

Abstract
Arsenic is a naturally existing toxin and carcinogen. As(III) S-adenosylmethionine methyltransferases (AS3MT in mammals and ArsM in microbes) methylate As(III) three times in consecutive steps and play a central role in arsenic metabolism from bacteria to humans. Current assays for arsenic methylation are slow, laborious, and expensive. Here we report the development of two in vitro assays for AS3MT activity that are rapid, sensitive, convenient, and relatively inexpensive and can be adapted for high-throughput assays. The first assay measures As(III) binding by the quenching of the protein fluorescence of a single-tryptophan derivative of an AS3MT ortholog. The second assay utilizes time-resolved fluorescence resonance energy transfer to directly measure the conversion of the AS3MT substrate, S-adenosylmethionine, to S-adenosylhomocysteine catalyzed by AS3MT. These two assays are complementary, one measuring substrate binding and the other catalysis, making them useful tools for functional studies and future development of drugs to prevent arsenic-related diseases.
Keywords: Arsenic, Methylation, AS3MT, High-throughput screens
Arsenic is considered the most ubiquitous environmental toxin and carcinogen, and, consequently, the U.S. Environmental Protection Agency and Agency for Toxic Substances and Disease Registry rank arsenic at the top of the U.S. Priority List of Hazardous Substances (http://www.atsdr.cdc.gov/SPL/index.html). In humans exposure is a major contributor to arsenic-related diseases [1,2], including bladder, lung, and skin cancers [3]. The primary sources of dietary arsenic are food [4] and drinking water [5]. As a consequence of the environmental pervasiveness of arsenic, detoxifying systems are found in nearly every organism, from bacteria to humans [6]. A common means of microbial arsenic detoxification is by methylation catalyzed by As(III) S-adenosylmethionine (SAM)1 methyltransferases, enzymes termed ArsM [7,8]. The enzyme was originally identified in mammals, in which it was called Cyt19 or AS3MT [9]. Methylation by AS3MT was originally considered a detoxification mechanism but is now thought to transform inorganic arsenic (As(III)) into the more toxic and carcinogenic trivalent methylated species methylarsenite (MAs(III)) and dimethylarsenite (DMAs(III)) [10,11].
Identification of inhibitors or activators of AS3MT will be useful for the development of drugs to prevent arsenic-related diseases. To date there is no convenient, rapid assay for AS3MT activity that can be adapted for high-throughput assays for drug development. Current assays of As(III) methyltransferase activity are slow and laborious, making them unsuited to high-throughput screening for inhibitors. Postreaction assays of AS3MT activity utilize radioactive substrates [12,13] or expensive instrumentation such as high-performance liquid chromatography (HPLC) coupled with hydride generation–atomic absorption spectroscopy [14], electro-spray ionization tandem mass spectrometry detection, or inductively coupled plasma mass spectroscopy (ICP–MS) [15]. Moreover, these assays use high amounts of enzyme and substrates with long reaction times and are difficult to apply to detailed enzymological studies.
We have developed two new assays for analysis of AS3MT activity that are better, faster, and cheaper than current assays. Both assays utilize small amounts of enzyme and short reaction times. The first assay employs As(III)-induced fluorescence quenching of an engineered single-tryptophan derivative of an AS3MT ortholog from the eukaryotic alga Chlamydomonas reinhardtii termed either CrArM or CrAS3MT. This real-time assay for substrate binding is based on the effect of As(III) on a single-tryptophan derivative of a thermophilic ortholog [16], but the current assay has the advantage that it works at room temperature in microtiter plates. The second assay utilizes time-resolved fluorescence resonance energy transfer (TR–FRET) between a Tb3+-labeled cryptate coupled to an S-adenosylhomocysteine (SAH) antibody and a fluorescently labeled SAH to detect formation of the SAH product of the AS3MT reaction. This is the first rapid assay for detection and quantification of SAH generation within the first few minutes of reaction. These assays are complementary and provide the basis for future high-throughput screens for inhibitors and activators of AS3MT.
Materials and methods
Reagents
SAM was purchased from Cayman Chemical Co. (Ann Arbor, MI, USA). MTS reagents were purchased from Biotium, Inc. (Hayward, CA, USA). Other reagents were purchased from Sigma–Aldrich (St. Louis, MO, USA). A stock solution of tris(2-carboxyethyl)phosphine (TCEP) was prepared at 0.5 M and adjusted to pH 7.0. MAs(V), 4-aminobenzenearsonic acid (p-arsanilic acid or pASA(V)), 4-hydroxy-3-nitrobenzenearsonic (roxarsone or Rox(V)), and (4-nitrophenyl)arsonic acid (nitarsone or Nit(V)) were reduced to trivalent MAs(III), pASA(III), Rox(III), and Nit(III) and adjusted to pH 6.5, as described [17]. The products of reduction were analyzed by HPLC coupled to ICP–MS, as described below. The products were simultaneously analyzed for both arsenic and sulfur, and no sulfur eluted with the arsenic, demonstrating that the reduction did not produce thioarsenicals.
DNA manipulation and mutagenesis
Wild-type CrAS3MT [18] was used as the starting point for construction of a single-tryptophan derivative. First, the codon for Trp332 was changed to a tyrosine codon by site-directed mutagenesis using a QuikChange mutagenesis kit (Stratagene, La Jolla, CA, USA). The forward and reverse primers were 5′-GGTGGGCGAGTCCTATCTGGCACCCCACTT-3′ and 5′-AAGTGGGGTGCCAGATAGGACTCGCCCACC-3′, respectively. A single-tryptophan derivative, Y72W, was constructed by site-directed mutagenesis of the W332Y CrAS3MT in which the codon for Tyr72 was changed to a tryptophan codon using primers 5′-CACAGAGGTGAAGGAGAAGTTCTGGGGCTGCGGAAAC-3′ and 5′-GTTTCCGCAGCCCCAGAACTTCTCCTTCACCTCTGTG-3′. The Y72W/W332Y CrAS3MT construct has a single tryptophan residue near the As(III) binding site to serve as a fluorescence reporter of As(III) binding.
Purification of AS3MT enzymes
AS3MT enzymes with a C-terminal histidine tag were purified by Ni-NTA chromatography from cells of Escherichia coli, as described previously [19,20].
Assays of fluorescence quenching
Protein fluorescence measurements were assayed in 384-well microtiter plates with a total volume of 15 μ of a buffer consisting of 50 mM MOPS–KOH, pH 7.5, containing 0.15 M KCl and 5 μM Y72W CrAS3MT. Substrates were added to the assay buffer to initiate the reaction. Where indicated, purified CrAS3MT was preincubated with the thiol-modifying reagent methylmethanethiosulfonate (MMTS) for 15 min before the reaction with As(III) was initiated. Protein fluorescence was determined with a Synergy H4 Hybrid Multi-Mode microplate reader (BioTek Instruments, Winooski, VT, USA) with excitation and emission wavelengths set at 295 and 345 nm, respectively.
TR–FRET assay of AS3MT activity
AS3MT activity was assayed with an EPIgeneous Methyltransferase Assay kit (Cisbio Bioassays, Bedford, MA, USA) by measuring the conversion of SAM to SAH according to the manufacturer’s directions. The reaction has two steps: (1) the enzymatic reaction, which converts SAM to SAH, and (2) the detection step that quantifies SAH production. An anti-SAH antibody was labeled with a terbium cryptate, which fluoresces at 620 nm when excited at 337 nm. When the antibody binds a proprietary SAH-d2-labeled tracer, there is energy transfer with emission at 665 nm. The SAH released by the enzymatic reaction competes with labeled SAH-d2, leading to a decrease in the homogeneous time-resolved fluorescence (HTRF) signal. The two steps were carried out sequentially in the same well of a low-volume 384-well microtiter plate, with a total volume of 20 μl (10 μl for the enzymatic step and 10 μl for the detection step) in a buffer consisting of 50 mM MOPS, pH 7.5, containing 0.15 M KCl, 10 μM SAM, 10 μM As(III), 20 μM TCEP, and inhibitors, as indicated. Purified CrAS3MT or hAS3MT at 1 μlM, final concentration, was added to initiate the reaction. The reactions were carried out at either room temperature or 37 °C, as indicated, for 5 min in most assays or, when noted, as a time course. The reaction was terminated by addition of the proprietary detection reagent, followed by SAH-d2 and anti-SAH-Lumi4-Tb reagents. The plates were incubated for 1 h, and fluorescence was measured at both 665 and 620 nm with excitation at 337 nm in a Synergy H4 Hybrid Multi-Mode microplate reader. The HTRF was calculated from the ratio of emission at 665 and 620 nm. The concentration of SAH was calculated from a calibration curve of the HTRF with known concentrations of SAH.
The performance parameters, including the signal-to-background ratio, signal-to-noise ratio, and coefficient of variation, were determined at both 665 and 620 nm (Table 1). The robustness of the assay was evaluated from the processed coefficient of variation and the Z′ factor, a dimensionless statistical value of the quality of the assay, both of which were calculated from the 665 nm/620 nm HTRF ratio. The values for the statistical parameters meet the requirements for high-throughput screening assays [21].
Table 1.
Performance and robustness parameters.
| Performance parameters
|
Robustness parameters
|
||||
|---|---|---|---|---|---|
| Signal to background | Signal to noise | CV at each wavelength | CV from HTRF | Z’ factor | |
| Wavelength | |||||
| 620 nm | 76.3 ± 12.4 | 23.8 | 0.06 | ||
| 655 nm | 18.8 ± 3.8 | 19.0 | 0.11 | ||
| No enzyme | 0.100 | ||||
| +CrAS3MT | 0.033 | 0.78 | |||
The signal-to-background ratio, signal-to-noise ratio, and first coefficient of variation (CV) were measured and calculated at both wavelengths. The second CV and the Z’ factor were calculated from the 665 nm/620 nm HTRF ratio.
HPLC–ICP–MS assay of AS3MT activity
Methylation of As(III) was assayed in a buffer consisting of 50 mM MOPS, pH 7.5, containing 0.15 M KCl, 0.5 mM SAM, 10 μM As(III), 1 mM TCEP [20]. Inhibitors were added as indicated. The reaction was initiated by addition of purified CrAS3MT, 1 μM final concentration. The reactions were carried out at room temperature for 2.5 h. The assay was terminated by adding H2O2 at 6% final concentration to release all bound arsenic by oxidation to pentavalency, and the solution was passed through a 3-kDa cutoff Amicon Ultrafilter (Millipore, Billerica, MA, USA) to remove protein. The filtrate was speciated by HPLC (PerkinElmer Series 2000) using a C18 reversed-phase column eluted with a mobile phase consisting of 3 mM malonic acid, 5 mM tetrabutylammonium hydroxide, and 5% (v/v) methanol (pH 5.9) with a flow rate of 1 ml/min, and arsenic content was determined by ICP–MS using an ELAN DRC-e spectrometer (PerkinElmer, Waltham, MA, USA).
Homology models of AS3MT structures
The hAS3MT homology model of hAS3MT built on the structure of PhAs(III)-bound CmArsM (PDB ID: 4KW7) has been reported [20]. A similar model for CrAS3MT was built on the CmArsM structure with bound Rox(III) (PDB ID: 4RSR) using a fully automated protein structure homology modeling server SWISS-MODEL (http://swissmodel.expasy.org/) [22]. The model quality was estimated based on the QMEAN scoring function [23]. The QMEAN score of 0.65 was within the acceptable range. The CrAS3MT model structure with residues 46–349 (residue numbers based on the CrAS3MT sequence) from PhAs(III)-bound CmArsM was used as a template, and the final homology model incorporated 299 of 328 residues. In silico docking with SAM was carried out using the PATCHDOCK server [24]. The docked CrAS3MT model with SAM was superimposed with the As(III)-bound structure of CmArsM (PDB ID: 4FSD) to acquire the arsenic atom in the As(III) binding site of CrS3MT [25]. PyMOL version 1.3 was used to visualize the structural models [26,27].
Results
A fluorescence assay of As(III) binding by AS3MT
AS3MT orthologs share structural similarity with one another. We recently cloned and expressed a close ortholog of hAS3MT from the common soil alga C. reinhardtii that is very active at room temperature and expresses well in E. coli, which makes it a better target for assay developing than the human enzyme [18]. Homology models of hAS3MT and CrAS3MT based on the crystal structure of an ortholog from the alga Cyanidioschyzon merolae indicate that both CrAS3MT and hAS3MT have SAM and As(III) binding sites [20,28] (Fig. 1). The As(III) binding site in each AS3MT contains the four conserved cysteine residues that have been shown to be involved in catalysis [16]. In CmAS3MT, conserved cysteine residue Cys74 moves during the catalytic cycle, which suggested that engineering a tryptophan residue at position 72 in CrAS3MT, which is next to the equivalent cysteine residues, might produce a spectroscopic real-time assay of As(III) binding as it did in CmAS3MT [16]. The effect of inorganic As(III), the related metalloid Sb(III), and the aromatic arsenical PhAs(III) on the fluorescence of Y72W was examined (Fig. 2). Sb(III) produced the most quenching at the lowest concentration, followed by PhAs(III) and As(III). As(V), which is not a substrate of AS3MT enzymes, had no effect on the fluorescence of Y72W. These results suggest that binding of trivalent metalloids produces a conformational change that can be reported by the tryptophan residue at position 72. Importantly, this assay has been adapted to microtiter plates, which can be modified for rapid high-throughput screening for potential inhibitors.
Fig.1.
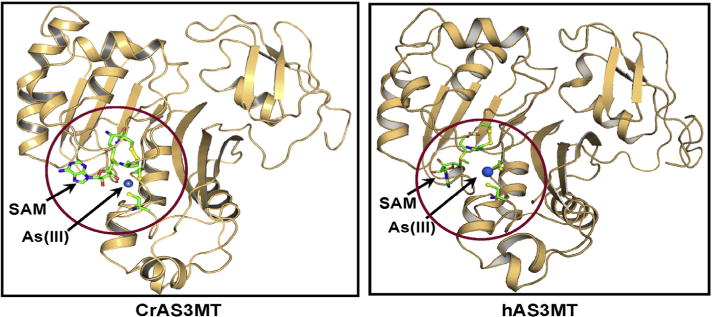
Comparison of the homology structural models of human and C. reinhardtii AS3MTs. Structural models of human and C. reinhardtii AS3MTs were constructed based on the structure of the PhAs(III)-bound CmArsM ortholog (PDB ID: 4KW7) and the Rox(III)-bound CmArsM ortholog (PDB ID: 4RSR). The circled view shows SAM (in ball-and-stick) occupying its binding site with its methyl group poised to be donated to the arsenic atom. The locations of the four conserved cysteine residues are shown in yellow ball-and-stick with As(III) bound to Cys156 and Cys206 of hAS3MT and Cys170 and Cys220 of CrAS3MT.
Fig.2.
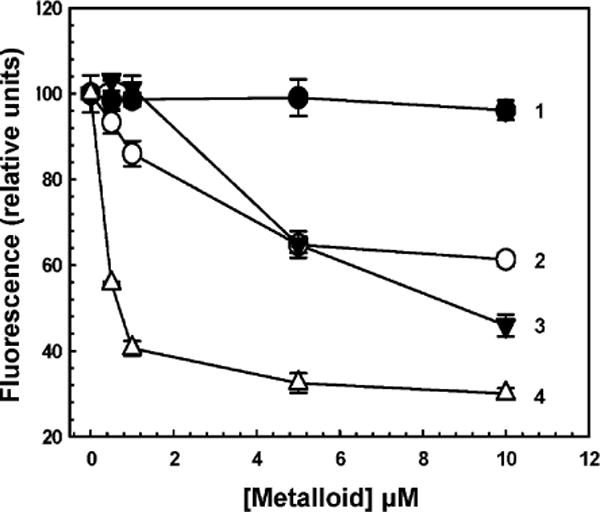
Quenching of CrAS3MT Y72W reveals metalloid binding. Protein fluorescence was assayed at 23 °C after 2 min with excitation and emission wavelengths of 295 and 345 nm, respectively, as described under Materials and methods. Metalloids were added at the indicated concentrations to 5 μM CrAS3MT. Additions: (●) As(V), (○) As(III), (▼) PhAs(III), (△) Sb(III). The data are the mean ± SE (n = 3).
The effects of two types of inhibitors of AS3MT activity, a thiol reagent and a SAH analog, on ligand-induced quenching of Y72W were examined. MMTS is a small sulfhydryl-reactive compound, modifying cysteine thiolates to the dithiomethane (–S–S–CH3). MMTS quenched the fluorescence of Y72W. MMTS had no effect on the fluorescence of tryptophan alone (Fig. 3A, curve 1). Quenching was dependent on the concentration of MMTS, with half-maximal inhibition at approximately 50 μM (Fig. 3A, curve 2). This is 10-fold molar excess of MMTS over enzyme but, with only four cysteines in the enzyme, it is only 2.5-fold over the effective active site cysteine thiolate concentration. These results suggest that binding of MMTS to the cysteine residues that form the As(III) binding site produces a conformational change similar to that produced by binding of As(III) itself. To examine whether binding of MMTS competed with As(III) binding, Y72W was incubated with 20 μM MMTS, a concentration nearly equimolar with the active site thiols. At this concentration protein fluorescence was not substantially quenched, suggesting that the conformation of the enzyme was not greatly altered. When 20 μM MMTS was followed by the addition of various concentrations of As(III), a significant reduction in quenching was observed (Fig. 3B). This result suggests that MMTS inhibited As(III) binding before it produced a substantial conformational change by itself, perhaps because not all four conserved cysteine residues in the active site are derivatized.
Fig.3.
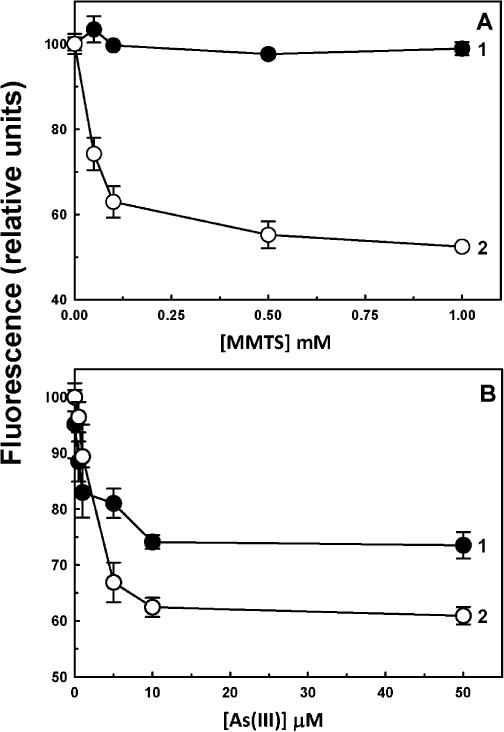
Effect of MMTS on the binding of As(III) to CrAS3MT. (A) MMTS quenches the fluorescence of 5 μM CrAS3MT Y72W (curve 2) but not that of free tryptophan (curve 1). (B) MMTS reduces the quenching of Y72W by As(III): curve 1, As(III) pretreated with 20 μM MMTS for 15 min; curve 2, As(III) in the absence of MMTS. As(III) was added to initiate the reaction, and fluorescence was measured after 2 min. The data are the mean ± SE (n = 3).
Sinefungin, an antibiotic produced by Streptomyces, is a nucleoside analog of SAH that inhibits SAM methyltransferases [29]. Sinefungin did not quench the fluorescence of Y72W, even at 0.1 mM (Fig. 4, curve 1), compared with As(III) in the absence of sinefungin (Fig. 4, curve 2). Prior addition of sinefungin had no effect on As(III)-induced fluorescence quenching (Fig. 4, curve 3). Thus, it appears that the two binding sites are independent. This was a critical result. Because binding of inhibitors at the SAM binding site does not inhibit binding of As(III) to its site, this assay can be used for screening of compounds that specifically prevent arsenic binding, a long-term goal of this study.
Fig.4.
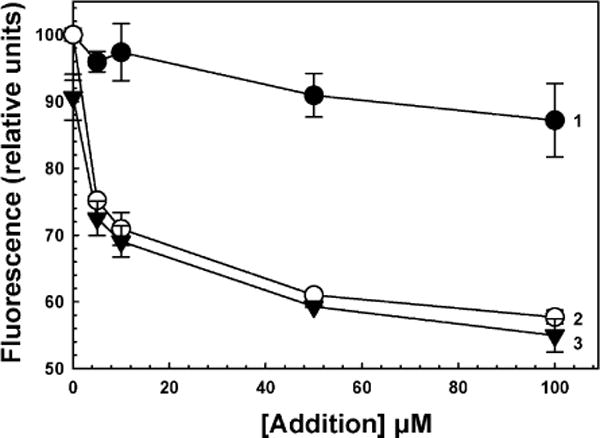
Effect of sinefungin on quenching of CrAS3MT Y72W fluorescence. As(III) was added to initiate the reaction, and fluorescence was measured after 2 min. Curve 1, plus indicated concentrations of sinefungin; curve 2, plus indicated concentrations of As(III); curve 3, 0.1 mM sinefungin followed by the indicated concentrations of As(III). The data are the mean ± SE (n = 3).
A fluorescence assay for AS3MT activity
Current assays of AS3MT activity are complex, time consuming, expensive, and not conducive to high-throughput screening. We adapted a commercial methyltransferase assay kit to the measurement of AS3MT activity of purified enzymes by FRET. This assay cannot be used in cell-based assays because all SAM methyltransferases produce SAH, so the signal cannot be attributed to a specific enzyme. The advantage of this method in vitro is that it is rapid and can be performed in microtiter plates. Production of SAH was nearly linear over an hour with wild-type hAS3MT (Fig. 5). No SAH was generated by hAS3MT mutants with serine substitutions of conserved residue Cys32 or Cys61 or with the C32S/C61S double mutant, which are unable to methylate As(III) [20]. Thus SAH production measured by the FRET assay correlates with As(III) methylation.
Fig.5.
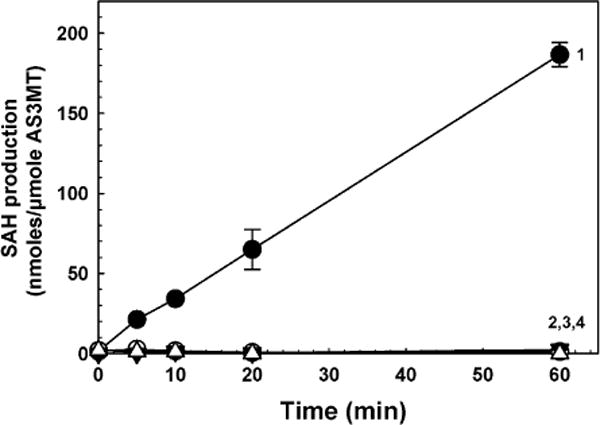
Time course of wild-type and mutant hAS3MTs. Methylation activity was assayed by production of SAH, which was determined with an EPIgeneous Methyltransferase Assay kit. Curve 1, wild-type hAS3MT; curve 2, hAS3MT C32S; curve 3, hAS3MT C61S; curve 4, hAS3MT C32S/C61S double mutant. Each purified enzyme was added at 1 lM, final concentration, and As(III) and SAM were 10 μM. The data are the mean ± SE (n = 3).
AS3MT orthologs methylate trivalent inorganic As(III) and the methylated species MAs(III) [16,20]. Recently an ortholog was shown to have low methylation activity with the trivalent aromatic arsenical PhAs(III) [30]. Other trivalent aromatic arsenicals such as the reduced forms of roxarsone, nitarsone, and pASA have not been tested as substrates. The ability of CrAS3MT to use these inorganic and organic arsenicals in the production of SAH was examined (Fig. 6A). Highest activity was observed with As(III) and MAs(III), and no activity with pentavalent As(V). Only low activity was observed with PhAs(III) and pASA(III) and essentially none with Rox(III) or Nit(III). Activity was determined as a function of As(III) (Fig. 6B). Activity could be detected at 10−8 molar As(III), with maximal activity at approximately 10−6 molar and half-maximal enzyme activity at approximately 0.23 μM As(III).
Fig.6.
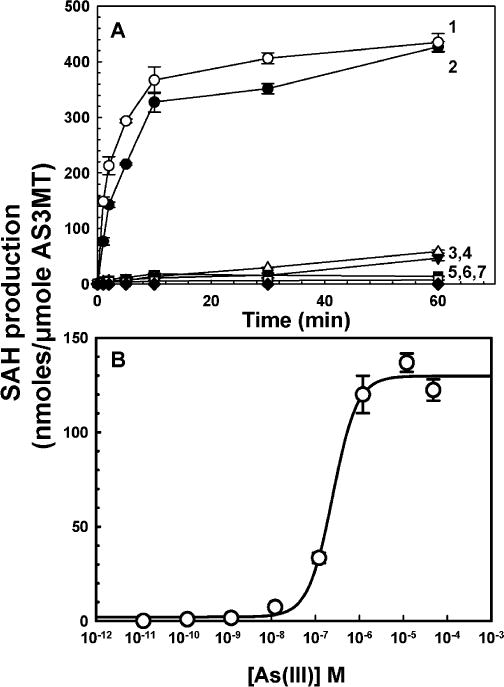
Methyltransferase activity with different arsenical substrates. (A) Methyltransferase activity was assayed with 1 μM CrAS3MT. Each arsenical substrate was added at a final concentration of 10μM, and SAM was added at 10μM. Curve 1, MAs(III); curve 2, As(III); curve 3, pASA(III); curve 4, PhAs(III); curve 5, Rox(III); curve 6, Nit(III); curve 7, As(V). (B) Concentration dependence of As(III) methylation. Activity was assayed for 5 min with 1 μM CrAS3MT. The data were fitted with SigmaPlot (Systat Software, San Jose, CA, USA), and the As(III) concentration to achieve half-maximal enzyme activity was calculated to be 0.23 μM. The data are the mean ± SE (n = 3).
The effects of inhibitors on CrAS3MT were examined on both the formation of SAH measured by TR-FRET and the formation of methylated species by HPLC–ICP–MS. Sinefungin inhibits SAM methyltransferases with IC50 values in the range of 0.1–20 μM [31–33]. To demonstrate that sinefungin inhibited arsenic methylation by CrAS3MT, the effect of sinefungin on the production of methylated species was examined by HPLC–ICP–MS. Sinefungin significantly inhibited formation of DMAs(V) by CrAS3MT (Fig. 7). Using the TR-FRET assay, at 10 μM SAM, sinefungin inhibited SAH production by CrAS3MT with an IC50 of 0.5 lM, a value that is well within the range of its effect on other SAM methyltransferases (Fig. 8A). NEM is a nonspecific inhibitor that affects enzyme activity by modifying cysteine residues. The effect of NEM on formation of methylated species was examined by HPLC–ICP–MS. At 0.15 mM, NEM significantly decreased DMAs(V) production, and 1.5 mM NEM completely inhibited arsenic methylation by CrAS3MT (Fig. 7). This correlated well with the results of the TR-FRET assay, in which NEM inhibited SAH production by CrAS3MT with an IC50 of 0.14 mM (Fig. 8B).
Fig.7.
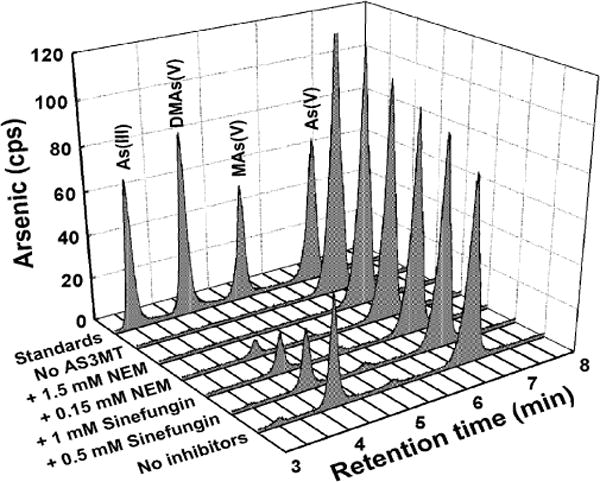
Effects of sinefungin and NEM on arsenic methylation by CrAS3MT. The indicated concentrations of sinefungin and NEM were added to assay buffer. As(III) was 10 μM, and SAM was 0.5 mM. 1 μM CrAS3MT was added to initiate the reaction, which was terminated by addition of 6% (v/v) H2O2 after 2.5 h at room temperature to oxidize all arsenic species, which were speciated by HPLC–ICP–MS.
Fig.8.
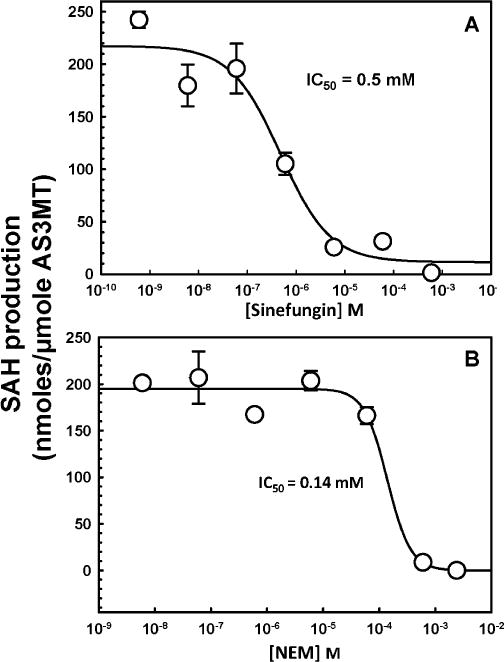
Effects of sinefungin and NEM on SAH production by CrAS3MT. The indicated concentrations of (A) sinefungin or (B) NEM were added to the assay buffer. As(III) and SAM were added at 10 μM, final concentration. 1 μM CrAS3MT was added to initiate the reaction, which was terminated after 5 min. The data were fitted with SigmaPlot, and the concentrations of inhibitor required to achieve half-maximal inhibition were calculated to be 0.5 μM for sinefungin and 0.14 mM for NEM. The data are the mean ± SE (n = 3).
MTS reagents are nonspecific cysteine modifiers that react preferentially with solute-exposed cysteine residues in proteins [34]. Four MTS reagents with different sizes and charges were used as probes of the environment surrounding the As(III) binding site. The neutral MMTS is the smallest cysteine-modifying reagent. MTSES is negatively charged, and MTSET and MTSEA are positively charged. MMTS, which has the ability to penetrate into proteins, showed the highest inhibition of the four, with an IC50 of 43 μM (Fig. 9). The bulky positively charged MTSEA and MTSET reagents inhibited less, with IC50 values of 0.24 and 0.22 mM, respectively. The bulky negatively charged MTSES reagent had the lowest effect, with an IC50 of 0.32 mM. Thus small, neutral reagents can enter the As(III) binding site and react with critical cysteine residues. Bulkier reagents are less able to penetrate, and positively charged reagents can access the binding site preferentially over a negatively charged one. These results indicate that the substrate binding site is solvent exposed but has size and charge restrictions.
Fig.9.
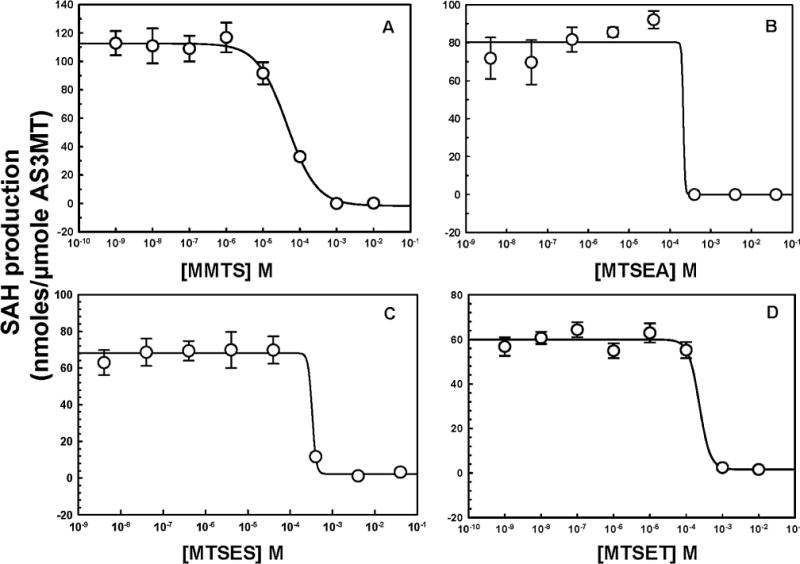
Inhibition of CrAS3MT activity by MTS thiol-reactive reagents. The indicated concentrations of MTS reagents were added to the assay buffer, and 1 μM CrAS3MT was added to initiate the reaction, which was terminated after 5 min. As(III) and SAM were added at 10 μM, final concentration. Additions: (A) MMTS, (B) MTSEA, (C) MTSES, (D) MTSET. The data were fitted with SigmaPlot, and the concentrations of inhibitor required to achieve half-maximal inhibition were calculated to be 43 μM for MMTS, 0.24 mM for MTSEA, 0.32 mM for MTSES, and 0.22 mM for MTSET. The data are the mean ± SE (n = 3).
Discussion
Arsenic is the most ubiquitous environmental toxin, and nearly every organism has an arsenic detoxifying system [7]. A common microbial resistance mechanism is methylation by As(III) SAM methyltransferases [8]. Mammals have a liver ortholog, AS3MT [35]. The increased toxicity of arsenic in AS3MT-knockout mice indicates that the function of this enzyme is to detoxify arseni [36], but in doing so converts inorganic arsenic into more toxic and carcinogenic methylated species [11,13]. To understand the role of AS3MT in arsenic-related diseases requires elucidation of its enzymatic mechanism. Current methods for assaying human AS3MT and orthologs lack robustness and accuracy and are not conducive to kinetic analysis.
The two new assays described here have a number of advantages over current methods. First, and most importantly, the assays can measure either binding activity or enzymatic activity in seconds to minutes, which allows for kinetic analysis. Second, the enzymatic assay is sufficiently sensitive to measure production of nanomolar SAH. Third, the assay of tryptophan fluorescence can measure binding in real time. Fourth, both assays are straightforward mix-and-measure assays that do not require extra steps or expensive instrumentation. Fifth, both assays utilize small amounts of enzyme and reagents in small reaction volumes (10–15 μl). Sixth, the assays use microtiter dishes and can be adapted to high-throughput screening for modulators of activity. High-throughput assays have been useful for screening for inhibitors of other SAM methyltransferases such as DNA methyltransferase I and protein methyltransferase SET7/9, both of which have been implicated in human cancers. The newly developed assays reported here will provide the first useful tools for screening for activators or inhibitors of human AS3MT that could lead to the discovery of drugs to prevent or ameliorate arsenic-related diseases.
Acknowledgments
This work was supported by Bankhead-Coley Program Grant 4BF01 from the State of Florida Department of Health and NIH Grant R01 ES023779 from the National Institute of Environmental Health Sciences to B.P.R. W.X. was supported by a Visiting Scholarship from the Chinese Academy of Sciences.
Footnotes
Abbreviations used: SAM, S-adenosylmethionine; SAH, S-adenosylhomocysteine; sinefungin, 5′-deoxy-5′-(1,4-diamino-4-carboxybutyl)adenosine; AS3MT, As(III) SAM methyltransferase; TR-FRET, time-resolved fluorescence resonance energy transfer; As(III), arsenite; MAs(III), methylarsenite; DMAs(III), dimethylarsenite; Rox(III), reduced roxarsone (3-nitro-4-hydroxyphenylarsonic acid); Nit(III), reduced nitarsone (p-nitrophenylarsonic acid); pASA(III), reduced p-arsanilic acid; PhAs(III), phenylarsenite or phenylarsine oxide; NEM, N-ethylmaleimide; MTS, methanethiosulfonate; MMTS, methylmethanethiosulfonate; MTSEA, ethylaminomethanethiosulfonate; MTSET, ethyl(trimethylammonium)methanethiosulfonate; MTSES, 2-sulfonatoethyl methanethiosulfonate; TCEP, tris(2-carboxyethyl)phosphine.
References
- 1.Jomova K, Jenisova Z, Feszterova M, Baros S, Liska J, Hudecova D, Rhodes CJ, Valko M. Arsenic: toxicity, oxidative stress and human disease. J Appl Toxicol. 2011;31:95–107. doi: 10.1002/jat.1649. [DOI] [PubMed] [Google Scholar]
- 2.Hughes MF, Beck BD, Chen Y, Lewis AS, Thomas DJ. Arsenic exposure and toxicology: a historical perspective. Toxicol Sci. 2011;123:305–332. doi: 10.1093/toxsci/kfr184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oberoi S, Barchowsky A, Wu F. The global burden of disease for skin, lung, and bladder cancer caused by arsenic in food. Cancer Epidemiol Biomarkers Prev. 2014;23:1187–1194. doi: 10.1158/1055-9965.EPI-13-1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu YG, Williams PN, Meharg AA. Exposure to inorganic arsenic from rice: a global health issue? Environ Pollut. 2008;154:169–171. doi: 10.1016/j.envpol.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 5.Staff NRC. Arsenic in Drinking Water, Washington, National Academies Press, DC. 1999 [PubMed] [Google Scholar]
- 6.Rensing C, Rosen BP. Heavy metals cycles (arsenic, mercury, selenium, others) In: Schaechter M, editor. Encyclopedia of Microbiology. Elsevier; Oxford, UK: 2009. pp. 205–219. [Google Scholar]
- 7.Zhu YG, Yoshinaga M, Zhao FJ, Rosen BP. Earth abides arsenic biotransformations. Annu Rev Earth Planet Sci. 2014;42:443–467. doi: 10.1146/annurev-earth-060313-054942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ye J, Rensing C, Rosen BP, Zhu YG. Arsenic biomethylation by photosynthetic organisms. Trends Plant Sci. 2012;17:155–162. doi: 10.1016/j.tplants.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liscombe DK, Louie GV, Noel JP. Architectures, mechanisms and molecular evolution of natural product methyltransferases. Nat Prod Rep. 2012;29:1238–1250. doi: 10.1039/c2np20029e. [DOI] [PubMed] [Google Scholar]
- 10.Styblo M, Drobna Z, Jaspers I, Lin S, Thomas DJ. The role of biomethylation in toxicity and carcinogenicity of arsenic: a research update. Environ Health Perspect. 2002;110(Suppl 5):767–771. doi: 10.1289/ehp.110-1241242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomas DJ, Rosen BP. Arsenic methyltransferases. In: Kretsinger RH, Uversky VN, Permyakov EA, editors. Encyclopedia of Metalloproteins. Springer; New York: 2013. pp. 140–145. [Google Scholar]
- 12.Wood TC, Salavagionne OE, Mukherjee B, Wang L, Klumpp AF, Thomae BA, Eckloff BW, Schaid DJ, Wieben ED, Weinshilboum RM. Human arsenic methyltransferase (AS3MT) pharmacogenetics: gene resequencing and functional genomics studies. J Biol Chem. 2006;281:7364–7373. doi: 10.1074/jbc.M512227200. [DOI] [PubMed] [Google Scholar]
- 13.Drobna Z, Waters SB, Devesa V, Harmon AW, Thomas DJ, Styblo M. Metabolism and toxicity of arsenic in human urothelial cells expressing rat arsenic (+3 oxidation state)-methyltransferase. Toxicol Appl Pharmacol. 2005;207:147–159. doi: 10.1016/j.taap.2004.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Styblo M, Yamauchi H, Thomas DJ. Comparative in vitro methylation of trivalent and pentavalent arsenicals. Toxicol Appl Pharmacol. 1995;135:172–178. doi: 10.1006/taap.1995.1220. [DOI] [PubMed] [Google Scholar]
- 15.McKnight-Whitford A, Chen B, Naranmandura H, Zhu C, Le XC. New method and detection of high concentrations of monomethylarsonous acid detected in contaminated groundwater. Environ Sci Technol. 2010;44:5875–5880. doi: 10.1021/es100273b. [DOI] [PubMed] [Google Scholar]
- 16.Marapakala K, Qin J, Rosen BP. Identification of catalytic residues in the As(III) S-adenosylmethionine methyltransferase. Biochemistry. 2012;51:944–951. doi: 10.1021/bi201500c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J, Sun S, Li CZ, Zhu YG, Rosen BP. Biosensor for organoarsenical herbicides and growth promoters. Environ Sci Technol. 2014;48:1141–1147. doi: 10.1021/es4038319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yin XX, Chen J, Qin J, Sun GX, Rosen BP, Zhu YG. Biotransformation and volatilization of arsenic by three photosynthetic cyanobacteria. Plant Physiol. 2011;156:1631–1638. doi: 10.1104/pp.111.178947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Packianathan C, Pillai JK, Riaz A, Kandavelu P, Sankaran B, Rosen BP. Crystallization and preliminary X-ray crystallographic studies of CrArsM, an arsenic(III) S-adenosylmethionine methyltransferase from Chlamydomonas reinhardtii. Acta Crystallogr F Struct Biol Commun. 2014;70:1385–1388. doi: 10.1107/S2053230X14018469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dheeman DS, Packianathan C, Pillai JK, Rosen BP. Pathway of human AS3MT arsenic methylation. Chem Res Toxicol. 2014;27:1979–1989. doi: 10.1021/tx500313k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 22.Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T. The SWISS-MODEL repository and associated resources. Nucleic Acids Res. 2009;37:D387–392. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27:343–350. doi: 10.1093/bioinformatics/btq662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33:W363–367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 26.DeLano WL. The PyMOL User’s Manual, San Carlos, DeLano Scientific, CA. 2001 [Google Scholar]
- 27.Peters B, Moad C, Youn E, Buffington K, Heiland R, Mooney S. Identification of similar regions of protein structures using integrated sequence and structure analysis tools. BMC Struct Biol. 2006;6:4. doi: 10.1186/1472-6807-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ajees AA, Marapakala K, Packianathan C, Sankaran B, Rosen BP. Structure of an As(III) S-adenosylmethionine methyltransferase: insights into the mechanism of arsenic biotransformation. Biochemistry. 2012;51:5476–5485. doi: 10.1021/bi3004632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nolan LL. Molecular target of the antileishmanial action of sinefungin. Antimicrob Agents Chemother. 1987;31:1542–1548. doi: 10.1128/aac.31.10.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marapakala K, Packianathan C, Ajees AA, Dheeman DS, Sankaran B, Kandavelu P, Rosen BP. A disulfide-bond cascade mechanism for arsenic(III) S-adenosylmethionine methyltransferase. Acta Crystallogr D Biol Crystallogr. 2015;71:505–515. doi: 10.1107/S1399004714027552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cole PA. Chemical probes for histone-modifying enzymes. Nat Chem Biol. 2008;4:590–597. doi: 10.1038/nchembio.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schluckebier G, Kozak M, Bleimling N, Weinhold E, Saenger W. Differential binding of S-adenosylmethionine S-adenosylhomocysteine and Sinefungin to the adenine-specific DNA methyltransferase M.TaqI. J Mol Biol. 1997;265:56–67. doi: 10.1006/jmbi.1996.0711. [DOI] [PubMed] [Google Scholar]
- 33.Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Discovery. 2009;8:724–732. doi: 10.1038/nrd2974. [DOI] [PubMed] [Google Scholar]
- 34.Karlin A, Akabas MH. Substituted-cysteine accessibility method. Methods Enzymol. 1998;293:123–145. doi: 10.1016/s0076-6879(98)93011-7. [DOI] [PubMed] [Google Scholar]
- 35.Lin S, Shi Q, Nix FB, Styblo M, Beck MA, Herbin-Davis KM, Hall LL, Simeonsson JB, Thomas DJ. A novel S-adenosyl-L-methionine:arsenic(III) methyltransferase from rat liver cytosol. J Biol Chem. 2002;277:10795–10803. doi: 10.1074/jbc.M110246200. [DOI] [PubMed] [Google Scholar]
- 36.Drobna Z, Naranmandura H, Kubachka KM, Edwards BC, Herbin-Davis K, Styblo M, Le XC, Creed JT, Maeda N, Hughes MF, Thomas DJ. Disruption of the arsenic (+3 oxidation state) methyltransferase gene in the mouse alters the phenotype for methylation of arsenic and affects distribution and retention of orally administered arsenate. Chem Res Toxicol. 2009;22:1713–1720. doi: 10.1021/tx900179r. [DOI] [PMC free article] [PubMed] [Google Scholar]