sensitivity (i.e. the lowest contrast threshold) at much brighter background light than WT control mice. However, Nckx1−/− mice were still able to resolve the gratings at substantially lower light levels than Gnat1−/− mice, whose vision is mediated solely by cone photoreceptors. These results suggest that although removal of NCKX1 desensitizes dim light vision, the mutant rods still contribute to the overall visual function. We did not observe a statistically significant difference in the contrast threshold in bright light between WT, Nckx1−/− and Gnat1−/− mice, suggesting that the function of cones is not compromised by the deletion of NCKX1.
Figure 4.

Suppression of rod-driven vision in Nckx1−/− mice. (A) Contrast threshold as a function of mean background luminance for WT control (black, n = 4), Nckx1−/− (red, n = 5) and Gnat1−/− (blue, n = 4) mice (mean ± SEM). (B) Insets show responses recorded from dark-adapted retinas perfused with Ames medium to flashes of light ranging from 1 to 2000 photons µm−2 for WT control mice (black) and from 1 to 15,600 photons µm−2 for Nckx1−/− mice (red). Population-averaged b-wave amplitudes as a function of flash energy for WT control (black, n = 4), Nckx1−/− (red, n = 4) and Gnat1−/− (blue, n = 3 retinas from two mice) mice (mean ± SEM). Smooth lines plot Equation 1 with the following parameter values, WT: E1/2,1 = 5.0 photons µm−2; Rmax,1 = 700 µV; n1 = 1.7; E1/2,2 = 920 photons µm−2; Rmax,2 = 270 µV; n2 = 2, Nckx1−/−: E1/2,1 = 41 photons µm−2, Rmax,1 = 517 µV; n1 = 1.2; E1/2,2 = 1400 photons µm−2; Rmax,2 = 113 µV; n2 = 1.2, and Gnat1−/−: E1/2,2 = 6100 photons µm−2; Rmax,2 = 413 µV; n2 = 1.5. (C) Representative responses to sinusoidal 100% contrast 0.5, 3 and 6 Hz flicker stimulus recorded from dark-adapted WT control (black) and Nckx1−/− (red) retinas. The mean intensity of flickering light was 18 and 8.5 photons µm−2 s−1 for control and Nckx1−/− mice, respectively. Y-axis scale is shown in 10-µV increments (major ticks) and x-axis scale is shown in 1-s increments (major ticks). (D) Fundamental amplitude Rf normalized with the Rf at 0.5 Hz is plotted as a function of flicker frequency (f) for WT control (black) and Nckx1−/− (red) mice (mean ± SEM).
We next used ERG recordings from dark-adapted mice to test the role of NCKX1 in rod-mediated signaling. The insets in Figure 4B exemplify responses to short test flashes of varying energy recorded from WT control and Nckx1−/− mice. Notably, the scotopic a-waves from Nckx1−/− mice were much smaller compared with WT control mice as will be described in more detail later. In order to quantify the sensitivity of rod-mediated signaling, we fitted a sum of two Hill functions (Equation 1) describing the rod and cone contributions to the b-wave amplitude data [Fig. 5B (38)]. The sensitivity of the rod pathway is described by the smaller half-saturating flash energy (E1/2,1). Consistent with our behavior results, E1/2,1 in Nckx1−/− mice was 0.9 log-units larger than that in WT controls but 2.2 log-units smaller than that in Gnat1−/− mice, demonstrating that the sensitivity of rod-to-rod bipolar cell signaling is decreased by the deletion of NCKX1. However, Ncxk1−/− rods still mediated a signaling pathway more sensitive than that driven by cones alone.
Figure 5.

Light responses from Nckx1−/− mouse rods. Representative responses recorded from dark-adapted WT controls (A) and Nckx1−/− (B) retinas to flashes of light ranging from 4 to 1800 photons µm−2 for control and from 8 to 460 photons µm−2 per rod for Nckx1−/− mice. Insets show responses from a single WT rod (A, flash energy from 8 to 1100 photons µm−2 per rod) and from several Nckx1−/− rods drawn in the recording pipette (B, flash energy 150 photons µm−2 per rod, mean response from seven groups of cells). (C) Responses to the flash delivering 8 photons µm−2 per rod normalized with the maximal response amplitude shown for WT control (black, n = 6 retinas from three mice) and Nckx1−/− (red, n = 6 retinas from four mice) mice (mean ± SEM). (D) Normalized response amplitudes plotted as a function of flash energy for WT control (black, n = 6 retinas from three mice) and Nckx1−/− (red, n = 6 retinas from four mice) mice (mean ± SEM). (E and F) Representative responses from dark-adapted WT mouse retinas perfused with 0.2% DMSO- (E, vehicle control) and 0.1 mm KB-R7943-containing (F) Ringer to flashes of light ranging from 7 to 1400 photons µm−2. The insets in E and F show responses to a dim flash in vehicle control (black and red) and KB-R7943 solution (blue and green) that have been normalized with the saturated response amplitude and flash energy (in photons µm−2) for WT and Nckx1−/− mouse retinas, respectively.
To test the effect of NCKX1 removal on the temporal resolution of rod vision, we recorded the ERG responses to dim flicker stimuli from WT and Nckx1−/− mice. Rods in control mice could easily follow the time course of the stimulus at 3 Hz and showed a robust flicker response at 6 Hz (Fig. 4C, left panels). In contrast, the rod flicker response in Nckx1−/− mice was near saturation already at 3 Hz and was completely blocked at 6 Hz (Fig. 4C, right panels). The inability of Nckx1−/− rods to follow flicker stimulation normally can also be seen from the rapid decline in their fundamental response amplitude, measured from peak-to-peak (Rf, see Materials and Methods) (Fig. 4D). These results are consistent with the notion that the slow Ca2+ extrusion in NCKX1-deficient rods delays the feedback to the phototransduction cascade and results in abnormally slow recovery of the rod photoresponse.
Rod photoresponses are 100-fold smaller in NCKX1-deficient mice
Our results so far demonstrate that NCKX1 is important for long-term survival of rods and the function of rod-mediated vision. However, Nckx1−/− rods still contribute to mouse vision. To understand how the removal of NCKX1 affects the function of rod photoreceptors, we first recorded from single rod photoreceptors by using the well-established suction electrode method. Whereas rods from WT control mice routinely gave 10–15 pA maximal responses (inset in Fig. 5A), individual Nckx1−/− mouse rods failed to produce detectable responses. However, drawing several rod outer segments into the pipette yielded consistently a very small (∼0.5 pA) but detectable maximal response (inset in Fig. 5B), suggesting that each mutant rod has a very small residual transduction channel current that can be suppressed by light. This result is reminiscent of the case of Cngβ1−/− mice where the majority of rods failed to respond to light and in those that did, the CNG channel current was reduced by ∼10-fold (32,39).
We next turned to transretinal ERG recordings to better quantify the effect of NCKX1 removal on rod physiology. We isolated the photoreceptor component of the ERG signal by using DL-AP4 and BaCl2 in the perfusion to block synaptic transmission and to remove its glial component, respectively. In these conditions, photoreceptor responses of the dark-adapted retina are dominated by rods and show kinetics and sensitivity comparable to these from single-cell recordings (40). Figure 5A illustrates a family of flash responses recorded from a WT control mouse retina. A similar experiment from a representative Nckx1−/− mouse retina revealed a dramatically reduced maximal response amplitude (Rmax) of the rods (Fig. 5B). Our average results demonstrated a 100-fold reduction of the rod Rmax in Nckx1−/− as compared with WT control mice (see Table 1). The dim flash responses of Nckx1−/− mouse rods also appeared slower than those of WT controls as exemplified by 88% larger time-to-peak (tp) and 175% longer integration time (Ti) (see Table 1). These results showing dramatically smaller and slower rod responses in the absence of NCXK1 are consistent with the behavior and ERG b-wave experiments demonstrating less sensitive and slower rod-mediated vision in the Nckx1−/− mice (Fig. 4).
Table 1.
Rod parameter values from transretinal ERG recordings of WT control and Nckx1−/− as well as from WT mouse retinas perfused with Ringer containing the NCX/NCKX inhibitor KB-R7943 (WT/Blocker, see Materials and Methods) and its vehicle (WT/DMSO)
| Parameter | Rmax | tp | Ti | SF,D | E1/2, a-wave |
|---|---|---|---|---|---|
| Control | 582 ± 50 | 129 ± 4 | 203 ± 20 | 1.1 ± 0.06 | 61 ± 4 |
| Nckx1−/− | 5.8 ± 0.4 | 243 ± 22 | 559 ± 51 | 2.8 ± 0.3 | 26 ± 4 |
| WT (DMSO) | 239 ± 28 | 143 ± 8 | NA | 0.6 ± 0.06 | NA |
| WT (Blocker) | 108 ± 15 | 231 ± 11 | NA | 1.0 ± 0.06 | NA |
All values are mean ± SEM, six retinas from three WT control, six retinas from four Nckx1−/−, four retinas from two WT mice perfused with DMSO-containing Ringer and three retinas from two WT mice perfused with KB-R7943-containing Ringer.
Rmax, the maximal rod response amplitude measured at the plateau after the initial peak; tp, time for a dim flash response (E = 4 or 8 photons µm−2 rod−1) to reach the peak amplitude; Ti, integration time defined as the integrated area of the dim flash response divided by its amplitude; SF,D, fractional sensitivity defined as dim flash response amplitude divided by flash energy (in photons µm−2) and Rmax; E1/2, half-saturating flash energy (see Materials and Methods). All parameter values between WT control and Nckx1−/− retinas as well as between WT retinas perfused with vehicle (0.2% DMSO) control solution and Blocker solution are statistically different (two-tailed Student's t-test, P < 0.006).
The size of the circulating current is determined by the number of CNG channels on the plasma membrane as well as the level of free cGMP that gates these channels open. Because the reduction of CNG channel expression in Nckx1−/− mice cannot fully explain the degree to which the circulating current was reduced, we also measured the level of cGMP in dark-adapted WT and Nckx1−/− retinas at 4 weeks using an enzyme-linked immunosorbant assay. We found the level of cGMP in the dark-adapted Nckx1−/− retinas to be significantly reduced compared with WT (31 ± 5 versus 71 ± 14 pmol/mg protein, mean ± SD, P ≤ 0.018). Assuming that these changes in total cGMP concentration reflect a reduction in free cGMP, and considering the cooperative binding of three cGMP molecules to the CNG channel (41–43), this 2.3-fold reduction in cGMP levels would be expected to produce a substantial additional reduction in the dark current. We also compared the effect of increasing cGMP concentration between Nckx1−/− and WT control rods on their maximal transretinal ERG a-wave amplitudes (in the presence of DL-AP4 and Barium) by using the PDE antagonist 3-isobutyl-1-methylxanthine (IBMX). Consistent with the lower baseline cGMP of Nckx1−/− rods, IBMX exposure (300 µm) increased their maximal response amplitude 2.2-fold more than that in WT control rods [4.9 ± 0.6-fold (n = 4) versus 2.2 ± 0.03-fold (n = 3) increase of Rmax, mean ± SEM]. Together, these findings provide an explanation for the bulk of the reduction in the circulating current.
Surprisingly, normalization of the responses to identical dim flashes from WT and Nckx1−/− mouse rods with their respective Rmax revealed that the fractional sensitivity was 2.5-fold higher in NCKX1-deficient rods (Fig. 5C and Table 1). Consistent with this, the half-saturating flash energy (E1/2) was also over 2-fold lower for Nckx1−/− rods compared with WT controls (Fig. 5D) (see also Table 1). Thus, although the maximal response amplitude of Nckx1−/− rods was severely reduced, their fractional sensitivity actually increased compared with WT controls. This result is consistent with the assumed role of NCXK1 in Ca2+ extrusion and indicates that the slowed Ca2+ feedback to the phototransduction cascade in the absence of NCKX1 yielded larger rod response and resulted in higher fractional sensitivity. This finding is reminiscent of the GCAPs knockout mice, where lack of a key component of the rod Ca2+ feedback, the GCAPs-stimulated synthesis of cGMP by guanylyl cyclases, results in several fold higher rod single photon response and fractional sensitivity (44).
In order to investigate whether other Ca2+ exchanger mechanisms in addition to NCKX1 modulate rod physiology, we compared the effect of the NCX inhibitor KB-R7943 that has been shown also to inhibit NCKX1 and NCKX2 (45,46), on light responses of WT and Nckx1−/− mice. We found that KB-R7943 slowed down and decreased the maximal amplitude of WT mouse rod flash responses (Fig. 5E, F and Table 1). Furthermore, similar to NCKX1 deletion, application of KB-R7943 increased the fractional sensitivity of WT rods (inset in Fig. 5E and Table 1). Notably, application of KB-R7943 to the Nckx1−/− retina did not affect much the maximal response amplitude or sensitivity of rods and actually accelerated their flash response kinetics (inset in Fig. 5F). Longer exposures to KB-7943 often gradually decreased response amplitudes, suggesting that 0.1 mm KB-7943 together with 0.2% DMSO in the Ringer might have some mild toxic effect on photoreceptor function. Together these results suggest that KB-R7943 can suppress NCKX1 function in vivo and that aside from NCKX1, no other KB-R7943-dependent Ca2+ extrusion mechanisms contribute to the regulation of dark-adapted rod sensitivity or flash response kinetics and amplitude.
Weber-like rod light adaptation is preserved in NCKX1-deficient mice
If NCKX1 is the only mechanism that can extrude Ca2+ from the rod outer segment, its deletion should remove the Ca2+-mediated feedback known to be critical for rod background light adaptation. For example, removal of the Ca2+ sensor proteins GCAPs that modulate cGMP synthesis compresses the range of background light levels over which rods can operate (44). To evaluate the effect of NCKX1 deletion of rod background adaptation, we compared the sensitivity of WT control and Nckx1−/− rods with short 1-ms test flashes delivered 5 s after the onset of a 10-s step of steady background light (Fig. 6A and B). Surprisingly, we found that rod sensitivity in background light (sF), normalized to its dark-adapted value (sF,D), could still be described by Weber-Fechner function (Equation 2) in Nckx1−/− retinas (Fig. 6C). This result is in contrast to previously published results with GCAPs−/− mice and indicates that over the 5 s of background light exposure, the level of Ca2+ in the rod outer segments declined and mediated normal background adaptation in Nckx1−/− rods. The deletion of NCKX1 shifted the rod operating range to dimmer background light intensities as exemplified by the ∼2-fold decrease of the sensitivity-halving background light intensity (I0). However, this shift could be easily explained by the higher fractional sensitivity and larger integration time of Nckx1−/− rods.
Figure 6.

Preserved Weber-like adaptation in Nckx1−/− mouse rods. Representative responses to 10-s steps of light with a flash delivered 5 s after the step onset from WT control (A) and Nckx1−/− (B) retinas. The step intensities ranged from 20 to 5300 and 2700 photons µm−2 s−1 in control and Nckx1−/− mice, respectively, whereas flash energies were increased from 4–8 photons µm−2 used with dim backgrounds up to 386 photons µm−2 with the brightest background light. (C) Normalized sensitivity (sF/sF,D, see Materials and Methods) plotted as a function of background light intensity for WT control (black, n = 6 retinas from three mice) and Nckx1−/− (red, n = 6 retinas from four mice) mice (mean ± SEM). Smooth lines plot Equation 2 with I0 of 41 and 85 photons µm−2 s−1, for WT and Nckx1−/− retinas, respectively. The dashed line here and in D represents the theoretical decrease in sensitivity of Nckx1−/− rods in the absence of adaptation (Equation 3). (D) Normalized sensitivity (sF/sF,D) plotted as a function of background light intensity for Nckx1−/− mice in Ames perfusion (triangles). Smooth red line plots a function I0n (I0n + IBn)−1 with I0 = 155 and n = 0.7. Insets show the flash responses (red) from which the sensitivity has been extracted in darkness and in backgrounds of 800 and 2700 photons µm−2 s−1. The scale bars are 2 µV (vertical) and 1 s (horizontal). The corresponding cone responses, determined by using double-flash technique (see Materials and Methods), are shown in gray. Sensitivity data are also plotted for one Nckx1−/− retina perfused with Ames containing 0.1 mm KB-7943 (green circles). Dark-adapted dim flash response for an Nckx1−/− retina exposed to KB-7943 (green trace) is shown for comparison with a dim flash response from an Nckx1−/− retina perfused with Ames without KB-7943 (red trace).
One potential concern with these results is that owing to the extremely small responses of NCKX1-deficient rods, a cone component to the overall transretinal response could affect our adaptation analysis particularly at higher background light intensities. However, independent cone recordings from Nckx1−/− retinas (see Fig. 7C below) revealed that cones would not contribute to the measured sensitivity for all but the two brightest background light intensities (720 and 2400 photons µm−2 s−1) shown in Figure 6C. In addition, because the background light used in this experiment is too dim to desensitize the cones, their sensitivity should not be affected by the increasing background light used here and thus the observed decline in sensitivity of Nckx1−/− retinas reflects adaptation of their rods.
Figure 7.

Cone survival and function in aging Nckx1−/− mice. (A) Frozen retinal sections prepared from the indicated mice were stained for PNA (red) and cone shortwave opsin (S-op, green). DAPI-stained nuclei are shown in blue. Scale bar = 25 μm. Cone flash response families recorded from dark-adapted 2 m.o. WT control (B), 2 m.o. Nckx1−/− (C) and 1 y.o. Nckx1−/− (D) mouse retinas perfused with Ames medium supplemented with 50 µm DL-AP4 and 100 µm Barium. Cone responses were isolated by double-flash technique with a constant probe flash of 65,000 photons (505 nm) µm−2 and 250-ms inter-stimulus interval between probe and test flashes (see Materials and Methods for details). Test flashes ranged from 700 to 27,000 photons µm−2 and from 170 to 27,000 photons µm−2 in control and Nckx1−/− mice, respectively. (E) Normalized (R/Rmax) response amplitudes plotted as a function of flash energies for young WT (black, n = 4), young Nckx1−/− (red, n = 3) and old Nckx1−/− (green, n = 3) mouse cones. Inset shows the maximal cone response amplitudes for these mice. Contrast sensitivity (F) and visual acuity (G) under photopic conditions are shown for young WT (black), young Nckx1−/− (red) and old Nckx1−/− (green) mice. Spatial frequency and temporal frequency were 0.128 c/d and 0.75 Hz, respectively, in E and contrast sensitivity was 100% in F (mean ± SEM). (H) Dissociated retinal cells were double-labeled with mouse cone arrestin antibody to label cones (red) and with NCKX1 (green). One representative cone cell is shown. No NCKX1 staining was associated with cones. Scale bar = 10 µm.
To confirm that Weber adaptation of rods is preserved in the absence of NCKX1, we did a set of experiments from 3-week-old Nckx1−/− mouse retinas perfused with Ames medium (containing 40 µm DL-AP4 and 100 µm BaCl2) in order to obtain as large responses as possible. Indeed, in these experiments, the maximal rod response amplitude of Nckx1−/− rods was on average 28 µV (n = 4), 5-fold higher than in Ringer solution (Fig. 5). Thus, it was possible to use dimmer flashes in light adaptation experiments facilitating extraction of pure rod responses that would not be contaminated by cones. We also recorded dark-adapted cone responses to identical flashes used in each background by using double-flash technique. Comparison of these cone responses to the flash responses recorded during background lights revealed that we could measure the sensitivity of rods up to ∼2700 photons µm−2 s−1 without a significant cone contribution (see insets in Fig. 6D). Even under these conditions rod background light adaptation was not compromised by removal of NCKX1 (Fig. 6D). Overall, we conclude that light adaptation of rods is not significantly compromised by deletion of NCKX1. This surprising result suggests that Ca2+ can somehow be extruded from the rod outer segment even in the absence of NCXK1.
To study the nature of this NCKX1-independent mechanism, we tested the effect of NCX/NCKX blocker KB-7943 (see above) to the Nckx1−/− rod light adaptation. These experiments were complicated by the occasional gradual decrease of rod responses in KB-7943 perfusion. Figure 6D (green circles) shows one example from a retina where sensitivity declined only minimally during the experiment. Despite the presence of KB-7943, Weber adaptation in the Nckx1−/− retina appeared to be preserved. Hence, KB-7943-sensitive mechanisms other than NCKX1, such as NCKX2 and NCXs, are likely not contributing to the rod light adaptation or Ca2+ extrusion from the rod outer segments.
Cone-mediated vision is not compromised in NCKX1-deficient mice
To evaluate the effect of the slow progressive rod degeneration in Nckx1−/− mice on the survival of their cones, retinal sections were stained for the cone markers peanut agglutinin (PNA) and cone S-opsin (S-op). Although the cone outer segments became progressively shorter in older mice, cone numbers appeared to be maintained (Fig. 7A), indicating the absence of detectable cone degeneration. To directly evaluate whether the slow degeneration of rods affects cone function in aging NCKX1-deficient retinas, we compared the responses of young (2 months old) and old (1 year old) Nckx1−/− and WT control mice by conducting transretinal ERG recordings. Cone responses to varying flash strengths isolated with double-flash technique (see Materials and Methods) from representative retinas of young WT (black) and Nckx1−/− (red) mice demonstrated that cones respond robustly to light stimulation even in the slowly degenerating Nckx1−/− retina (Fig. 7B–E). Surprisingly, cone responses from the Nckx1−/− retinas appeared larger in amplitude than those of WT control mice and this difference was statistically significant (P = 0.037, two-tailed t-test; see inset of Fig. 7E). Figure 7D and the inset in Figure 7E (green) show that cone response amplitudes in 1-year-old mice remained similar to those in young Nckx1−/− mice. The sensitivity of cones also appeared normal in the absence of NCKX1 and was not affected by age (Fig. 7E). As removal of NCKX1 appeared to increase cone response amplitudes, we looked for a possible expression of NCKX1 in cones by staining dissociated photoreceptor cells from WT retinas with cone arrestin (mCAR) and NCKX1 antibodies (Fig. 7H). NCKX1 did not co-localize with cone arrestin, confirming the rod-specific expression of NCKX1 in the mouse retina.
We next tested whether removal of NCKX1 compromises the cone-driven daytime vision of mice by performing OptoMotory behavior test under photopic light conditions (see Materials and Methods) from young WT and Nckx1−/− mice as well from 1-year-old Nckx1−/− mice. We did not find any statistically significant differences in contrast sensitivity (Fig. 7F) or visual acuity (Fig. 7G) between young WT and young or old Nckx1−/− mice. Overall, our results indicate that removal of NCKX1 does not compromise cone-mediated vision in mice up to 1 year of age, even when rod degeneration is already evident. These data are consistent with the phenotype of congenital stationary night blindness in humans with mutant NCKX1 (28).
Discussion
To date, NCKX1 is the only known Ca2+ extrusion mechanism in the rod outer segment plasma membrane. Indeed, as we show here, NCKX1 plays an important role in setting the sensitivity and temporal properties of rod signaling. Equally important, rod background light adaptation is not compromised by the deletion of NCKX1, indicating the presence of a previously unsuspected NCKX1-independent Ca2+ clearance mechanism in rod outer segments. The morphological and functional phenotype of our Nckx1−/− mice is comparable with the CSNB disease caused by mutation in the human NCKX1 (28), demonstrating the etiological significance and therapeutic potential of these mice. Finally, our results provide support for the previously suggested interaction between NCKX1 and CNG channels (47–50) in intact mouse rods and demonstrate that this interaction is important for normal channel function.
NCKX1 and the regulation of rod phototransduction
Extensive studies with bovine rod outer segments suggest that NCKX1 is the rod-specific Ca2+ extrusion mechanism (30,51–53). However, it was not clear from these studies how much NCKX1 regulates the physiology of intact mammalian rods and whether NCKX1 is the only mechanism responsible for Ca2+ extrusion from their outer segments. We demonstrate here that rods without NCKX1 are viable and contribute to mouse dim light vision and rod-mediated synaptic transmission even though the absolute rod response amplitudes are dramatically decreased by the deletion of NCKX1 (Figs 4 and 5). It is notable that the very small responses of rods that should not be transmitted to the rod bipolar cells (54,55) are able to support rod-mediated vision. Although this might be due to some developmental compensation in the Nckx1−/− mice, it is also possible that their small rod signals are transmitted through gap junctions to cone bipolar cells (which do not have a threshold) and integrated by them to produce robust ganglion cell responses and visual behavior (56–58). Future work by using, e.g. multi-electrode array retina recordings together with pharmacology and genetics, could reveal the exact mechanism by which such a small rod signals can be transmitted through the retina. Closer examination of rod photoreceptor responses revealed that removal of NCKX1 delayed their recovery and increased the fraction of channels closed by rhodopsin activation (Fig. 5). Interestingly, fractional dim flash responses of Nckx1−/− mouse rods (Fig. 5C) are reminiscent of those from GCAPs−/− mice that lack the dominant Ca2+-feedback mechanism (44). We found qualitatively similar effects of acute inhibition of Ca2+ extrusion by KB-R7943 as compared with the genetic removal of NCKX1 or GCAPs [Fig. 5, Table 1 and (44)]. Thus, NCKX1 plays a key role in mediating the fast reduction of Ca2+ concentration upon light stimulation that is critical for the timely recovery of the rod response. However, in contrast to GCAPs−/− mice, the rods in Nckx1−/− mice exhibit normal background light adaptation (Fig. 6). Hence, Ca2+ can apparently be extruded from the rod outer segments by an unknown NCKX1-independent mechanism within several seconds after the onset of background light. However, this mechanism is too slow to provide normal feedback to the phototransduction cascade in the time scale of dim flash responses (Figs 4C and 5C). The nature of this NCKX1-independent mechanism for Ca2+ extrusion from rod outer segments is currently unknown. Three possibilities exist: (1) Ca2+ is sequestered into intracellular stores (e.g. outer segment intradiscal space), (2) Ca2+ diffuses to the inner segment through the connecting cilium and (3) some yet-unknown Ca2+ extrusion mechanism exist in the mouse rod outer segment. The failure of the Ca2+ exchange inhibitor KB-R7943 to slow down the responses or compromise light adaptation of Nckx1−/− rods (inset in Figs 5F and 6D) implies that such Ca2+ extrusion is unlikely to be mediated by NCX or NCKX mechanisms. Future molecular, genetic and pharmacology experiments together with direct Ca2+ measurements from rod outer and inner segments should resolve the nature of this NCKX1-independent Ca2+ extrusion mechanism.
Structural and functional dependence between NCKX1 and the CNG channel
In a previous study using commercial antibodies, NCKX1 (Slc24a1) was reported to be localized in the inner segment, the outer and inner nuclear layers and ganglion cells in both the P13 and adult mouse eyes, but undetectable in rod outer segments (28). However, this labeling pattern is most likely due to nonspecific labeling, as in our hands the commercial antibodies produced similar staining patterns in wild type and Nckx1−/− retinal sections. In contrast, our immunofluorescence studies show a NCKX1 staining pattern that outlines the rod outer segments and is undetectable in other layers of the retina. This result is in agreement with earlier immunoelectron microscopy studies on bovine NCKX1 that indicate its localization to the rod outer segment plasma membrane (30). Thus, NCKX1 distribution is similar to that of the CNG transduction channels and its location is consistent with its presumed role of extruding Ca2+ from the rod outer segments.
In the absence of NCKX1, the level of CNG channel α and β-subunits decreased, as did GARP2, suggesting that the complex offered structural stability. This phenomenon has been previously observed for other phototransduction proteins, such as Gβ5 and RGS9 (59), PDE6γ and PDEαβ (60) and rod transducin βγ subunits (61). The similar effect of the absence of NCKX1 and CNG channel on the outer segment disk structure (32) suggests that the complex, and/or GARP2, is required for the maintenance of disc morphology. Overall, our results support previous biochemical evidence for the interaction between NCKX1 and CNG channels and further suggest that this interaction is critical for the proper function of CNG channels in mammalian rod photoreceptors.
The deletion of NCKX1 reduced rod sensitivity by 100-fold, a phenotype similar to CNGβ1−/− rods, although the underlying mechanism appears to be different. In the latter case, the absence of CNGβ1 profoundly affected the number of functional channels at the outer segment plasma membrane, thus reducing the amount of inward current. Although we found a 2-fold downregulation of CNG channel expression in Nckx1−/− mouse retina, its robust residual expression and its normal localization pattern (Fig. 3A and B) suggest additional mechanisms contributing to the dramatic reduction of rod signals. These channels appear to be functional, inasmuch as inhibition of PDE by IBMX increased the amount of circulating current in Nckx1−/− rods. Instead, the basis for the reduced current appears to be lowered cGMP levels in the Nckx1−/− retina. Decreased level of cGMP is consistent with the known inhibitory role of GCAPs on the cyclase under elevated [Ca2+]i owing to impaired Ca2+ extrusion. This reduction of cGMP, in turn, is expected to greatly impact the channel's conductance as the cooperativity of the channel activation by cGMP is 3 (41–43).
Importance of NCKX1 for the survival of rod photoreceptors
Mutations that disrupt the balance of cGMP synthesis/hydrolysis reactions leading to excessive number of open CNG channels are thought to kill photoreceptors through increased CNG-mediated Ca2+ influx (1,4,9,10,13,62,63). Reduction of Ca2+ influx into the outer segments through mutant CNG channels is also correlated with cell death (16,39,64). However, whether direct disruption of Ca2+ extrusion from the mammalian rod outer segments affects the long-term survival of rods had not been tested. If NCKX1 is the only Ca2+ extrusion mechanism in the rod outer segment, we expected its deletion to cause increased Ca2+ and severe rod degeneration. Hence, it was surprising that NCKX1 deletion resulted only in subtle morphological changes and slow retinal degeneration (Fig. 2) with preserved cone function even in 1-year-old Nckx1−/− mice (Fig. 7). The lack of severe degeneration of NCKX1-deficient rods suggests that these cells did not experience largely abnormal [Ca2+]i in their outer segments. As discussed earlier, one mechanism by which rods may achieve this is the NCKX1-independent regulation of [Ca2+]i. In addition, the reduced expression of transduction channels in Nckx1−/− rods, together with the reduced inward current, demonstrates a compensatory mechanism that would decrease Ca2+ influx when its extrusion is compromised. In summary, we believe that lowered channel expression, lowered channel conductance and NCKX1-independent extrusion help to prevent excessive accumulation of Ca2+ in Nckx1−/− rods. This might explain the slower than expected rod degeneration and the preservation of cone-mediated vision in mice lacking NCKX1.
Nckx1−/− mice as a model for human CSNB disease
Congenital stationary night blindness is a genetically heterogeneous disorder. Some of the associated mutations affect proteins involved in the synaptic transmission from rods to rod bipolar cells [TRPM1, CACNA1F, NYX (65–67)] and others are linked to phototransduction proteins [RHO, PDE6B, GNAT1 (68–70)]. A mutation in the human NCKX1 gene (SLC24A1), expected to suppress the exchanger activity, was also recently linked to CSNB disease (28). It should be emphasized that the CSNB diagnosis is based on long-term preservation of photopic vision and does not preclude slow rod degeneration. Indeed, Oguchi patients that harbor mutations in the ARR1 or GRK1 genes are diagnosed with CSNB despite ongoing rod cell death caused by constitutive transducin signaling (9,10,71). Our Nckx1−/− mouse results are consistent with the stationary nature of this disorder and suggest that this is due both to suppressed Ca2+ influx in the absence of NCKX1 and some unknown NCKX1-independent extrusion of Ca2+ from the rod outer segment. Thus, the Nckx1−/− mouse provides an excellent model for CSNB that should be useful for development and testing of therapeutic strategies to prevent this type of night blindness.
Materials and Methods
Animals
All experiments were conducted with adult mice [free of rd8 mutation (72)] of either sex. The Nckx1−/− mice were on a C57BL/6 background (PRID: MGI:3709293), and wild-type littermates were used as controls. For convenience, cone functional experiments were conducted with mice lacking the α subunit of rod transducin [Gnat1−/− (73)], which prevents rods from generating light responses but does not affect the morphology and function of cones (74). The mice used did not have rd8 mutation. Animals were kept in 12/12 h dark/light cycle with free access to water and food. All experimental protocols were in accordance with the Guide for the Care and Use of Laboratory Animals and were approved by the institutional Animal Studies Committee at Washington University.
Generation of NCKX1-deficient mouse
NCKX1 knockout mice (Nckx1−/−) were generated by Taconic Biosciences, Inc. following the strategy outlined in Figure 1A. Male chimeras were bred with C57BL/6 females to produce Nckx1+/− mice. These heterozygous mice were crossed to obtain Nckx1+/+ and Nckx1−/− mice that were used for experiments. All genotyping was performed by Transnetyx, Inc., and genotyping protocols are available upon request for the strain name NCKX1.
Generation of Cngβ1-deficient mice
CNGβ1 knockout mice (Cngβ1−/−) were generated by insertion of neoloxP cassette into intron 19 of the CNGβ1 gene (75). The insertion disrupted the splicing site and abrogated protein expression.
Morphology
Mice were euthanized by CO2 inhalation followed by cervical dislocation before eye enucleation. The whole eye was placed in 1/2 Karnovsky buffer (2.5% glutaraldehyde, 2% formaldehyde in 0.1 m cacodylate buffer, pH 7.2), and the cornea and lens were removed. The remaining eyecup was further fixed overnight in 1/2 Karnovsky buffer at 4°C, rinsed in 0.1 m cacodylate buffer and prepared into epoxy resin blocks as previously described (76). The central retina was sectioned along the superior–inferior axis at 1 micron thickness for light microscopy and 60 nm for TEM.
Immunofluorescence
The eyecup was prepared as described earlier in a different fixation buffer (4% formaldehyde, 0.5% glutaraldehyde in 0.1 m cacodylate buffer, pH 7.2) for 1 h and rinsed three times, 10 min each, in 0.1 m cacodylate buffer. The eyecups were then cryoprotected in 30% sucrose overnight at 4°C and then embedded in O.C.T™ (Tissue-Tech®) and sectioned at 10 μm thickness in a cryostat (CM 3050 S, Leica Microsystems) and stored at −80°C. The thawed sections were air-dried and blocked with 2% bovine serum albumin, 2% goat serum and 0.3% Triton X-100 in phosphate-buffered saline (PBS) for 1 h and then incubated with the following mouse monoclonal antibodies: NCKX1 8H6 (1:20, generated from a mouse immunized with a GST-fusion protein containing amino acids 700–760 of mouse NCKX1), GARP 4B1 antibody [1:20 (33)] and rhodopsin 1D4 [1:1000 (77)] or the following rabbit polyclonal antibodies: GFAP (1:500, Millipore AB5804), cone S-opsin [1:100 (78)]. The sections were incubated overnight at 4°C, rinsed and incubated for 1 h with a fluorescein- or Texas Red-labeled secondary antibody (1:400, Vector Laboratories). The sections were further rinsed and mounted in Vectashield containing DAPI (Vector Laboratories). Rhodamine-conjugated PNA (Vector Laboratories) was diluted 1:100 in buffer (PBS containing 1 mm CaCl2, 1 mm MgCl2, 1 mg/ml BSA) and incubated on the slides for 1 h at room temperature. For immunofluorescence on dissociated cells, isolated retinas were chopped in PBS. Dissociated cells were placed on gelatin-coated glass slides and fixed with 4% formaldehyde in PBS for 5 min. The cells were rinsed in PBS and processed for immunofluorescence as described for retinal sections. The primary antibodies used were NCKX1 8H6 and a rabbit antibody against mouse cone arrestin [LUMI-J, 1:100 dilution (79,80)]. The images were obtained using an LSM 5 confocal microscope (Zeiss Microscope). Images were first obtained for WT control sections, and the identical settings were then used to obtain images from Nckx1−/− sections.
Protein expression assays
The expression levels of rhodopsin were measured by spectroscopy. Mice were dark-adapted overnight, and the retinas were dissected under infrared light. Retinas were solubilized in 200 μl 1% dodecylmaltoside in PBS containing complete mini protease inhibitor (Roche Applied Sciences) for 3 h at 4°C, followed by 4000 rpm centrifugation for 3 min to remove particulates. The supernatant was transferred to a quartz cuvette to measure the UV/visible (270–700 nm) absorption spectra using spectrophotometer DU640 (Beckman Coulter). The concentration of rhodopsin was calculated by the difference in absorbance before and after full bleach by white light and its extinction coefficient (40,600 M−1 cm−1). The value was normalized to the total protein concentration estimated by the absorbance at 280 nm.
The expression levels of the other phototransduction proteins were quantified by western blotting. Each isolated retina was homogenized in 150 μl buffer (150 mm NaCl, 50 mm Tris pH 8.0, 0.1% NP-40, 0.5% deoxycholic acid) containing 0.1 mm PMSF and complete mini protease inhibitor. DNase I (30U, Roche Applied Sciences) was then added and incubated at room temperature for 30 min. The total protein amount of each sample was determined by the BCATM Protein Assay Kit (Thermo Scientific). An equal amount of retinal homogenate from each sample was electrophoresed on 4–12% Bis–Tris SDS–PAGE Gel (Invitrogen) followed by transfer to nitrocellulose membrane (Whatman™, GE Healthcare Life Sciences) and incubated overnight with the following primary antibodies: rabbit anti-PDE polyclonal antibody (1:1000, Cytosignal, PAB-06800), rabbit anti-ROS-GC1 polyclonal antibody (1:500, Santa Cruz, sc50512), mouse Anti-Gtα antibody (1:5000, EMD4Biosciences, 371740), rabbit polyclonal anti-GCAP1 and GCAP2 antibodies [1:2000 and 1:1000, respectively (81)], rabbit anti-Recoverin antibody (1:1000), mouse anti-GARP 4B1 antibody (1:500), mouse anti-CNGα antibody PMc 1D1 [1:500 (37)], mouse NCKX1 8H6 antibody (1:1000) and mouse anti-Actin antibody (1:5000, Millipore, MAB1501). The membranes were then incubated with fluorescently labeled secondary antibodies (1:10,000, LI-COR biosciences, 926–31081) at room temperature for 1 h and detected by Odyssey infrared imaging system. The intensity of individual bands was quantified using Image J. Quantification of Western data was obtained using at least three independent sets of experiments (n ≥ 3).
cGMP enzyme-linked immunosorbant assay
Mice were dark-adapted overnight and killed as described earlier. Retinas were isolated under infrared light and immediately frozen in liquid nitrogen. The frozen retinas were homogenized in 150 μl cold 6% trichloroacetic acid followed by six times repeated extraction with water-saturated ether. The aqueous fraction was dried in a vacuum centrifuge. The total cGMP was determined by cyclic GMP XP® Assay Kit (Cell Signaling #4360S) following manufacturer's protocol.
Behavior
Contrast thresholds of 2- to 3-month-old age-matched WT control, Nckx1−/− and Gnat1−/− female mice were determined as described previously by using a commercial Optomotry system [Cerebral Mechanics (82) (61)] at different background light levels ranging from −7.2 to 1.9 lg(Cd m−2). Temporal (ft) and spatial (fs) frequency as well as speed of the moving gratings (vD) were 0.75 Hz, 0.128 cyc deg−1 and 5.4 deg s−1 in all experiments and background light conditions. All mice were dark-adapted (>12 h) before experiments and contrast thresholds were determined starting from the dimmest background. At the background range where contrast threshold decreased with the steepest slope with respect to background luminance, the measurement was repeated in each mouse with dimmer background after using ∼1 log-unit brighter background to ensure that light adaptation did not have a significant effect on the measured contrast thresholds. Contrast sensitivity and visual acuity were also determined under photopic conditions in young WT and KO mice as well as in 1-year-old KO mice. In these experiments, mean background light intensity and vD were 1.9 lg(Cd m−2) and 12 deg s−1, respectively. In contrast sensitivity experiments, fs was set to 0.128 cyc deg−1 and in visual acuity experiments contrast was set to 100%.
Single-cell recordings
Rod photoreceptor outer segment current was recorded with a suction electrode from retinal pieces perfused with Locke's solution as described previously (78). The perfusion solution contained (in mm): NaCl, 112; KCl, 3.6; MgCl2, 2.4; CaCl2, 1.2; HEPES, 10; NaHCO3, 20; Na2-succinate, 3; Na-glutamate, 0.5; glucose, 10. In addition, the solution was supplemented with 0.1% of MEM vitamins and amino acids (Sigma–Aldrich), and equilibrated with 95%O2/5%CO2 at 37°C. The recording electrode contained (in mm): NaCl, 140; KCl, 3.6; MgCl2, 2.4; CaCl2, 1.2; HEPES, 3; glucose, 10. The pH was adjusted to 7.4 with NaOH. Light stimulation and calibration were performed as in transretinal ERG recordings (see below).
Transretinal ERG
Transretinal ERG recordings were performed from >12 h dark-adapted mice. Animals were euthanized by CO2 inhalation followed by cervical dislocation under dim red light. Retinas were dissected under IR light by using a dissection microscope equipped with IR converters, placed photoreceptor-side up on black filter paper (HABG01300, Millipore), and set on a custom-build specimen holder similar to that described previously (83,84). In b-wave flicker and also some a-wave experiments, retinas were perfused at 3–5 ml min−1 with bicarbonate-buffered Ames medium (Sigma–Aldrich) bubbled with 95%O2/5%CO2 at 37°C. Rod photoreceptor responses were recorded from retinas perfused with HEPES-buffered Ringer's solution containing (in mm): Na+, 133.9; K+, 3.3; Mg2+, 2.0; Ca2+, 1.0; Cl−, 143.2; glucose, 10.0; EDTA, 0.01; HEPES, 12.0, buffered to pH 7.5 with NaOH. The solution was supplemented with 0.72 g/l Leibovitz culture medium L-15 (Sigma–Aldrich) to improve retina viability and with 40 µm DL-AP4 (Tocris Biosciences) and 100 µm BaCl2 to isolate the photoreceptor component of the ERG signal. The temperature of the retina was maintained at 37°C by circulating the perfusion tubing through a heated ceramic peace just before the retina and by measuring it with a thermocouple (Physitemp Instruments, Inc., IT-18) immersed in the perfusion right after the retina. Signal was amplified by a differential amplifier (DP-311, Warner Instruments) and tunable active filter (Krohn-Hite Corporation, model 3382), low-pass filtered at 300 Hz (8-pole Bessel, Krohn-Hite Corporation, model 3382) and sampled at 10 kHz with 0.03 µV resolution by using a digitizer (1440A Digidata, Molecular Devices) and pCLAMP 10 software (Molecular Devices). Light stimulation was provided by a custom-build LED system that produced a homogenous light spot overfilling the effective measurement area of 0.5 mm in diameter at the central retina. LED light was aligned on the retina by using an optical cable (Newport, 77536) and optics of an inverted microscope. Light intensity and the length of light flashes (1 ms) and light steps were controlled by an LED driver (Thorlabs, LDC210C) and neutral density filters. The peak wavelength of the LED light spectrum was 505 nm (Rebel star, SR-01-E0070), and the total light power was measured by calibrated optometer (UDT Instruments, Model 211) near the plane of the retina. The intensity was then calculated based on the light spot area at the plane of the retina (2.35 mm in diameter) and converted to a number of 505 nm photons µm−2 s−1. An inhibitor of Ca2+ exchangers, KB-R7943 mesylate (Tocris Bioscience) or IBMX (Sigma–Aldrich), was used in a set of experiments. A 50 mm KB-R7943 or 30 mm IBMX stock solution was prepared in DMSO. These stock solutions were added to Ringer solution to achieve 0.1 mm (0.2%) or 0.3 mm (0.1%) final concentration in KB-R7943 and IBMX experiments, respectively. In KB-R7943 experiments, retinas were incubated for 30–60 min in KB-R7943 Ringer before recordings that were conducted under perfusion also containing KB-R7943 (0.1 mm). Control recordings were made from retinas perfused with Ringer containing 0.2% or 0.1% DMSO in KB-R7943 and IBMX experiments, respectively. A sum of two Hill functions
| (1) |
was fitted to the b-wave amplitude (R) data of the WT and Nckx1−/− mouse retinas. There, E is the flash energy (in photons µm−2), E1/2,1, E1/2,2 are the half-saturating flash energies, Rmax,1, Rmax, 2 are the maximal b-wave amplitudes, and n1 and n2 are the Hill-coefficients for the presumed rod- and cone-driven responses (38). The sum Rmax,1 + Rmax,2 was set equal to the maximal b-wave amplitude measured with the brightest flash used. In case of Gnat1−/− mice or when recording photoreceptor responses (Ringer's solution with blockers), only one term of the right-hand side of the equation was used as the responses in those conditions were dominated by only one photoreceptor type. In some transretinal experiments, cone responses were isolated using double-flash technique from WT mouse retinas (40). We used 65,000 photons (505 nm) µm−2 pre-flash and various test flashes to elicit cone responses with 250 ms inter-stimulus interval. In flicker ERG experiments, the fundamental amplitude (Rf) was determined from the peak-to-peak amplitude of the ERG response at >2 s after the flicker stimulus onset when the amplitudes had reached a stable value. Mean values of 100% contrast sinusoidal flicker stimulus intensities are given in photons (505 nm) µm−2 s−1, where Imax is the maximal intensity at the peak of the sinusoidal stimulus. In light adaptation experiments, a Weber-Fechner function
| (2) |
was fitted to the sensitivity data recorded from WT control and Nckx1−/− mouse retinas. There, sF is the sensitivity of the rods to dim flash of light defined as the response amplitude divided by the flash energy (R/E), sF,D is the sensitivity in darkness, I is the intensity of background light in photons (505 nm) µm−2 s−1 and I0 is the sensitivity-halving background light intensity. The dark-adapted fractional sensitivity, SF,D, represents the sensitivity in darkness, sF,D, normalized to the corresponding maximal response Rmax. Light adaptation data in Nckx1−/− mice were compared with a theoretical function that describes the decay of sensitivity as a function of background light in the absence of any light adaptation [all the active (Ca2+)-feedback mechanisms removed (75)]
| (3) |
where Rmax is the maximal saturated response amplitude and Ti is the integration time of a dark-adapted dim flash response (integrated area between the baseline and a dim flash response divided by the peak amplitude).
Statistical analysis
Unless noted otherwise, two-tailed unpaired Student's t-test was used to test for the significance of difference in the mean values of two sample groups. P-values of <0.05 were considered to be statistically significant.
Study approval
The maintenance and treatment of the animals followed the protocols approved by Washington University Animal Studies Committee.
Funding
This work was supported by National Institutes of Health (EY019312 and EY021126 to V.J.K., EY012155 to J.C., EY002422 to R.M., EY002687 to the Department of Ophthalmology and Visual Sciences at Washington University); and by Research to Prevent Blindness.
Acknowledgements
We thank Janis Lem for the Gnat1−/− animals, C. –K. Chen for the recoverin antibody, Cheryl Craft for the cone arrestin antibody, Paul Schnetkamp for the GST-NCKX1 fusion protein and Laurie Molday for technical assistance in generating the NCKX 8H6 monoclonal antibody.
Conflict of Interest statement. None declared.
References
- 1.Farber D.B. (1995) From mice to men: the cyclic GMP phosphodiesterase gene in vision and disease. The proctor lecture. Invest. Ophthalmol. Vis. Sci., 36, 263–275. [PubMed] [Google Scholar]
- 2.Sokal I., Li N., Surgucheva I., Warren M.J., Payne A.M., Bhattacharya S.S., Baehr W., Palczewski K. (1998) GCAP1 (Y99C) mutant is constitutively active in autosomal dominant cone dystrophy. Mol. Cell, 2, 129–133. [DOI] [PubMed] [Google Scholar]
- 3.Rapp L.M., Williams T.P. (1980) The role of ocular pigmentation in protecting against retinal light damage. Vision Res., 20, 1127–1131. [DOI] [PubMed] [Google Scholar]
- 4.Dizhoor A.M., Woodruff M.L., Olshevskaya E.V., Cilluffo M.C., Cornwall M.C., Sieving P.A., Fain G.L. (2008) Night blindness and the mechanism of constitutive signaling of mutant G90D rhodopsin. J. Neurosci., 28, 11662–11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dowling J.E., Wald G. (1960) The biological function of Vitamin A acid. Proc. Natl Acad. Sci. USA, 46, 587–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fan J., Woodruff M.L., Cilluffo M.C., Crouch R.K., Fain G.L. (2005) Opsin activation of transduction in the rods of dark-reared Rpe65 knockout mice. J. Physiol., 568, 83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Redmond T.M., Yu S., Lee E., Bok D., Hamasaki D., Chen N., Goletz P., Ma J.X., Crouch R.K., Pfeifer K. (1998) Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat. Genet., 20, 344–351. [DOI] [PubMed] [Google Scholar]
- 8.Fain G.L., Lisman J.E. (1993) Photoreceptor degeneration in vitamin A deprivation and retinitis pigmentosa: the equivalent light hypothesis. Exp. Eye Res., 57, 335–340. [DOI] [PubMed] [Google Scholar]
- 9.Chen C.K., Burns M.E., Spencer M., Niemi G.A., Chen J., Hurley J.B., Baylor D.A., Simon M.I. (1999) Abnormal photoresponses and light-induced apoptosis in rods lacking rhodopsin kinase. Proc. Natl Acad. Sci. USA, 96, 3718–3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen J., Simon M.I., Matthes M.T., Yasumura D., LaVail M.M. (1999) Increased susceptibility to light damage in an arrestin knockout mouse model of Oguchi disease (stationary night blindness). Invest. Ophthalmol. Vis. Sci., 40, 2978–2982. [PubMed] [Google Scholar]
- 11.Fain G.L. (2006) Why photoreceptors die (and why they don't). Bioessays, 28, 344–354. [DOI] [PubMed] [Google Scholar]
- 12.Semple-Rowland S.L., Lee N.R., Van Hooser J.P., Palczewski K., Baehr W. (1998) A null mutation in the photoreceptor guanylate cyclase gene causes the retinal degeneration chicken phenotype. Proc. Natl Acad. Sci. USA, 95, 1271–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang R.B., Robinson S.W., Xiong W.H., Yau K.W., Birch D.G., Garbers D.L. (1999) Disruption of a retinal guanylyl cyclase gene leads to cone-specific dystrophy and paradoxical rod behavior. J. Neurosci., 19, 5889–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dryja T.P., Finn J.T., Peng Y.W., McGee T.L., Berson E.L., Yau K.W. (1995) Mutations in the gene encoding the alpha subunit of the rod cGMP-gated channel in autosomal recessive retinitis pigmentosa. Proc. Natl Acad. Sci. USA, 92, 10177–10181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huttl S., Michalakis S., Seeliger M., Luo D.-G., Acar N., Geiger H., Hudl K., Mader R., Haverkamp S., Moser M. et al. (2005) Impaired channel targeting and retinal degeneration in mice lacking the cyclic nucleotide-gated channel subunit CNGB1. J. Neurosci., 25, 130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu J., Morris L., Fliesler S.J., Sherry D.M., Ding X.-Q. (2011) Early-onset, slow progression of cone photoreceptor dysfunction and degeneration in CNG channel subunit CNGB3 deficiency. Invest. Ophthalmol. Vis. Sci., 52, 3557–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lisman J., Fain G. (1995) Support for the equivalent light hypothesis for RP. Nat. Med., 1, 1254–1255. [DOI] [PubMed] [Google Scholar]
- 18.Fain G.L., Lisman J.E. (1999) Light, Ca2+, and photoreceptor death: new evidence for the equivalent-light hypothesis from arrestin knockout mice. Invest. Ophthalmol. Vis. Sci., 40, 2770–2772. [PubMed] [Google Scholar]
- 19.Schnetkamp P.P., Jalloul A.H., Liu G., Szerencsei R.T. (2014) The SLC24 family of K(+)-dependent Na(+)-Ca(2)(+) exchangers: structure-function relationships. Curr. Top. Membr., 73, 263–287. [DOI] [PubMed] [Google Scholar]
- 20.Sampath A.P., Matthews H.R., Cornwall M.C., Fain G.L. (1998) Bleached pigment produces a maintained decrease in outer segment Ca2+ in salamander rods. J. Gen. Physiol., 111, 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sampath A.P., Matthews H.R., Cornwall M.C., Bandarchi J., Fain G.L. (1999) Light-dependent changes in outer segment free-Ca2+ concentration in salamander cone photoreceptors. J. Gen. Physiol., 113, 267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gray-Keller M.P., Detwiler P.B. (1994) The calcium feedback signal in the phototransduction cascade of vertebrate rods. Neuron, 13, 849–861. [DOI] [PubMed] [Google Scholar]
- 23.Nakatani K., Yau K.W. (1988) Calcium and light adaptation in retinal rods and cones. Nature, 334, 69–71. [DOI] [PubMed] [Google Scholar]
- 24.Matthews H.R., Murphy R.L., Fain G.L., Lamb T.D. (1988) Photoreceptor light adaptation is mediated by cytoplasmic calcium concentration. Nature, 334, 67–69. [DOI] [PubMed] [Google Scholar]
- 25.Arshavsky V.Y., Burns M.E. (2012) Photoreceptor signaling: supporting vision across a wide range of light intensities. J. Biol. Chem., 287, 1620–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paquet-Durand F., Beck S., Michalakis S., Goldmann T., Huber G., Muhlfriedel R., Trifunovic D., Fischer M.D., Fahl E., Duetsch G. et al. (2011) A key role for cyclic nucleotide gated (CNG) channels in cGMP-related retinitis pigmentosa. Hum. Mol. Genet., 20, 941–947. [DOI] [PubMed] [Google Scholar]
- 27.Woodruff M.L., Olshevskaya E.V., Savchenko A.B., Peshenko I.V., Barrett R., Bush R.A., Sieving P.A., Fain G.L., Dizhoor A.M. (2007) Constitutive excitation by Gly90Asp rhodopsin rescues rods from degeneration caused by elevated production of cGMP in the dark. J. Neurosci., 27, 8805–8815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Riazuddin S.A., Shahzadi A., Zeitz C., Ahmed Z.M., Ayyagari R., Chavali V.R., Ponferrada V.G., Audo I., Michiels C., Lancelot M.E. et al. (2010) A mutation in SLC24A1 implicated in autosomal-recessive congenital stationary night blindness. Am. J. Hum. Genet., 87, 523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X.F., Kiedrowski L., Tremblay F., Fernandez F.R., Perizzolo M., Winkfein R.J., Turner R.W., Bains J.S., Rancourt D.E., Lytton J. (2006) Importance of K+-dependent Na+/Ca2+-exchanger 2, NCKX2, in motor learning and memory. J. Biol. Chem., 281, 6273–6282. [DOI] [PubMed] [Google Scholar]
- 30.Reid D.M., Friedel U., Molday R.S., Cook N.J. (1990) Identification of the sodium-calcium exchanger as the major ricin-binding glycoprotein of bovine rod outer segments and its localization to the plasma membrane. Biochemistry, 29, 1601–1607. [DOI] [PubMed] [Google Scholar]
- 31.Eisenfeld A.J., Bunt-Milam A.H., Sarthy P.V. (1984) Muller cell expression of glial fibrillary acidic protein after genetic and experimental photoreceptor degeneration in the rat retina. Invest. Ophthalmol. Vis. Sci., 25, 1321–1328. [PubMed] [Google Scholar]
- 32.Zhang Y., Molday L.L., Molday R.S., Sarfare S.S., Woodruff M.L., Fain G.L., Kraft T.W., Pittler S.J. (2009) Knockout of GARPs and the beta-subunit of the rod cGMP-gated channel disrupts disk morphogenesis and rod outer segment structural integrity. J. Cell Sci., 122, 1192–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poetsch A., Molday L.L., Molday R.S. (2001) The cGMP-gated channel and related glutamic acid-rich proteins interact with peripherin-2 at the rim region of rod photoreceptor disc membranes. J. Biol. Chem., 276, 48009–48016. [DOI] [PubMed] [Google Scholar]
- 34.Ardell M.D., Makhija A.K., Oliveira L., Miniou P., Viegas-Pequignot E., Pittler S.J. (1995) cDNA, gene structure, and chromosomal localization of human GAR1 (CNCG3L), a homolog of the third subunit of bovine photoreceptor cGMP-gated channel. Genomics, 28, 32–38. [DOI] [PubMed] [Google Scholar]
- 35.Colville C.A., Molday R.S. (1996) Primary structure and expression of the human beta-subunit and related proteins of the rod photoreceptor cGMP-gated channel. J. Biol. Chem., 271, 32968–32974. [DOI] [PubMed] [Google Scholar]
- 36.Chen T.Y., Peng Y.W., Dhallan R.S., Ahamed B., Reed R.R., Yau K.W. (1993) A new subunit of the cyclic nucleotide-gated cation channel in retinal rods. Nature, 362, 764–767. [DOI] [PubMed] [Google Scholar]
- 37.Cook N.J., Molday L.L., Reid D., Kaupp U.B., Molday R.S. (1989) The cGMP-gated channel of bovine rod photoreceptors is localized exclusively in the plasma membrane. J. Biol. Chem., 264, 6996–6999. [PubMed] [Google Scholar]
- 38.Herrmann R., Lobanova E.S., Hammond T., Kessler C., Burns M.E., Frishman L.J., Arshavsky V.Y. (2010) Phosducin regulates transmission at the photoreceptor-to-ON-bipolar cell synapse. J. Neurosci., 30, 3239–3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huttl S., Michalakis S., Seeliger M., Luo D.G., Acar N., Geiger H., Hudl K., Mader R., Haverkamp S., Moser M. et al. (2005) Impaired channel targeting and retinal degeneration in mice lacking the cyclic nucleotide-gated channel subunit CNGB1. J. Neurosci., 25, 130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heikkinen H., Nymark S., Koskelainen A. (2008) Mouse cone photoresponses obtained with electroretinogram from the isolated retina. Vision Res., 48, 264–272. [DOI] [PubMed] [Google Scholar]
- 41.Haynes L.W., Kay A.R., Yau K.W. (1986) Single cyclic GMP-activated channel activity in excised patches of rod outer segment membrane. Nature, 321, 66–70. [DOI] [PubMed] [Google Scholar]
- 42.Zimmerman A.L., Baylor D.A. (1986) Cyclic GMP-sensitive conductance of retinal rods consists of aqueous pores. Nature, 321, 70–72. [DOI] [PubMed] [Google Scholar]
- 43.Ruiz M., Brown R.L., He Y., Haley T.L., Karpen J.W. (1999) The single-channel dose-response relation is consistently steep for rod cyclic nucleotide-gated channels: implications for the interpretation of macroscopic dose-response relations. Biochemistry, 38, 10642–10648. [DOI] [PubMed] [Google Scholar]
- 44.Mendez A., Burns M.E., Sokal I., Dizhoor A.M., Baehr W., Palczewski K., Baylor D.A., Chen J. (2001) Role of guanylate cyclase-activating proteins (GCAPs) in setting the flash sensitivity of rod photoreceptors. Proc. Natl Acad. Sci. USA, 98, 9948–9953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iwamoto T., Watano T., Shigekawa M. (1996) A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+ exchange in cells expressing NCX1. J. Biol. Chem., 271, 22391–22397. [DOI] [PubMed] [Google Scholar]
- 46.Altimimi H.F., Szerencsei R.T., Schnetkamp P.P. (2013) Functional and structural properties of the NCKX2 Na(+)-Ca (2+)/K (+) exchanger: a comparison with the NCX1 Na (+)/Ca (2+) exchanger. Adv. Exp. Med. Biol., 961, 81–94. [DOI] [PubMed] [Google Scholar]
- 47.Bauer P.J., Drechsler M. (1992) Association of cyclic GMP-gated channels and Na(+)-Ca(2+)-K+ exchangers in bovine retinal rod outer segment plasma membranes. J. Physiol., 451, 109–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schwarzer A., Schauf H., Bauer P.J. (2000) Binding of the cGMP-gated channel to the Na/Ca-K exchanger in rod photoreceptors. J. Biol. Chem., 275, 13448–13454. [DOI] [PubMed] [Google Scholar]
- 49.Kang K., Bauer P.J., Kinjo T.G., Szerencsei R.T., Bonigk W., Winkfein R.J., Schnetkamp P.P. (2003) Assembly of retinal rod or cone Na(+)/Ca(2+)-K(+) exchanger oligomers with cGMP-gated channel subunits as probed with heterologously expressed cDNAs. Biochemistry, 42, 4593–4600. [DOI] [PubMed] [Google Scholar]
- 50.Molday R.S., Molday L.L. (1998) Molecular properties of the cGMP-gated channel of rod photoreceptors. Vision Res., 38, 1315–1323. [DOI] [PubMed] [Google Scholar]
- 51.Cook N.J., Kaupp U.B. (1988) Solubilization, purification, and reconstitution of the sodium-calcium exchanger from bovine retinal rod outer segments. J. Biol. Chem., 263, 11382–11388. [PubMed] [Google Scholar]
- 52.Reilander H., Achilles A., Friedel U., Maul G., Lottspeich F., Cook N.J. (1992) Primary structure and functional expression of the Na/Ca,K-exchanger from bovine rod photoreceptors. EMBO J., 11, 1689–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schnetkamp P.P. (1986) Sodium-calcium exchange in the outer segments of bovine rod photoreceptors. J. Physiol., 373, 25–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Field G.D., Rieke F. (2002) Nonlinear signal transfer from mouse rods to bipolar cells and implications for visual sensitivity. Neuron, 34, 773–785. [DOI] [PubMed] [Google Scholar]
- 55.Sampath A.P., Rieke F. (2004) Selective transmission of single photon responses by saturation at the rod-to-rod bipolar synapse. Neuron, 41, 431–443. [DOI] [PubMed] [Google Scholar]
- 56.Schneeweis D.M., Schnapf J.L. (1995) Photovoltage of rods and cones in the macaque retina. Science, 268, 1053–1056. [DOI] [PubMed] [Google Scholar]
- 57.Ribelayga C., Cao Y., Mangel S.C. (2008) The circadian clock in the retina controls rod-cone coupling. Neuron, 59, 790–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abd-El-Barr M.M., Pennesi M.E., Saszik S.M., Barrow A.J., Lem J., Bramblett D.E., Paul D.L., Frishman L.J., Wu S.M. (2009) Genetic dissection of rod and cone pathways in the dark-adapted mouse retina. J. Neurophysiol., 102, 1945–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen C.K., Burns M.E., He W., Wensel T.G., Baylor D.A., Simon M.I. (2000) Slowed recovery of rod photoresponse in mice lacking the GTPase accelerating protein RGS9–1. Nature, 403, 557–560. [DOI] [PubMed] [Google Scholar]
- 60.Baehr W., Devlin M.J., Applebury M.L. (1979) Isolation and characterization of cGMP phosphodiesterase from bovine rod outer segments. J. Biol. Chem., 254, 11669–11677. [PubMed] [Google Scholar]
- 61.Kolesnikov A.V., Rikimaru L., Hennig A.K., Lukasiewicz P.D., Fliesler S.J., Govardovskii V.I., Kefalov V.J., Kisselev O.G. (2011) G-protein betagamma-complex is crucial for efficient signal amplification in vision. J. Neurosci., 31, 8067–8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sokal I., Li N., Surgucheva I., Warren M.J., Payne A.M., Bhattacharya S.S., Baehr W., Palczewski K. (1998) GCAP1 (Y99C) mutant is constitutively active in autosomal dominant cone dystrophy. Mol. Cell, 2, 129–133. [DOI] [PubMed] [Google Scholar]
- 63.Semple-Rowland S.L., Lee N.R., Van Hooser J.P., Palczewski K., Baehr W. (1998) A null mutation in the photoreceptor guanylate cyclase gene causes the retinal degeneration chicken phenotype. Proc. Natl Acad. Sci. USA, 95, 1271–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dryja T.P., Finn J.T., Peng Y.W., McGee T.L., Berson E.L., Yau K.W. (1995) Mutations in the gene encoding the alpha subunit of the rod cGMP-gated channel in autosomal recessive retinitis pigmentosa. Proc. Natl Acad. Sci. USA, 92, 10177–10181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bech-Hansen N.T., Naylor M.J., Maybaum T.A., Sparkes R.L., Koop B., Birch D.G., Bergen A.A., Prinsen C.F., Polomeno R.C., Gal A. et al. (2000) Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat. Genet., 26, 319–323. [DOI] [PubMed] [Google Scholar]
- 66.Strom T.M., Nyakatura G., Apfelstedt-Sylla E., Hellebrand H., Lorenz B., Weber B.H., Wutz K., Gutwillinger N., Ruther K., Drescher B. et al. (1998) An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat. Genet., 19, 260–263. [DOI] [PubMed] [Google Scholar]
- 67.Audo I., Kohl S., Leroy B.P., Munier F.L., Guillonneau X., Mohand-Said S., Bujakowska K., Nandrot E.F., Lorenz B., Preising M. et al. (2009) TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet., 85, 720–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dryja T.P., Berson E.L., Rao V.R., Oprian D.D. (1993) Heterozygous missense mutation in the rhodopsin gene as a cause of congenital stationary night blindness. Nat. Genet., 4, 280–283. [DOI] [PubMed] [Google Scholar]
- 69.Gal A., Orth U., Baehr W., Schwinger E., Rosenberg T. (1994) Heterozygous missense mutation in the rod cGMP phosphodiesterase beta-subunit gene in autosomal dominant stationary night blindness. Nat. Genet., 7, 551. [DOI] [PubMed] [Google Scholar]
- 70.Dryja T.P., Hahn L.B., Reboul T., Arnaud B. (1996) Missense mutation in the gene encoding the alpha subunit of rod transducin in the Nougaret form of congenital stationary night blindness. Nat. Genet., 13, 358–360. [DOI] [PubMed] [Google Scholar]
- 71.Yamamoto S., Sippel K.C., Berson E.L., Dryja T.P. (1997) Defects in the rhodopsin kinase gene in the Oguchi form of stationary night blindness. Nat. Genet., 15, 175–178. [DOI] [PubMed] [Google Scholar]
- 72.Mattapallil M.J., Wawrousek E.F., Chan C.C., Zhao H., Roychoudhury J., Ferguson T.A., Caspi R.R. (2012) The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest. Ophthalmol. Vis. Sci., 53, 2921–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Calvert P.D., Krasnoperova N.V., Lyubarsky A.L., Isayama T., Nicolo M., Kosaras B., Wong G., Gannon K.S., Margolskee R.F., Sidman R.L. et al. (2000) Phototransduction in transgenic mice after targeted deletion of the rod transducin alpha -subunit. Proc. Natl Acad. Sci. USA, 97, 13913–13918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nikonov S.S., Kholodenko R., Lem J., Pugh E.N. Jr. (2006) Physiological features of the S- and M-cone photoreceptors of wild-type mice from single-cell recordings. J. Gen. Physiol., 127, 359–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen J., Woodruff M.L., Wang T., Concepcion F.A., Tranchina D., Fain G.L. (2010) Channel modulation and the mechanism of light adaptation in mouse rods. J. Neurosci., 30, 16232–16240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Concepcion F., Chen J. (2010) Q344ter mutation causes mislocalization of rhodopsin molecules that are catalytically active: a mouse model of Q344ter-induced retinal degeneration. PLoS One, 5, e10904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.MacKenzie D., Arendt A., Hargrave P., McDowell J.H., Molday R.S. (1984) Localization of binding sites for carboxyl terminal specific anti-rhodopsin monoclonal antibodies using synthetic peptides. Biochemistry, 23, 6544–6549. [DOI] [PubMed] [Google Scholar]
- 78.Shi G., Yau K.W., Chen J., Kefalov V.J. (2007) Signaling properties of a short-wave cone visual pigment and its role in phototransduction. J. Neurosci., 27, 10084–10093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhu X., Li A., Brown B., Weiss E.R., Osawa S., Craft C.M. (2002) Mouse cone arrestin expression pattern: light induced translocation in cone photoreceptors. Mol. Vis., 8, 462–471. [PubMed] [Google Scholar]
- 80.Chan S., Rubin W.W., Mendez A., Liu X., Song X., Hanson S.M., Craft C.M., Gurevich V.V., Burns M.E., Chen J. (2007) Functional comparisons of visual arrestins in rod photoreceptors of transgenic mice. Invest. Ophthalmol. Vis. Sci., 48, 1968–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hoyo N.L., Lopez-Begines S., Rosa J.L., Chen J., Mendez A. (2014) Functional EF-hands in neuronal calcium sensor GCAP2 determine its phosphorylation state and subcellular distribution in vivo, and are essential for photoreceptor cell integrity. PLoS Genet., 10, e1004480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Umino Y., Solessio E., Barlow R.B. (2008) Speed, spatial, and temporal tuning of rod and cone vision in mouse. J. Neurosci., 28, 189–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nymark S., Haldin C., Tenhu H., Koskelainen A. (2006) A new method for measuring free drug concentration: retinal tissue as a biosensor. Invest. Ophthalmol. Vis. Sci., 47, 2583–2588. [DOI] [PubMed] [Google Scholar]
- 84.Vinberg F., Kolesnikov A.V., Kefalov V.J. (2014) Ex vivo ERG analysis of photoreceptors using an in vivo ERG system. Vision Res., 101, 108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]