

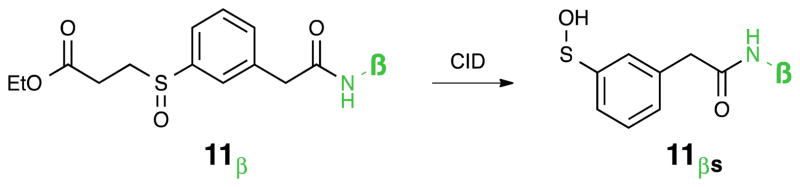
Abstract
The cross-linking Mass Spectrometry (XL-MS) technique has enormous potential for studying the interactions between proteins, and it can provide detailed structural information about the interaction interfaces in large protein complexes. Such information has been difficult to obtain by conventional structural methods. One of the primary impediments to the wider use of the XL-MS technique is the extreme challenge in sequencing cross-linked peptides because of their complex fragmentation patterns in MS. A recent innovation is the development of MS-cleavable cross-linkers, which allows direct sequencing of component peptides for facile identification. Sulfoxides are an intriguing class of thermally-cleavable compounds that have been shown to fragment selectively during low-energy collisional induced dissociation (CID) analysis. Current CID-cleavable cross-linkers create fragmentation patterns in MS2 of multiple peaks for each cross-linked peptide. Reducing the complexity of the fragmentation pattern in MS2 facilitates subsequent MS3 sequencing of the cross-linked peptides. The first authentic identical mass linker (IML) has now been designed, prepared, and evaluated. Multistage tandem mass spectrometry (MSn) analysis has demonstrated that the IML cross-linked peptides indeed yield one peak per peptide constituent in MS2 as predicted, thus allowing effective and sensitive MS3 analysis for unambiguous identification. Selective fragmentation for IML cross-linked peptides from the 19S proteasome complex was observed, providing a proof-of-concept demonstration for XL-MS studies on protein complexes.
Keywords: CID-cleavable cross-linker, isobaric, proteomics, sulfoxide elimination
Protein assemblies play a central role in the function of the cell. Determining the structure of protein complexes is a daunting challenge that often relies on a combination of methods to construct multi-protein structural models.1 Recently, cross-linking Mass Spectrometry (XL-MS) analysis has been recognized as a valuable tool for the structural analysis of protein assemblies.2 Identification of cross-linked peptides provides amino acid level resolution information about protein physical contacts that can be used to assemble three-dimensional models of interacting proteins.2,3 The advantages of XL-MS studies include small sample size, speed of sample preparation, and speed of data acquisition. One of the most significant challenges of cross-linking studies is the difficulty in unambiguously sequencing low abundance cross-linked peptides in complex mixtures.
One effective way to address the challenge of interpreting cross-linked sample data is to strategically design the linker to simplify MS sequencing of cross-linked peptides. Our group has demonstrated such a simplification through the design of CID-cleavable protein cross-linkers as outlined in Figure 1. Other groups have also reported CID-cleavable cross-linkers.4,5 DSSO (1) contains a sulfoxide group that cleaves in the MS instrument at lower collision energy than the peptide backbone.6,7 This selective fragmentation separates the two linked peptide chains, allowing simplified sequencing in MS3 using conventional database searching tools.3,7 In combination with new bioinformatics tools, DSSO has been proven as a very effective protein cross-linker for elucidating structures of protein complexes because of its size, structural simplicity, and robust cleavable bonds.3,7 In addition, two new derivatives of DSSO have recently been developed for cross-linking studies, and their successful applications in probing protein-protein interactions further demonstrate the robustness of sulfoxide-containing CID-cleavable reagents.8
Figure 1.

Representation of the detection of a DSSO (1) interlinked peptide in MS1 and its fragmentation in MS2. The four-peak pattern in MS2 results from fragmentation on both sides of the sulfoxide. Subsequent elimination of water is usually observed with the sulfenic acid derivatives.
The fragmentation pattern of DSSO interlinked peptides is more complex than desired.9,10 Although the DSSO molecule itself is symmetric, the fragmentation event is not, and this asymmetry leads each peptide chain in the cross-linked structure to produce two different daughter peaks in the MS2. A representative four-peak MS2 pattern of a DSSO cross-linked peptide (α-β) after CID-cleavage is illustrated in Figure 1. Each peptide (α and β) produces two MS2 peaks: an alkene fragment and a sulfenic acid modified fragment.7 Although the four-peak pattern is effective, it does require MS3 sequencing of at least three of the four MS2 fragments to ensure unambiguous identification of an inter-linked peptide in our current workflow. This increases MSn analysis time and leads to a fewer peptides being sequenced in a given experiment. In addition, the higher the number of MS2 fragment ions, the lower the sensitivity observed for MS3 sequencing. These limitations that are common to all published CID-cleavable linkers create a need for new linker designs.4,5 An appropriate new linker design would lead to one peak per peptide in MS2, facilitating targeted peak selection during MS3 analysis, improving sensitivity, and decreasing cycle time.
Our group envisioned a new derivative of DSSO that would result in equivalent mass modifications on all peptide fragments in MS2, thereby decreasing the number of peaks per cross-linked peptide in MS2 and increasing the signal-to-noise ratio for these peaks. The Ranish group previously reported the CID-cleavable BDRG cross-linker11 to address this problem. It is structurally complex, and fragments under CID to produce two peaks (one for each peptide component) that are one Dalton apart, thus allowing the observation of a pseudo two-fragment MS2 pattern in a low-resolution instrument. This predictable pattern makes it easier to process MS2 peaks, but it does not solve the complexity problem in MS2 and can compromise the peptide identification accuracy. Therefore, it is desirable to develop a true equal-mass linker that would generate identical (isobaric) fragments in the MS2, and lead to only two predominant peaks in the MS2. Such an identical mass linker (IML) was the goal of our investigation. Herein we report the design and evaluation of new CID-cleavable identical mass cross-linkers based on the sulfoxide functional group to facilitate MS2 and MS3 sequencing.
Results and Discussion
Design and Synthesis of a double-fragmentation cross-linker
Two approaches to solving the problem of four-peak MS2 were explored: one double fragmentation event, where two identical fragments would split from a central core, and one single fragmentation event, where two structurally different (yet isobaric), fragments would result. Our efforts began with the double fragmentation approach. It was hypothesized that a symmetrical linker with two cleavable sites would allow for identical mass modifications on both peptides in MS2. To this end, aryl disulfoxide 2 was designed with the goal of inducing two simultaneous fragmentation events during MS2, resulting in peptides with identical alkene modifications (Figure 2).12 If the desired fragmentation occurred, then MS2 would have two peaks, corresponding to the two formerly interlinked peptides (α and β represent different peptides in Figure 2). Unfortunately, when peptides interlinked by aryl disulfoxide 2 were exposed to varying levels of CID energy in a Thermo Scientific LTQ Orbitrap XL mass spectrometer, only one of the two sulfoxide groups fragmented per CID event, resulting in a four-peak pattern similar to DSSO (Figure 2). The MS3 of the sulfenic acid piece showed further fragmentation to the alkene, but did not cleave the peptide backbone. Increasing the energy of the fragmentation event did not significantly alter the outcome. Although the result is consistent with the physics of the CID step, we were surprised that the selectivity for single bond cleavage was high enough to render the symmetric design of disulfoxide 2 ineffective.
Figure 2.

Top: a proposed double-fragmentation event in CID would lead to only two peptide-containing peaks in the MS2. Bottom: aryl disulfoxide linker cleaves only one bond at a time in a CID step, creating a four-peak pattern in MS2.
Design and synthesis of asymmetric single fragmentation cross-linkers
The failure of the double fragmentation strategy stimulated a new design principle for identical mass linkers. While the desired double fragmentation of cross-linker 2 did not occur, it was observed that the fragmentation occurred selectively on the aliphatic side of the sulfoxide group. This simple feature for controlling the direction of fragmentation of the sulfoxide was incorporated into the design of the new IML (3). The design concept is outlined in Figure 3. Unlike prior cross-linkers, the new IML designs depend upon an asymmetric cross-linker and a selective sulfoxide cleavage. Cross-linking α- and β-peptides leads to two different structures, 4 and 4′. They should have similar mobility in the LC and are likely to elute together. They have the same elemental composition, and will result in MS1 peaks of the same mass. The selective cleavage in the CID step will produce an α-peptide alkene 4α and an α-peptide sulfenic acid 4′α. Similarly, the β-peptide will be represented by a sulfenic acid 4β and an alkene 4′β. If the two sides of the cross-linker are carefully balanced, the sulfenic acid and alkene will have identical formulas and thus converge in MS2. Funneling both structures into MS3 will result in a superimposable fragmentation pattern that could be directly sequenced. At each step along the way the molecular structures will be more complex than for DSSO, but the resulting MS spectra will be simpler because the components are isobaric.
Figure 3.

The identical mass linker (IML) design includes a CID-cleavable sulfoxide, lysine-reactive NHS esters, and identical formula modifications after fragmentation, allowing for one peak per peptide in MS2.
Initial investigations taught us that the thermal stability of the sulfoxide group could not be taken for granted. An effective sulfoxide bond must survive storage and crosslinking, only cleaving during the low energy CID step in the LCMS. To address this concern, control compounds 5 and 6 were prepared to determine the stability of relevant sulfoxide structures (Figure 4). Both compounds 5 and 6 contain a secondary aromatic sulfoxide, but in one case it is benzylic and in the other case it is aliphatic. The two thioether model systems were prepared, oxidized with one equivalent of m-CPBA, and the resulting sulfoxides were monitored by 1H NMR spectroscopy. Alkyl substituted compound 5 showed a small amount of elimination over the course of a week. However, aryl substituted compound 6 completely eliminated over the course of 24 hours. The latter outcome is surprising, and we note that stable secondary benzylic sulfoxides are known in the literature, but the addition of the ester group appears to accelerate the elimination reaction.13 This data demonstrated that benzylic sulfoxides should be avoided in the IML designs, but that aliphatic sulfoxides are promising.
Figure 4.
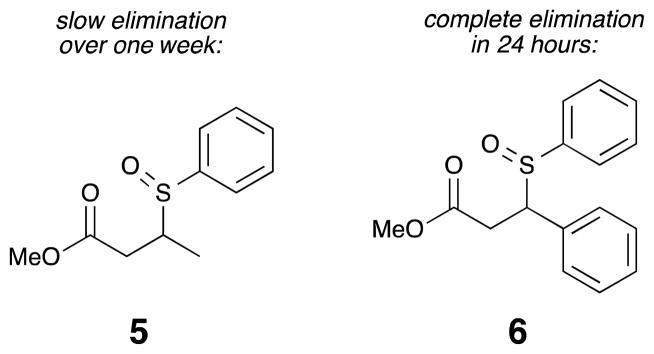
Elimination is slower for α-alkyl sulfoxides than for α-aryl sulfoxides.
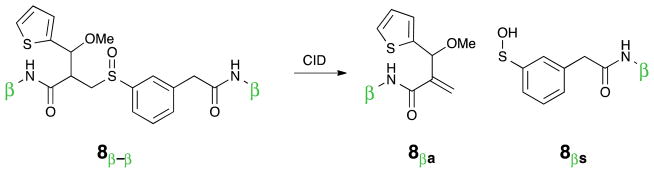
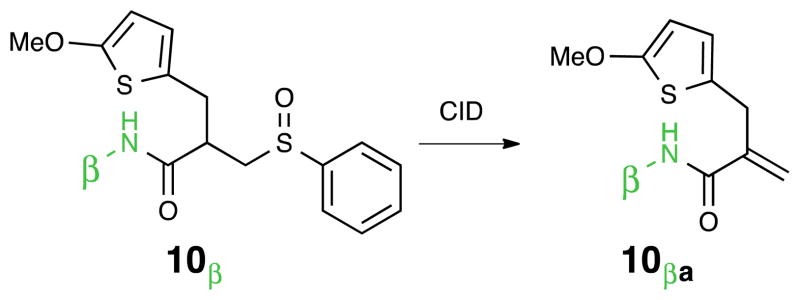
Over the course of this project several proposed IML structures were designed, synthesized, and evaluated. The three IMLs structures are shown in Figure 5 as compounds 7, 8, and 9. In each design, a sulfur atom was incorporated as a stable thiophene moiety, to balance the sulfenic acid, and a methylene group was added to avoid labile benzylic sulfoxides, vida supra. In order to understand the CID-induced fragmentation of these structures, model sulfoxides 10 and 11 were prepared. They were designed to produce the same two molecular fragments as IML 3 (9), but each of these isobaric molecular fragments can be tracked in the MS separately. Sulfoxides 7–11 define the cross-linkers and model compounds that were evaluated in this investigation.
Figure 5.

Three identical mass linkers (7, 8, & 9) and two control compounds (10, 11) were prepared for these studies.
The common building block for these cross-linkers is thiophenol 15, the preparation of which is outlined in Scheme 1. The route closely follows literature precedent.14 3-Hydroxycinnamic acid 12 was converted to the methyl ester with sulfuric acid in methanol before reduction to the dihydrocinnamate 13. The resulting phenol was combined with N,N,-dimethylcarbamoyl chloride in the presence of DABCO, followed by thermal rearrangement to the dimethyl thiocarbamate at 300 °C. Subsequent hydrolysis with KOH in methanol and tetrahydrofuran yielded the free thiol carboxylic acid 15 (Scheme 1).15
Scheme 1.

Synthesis of thiol building block 15
The thiophene half (18) for IML 1 (7) was prepared by lithiation of 2-methoxythiophene 16 and alkylation with allyl bromide. The ester was introduced using a Grubbs cross-metathesis with methyl acrylate (Scheme 2).16,17 These sequences provided enough material for compound characterization and MS evaluation; as a result, the yields for individual steps were not optimized. Thiophene conjugate acceptor 18 was coupled to thiophenol 15 with triethylamine in methanol to form the diester. Subsequent hydrolysis with lithium hydroxide led to the desired diacid 19. Preformed NHS•TFA was used to activate the diacid to form the di-NHS ester.18,19 This procedure led to fewer side products and higher yields than the traditional EDCI coupling. Oxidation to sulfoxide 7 (IML 1) with m-CPBA proceeded uneventfully.10 Subsequent cross-linking studies on IML 1 showed that the compound decomposed (through loss of water) to 21 prior to MS analysis (Scheme 2). This unexpected instability stimulated the design of the two new linkers IML 2 (8) and IML 3 (9), which feature α-branching in place of β-branching to avoid the undesired rearrangement.
Scheme 2.

Synthesis of IML 1 (7)
IML 2 (8) was comprised of the thiophenol fragments 15 and a thiophene fragment with a benzylic methoxy substituent (24). The desired thiophene was rapidly synthesized by a Baylis-Hillman reaction of 2-thiophenecarboxaldehyde 22 and methyl acrylate.20 Methylation of the resulting alcohol using silver oxide and methyl iodide21 was followed by lithium hydroxide hydrolysis of the ester to deliver the carboxylic acid 24. Direct Michael addition of thiols to acrylic acids with catalytic TBAF·3H2O has been reported.22 We incorporated this step in the sequence to avoid the hydrolysis step on the methyl ester since the hydrolysis step led to large amounts of elimination side product in the synthesis of IML 1 (7). This approach decreased the step count and circumvented thiophenol elimination, but resulted in modest yields. The TBAF·3H2O catalyzed reaction produced diacid 25, which was further elaborated to the di-NHS ester and oxidized to the sulfoxide 8 (IML 2, Scheme 3). IML 2 was stable enough for cross-linking and MS evaluation.
Scheme 3.

Synthesis of IML 2 (8)
IML 3 differed from IML 2 only in the position of the methoxy group. In this case, the methoxy resides on the C2 position of the thiophene instead of on the benzylic position. Preparation of IML 3 began with lithiation and carbonylation of 2-methoxythiophene (26) in Scheme 4.23 A Knoevenagel condensation with diethyl malonate introduced the required ester, and subsequent reduction with sodium borohydride selectively removed the reactive alkene to give diester 28.24,25 The esters were hydrolyzed with potassium hydroxide. A decarboxylative Mannich addition led to the desired acrylic acid 29.25 Thiophenol 15 and acrylic acid 29 were combined using the TBAF·3H2O catalyzed reaction22 to produce the diacid, and further elaboration to the di-NHS ester was carried out using the NHS•TFA reagent. Careful oxidation with m-CPBA produced the sulfoxide 9.
Scheme 4.

Synthesis of IML 3 (9)
Two model sulfoxides were prepared, NHS esters 10 and 11, to aid in understanding the MS behavior of IML 3. The synthetic routes are presented in the Scheme 5. Thiophenol 15 was added to thiophene 29 under TBAF•H2O conditions to produce adduct 31 in modest yield. NHS ester formation and oxidation generated the target sulfoxide 10. Preparation of sulfoxide 11 began with thiol 15. Conjugate addition to ethyl acrylate gave adduct 32 in modest yield. NHS ester formation used the NHS•TFA reagent, and subsequent oxidation delivered sulfoxide 11. With the successful preparation of all three cross-linkers and the two model sulfoxides, they were ready to be evaluated in the peptide and protein LCMSn experiments.
Scheme 5.

Synthesis of Sulfoxides 10 & 11
MS Evaluation of IML Agents
The cross-linking behavior of IML 1 (7) was examined using model protein cytochrome C.26 Analysis of cross-linked cytochrome C by SDS-PAGE revealed that IML1 exhibits comparable cross-linking efficiency to DSSO, but MS data showed no sulfoxide fragmentation during CID. The unused cross-linker was recovered and analyzed by ESI+ MS, showing loss of water. 1H NMR showed the mixture of IML 1 diastereomers had coalesced to a single compound. It is hypothesized that a Pummerer-like rearrangement occurred, leading to an α,β-unsaturated cross-linker 21 that is not CID-cleavable (Scheme 2). The next two IML designs included α-branching instead of β-branching to avoid the undesired rearrangement.
The cross-linking ability of IML 2 and IML 3 was compared to that of DSSO. First, an SDS-PAGE analysis of model protein (cytochrome C and lysozyme) cross-linking was explored. The gel in Figure 6 shows both IML linkers exhibit considerable cross-linking activity. In the cyto-chrome C case, they were both comparable to DSSO; in the lysozyme case it appears they may have induced higher order aggregate species. With proof of protein cross-linking, IML cross-linked samples were subjected to MS analysis.
Figure 6.

SDS-PAGE analysis of cytochrome C (left) and lysozyme (right) with DSSO, IML 2, and IML 3 shows comparable small protein cross-linking.
The desired fragmentation (top) and example MS2 fragmentation of IML 2 cross-linked sample (bottom) are shown in Figure 7. Initial testing of IML 2 with synthetic peptide Ac-IR7 (Acetyl-IEAEKGR) in the MS instrument showed significant loss of the methoxy group during CID (Figure 7). The peak with m/z 595.752+ represents a dead end-modified peptide10 that has lost the methoxy substituent during the first CID and has not undergone sulfoxide elimination. This dead end linker could be bonded to the peptide on either side, so it is likely the peak represents a mixture of both possible peptide linkages. (Further detail of this experiment is presented in Supporting Information.) The complexity of the MS data for IML 2 arose from competitive cleavage of the desired sulfoxide and the undesired benzylic methoxy group in 8. This problem led to pri-oritization of IML 3 as the new target.
Figure 7.
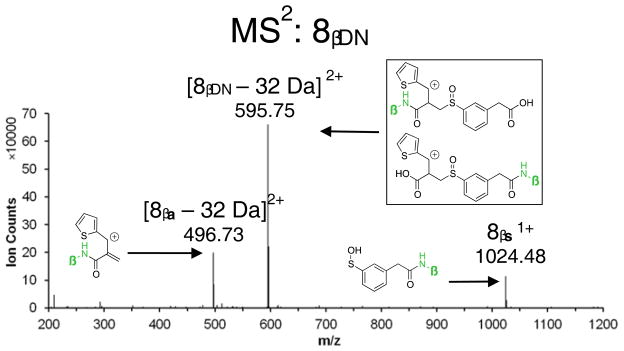
IML 2 suffers from loss of the methoxy substituent at similar energies to sulfoxide fragmentation. (DN = dead end)
The challenges associated with interpreting the m/z 595.75 peak from IML 2 cross-linking experiments (Figure 7 and Supporting Information) led us to prepare model compounds to mimic the behavior each side of the IML 3 cross-linker. Compound 10 mimics the thiophene half, while compound 11 mimics the phenyl portion. In each case, the CID sulfoxide elimination should generate one component of the full IML 3 CID process, and these individual components can be directly analyzed in MS3 without complications from other peptidic components.
CID fragmentation of model compound 10 generated the alkene-modified component of the fragmentation products (Figure 8). Compound 10-modified Ac-IR7 (β) appeared as a 1+ ion in MS1 (m/z 1150.50) and MS2 (m/z 1024.48) (Figure 8). The resulting 10βa peak was sequenced in the MS3 step without difficulty.
Figure 8.
MS2 and MS3 spectra of Ac-IR7 modified by alkene control compound 10, i.e. 10βa.
Model compound 11 was use to probe the sulfenic acid fragmentation products. The 11-modified Ac-IR7 (β) appeared as a 1+ ion in MS1 (m/z 1124.54) and MS2 (m/z 1024.48) (Figure 9). The MS2 peak representing the 1+ ion of thiophenol-modified Ac-IR7 was successfully sequenced in MS3 (Figure 9). As expected the MS2 peaks generated from IR7-10 and IR7-11 had identical m/z 1024.48, satisfying our expectations for the design of the IML 3.
Figure 9.
MS2 and MS3 spectra of Ac-IR7 modified by sulfenic acid control compound 11
Testing of IML 3 began with peptide cross-linking of Ac-TR9 (Acetyl-TTSYKVTIR) and Ac-IR7 (Acetyl-IEAEKGR). As expected, an IML 3 interlink showed identical mass modifications in MS2, achieving proof of concept for the desired two-peak pattern in MS2 (Figure 10). As predicted, the MS2 of Ac-IR7 is identical for IML 3 and for both control compounds 10 and 11 (Figures 8–10). The extraordinarily clean MS2 for IML 3 cross-linked peptides will dramatically simplify the XL-MS analysis of more complex proteins.
Figure 10.

The simplified MS2 pattern with IML 3 cross-linked to two different peptides is shown. Only two peaks are observed in the MS2.
IML 3 cross-linker was next evaluated in the analysis of a protein complex. IML 3 was mixed with a purified sample of the multi-subunit yeast 19S complex (~1MDa) in order to demonstrate its utility as a tool for structural studies. Since the 19S has been thoroughly interrogated with DSSO in our studies, we were able to compare to a previously identified cross-link.3 In this case, IML 3 was used to successfully identify an Rpt4–Rpt5 interlink with clear MS2 and MS3 data (Figure 11). The MS2 shows two peaks representing two interlinked peptides (m/z 488.262+ and m/z 626.332+). Each of these was selected for MS3 and, after undergoing CID, led to peptide sequence data. It is worth noting that in an actual multi-protein analysis, the MS2 spectrum contain a number of small peaks from impurities in the peptide digest sample. The two interlinked peptides were identified and automatically sequenced in this sample because of the improved simplicity in the MS2 spectrum.
Figure 11.

MSn data of a representative IML 3 interlinked peptide of the 19S proteasome complex
3. Conclusion
Three different identical mass linkers (IML) for protein cross-linking studies have been prepared and evaluated. The first one, IML 1, was chemically unstable. IML 2 led to fragmentation patterns complicated by the loss of a labile methoxy group. The third linker, IML 3, showed both good chemical stability and clean fragmentation patterns in LCMSn. IML 3 led to much simpler MS2 patterns upon CID, facilitating the selection of meaningful peaks in the MS3 analysis. These proof-of-concept experiments have elucidated important design criteria for CID cleavable sulfoxide cross-linkers. Foremost among these criteria is the use of primary sulfoxides for their balance of thermal stability and CID lability. The IML strategy, where the cross-linker is designed to produce identical mass and formula components upon CID, is a promising approach to improve the analysis of protein-protein interactions by cross-linking and LCMSn methods.
Experimental Section
General Information
1H NMR and 13C NMR spectra were recorded at ambient temperature at 500 MHz and 125 MHz, respectively, on a Bruker DRX500 NMR instrument. 1H and 13C NMR data is reported as follows: chemical shifts are reported in ppm on a δ scale and referenced to internal tetramethylsilane or residual solvent (TMS: δ 0.00; CHCl3: δ 7.27(1H), 77.23 (13C)), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, qu = quintet, m = multiplet), coupling constants (Hz), and integration. Infrared (IR) spectra were obtained using a FT-IR spectrometer. Accurate mass spectra were acquired on a Waters LCT Premier quadrupole time-of-flight spectrometer and were obtained by peak matching. Gas chromatography/mass spectrometry (GC/MS) was performed with a Thermo-Finnigan Trace Mass Spectrometer Plus quadrupole system with a fused silica capillary column (30 m × 0.32 mm × 0.25 mm) wall-coated with DB-5 (J & W Scientific) using electron ionization (70 eV) or a Waters GCT Premier orthogonal acceleration time-of-flight spectrometer using chemical ionization. Melting points are uncorrected and were obtained using a Büchi 510 melting point apparatus. Analytical thin layer chromatography was performed on EMD Chemicals Inc. silica gel 60 F254 plates. Liquid chromatography was performed using forced flow (flash chromatography) of the indicated solvent system on Sorbent Technologies silica gel (SiO2) 60 (230–400 mesh). Unless otherwise noted, all reactions were carried out under an atmosphere of argon in flame-dried glassware. Solvents were distilled from CaH2 or filtered through alumina before use.27
General procedure 1: Ester Hydrolysis
Starting ester was dissolved in 2:1 or 4:1 THF:H2O, depending on solubility. The mixture was cooled to 0 °C and lithium hydroxide (98%, 5 equiv.) dissolved in minimal H2O was added slowly. The reaction was monitored by thin layer chromatography, while stirring vigorously. Upon completion, the mixture was diluted with diethyl ether and water before the layers were separated. Diethyl ether was added to the resulting aqueous layer, followed by addition of HCl to pH 1. The layers were separated and the aqueous layer was extracted with diethyl ether (2×). Organics were combined, washed with brine, dried over Na2SO4, filtered and evaporated in vacuo.
General procedure 2: TBAF-Promoted Michael Additions
Acrylic acid (0.28 mmol), thiophenol (0.51 mmol), and THF (1 mL) were combined in a round bottom flask to which TBAF•3H2O (0.06 mmol) was added at rt. The flask was fitted with a cold-water condenser and the mixture was heated to 50° C for 16 h. The mixture was cooled to rt, evaporated in vacuo, diluted with EtOAc and washed with 1N HCl (2×). The organic layer was washed with brine, dried over Na2SO4, filtered, and evaporated in vacuo to yield the crude product. In some cases, this reaction was run neat.22
General procedure 3: NHS Ester Preparation
Crude diacid (0.08 mmol) was dissolved in dry dichloromethane (0.62 mL) in a flame dried round bottom flask under argon. NHS•TFA (0.41 mmol) was added before a slow addition of triethylamine (0.49 mmol) at 0° C. The mixture was left to warm slowly overnight while stirring under argon. After 16 h, the mixture was diluted with dichloromethane and washed with water (2×). The organic layer was dried with Na2SO4, filtered, and evaporated in vacuo.
General procedure 4: m-CPBA-Oxidation to Sulfoxide
Di-NHS ester (0.026 mmol) was dissolved in CDCl3 (1 mL) and m-CPBA (77% w/w, 0.026 mmol) was added slowly while monitoring by LRMS ESI + and 1H NMR. When this reaction was run on larger scale than 30 mg starting material, the solution was cooled in an ice bath prior to m-CPBA addition. Once the reaction was complete, the solution was diluted with dichloro-methane (2 mL) and washed with 10% sodium bicarbonate solution (3 × 2 mL). The organic layer was dried over Na2SO4, filtered, and evaporated in vacuo to afford the product.
Experimental Procedures
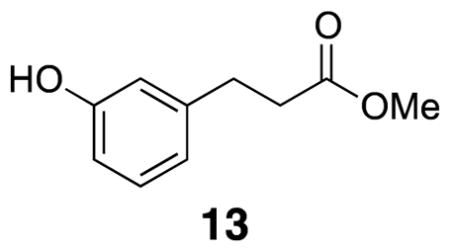
Methyl 3-(3-hydroxyphenyl)propanoate (13)
Acetyl chloride (4.39 mL, 61.5 mmol), m-hydroxycinnamic acid (12) (10.0 g, 60.9 mmol), and methanol (300 mL) were combined in a round bottom flask, fitted with a cold water condenser, and heated to 70 °C for 4.5 h. The reaction mixture was cooled to room temperature and the solvent was removed and flushed with CH2Cl2. The crude methyl ester was dissolved in ethanol (206 mL) and acetic acid (11.0 mL) before addition of Pd/C (5% by wt, 6.50 g, 3.05 mmol). The headspace was evacuated and a balloon of H2 gas was attached to the flask. The reaction stirred for two days and there was no change in Rf. The reaction mixture was filtered through Celite and solvent was removed in vacuo. The crude product was purified by flash column chromatography (30% EtOAc/hexanes) to yield desired methyl ester 13 (8.82 g, 80%). Spectra were identical to previously reported data.28
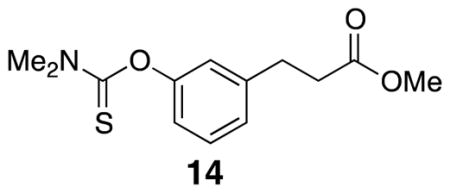
Methyl 3-(3-((dimethylcarbamothioyl)oxy)phenyl)propanoate (14)
Methyl ester 13 (5.00 g, 27.7 mmol) was combined with dimethylthiocarbamoyl chloride (10.3 g, 83.3 mmol) and DABCO (9.3 g, 82.9 mmol) in DMF (14 mL) in a round bottom flask. The mixture was heated to 80 °C for 18 h before the addition of EtOAc, H2O, and brine. The layers were separated and the organic layer was washed with brine. The aqueous layer was extracted with EtOAc and the organic layer was washed with brine. The organic layers were combined, dried over Na2SO4, and the solvent was removed in vacuo. The crude material was purified by flash column chromatography (20% EtOAc/hexanes) to afford the desired product (4.60 g, 62%). Spectra were comparable to previously reported data.28 1H NMR (500 MHz, CDCl3) δ 7.31 (t, J = 7.7, 1H), 7.10 (d, J = 7.5, 1H), 6.93 (overlapping d, J = 7.5, 2H), 3.68 (s, 3H), 3.46 (s, 3H), 3.35 (s, 3H), 2.98 (t, J = 7.7, 2H), 2.66 (t, J = 7.7, 2H); 13C NMR (125 MHz, CDCl3) δ 30.9, 35.6, 38.9, 43.5, 51.9, 120.9, 122.9, 126.1, 129.4, 142.1, 154.3, 173.4, 188.0; IR (thin film) 3404, 2948, 1736, 1532, 1286 cm−1; Accurate Mass ES+ m / z calcd for C13H17NO3SNa (M+Na)+ 290.0827, found 290.0835.
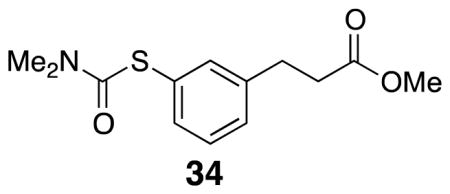
Methyl 3-(3-((dimethylcarbamoyl)thio)phenyl)propanoate (34)
Starting protected alcohol 14 (0.2 g, 0.748 mmol) was placed in a crimp-top vial and sealed under argon. The neat oil was placed in a 310 °C sand bath. After 20 min, the reaction was complete by TLC and the vial was allowed to cool to rt. The crude product was purified by flash column chromatography (20% EtOAc/hexanes) to yield dark yellow oil product 34 (91 mg, 45%). 1H NMR (500 MHz, CDCl3) δ 7.35–7.29 (m, 3H), 7.22 (d, J = 7.4, 1H), 3.08 (br s, 3H), 3.03 (br s, 3H), 2.96 (t, J = 8.0, 2H), 2.64 (t, J = 8.0, 2H); 13C NMR (125 MHz, CDCl3) δ 173.4, 167.1, 141.5, 135.7, 133.9, 129.4, 129.2, 129.0, 51.9, 37.1, 36.0, 30.9; IR (thin film) 2951, 2360, 1736, 1670, 1365 cm−1; Accurate Mass ES+ m / z calcd for C13H17NO3SNa (M+Na)+ 290.0827, found 290.0830.
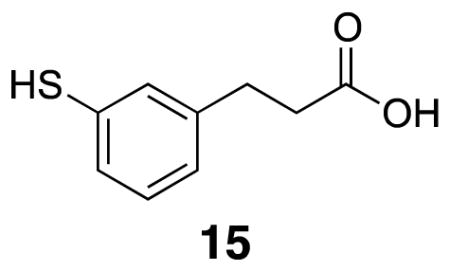
3-(3-Mercaptophenyl)propanoic acid 15
Dimethyl thiocarbamate 34 (0.20 g, 0.75 mmol) was combined with KOH (0.17 g) in methanol (5 mL) and THF (5 mL) in a sealed vial under argon. The vial was heated to 70 °C for five h. The vial was allowed to cool and CH2Cl2 and 5% HCl were added. The layers were separated before the aqueous layer was extracted three times with CH2Cl2. The organic layer was dried with Na2SO4, filtered, and the solvent was removed in vacuo. The crude product was purified by flash column chromatography (40% EtOAc/hexanes) to yield the product (86 mg, 63 %). m.p. 62–65 °C; 1H NMR (500 MHz, CDCl3) δ 7.18–7.10 (m, 3H), 6.98 (d, J = 7.2, 1H), 3.43 (s, 1H), 2.89 (t, J = 7.8, 2H), 2.66 (t, J = 7.8, 2H); 13C NMR (125 MHz, CDCl3) δ 179.4, 141.4, 131.2, 129.5, 129.4, 127.6, 35.5, 30.4; IR (thin film) 3055, 2931, 2862, 2557, 1693 cm−1; Accurate Mass ES− m / z calcd for C9H9O2S (M-H)− 181.0323, found 181.0323.
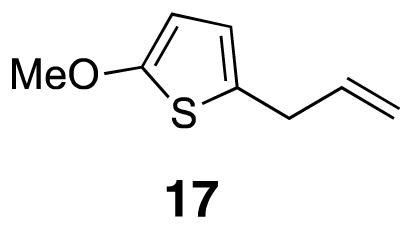
2-Allyl-5-methoxythiophene (17)
2-Methoxythiophene (16) (1.1 g, 1.0 mL, 9.9 mmol) was dissolved in dry diethyl ether (28 mL) in a flame dried round bottom flask. sec-Butyllithium (13.7 mL of [0.72] solution, 9.9 mmol) was added to the mixture dropwise at 0 °C. After 90 min, the reaction mixture was cooled to −78 °C allyl bromide (0.86 mL, 9.9 mmol) was added. The reaction was allowed to slowly warm and stir overnight. The reaction was cooled to 0 °C before addition of saturated ammonium chloride solution (14 mL). The layers were separated before the aqueous layer was extracted with diethyl ether (2×). The organic layers were combined, washed with brine, and dried over MgSO4 before being filtered and evaporated. The crude product was purified by flash column chromatography (pentane) to yield the product as a green oil (0.70 g, 46%). 1H NMR (500 MHz, CDCl3) δ 6.39 (d, J = 3.7, 1H), 6.00 (d, J = 3.7, 1H), 5.97–5.92 (m, 1H), 5.13 (d, J = 15.5, 1H), 5.07 (dt, J = 8.7, 1H), 3.40 (d, J = 5.5, 2H); 13C NMR (125 MHz, CDCl3) δ 164.9, 136.8, 129.3, 121.7, 116.3, 103.4, 60.4, 34.9; IR (thin film) 3008, 2831, 1562, 1512, 1215 cm−1; Accurate Mass (GCMS CI+) m / z calcd for C8H11OS (M+H)+ 155.0531, found 155.0528.
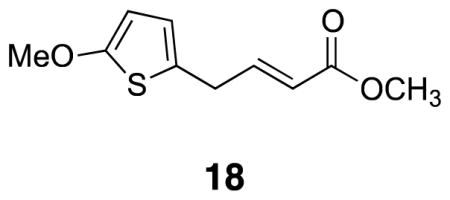
(E)-Methyl 4-(5-methoxythiophen-2-yl)but-2-enoate (18)
2-Allyl-5-methoxythiophene 17 (0.256 g, 1.66 mmol) was combined with methyl acrylate (4.15 mL) and Grubbs 2 catalyst (0.035 g, 0.0415 mmol) in a sealed vial under argon. The reaction was heated at 90 °C for eight hours. The reaction mixture was cooled to rt, evaporated, and placed on the high vacuum. The crude product was purified by flash column chromatography (20–40% CH2Cl2/pentane) to yield desired product (0.100 g, 28%). 1H NMR (500 MHz, CDCl3) δ 7.06–7.02 (dt, 1H, J = 6.6, 15.5), 6.42 (d, J = 3.7, 1H), 6.01 (d, J = 3.7, 1H), 5.88 (d, J = 15.5, 1H), 3.85 (s, 3H), 3.73 (s, 3H), 3.54 (d, J = 6.6, 2H); 13C NMR (125 MHz, CDCl3) δ 167.0, 165.4, 146.7, 126.0, 123.1, 122.3, 103.6, 60.5, 51.8, 33.0; IR (liquid) 2951, 2831, 1728, 1658, 1512 cm−1; Accurate Mass (GCMS CI+) m / z calcd for C10H12O3S (M+H)+ 213.0585, found 213.0589.
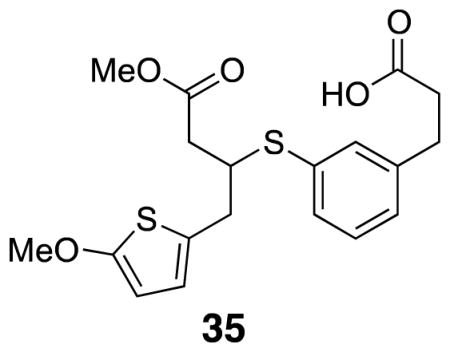
3-(3-((4-Methoxy-1-(5-methoxythiophen-2-yl)-4-oxobutan-2-yl)thio)phenyl)propanoic acid (35)
Dimethyl thiocarbamate 34 (0.10 g, 0.38 mmol) was combined with KOH (0.10 g, 1.8 mmol) in methanol (5.0 mL) and THF (5.0 mL) in a sealed vial under argon. The vial was heated to 70 °C for five hours. The vial was allowed to cool and 1M HCl (0.50 mL) was added. Methyl acrylate 18 (0.027 g, 0.13 mmol) and triethylamine (0.038 mL, 0.38 mmol) were added to the solution and the reaction mixture was left to stir at rt overnight. The reaction was extracted with CH2Cl2 (3×) and the organics were combined, dried over Na2SO4, filtered, and evaporated. The crude product was purified by flash column chromatography (20% EtOAc/hexanes) to yield desired product (0.017 g, 18%). 1H NMR (500 MHz, CDCl3) δ 7.31 (app d, J = 8.7, 2H), 7.23 (app d, J = 7.5, 2H), 7.11 (d, J = 7.6, 1H), 6.43 (d, J = 3.7, 1H), 5.99 (d, J = 3.7, 1H), 3.84 (s, 3H), 3.66 (s, 3H), 3.67–3.64 (overlapping m, 1H), 3.01 (dd, J = 15.0, 5.0, 1H), 2.98–2.86 (m, 3H), 2.71–2.60 (m, 3H), 2.53 (dd, J = 15.0, 5.0, 1H); 13C NMR (125 MHz, CDCl3) δ 177.7, 172.1, 165.4, 141.3, 133.8, 133.1, 131.3, 129.4, 127.9, 126.6, 124.0, 103.2, 60.4, 52.1, 46.3, 38.9, 35.7, 35.5, 30.6; IR (thin film) 2951, 1736, 1709, 1435, 1211 cm−1; Accurate Mass ES+ m / z calcd for C19H22O5S2Na (M+Na)+ 417.0806, found 417.0807.
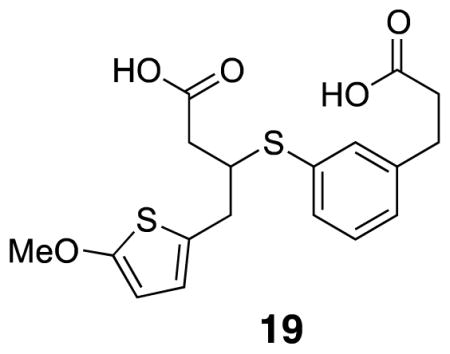
3-((3-(2-Carboxyethyl)phenyl)thio)-4-(5-methoxythiophen-2-yl)butanoic acid (19)
Mono-carboxylic acid 35 (0.017 g, 0.043 mmol) was dissolved in 4:1 THF/H2O (0.40 mL) and Li-OH•H2O was added and the mixture was stirred vigorously for 2.5 h. H2O and diethyl ether were added and the aqueous layer was washed twice more with diethyl ether. 1M HCl was added until pH 1 was reached, and the aqueous layer was extracted with diethyl ether (3×). The combined organic layers were washed with brine and evaporated. The crude product was redissolved in CH2Cl2, filtered through cotton and evaporated down to yield an orange oil (0.0060 g, 34%). 1H NMR (500 MHz, CDCl3) δ 7.35 (s, 1H), 7.30 (d, J = 7.7, 1H), 7.22 (t, J = 7.6, 1H), 7.11 (d, J = 7.5, 1H), 6.45 (d, J = 3.6, 1H), 6.00 (d, J = 3.7, 1H), 3.84 (s, 3H), 3.61 (quintet, J = 7.0, 1H), 3.03 (dd, J = 15.2, 6.5, 1H), 3.00–2.90 (m, 3H), 2.68 (t, J = 7.2, 2H), 2.64 (app dd, J = 11.3, 5.9, 1H), 2.53 (app dd, J = 16.1, 8.2, 1H); 13C NMR (125 MHz, CDCl3) δ 178.9, 177.6, 165.4, 141.3, 133.7, 133.6, 131.8, 129.4, 128.2, 126.5, 124.1, 103.3, 60.4, 46.7, 39.4, 36.0, 35.6, 30.6; IR (thin film) 3086, 2924, 2357, 1709, 1211 cm−1; Accurate Mass ES− m / z calcd for C18H19O5S2 (M-H)− 379.0674, found 379.0669.
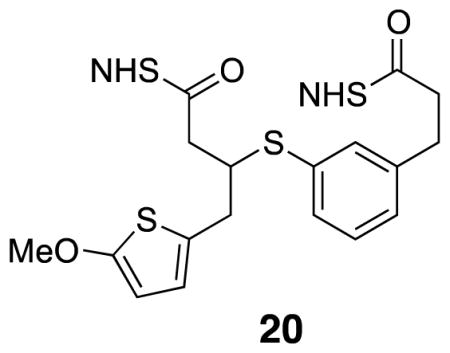
Di-NHS-ester 20
Diacid 19 (0.005 g, .013 mmol) was combined with CH2Cl2 (0.10 mL), tri-ethylamine (0.011 mL, 0.080 mmol) and NHS•TFA (0.017 g, 0.080 mmol) at rt under argon. After 3 h stirring at rt, TLC showed the reaction was complete. CH2Cl2 and H2O were added before the organic layer was washed with H2O (2×) and brine before it was filtered through cotton and evaporated. The crude product was purified by flash column chromatography (50% EtOAc/hexanes) to yield desired product (0.0060 g, 80%). 1H NMR (500 MHz, CDCl3) δ 7.45 (s, 1H), 7.39 (dd, J = 7.7, 12.8, 1H), 7.28 (t, J = 7.6, 1H), 7.18 (d, J = 7.5, 1H), 6.50 (d, J = 3.7, 1H), 6.01 (d, J = 3.7, 1H), 3.86 (s, 3H), 3.62 (quintet, J = 6.8, 1H), 3.08 (dd, 1H), 3.08–3.00 (m, 4H), 2.93 (dd, J = 8.0, 7.3, 2H), 2.93–2.75 (m, 9H); 13C NMR (125 MHz, CDCl3) δ 169.3, 169.2, 168.9, 168.0, 166.8, 165.6, 140.4, 134.3, 132.9, 132.6, 129.6, 128.6, 126.1, 125.8, 124.5, 103.4, 60.4, 46.1, 35.4, 35.2, 32.6, 30.4, 26.1, 25.8, 25.8, 25.7; IR (thin film) 2927, 1813, 1786, 1739, 1207 cm−1; Accurate Mass ES+ m / z calcd for C26H26N2O9S2 (M+Na)+ 597.0977, found 597.0980.
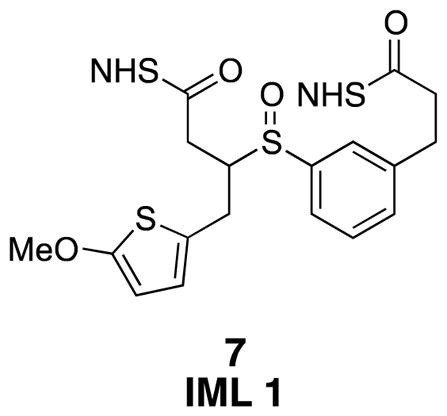
IML 1 (7)
Di-NHS-ester 20 (0.006 g, 0.01 mmol) was subjected to general procedure 4 to yield desired product as a mixture of diastereomers contaminated with grease (0.006 g crude after workup). 1H NMR (500 MHz, CDCl3) δ 7.51–7.30 (m, 4H), 6.45 (d, J = 3.8, 1H), 6.04 (d, J = 3.8, 1H), 3.88–3.83 (m, 2H), 3.63 (d, J = 6.5, 2H), 3.13–2.97 (m, 3H), 2.94–2.87 (m, 2H), 2.85 (s, 8H), 2.62 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 169.4, 169.3, 168.9, 153.0, 140.6, 140.5, 135.4, 134.1, 131.8, 130.6, 129.8, 129.7, 129.4, 127.42, 127.37, 126.1, 124.3, 124.2, 123.9, 123.0, 116.7, 103.8, 60.5, 41.2, 33.4, 32.6, 32.5, 30.5, 30.4, 30.2, 29.9, 26.1, 25.82, 25.76; IR (thin film) 2924, 2256, 1817, 1782, 1739 cm−1; Accurate Mass ES+ m / z calcd for C26H26N2O10S2Na (M+Na)+ 613.0927, found 613.0920.
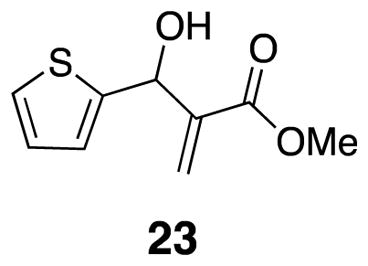
Methyl 2-(hydroxy(thiophen-2-yl)methyl)acrylate 23
Thiophene carboxaldehyde 22 (5 mL, 0.054 mmol) and methyl acrylate (6.24 mL, 0.070 mmol) were combined with DABCO (3.9 g, 0.035 mmol) in a round bottom flask and the mixture was sonicated for 24–48 h. The resulting mixture was loaded onto a silica gel column and the product was isolated by flash chromatography as yellow oil (7.14 g, 66%). Spectra were identical to previously reported data.20
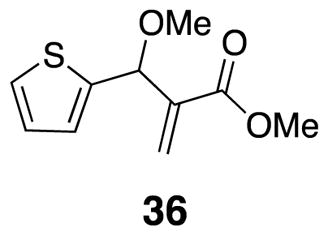
Methyl 2-(methoxy(thiophen-2-yl)methyl)acrylate (36)
Benzylic alcohol 23 (0.50 g, 2.5 mmol) was combined with Ag2O (1.2 g, 5.0 mmol) and methyl iodide (0.47 mL, 7.6 mmol) in CH2Cl2 (10 mL). The mixture was sealed in a capped vial and heated to 50 °C over 72 h.21 The reaction mixture was cooled, filtered through a silica plug with CH2Cl2 rinses, and evaporated in vacuo to afford the product as a clear colorless oil (0.374 g, 70%) 1H NMR (500 MHz, CDCl3) δ 7.27 (d, J = 5.5, 1H), 7.01 (d, J = 3.1, 1H), 6.95 (dd, J = 3.6, 4.8, 1H), 6.37 (s, 1H), 6.04 (s, 1H), 5.40 (s, 1H), 3.74 (s, 3H), 3.38 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 166.3, 143.6, 140.8, 126.8, 126.5, 125.8, 125.4, 57.3, 52.2; IR (thin film) 2993, 2951, 2827, 1724, 1631 cm−1; Accurate Mass ES+ m / z calcd for C10H12O3SNa (M+Na)+ 235.0405, found 235.0406.
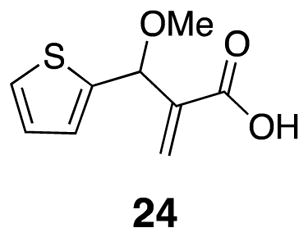
2-(Methoxy(thiophen-2-yl)methyl)acrylic acid (24)
Acid 24 was prepared from methyl ether 36 (0.374 g, 1.76 mmol) by general procedure 1. Crude product was purified by flash chromatography (30% EtOAc/hexanes) (0.1 g, 0.471 mmol) (300 mg, 86%) 1H NMR (500 MHz, CDCl3) δ 7.29 (d, J = 5.0, 1H), 7.02 (d, J = 3.3, 1H), 6.96 (t, J = 3.5, 1H), 6.53 (s, 1H), 6.15 (s, 1H), 5.37 (s, 1H), 3.38 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 171.1, 143.1, 140.2, 128.0, 126.8, 126.7, 126.0, 76.3, 57.3; IR (thin film) 2934, 2664, 1697, 1629, 1435 cm−1; Accurate Mass ES+ m / z calcd for C9H10O3SNa (M+Na)+ 221.0248, found 221.0240.
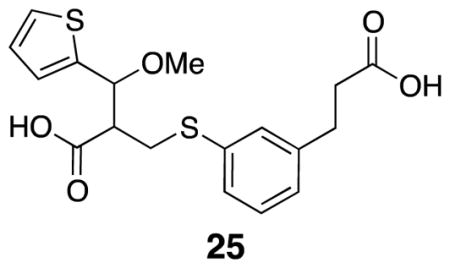
2-(((3-(2-Carboxyethyl)phenyl)thio)methyl)-3-methoxy-3-(thiophen-2-yl)propanoic acid (25)
Thiophenol 15 (0.054 g, 0.30 mmol), carboxylic acid 24 (0.039 g, 0.20 mmol), and THF (enough to dissolve) were combined with TBAF•H2O (0.012 g, 0.039 mmol) in a capped vial. The vial was evacuated and backfilled with argon before heating to 50 °C overnight. The solvent was removed in vacuo before the crude mixture was purified by flash column chromatography (60% EtOAc/hexanes) to yield desired product (0.047 g, 63%). The material was not clean; it was taken on to the next step without further purification. IR (thin film) 2930, 2566, 1708, 1594, 1420 cm−1; Accurate Mass ES+ m / z calcd for C18H20O5S2Na (M+Na)+ 403.0650, found 403.0656.
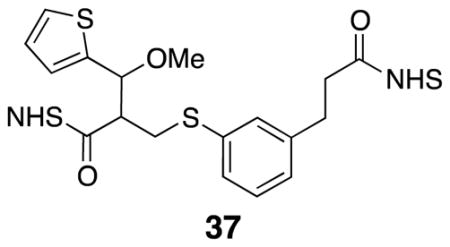
Di-NHS ester 37
Diacid 25 (0.031 g, 0.081 mmol) was subjected to general procedure 3. The crude product was purified by flash column chromatography (70% EtOAc/pentane) to yield desired product (0.031 g, 67%). 1H NMR (500 MHz, CDCl3) δ 7.38 (d, J = 4.7, 1H), 7.21–7.16 (m, 1H), 7.13 (d, J = 8.3, 1H), 7.07–7.01 (m, 4H), 5.01 (d, J = 5.3, 1H), 3.44 (dd, J = 13.9, 3.2, 1H), 3.38 (s, 3H), 3.32 (dd, J = 13.9, 9.9, 1H), 3.27–3.21 (m, 1H), 2.98–2.93 (m, 2H), 2.90–2.76 (m, 10H); 13C NMR (125 MHz, CDCl3) δ 169.3, 168.9, 168.0, 167.1, 141.5, 140.2, 135.5, 129.5, 129.1, 128.4, 127.3, 126.7, 126.6, 126.1, 79.2, 58.1, 51.5, 32.7, 32.6, 30.5, 30.0, 25.8; IR (thin film) 2940, 2255, 1813, 1784, 1739 cm−1; Accurate Mass ES+ m / z calcd for C26H26N2O9S2Na (M+Na)+ 597.0977, found 597.0980.
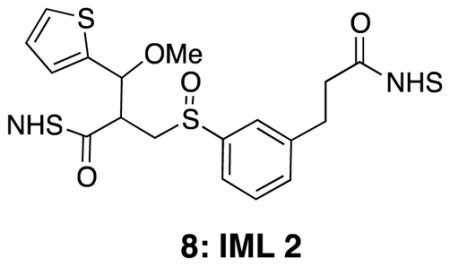
IML 2 (8)
Di-NHS ester 37 (0.015 g, 0.026 mmol) was subjected to general procedure 4 to yield desired product (0.013 g, 84%). 1H NMR (500 MHz, CDCl3) δ 7.53–7.29 (m, 4H), 7.03 (s, 1H), 7.01–6.96 (m, 1H), 5.17 (d, J = 4.3, 0.5H), 5.12 (d, J = 4.5, 0.5H), 3.60–3.22 (m, 6H), 3.20–3.00 (m, 1H), 3.12 (overlapping t, J = 7.6, 2H), 2.95 (t, J = 7.2, 2H), 2.83 (br s, 8H); 13C NMR (125 MHz, CDCl3) δ 169.27, 169.26, 169.23, 169.20, 169.1, 168.7, 167.8, 166.9, 166.6, 140.94, 140.93, 140.88, 140.6, 136.8, 135.5, 134.1, 131.8, 131.4, 130.6, 130.1, 130.0, 129.9, 129.8, 129.5, 127.6, 127.4, 127.0, 126.8, 126.42, 126.38, 126.30, 126.28, 126.18, 126.15, 124.33, 124.25, 123.9, 123.0, 122.9, 122.7, 79.1, 79.0, 76.4, 58.4, 58.3, 57.5, 54.8, 53.1, 46.1, 45.1, 32.6, 32.50, 32.47, 32.45, 30.5, 30.44, 30.38, 25.8; IR (thin film) 2939, 2251, 1812, 1781, 1738 cm−1. Accurate Mass ES+ m / z calcd for C26H26O10N2S2Na (M+Na)+ 613.0927, found 613.0925.
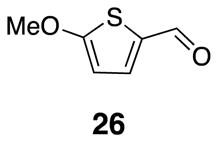
2-Methoxythiophene carboxaldehyde 26
2-Methoxythiophene (1.13 g, 9.89 mmol) was combined with diethyl ether (27.5 mL) in a flame-dried round bottom flask under argon. The reaction mixture was cooled to 0 °C before addition of s-butyllithium (13.7 mL, [0.72]) over 20 min. After one hour, the reaction mixture was cooled to −78 °C before slow addition of N-N-dimethylformamide (1.38 mL, 17.8 mmol). Once the addition was complete, the reaction mixture was left to warm slowly to rt and stir overnight. Crude product was purified by flash chromatography to yield 1.1 g (78%) of the desired product. Spectra were identical to previously reported data.23
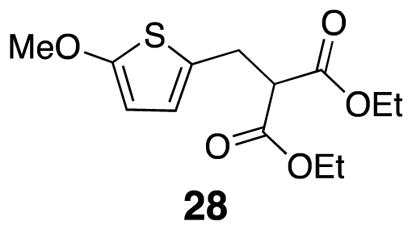
Diethyl 2-((5-methoxythiophen-2-yl)methyl)malonate (28)
2-Methoxythiophene-carboxaldehyde 26 (3.15 g, 22.2 mmol), cyclohexane (111 mL), diethyl malonate (3.48 mL, 22.8 mmol), piperidine (0.330 mL, 0.890 mmol), and benzoic acid (0.108 g, 0.890 mmol) were combined in a round bottom flask fitted with a Dean Stark trap, cold water condenser, and fitted with a calcium carbonate drying tube. The reaction mixture was heated in a 105 °C oil bath for 20 h. The reaction flask was allowed to cool to rt before the solvent was removed in vacuo. Diethyl ether was added to redissolve the mixture and the organic solution was washed with 10% hydrochloric acid solution (3×), saturated sodium bicarbonate solution (3×), and brine (1×) before it was filtered and the solvent was removed in vacuo. The product 27 (6.20 g, 98%) was not purified.24 The resulting crude product was dissolved in ethanol and cooled to 0° C. Sodium borohydride (0.429 g, 11.3 mmol) was added to the solution and gas evolved. After 45 min, the ice bath was removed and the solution was heated to 40° C in an oil bath. Reaction progress was monitored by TLC (20% EtOAc/hexanes). Once the reaction was complete, water was added before adding acetic acid to pH 4. Solids were filtered off and diethyl ether and water were added to the filtrate. The layers were separated, the organics washed with water and brine before drying with Na2SO4, filtering, and evaporating in vacuo. The crude product 28 was isolated as a yellow oil (5.26 g, 85%)24,25 1H NMR (500 MHz, CDCl3) δ 6.44 (d, J = 3.7, 1H), 5.96 (d, J = 3.8, 1H), 4.19 (qd, J = 7.2, 2.1, 4H), 3.83 (s, 3H), 3.59 (t, J = 7.6, 1H), 3.26 (d, J = 7.6, 2H), 1.25 (t, J = 7.1, 6H); 13C NMR (125 MHz, CDCl3) δ 168.7, 165.2, 126.2, 123.5, 103.3, 61.8, 60.4, 54.3, 29.7, 14.3; IR (thin film) 2939, 1731, 1561, 1506, 1208 cm−1; Accurate Mass ES+ m / z calcd for C13H18O5SNa (M+Na)+ 309.0773, found 309.0770.
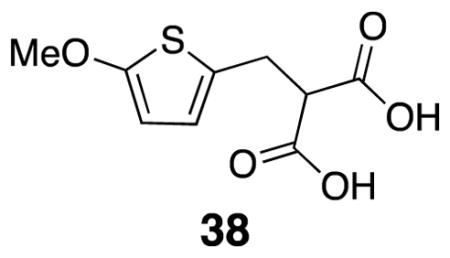
2-((5-Methoxythiophen-2-yl)methyl)malonic acid (38)
Diethyl ester 28 (5.26 g, 18.4 mmol) was dissolved in ethanol (47 mL) at rt. A solution of KOH (2.1 g, 36.7 mmol) in ethanol (20 mL) was added slowly to the mixture by addition funnel. After 150 min, the mixture was cooled to 0° C and diethyl ether was added to induce further crystallization. The solids were filtered off and rinsed with heptane. The isolated solids were dissolved in saturated bicarbonate solution and EtOAc was added. Concentrated HCl was added to pH 1 and the aqueous layer was extracted (3 x 75 mL). The organic layer was washed with brine, dried over Na2SO4, filtered, and evaporated in vacuo. The crude product was isolated as a white crystalline solid (5.63 g, 48%). m.p. 266–279 °C; 1H NMR (500 MHz, CD3OD) δ 6.50 (d, 1H, J = 3.8), 5.99 (d, 1H, J = 3.9), 3.85 (s, 3H), 3.73 (t, 1H, J = 7.3), 3.33 (app d, 2H, J = 7.3); 13C NMR (125 MHz, D2O) δ 178.4, 163.6, 129.9, 122.2, 103.9, 70.9, 60.5, 31.0; IR (thin film) 3354, 1576, 1512, 1332, 1201 cm−1; Accurate Mass ES− m / z calcd for C9H9O5S (M-H)− 229.0171, found 229.0174.
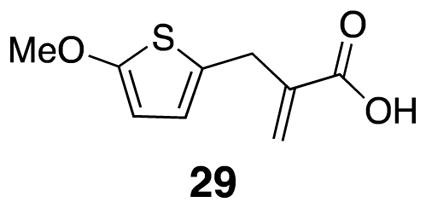
2-((5-Methoxythiophen-2-yl)methyl)acrylic acid (29)
Diacid 38 (1.89 g, 8.21 mmol) was combined with 37% formalin (5.96 mL) and diethylamine (1.44 mL) under argon. After three hours at room temperature, the mixture was heated to 100° C for 2 h. The mixture was cooled to rt, chloroform was added, and the organic layer was extracted with saturated bicarbonate solution (3 x 50 mL). Chloroform was added to the aqueous layer and concentrated HCl was added slowly, with stirring, to pH 1. The resulting mixture was extracted with chloroform (3×), dried over Na2SO4, filtered, and evaporated in vacuo. The product was isolated crude as a light brown solid (0.77 g, 47%),29 m.p. 59 °C–62 °C; 1H NMR (500 MHz, CDCl3) δ 6.45 (d, J = 3.7, 1H), 6.38 (s, 1H), 6.01 (d, J = 3.7, 1H), 5.74 (d, J = 1.0, 1H), 3.85 (s, 3H), 3.66 (s, 2H); 13C NMR (125 MHz, CDCl3) δ 171.5, 165.1, 138.9, 128.7, 126.9, 123.1, 103.3, 60.2, 32.2; IR (thin film) 2995, 1696, 1560, 1512, 1203 cm−1; Accurate Mass ES− m / z calcd for C9H9O3S (M-H) − 197.0272, found 197.0262.
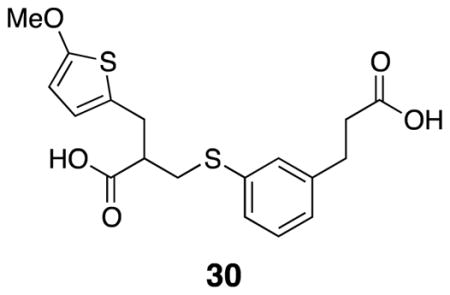
3-((3-(2-Carboxyethyl)phenyl)thio)-2-((5-methoxythiophen-2-yl)methyl)propanoic acid (30)
Acid 29 (0.77 g, 3.9 mmol), thiophenol 15 (0.76 g, 4.2 mmol), and THF (7 mL) were combined in a round bottom flask to which TBAF trihydrate (0.25 g, 0.78 mmol) was added at rt. The flask was fitted with a cold-water condenser and the mixture was heated to 50° C for 16 h. The mixture was cooled to rt, evaporated in vacuo, and diluted with EtOAc and washed with 1N HCl (2×). The organic layer was washed with brine, dried over Na2SO4, filtered, and evaporated in vacuo to yield dark red oil. The product was a mixture of thiophene starting material 29 and desired product 30 (crude: 1.48 g, quantitative). A small amount of desired product was purified for characterization by column chromatography (50% EtOAc/pentane). Pure product appeared as a white crystalline solid m.p. = 110 – 115° C. 1H NMR (500 MHz, CDCl3) δ 7.21–7.18 (m, 3H), 7.07–7.02 (m, 1H), 6.41 (d, J = 3.7, 1H), 5.97 (d, J = 3.7, 1H), 3.19 (dd, J = 7.5, 13.8, 1H), 3.11 (dd, J = 6.9, 14.8, 1H), 3.07 (dd, J = 6.0, 13.8, 1H), 3.02 (dd, J = 7.0, 15.2, 1H), 2.91 (t, J = 7.4, 2H), 2.86 (quintet, J = 6.7, 1H), 2.65 (app t, J = 7.1, 2H); 13C NMR (125 MHz, CDCl3) δ 179.5, 179.0, 165.2, 141.2, 135.4, 130.6, 129.5, 128.9, 127.5, 127.1, 126.1, 123.8, 103.4, 60.4, 47.3, 35.7, 34.9, 31.4, 30.7; IR (thin film) 2942, 1702, 1556, 1506, 1432 cm−1; Accurate Mass ES− m / z calcd for C18H19O5S2 (M-H)− 379.0674, found 379.0675.
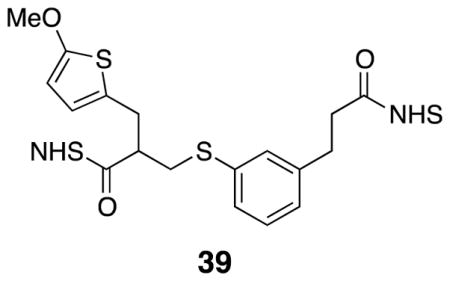
Di-NHS ester 39
Crude diacid 30 (1.48 g) was dissolved in dry dichloromethane (30 mL) in a flame dried round bottom flask under argon. NHS•TFA (4.12 g, 19.5 mmol) was added before a slow addition of triethylamine (3.24 mL, 23.4 mmol) at 0° C. The mixture was left to warm slowly overnight while stirring under argon. After 16 h, the mixture was diluted with dichloromethane and washed with water (2×). The organic layer was dried with Na2SO4, filtered, and evaporated in vacuo. The products were separated using flash column chromatography (50 % EtOAc/pentane – 70 % EtOAc/pentane), yielding di-NHS ester 39 (0.952 g, 43%). 1H NMR (500 MHz, CDCl3) δ 7.29 (s, 1H), 7.24 (overlapping d, 2H), 7.10 (d, 1H), 6.49 (d, J = 3.8, 1H), 6.00 (d, J = 3.8, 1H), 3.83 (s, 3H), 3.28 (dd, J = 7.4, 13.7, 1H), 3.24 (dd, J = 6.5, 15.2, 1H), 3.17–3.04 (m, 3H), 2.92 (d, J = 8.3, 3H), 2.83 (s, 8H); 13C NMR (125 MHz, CDCl3) δ 169.3, 169.2, 169.1, 168.0, 165.5, 140.4, 134.9, 130.7, 129.7, 129.4, 127.4, 124.8, 124.5, 103.5, 60.4, 44.9, 34.5, 32.6, 31.5, 30.5, 25.8, 25.8; IR (thin film) 2945, 1811, 1781, 1738, 1206 cm−1; Accurate Mass ES+ m / z calcd for C26H26N2O9S2Na (M+Na)+ 597.0977, found 597.0967.
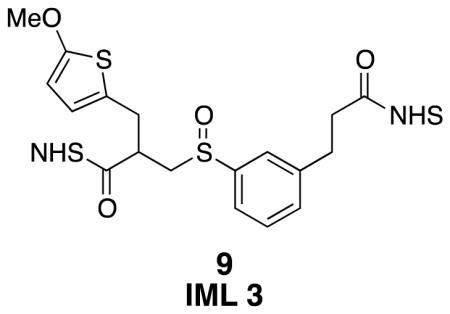
IML 3 (9)
Sulfoxide 9 was prepared from di-NHS ester 39 (0.030 g, 0.052 mmol) using general procedure 4. The isolated product appeared as an orange oil (27 mg, 87%). The product was characterized as a 1:0.7 mixture of diastereomers, with a very small amount of starting sulfide that was not consumed in the reaction. 1H NMR (500 MHz, CDCl3) δ 7.58–7.46 (m, 3H), 7.41–7.36 (m, 1H), 6.60 (d, J = 3.8, 0.6H), 6.50 (d, J = 3.8, 0.4H), 6.03 (d, J = 3.8, 0.6H), 5.99 (d, J = 3.8, 0.4H), 3.86 (s, 1.8H), 3.84 (s, 1.2H), 3.55–3.47 (m, 0.6H), 3.37–3.25 (m, 3.8H), 3.15 (t, J = 7.6, 2H), 3.08–3.01 (m, 0.6H), 2.97 (t, J = 7.6, 2H), 2.83 (br s, 8H); 13C NMR (125 MHz, CDCl3) δ 169.29, 169.25, 169.2, 168.9, 168.8, 168.5, 167.82, 167.80, 165.8, 165.7, 144.0, 143.5, 141.03, 140.96, 140.4, 131.9, 131.6, 131.1, 130.1, 129.9, 129.6, 127.5, 127.4, 126.0, 125.4, 124.9, 124.2, 124.1, 123.9, 123.8, 123.5, 122.9, 122.7, 103.7, 103.6, 60.39, 60.37, 60.3, 57.7, 56.5, 39.7, 39.2, 32.6, 32.5, 32.40, 32.39, 31.4, 30.5, 30.4, 29.9, 25.8, 25.8; IR (thin film) 1811, 1781, 1734, 1505, 1205 cm−1; Accurate Mass ES+ m / z calcd for C26H26N2O10S2Na (M+Na)+ 613.0927, found 613.0908.
Procedure for protein cross-linking and LCMS analysis
Cytochrome C and lysozyme were purchased from Sigma. Cross-linking of these proteins followed the same procedure as described previously.30
Cross-linked peptides were analyzed by LC MSn utilizing an LTQ-Orbitrap XL MS (Thermo Fisher, San Jose, CA) coupled on-line with an Easy-nLC 1000 (Thermo Fisher, San Jose, CA) as previously described.3,7 Each MSn experiment consists of one MS scan in FT mode (350–1400 m/z, resolution of 60,000 at m/z 400) followed by two data-dependent MS2 scans in FT mode (resolution of 7500) with normalized collision energy at 20% on the top two MS peaks with charges at 3+ or up, and three MS3 scans in the LTQ with normalized collision energy at 35% on the top three peaks from each MS2.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants RO1GM074830-06A1 and R21CA161807 to L.H., and Biomedical Informatics Training predoctoral fellowship to A.K. (5T15LM007443-10 to Pierre Baldi). The authors acknowledge Dr. John Greaves for his support of this project.
Abbreviations
- IML
identical mass linker
- CID
collision-induced dissociation
- TR9
Acetyl-TTSYKVTIR
- Ac-IR7
Acetyl-IEAEKGR
- XLMS
cross-linker mass spectrometry
- LCMS
liquid chromatography mass spectrometry
- DSSO
disuccinimidyl sulfoxide
- m-CPBA
meta-chloroperoxybenzoic acid
- DABCO
1,4-diazabicyclo[2.2.2]octane
- KOH
potassium hydroxide
- NHS•TFA
N-hydroxysuccinimide trifluoroacetic acid complex
- TBAF•3H2O
tetra-N-butylammonium fluoride trihydrate
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- ESI
electrospray ionization
- Rpt4–Rpt5
two interlinked proteasome regulatory subunits
Footnotes
Electronic supplementary information (ESI) available: General synthetic methods, experimental data, and spectral data are presented in pdf format.
References and notes
- 1.Robinson CV, Sali A, Baumeister W. Nature. 2007;450:973–982. doi: 10.1038/nature06523. [DOI] [PubMed] [Google Scholar]
- 2.Herzog F, Kahraman A, Boehringer D, Mak R, Bracher A, Walzthoeni T, Leitner A, Beck M, Hartl FU, Ban N, Malmström L, Aebersold R. Science. 2012;337:1348–1352. doi: 10.1126/science.1221483. [DOI] [PubMed] [Google Scholar]
- 3.Kao A, Randall A, Yang Y, Patel VR, Kandur W, Guan S, Rychnovsky SD, Baldi P, Huang L. Mol Cell Proteomics. 2012;11:1566–1577. doi: 10.1074/mcp.M112.018374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soderblom EJ, Goshe MB. Anal Chem. 2006;78:8059–8068. doi: 10.1021/ac0613840. [DOI] [PubMed] [Google Scholar]; Dreiocker F, Müller MQ, Sinz A, Schäfer M. J Mass Spectrom. 2010;45:178–189. doi: 10.1002/jms.1702. [DOI] [PubMed] [Google Scholar]; Petrotchenko EV, Serpa JJ, Borchers CH. Mol Cell Proteomics. 2011;10:M110.001420. doi: 10.1074/mcp.M110.001420. [DOI] [PMC free article] [PubMed] [Google Scholar]; Liu F, Wu C, Sweedler JV, Goshe MB. Proteomics. 2012;12:401–405. doi: 10.1002/pmic.201100352. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bhawal RP, Sadananda SC, Bugarin A, Laposa B, Chowdhury SM. Anal Chem. 2015;87:2178–2186. doi: 10.1021/ac503794s. [DOI] [PubMed] [Google Scholar]
- 5.Lu Y, Tanasova M, Borhan B, Reid GE. Anal Chem. 2008;80:9279–9287. doi: 10.1021/ac801625e. [DOI] [PubMed] [Google Scholar]
- 6.Politis A, Stengel F, Hall Z, Hernández H, Leitner A, Walzthoeni T, Robinson CV, Aebersold R. Nat Methods. 2014;11:403–406. doi: 10.1038/nmeth.2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kao A, Chiu CL, Vellucci D, Yang Y, Patel VR, Guan S, Randall A, Baldi P, Rychnovsky SD, Huang L. Mol Cell Proteomics. 2011;10:M110.002212. doi: 10.1074/mcp.M110.002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu C, Kandur W, Kao A, Rychnovsky S, Huang L. Anal Chem. 2014;86:2099–2106. doi: 10.1021/ac403636b. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kaake RM, Wang X, Burke AM, Yu C, Kandur W, Yang Y, Novitsky EJ, Second T, Duan J, Kao A, Guan S, Vellucci D, Rychnovsky SD, Huang L. Mol Cell Proteomics. 2014;13:3533–3543. doi: 10.1074/mcp.M114.042630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.In cross-linking studies, there are three common types of cross-linked peptides, i.e. dead-end, intralinked and interlinked peptides. Among them, interlinked peptides are most informative for mapping protein interaction interfaces between and within proteins.
- 10.Schilling B, Row RH, Gibson BW, Guo X, Young MM. J Am Soc Mass Spectrom. 2003;14:834–850. doi: 10.1016/S1044-0305(03)00327-1. [DOI] [PubMed] [Google Scholar]
- 11.Luo J, Fishburn J, Hahn S, Ranish J. Mol Cell Proteomics. 2012;11:M111.008318. doi: 10.1074/mcp.M111.008318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vellucci D. Strategies for the Analysis of Protein Interactions by Chemical Cross-linking and Tandem Mass Spectrometry. University of California; Irvine: 2010. [Google Scholar]
- 13.Eustache J, Bisseret P, Van De Weghe P. In: Comprehensive Organic Functional Group Transformations II. Taylor ARKJK, editor. Elsevier; Oxford: 2005. pp. 601–668. [Google Scholar]
- 14.Zhao C, Malecha JW, Noble SA, Duron SG, Lindstrom AK, Shiau AK. Aryl sulfonamide and sulfonyl compounds as modulators of PPAR and methods of treating metabolic disorders. 20050102356 20050407. US. 2005 Oct 20;
- 15.Krishnamurthy S, Aimino D. J Org Chem. 1989;54:4458–4462. [Google Scholar]
- 16.Donde Y, Nguyen JH. Substituted arylcyclopentenes as therapeutic agents. 7,713,968 B2. US Patent. 2010 May 11;
- 17.Forman GS, Tooze RP. J Organomet Chem. 2005;690:5863–5866. [Google Scholar]
- 18.Leonard NM, Brunckova J. J Org Chem. 2011;76:9169–9174. doi: 10.1021/jo201686e. [DOI] [PubMed] [Google Scholar]
- 19.Rao TS, Nampalli S, Sekher P, Kumar S. Tetrahedron Letters. 2002;43:7793–7795. [Google Scholar]
- 20.Coelho F, Almeida WP, Veronese D, Mateus CR, Rossi RC, Silveira, Gabriel PC, Pavam CH. Tetrahedron. 2002;58:7437–7447. [Google Scholar]
- 21.Kiren S, Hong X, Leverett CA, Padwa A. Org Lett. 2009;11:1233–1235. doi: 10.1021/ol900059e. [DOI] [PubMed] [Google Scholar]
- 22.Gao S, Tseng C, Tsai CH, Yao CF. Tetrahedron. 2008;64:1955–1961. [Google Scholar]
- 23.Batista RMF, Costa SPG, Belsley M, Lodeiro C, Raposo MMM. Tetrahedron. 2008;64:9230–9238. [Google Scholar]
- 24.Keenan RM, Weinstock J, Finkelstein JA, Franz RG, Gaitanopoulos DE, Girard GR, Hill DT, Morgan TM, Samanen JM. J Med Chem. 1993;36:1880–1892. doi: 10.1021/jm00065a011. [DOI] [PubMed] [Google Scholar]
- 25.Lu D, Vince R. Bioorg Med Chem Lett. 2007;17:5614–5619. doi: 10.1016/j.bmcl.2007.07.095. [DOI] [PubMed] [Google Scholar]
- 26.Cytochrome C has been extensively used by us and by other groups for evaluating cross-linking reagents because it is a small protein with a high number of lysine residues. Given its success in the past for cross-linking studies (ref. 7 & 30), we decided to use it as the model protein for characterizing our new cross-linking reagent.
- 27.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518–1520. [Google Scholar]
- 28.Chattopadhyay SK, Bandyopadhyay A, Pal BK. Tetrahedron Lett. 2007;48:3655–3659. [Google Scholar]
- 29.Liu X, Hu XE, Tian X, Mazur A, Ebetino FH. J Organomet Chem. 2002;646:212–222. [Google Scholar]
- 30.Vellucci D, Kao A, Kaake RM, Rychnovsky SD, Huang L. J Am Soc Mass Spectrom. 2010;21:1432–1445. doi: 10.1016/j.jasms.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.