

Abstract
The p38 mitogen-activated protein kinase cascade is required for the induction of a T helper type 17 (Th17) -mediated autoimmune response, which underlies the development and progression of several autoimmune diseases, such as experimental autoimmune encephalomyelitis, the animal model of multiple sclerosis (MS). However, the contribution of p38 phosphorylation to human Th cell differentiation has not been clarified. Here we demonstrate that the p38 signalling pathway is implicated in the generation of Th17 lymphocytes from human CD4+ CD27+ CD45RA+ naive T cells, both in healthy donors and in patients affected by the relapsing–remitting form of MS. Our data also indicate that p38 activation is essential for interleukin-17 release from central memory lymphocytes and committed Th17 cell clones. Furthermore, CD4+ T cells isolated from individuals with relapsing–remitting MS display an altered responsiveness of the p38 cascade, resulting in increased p38 phosphorylation upon stimulation. These findings suggest that the p38 signalling pathway, by modulating the Th17 differentiation and response, is involved in the pathogenesis of MS, and open new perspectives for the use of p38 inhibitors in the treatment of Th17-mediated autoimmune diseases.
Keywords: autoimmunity, experimental autoimmune encephalomyelitis/multiple sclerosis, neuroimmunology, signal transduction, T cells
Introduction
CD4+ T helper cells are key mediators of immunological homeostasis and modulate immune responses by generating different effector T helper subsets and regulatory T cells, depending on environmental cues such as specific stimuli and high concentrations of polarizing cytokines.1 For instance, the generation of pro-inflammatory T helper type 17 (Th17) lymphocytes from CD4 naive T cells requires activation through CD3, exposure to interleukin-6 (IL-6), IL-1β and transforming growth factor-β (TGF-β), and co-stimulation by CD28 and inducible T-cell co-stimulator. The differentiation of Th17 cells is then controlled by the activation of signal transducer and activator of transcription 3 and retinoid-related orphan receptor-γt (RORγt) and their commitment depends on IL-23.2–5 Interestingly, recent evidence indicates that activation of the p38 mitogen-activated protein kinase (MAPK) is a critical step for the generation and function of IL-17-releasing T cells in mice and shows that IL-17 production is regulated at the translational level by the p38 MAPK signalling pathway.6,7 In addition, recent evidence indicates an involvement of p38 signalling pathways in IL-17 release from human T cells upon glucose exposure.8 Noteworthy studies performed in humans showed apparent differences between human and mouse Th17 lymphocytes,9 suggesting that the signalling cascade that leads to Th17 differentiation could be significantly different between species.
The p38 kinase is a member of the MAPK superfamily, which plays an essential role in human lymphocyte development, proliferation and cytokine release.10–12 In lymphocytes, p38 activation is triggered by stress and by inflammatory signals, although an alternative pathway primed by T-cell receptor engagement has also been described.13 Confirming its central role in shaping the immune response, the p38 MAPK cascade has been shown to be required for the development and progression of experimental autoimmune encephalomyelitis (EAE), the animal model of multiple sclerosis (MS), through the induction of a Th17-mediated autoimmune response.14,15 Although committed Th17 cells are crucial for host defence against pathogens,16 these cells have also been associated with numerous human autoimmune diseases, including MS.17–20
Multiple sclerosis is an immune-mediated disorder, in which autoantigen-specific T cells infiltrate the central nervous system and carry out an immune reaction against self-structures, leading to progressive demyelinization and consequent nervous damage.21 Interleukin-17-releasing T-cell subsets have been detected in the brain of EAE mice,22 and several studies have confirmed the presence of Th17 lymphocytes at high frequency in the blood and the cerebrospinal fluid of patients affected by relapsing–remitting (RR) MS.23–25
The findings illustrated above, among others, indicate a possible link between the p38 signalling pathway and the breakdown of the immunological balance in MS. Also, pro-inflammatory cytokines such as tumour necrosis factor-α, IL-6 and IL-1β, which are leading actors of inflammation and play an important role in the pathogenesis of MS, have been shown to induce the activation of p38.26–28 Nevertheless, p38-dependent modulation of Th17 differentiation in humans still needs to be investigated, and little is known about the activation of the p38 signalling pathway in MS. With the current work we aimed to explore the role played by the p38 cascade in human lymphocyte commitment and to investigate the possible contribution of p38 activation to the pathogenesis of RR-MS. Our results indicate that p38 MAPK is indeed implicated in Th17 generation from CD4 naive T cells in humans, both in healthy donors and in patients with RR-MS, and that p38 phosphorylation is a critical mediator of IL-17 release by already committed Th17 lymphocytes. Importantly CD4 T cells from patients display an increased responsiveness of the p38 signalling pathway following stimulation. These observations suggest that the p38 cascade modulates the generation and function of human Th17 cells, possibly providing a critical contribution to the pathogenesis of MS.
Materials and methods
Patients
Patients with RR-MS were selected on the basis of their gender and age and were all female, between 26 and 60 years old, and met the revised McDonald criteria. The Expanded Disability Status Scale (EDSS) was used to discriminate the disease status and the group included individuals whose EDSS was in the range 1–3.
Patients with RR-MS had not undergone any treatment for at least 3 months before the present study (see Supplementary material, Table S1).
Blood sample collection and isolation
Heparinized peripheral blood was collected from healthy volunteers and patients affected by RR-MS, with the approval from the local Ethics Committee. Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-Hypaque density gradient (Amersham Pharmacia Biotech, Uppsala, Sweden). In some experiments, CD4+ T cells were further sorted using a MoFlo cell sorter (Beckman Coulter, Brea, CA) after staining with CD4, CD45RA, CD27, CCR6, CD161 antibodies for 30 min at 4° in 1% PBS containing 1% fetal calf serum (Lonza, Basel, Switzerland).
Flow cytometric analysis of cell phenotype
Isolated T cells were stained with the following anti-human monoclonal antibodies: CD45RA (BD Pharmingen, San Diego, CA, USA); CD27, CD4, interferon-γ (IFN-γ), Foxp3, IL-2 (eBioscience Inc., San Diego, CA, USA) IL-17 (BioLegend, London, UK) Live/Dead (Aqua, 405 nm emission; Invitrogen, Eugene, OR) and p38 (rabbit polyclonal anti-p38; Cell Signaling Technologies Inc., Danvers, MA, USA; Alexa Fluor 488 goat anti-rabbit immunoglobulin; Invitrogen). Samples were acquired on a CyAn (Coulter). Data were analysed using FlowJo software (TreeStar, Ashland, OR).
Cell culture and use of inhibitors
For experiments involving cell clones, cells were cultured in RPMI-1640 supplemented with 5% heat-inactivated fetal calf serum and 10% heat-inactivated human serum, 100 U/ml penicillin, 100 mg/ml streptomycin, 50 μg/ml gentamicin and 2 mm l-glutamine (all from Invitrogen) at 37° in a humidified 5% CO2 incubator. In the differentiation assays, X-VIVO 15 serum-free medium (Lonza) was used. In some experiments the p38 inhibitor BIRB796 was added to the culture at a final concentration of 500 nm. The inhibitor SB103580 was used at a concentration of 20 mm. A solution of 0·1% DMSO was used as control. Finally, the MAPK signal-integrating kinases (MNK) inhibitor was used at a concentration ranging from 1 to 10 μm. All inhibitors were added 30 min before the starting point of each experiment, and were not added again.
Cell clones
Th17 cell clones were generated on the basis of surface expression of CD4, CD161 and CCR6. PBMCs from two healthy donors were stained with anti-CD4, anti-CD161 and anti-CCR6 and CD4+ CCD161+ CCR6+ cells were sorted at 1 cell/well into 96-well culture plates containing 100 000/well irradiated autologous feeders in complete RPMI, 20 U/ml human recombinant IL-2 (Miltenyi Biotec, Bergisch Gladbach, Germany) and phytohaemagglutinin (PHA, Biochrom GmbH, Berlin, Germany).
T-helper differentiation assay
Naive CD4+ CD45RA+ CD27+ T cells were cultured in 96-well plates (Costar, Corning Inc., NY, USA) at a concentration of 5 × 104 cells per well in X-VIVO 15 serum-free medium (Lonza) in the presence of Dynabeads CD3/CD28 T Cell Expander (one bead per cell; Invitrogen) and the following cytokines: IL-1β (10 ng/ml), IL-6 (20 ng/ml), TGF-β1 (1 ng/ml), IL-23 (100 ng/ml), or IL-12 (10 ng/ml; R&D Systems, Abingdon, UK). After 5–6 days, cells were collected and restimulated for 6 hr with PMA and ionomycin (0·5 and 1 μg/ml, respectively, both from Sigma, St Louis, MO) for intracellular staining. Cell viability was determined using Aqua Live/Dead staining (Invitrogen). In some experiments the supernatants were collected and IL-17 concentration in culture was measured with the IL-17 ELISA kit (R&D Systems).
Staining of phosphorylated proteins by flow cytometry
The analysis of p38 (pT180/pY182) was performed directly ex vivo. Following surface staining for CD4 and CD8, PBMCs were fixed with warm Cytofix Buffer (BD Biosciences) at 37° for 10 min. Cells were then permeabilized with ice-cold Perm Buffer III (BD Biosciences) at 4° for 30 min and incubated with the anti-p38 antibody (pT180/pY182) (AlexaFluor 488; clone 36/p38; BD Pharmingen) for 30 min at room temperature.
Preparation of cell extracts and Western blot analysis
CD4+ T cells (2 × 106) were activated with PMA (0·5 μg/ml) and ionomycin (0·5 μg/ml), in the presence or absence of BIRB796. After 20 min, cells were harvested and lysed in 20 ml of buffer (50 mm HEPES, pH 7·4, 15 mm MgCl2, 150 mm NaCl, 15 mm EGTA, 10% glycerol, 1% Triton X-100, protease inhibitor mixture (Sigma-Aldrich, St Louis, MO, USA), 20 mm b-glycerophosphate, 2 mm dithiothreitol, 1 mm Na3VO4). Cells were tested by Western blot analysis to detect the levels of rabbit anti-phospho-p38 (Thr180 and Tyr182; Cell Signaling), anti-phospho-eIF4E (Ser209, BioSource International, Camarillo, CA), and mouse anti β-tubulin (Sigma-Aldrich). Proteins were separated on 12% SDS–PAGE gel and transferred to polyvinylidene fluoride Immobilon-P membranes (GE Healthcare UK Limited, Buckinghamshire, UK) using a semidry blotting apparatus (Bio-Rad, Hercules, CA). Membranes were saturated with 5% BSA (Sigma-Aldrich) in PBS containing 0·1% Tween-20 for 1 hr and incubated overnight at 4° with the primary antibodies. Secondary anti-mouse IgGs conjugated to horseradish peroxidase (Amersham Biosciences) were incubated with the membranes and immunostained bands were detected by chemiluminescence (Santa Cruz Biotechnology Inc., Dallas, TX, USA).
Real-time PCR
Total RNA was extracted with an RNeasy Micro kit (Qiagen, Germantown, MD, USA). A mixture containing random hexamers, oligo(dT), (Promega, Madison, WI, USA) and SuperScript II Reverse Transcriptase (Invitrogen) was used for cDNA synthesis. Transcripts were quantified by real-time PCR on a LightCycler 480 sequence detector (Roche, Basel, Switzerland) with Applied Biosystems predesigned TaqMan Gene and TaqMan Expression Master mix (Applied Biosystems, Foster City, CA). The following probes were used (Applied Biosystems assay identification numbers in parentheses): IL-17A (Hs00174383_m1), RORc (Hs01076112_m1). The result from each sample was analysed using the LightCycler 480 software 1.5.0 SP4, Roche. and normalized to the amount of ribosomal protein L34 (Hs00241560_m1).
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 4.00 (GraphPad Software, San Diego, CA). Data are presented as mean plus or minus standard error of mean (SEM). Significance was determined by two tailed unpaired Student’s t-test, and P ≤ 0·05 was considered statistically significant.
Results
p38 MAPK modulates Th17 generation and IL-17 release in vitro
p38 MAPK is a critical mediator of IL-17 release in mouse CD4+ T lymphocytes.6,7 To outline the contribution of the p38 signalling pathway on the induction of human Th17 differentiation, we performed a T-cell differentiation assay of naive CD4+ CD45RA+ CD27+ lymphocytes upon stimulation with anti-CD3 and anti-CD28 and the appropriate cytokines (TGF-β, IL-6, IL-1β, IL-23), in the presence or absence of the p38 inhibitor BIRB796. BIRB796 was selected for its ability to inhibit with great efficiency all four isoforms of p38 MAPK (p38α, p38β, p38γ, p38δ) and for its low toxicity on lymphocytes in culture. After 5 days of culture, cells were stimulated with PMA and ionomycin, and the release of IL-17 was assessed by flow cytometry. We were able to detect a sizeable frequency of IL-17-positive cells under polarizing conditions, and this frequency appeared to be significantly reduced in the presence of BIRB796 (Fig.1a,b). To rule out the risk that the decreased frequency of committed Th17 cells was the result of higher mortality caused by p38 inhibition, we measured by flow cytometry the viability of CD4 T cells at the end of the differentiation assay. The frequency of living cells after polarization was comparable in the presence and in the absence of BIRB796, so excluding the possibility that the compound was affecting cell survival (Fig.1c). BIRB796 was effective in blocking Th17 polarization even at a concentration lower than the commonly used one (500 nm), and the frequency of IL-17-releasing cells appeared to be regulated by the inhibitor in a dose-dependent manner (Fig.1d).
Figure 1.

p38 activation is essential for T helper type 17 (Th17) generation from human CD4+ naive T cells in vitro: (a) FACS-sorted CD4+ CD45RA+ CD27+ naive T cells were stimulated with anti-CD3 and anti-CD28 in the presence (lower panel) or absence (upper panel) of the p38 inhibitor BIRB796. A cocktail of cytokines composed of transforming growth factor-β (TGF-β), interleukin-1 β (IL-1β), IL-6 and IL-23 was added to the culture. After 5 days the cells were harvested and stimulated with PMA/ionomycin for 4 hr in the presence of brefeldin and then stained for surface markers. The expression of IL-17 was assessed by flow cytometry. Plots from a representative experiment out of eight are shown. (b) Graph represents the frequency of IL-17+ cells gated on live CD4+ cells. Error bar represents the SE from the mean of eight distinct experiments. (c) Graph represents the frequency of live CD4+ cells, gated on total lymphocytes. Error bar represents the SE from the mean of eight separate experiments. (d) FACS-sorted naive T cells were polarized into Th17, in the presence of different dilutions of BIRB796. The frequency of IL-17-releasing cells is shown. (e) Purified CD4+ naive T cells were polarized toward the Th17 profile, in the presence or absence of the p38 inhibitor SB203580. Representative plots from one experiment out of three are shown. **P < 0·01.
To further confirm the involvement of the p38 pathway in human Th17 commitment, we then tested the effect of a different p38 inhibitor, SB203580, on CD4 naive T-cell differentiation. Also SB203580 was able to reduce Th17 cell polarization after 5 days in culture, even though with slightly less efficiency than BIRB796 (Fig.1e). Finally, we measured by ELISA the release of IL-17 by isolated naive CD4 cells under polarizing conditions in the presence or absence of BIRB after 5 days. While uncommitted CD4 T cells did not release IL-17, Th17-polarized lymphocytes secreted significant amounts of the pro-inflammatory cytokine. Interleukin-17 release was significantly reduced when BIRB796 was used in the differentiation assay, so confirming the cytometric data (Fig.2a).
Figure 2.
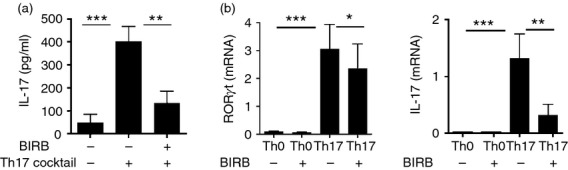
p38 activation modulates interleukin-17 (IL-17) release and RORC and IL-17 expression in vitro: (a) IL-17 levels in the supernatants from three distinct differentiation assays were assessed by ELISA. (b) FACS-sorted CD4+ CD45RA+ CD27+ naive T cells were stimulated with anti-CD3 and anti-CD28 in the presence or absence of the p38-inhibitor BIRB796. The cocktail of cytokines [transforming growth factor-β (TGF-β), interleukin-1 β (IL-1β), IL-6 and IL-23] was added to the culture where specified. After 5 days the cells were harvested and mRNA levels of the transcription factor RORγt (left panel) and of the cytokine IL-17A (right panel) were measured by quantitative RT-PCR. *P < 0·05, **P < 0·01; ***P < 0·001.
Since p38 MAPK is known to inhibit cytokine release, we asked whether p38 inhibition in our assay was blocking IL-17 release rather than modulating Th17 differentiation from CD4 naive T cells. Hence, we tested the levels of RORC, the transcription factor specific for the Th17 profile and whose expression is critical for the differentiation of IL-17-releasing cells. CD4+ naive T lymphocytes were therefore polarized into Th17 cells, in the presence or absence of BIRB796, and the expression of RORC and IL-17 was assessed by real-time PCR. After 5 days in culture, the mRNAs of both proteins were significantly reduced in the presence of the p38 inhibitor (Fig.2b), so indicating that the p38 cascade is essential for Th17 generation from CD4+ cells in vitro.
p38 inhibition does not affect IFN-γ and Foxp3 expression in culture, but significantly increase IL-21 release from Th17-polarized lymphocytes
Having shown that BIRB796 interferes with Th17 generation in vitro, we evaluated the possibility that p38 inhibition was redirecting the differentiation of CD4+ naive T cells toward cytokine profiles other than the Th17 one. We consequently measured the expression of Foxp3 and the expression of IFN-γ and IL-2 by naive CD4+ CD27+ CD45RA+ cells isolated from five donors after Th17 polarization in the presence of BIRB796. The fraction of Foxp3+ and IFN-γ+ lymphocytes was not significantly modified by the presence of the inhibitor, suggesting that p38 inhibition does not redirect lymphocyte polarization toward the Th1 or the regulatory T profile under these conditions. Of note, the compound caused a slight inhibition of cytokine expression, even if not significant (Fig.3a,b). However, this compound does not cause a general inhibition of cytokine expression because we noticed an increase of IL-2-positive CD4 T cells in the presence of BIRB796, even if this increase was not statistically significant (Fig.3c).
Figure 3.
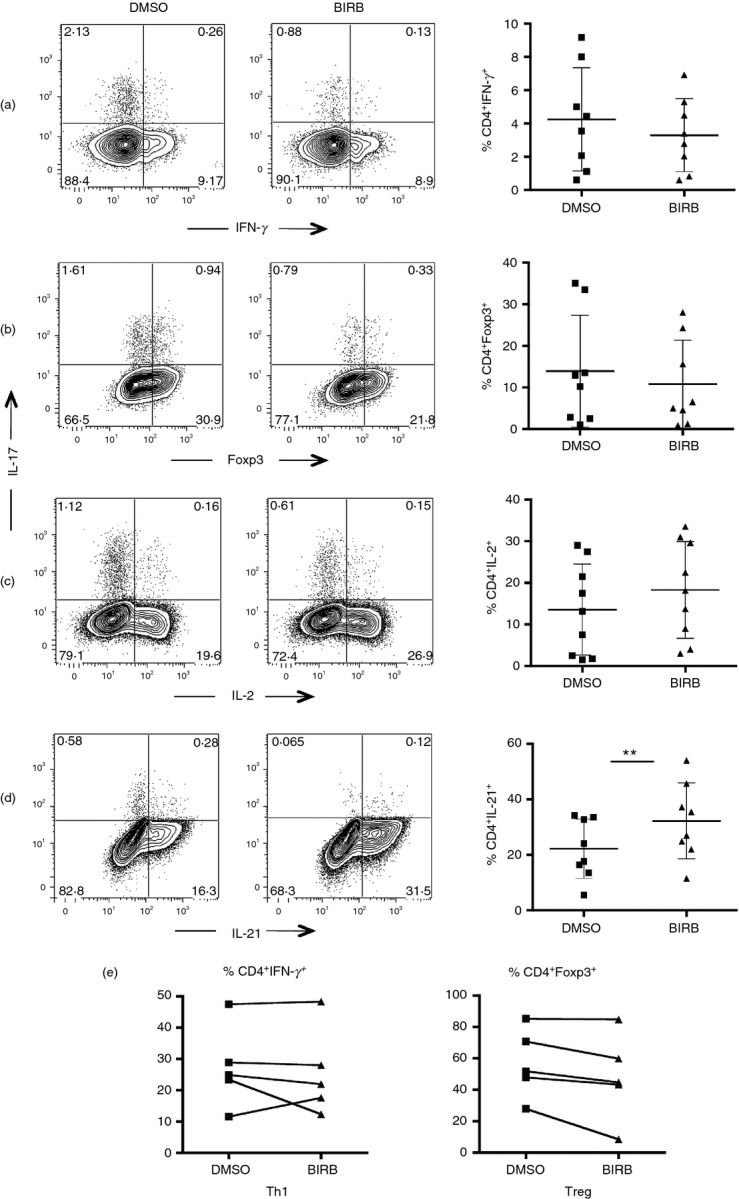
p38 inhibition does not affect interferon-γ (IFN-γ) and Foxp3 expression in culture and significantly increases interleukin-21 (IL-21) release: FACS-sorted CD4+ naive T lymphocytes were polarized into T helper type 17 (Th17) cells with and without BIRB796. After 5 days cells were harvested and stimulated with PMA/ionomycin for 4 hr. CD4 T lymphocytes were then stained to test the expression of IFN-γ (a) and Foxp3 (b). Also the expression of IL-2 (c) and IL-21 (d) after 5 days was evaluated by flow cytometry. Bar graphs on the right of each plot represent the frequency of IFN-γ+, Foxp3+, IL-2+ and IL-21+ cells gated on live CD4+ lymphocytes. Results from eight distinct experiments (a–c) and six distinct experiments (d) are shown. (e) FACS-sorted naive cells were polarized into Th1 and regulatory T cells, in the presence or absence of BIRB796. Graphs from five independent experiments are shown.
We then measured by flow cytometry the effect of BIRB796 on IL-21 expression in Th17-polarized cells. Interleukin-21 has been previously associated with IL-17 release from CD4+ lymphocytes as it induces Th17 generation from CD4+ naive T cells in mice.29 Here we show for the first time that p38 inhibition significantly increases the fraction of IL-21-positive T cells in human CD4+ cells under Th17 differentiating conditions, even though this increase does not restore IL-17 levels in the presence of BIRB796 (Fig.3d).
Finally, we explored the effect of the p38 signalling pathway on the differentiation of CD4 T cells toward the Th1 and Treg profile. We therefore polarized CD4+ CD27+ CD45RA+ T cells respectively with IL-12, TGF-β, and IL-2: this cytokine cocktail is known to induce IFN-γ release or Foxp3 expression. After 5 days in culture, cytokine release was measured by flow cytometry. The presence of BIRB in the cultures did not modify Th1 polarization, as these lymphocytes acquired the capacity to release IFN-γ despite inhibition of p38 activity (Fig.3e, left panel). On the other hand, the frequency of Foxp3+ Treg cells was decreased in three donors in the presence of BIRB, so confirming previous reports that describe p38 as a key regulator of Foxp3+ T-cell commitment;30 nevertheless, in our case the difference was not significant, as the observed decrease appeared to be donor-dependent (Fig.3e, right panel).
p38 activation is required for IL-17 release by already committed Th17 lymphocytes
Our results show that the p38 signalling pathway is involved in the polarization of human antigen-inexperienced naive CD4 cells into Th17 lymphocytes. However, in adults the lymphocyte compartment is largely composed of memory cells. Central memory lymphocytes are a heterogeneous group of T cells that maintain the capability to be polarized toward different possible cytokine profiles. Given the prominent role of these cells in the effector phase of the immune response, we set out to investigate the involvement of p38 signalling in the regulation of IL-17 release also by differentiated lymphocytes. Hence, CD4+ CDRA-CD27+ central memory T cells, sorted to purity from three different healthy donors, were cultured under IL-17 differentiation conditions in the presence or absence of BIRB796. Notably, p38 inhibition significantly reduced IL-17 release from CD4 cells after 5 days in culture (Fig.4a,b). BIRB796 reduced IL-17 release in a dose-dependent manner (Fig.4c), as previously observed for naive T cells. To confirm these findings, we generated CD4+ IL-17+ cell clones from the peripheral blood of healthy donors, and we then tested their capability to release IL-17 in the presence or absence of BIRB796. CD4+ CCR6+ CD161+ T cells were sorted from PBMCs of two healthy donors and then expanded in culture in the presence of IL-2 and PHA. After 15 days, 11 cell clones were stimulated with aCD3 and aCD28 and tested for their capability to release IL-17 in the presence or absence of the p38 inhibitor. Secretion of IL-17 was partially but significantly reduced following p38 blocking, so confirming the contribution of p38 in this process (Fig.4d,e).
Figure 4.
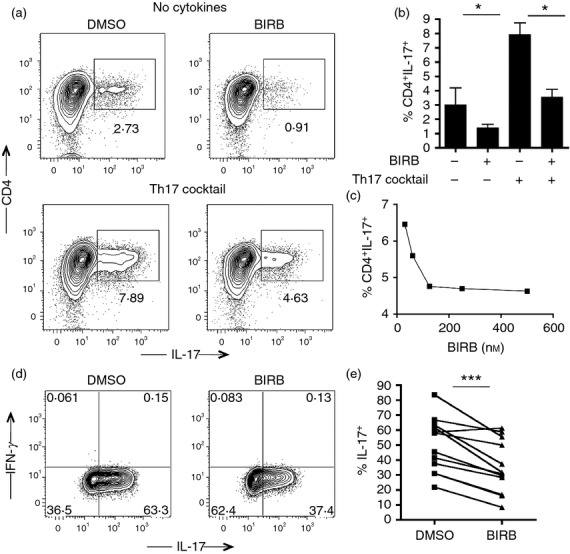
p38 inhibition modulates interleukin-17 (IL-17) release from central memory T cells and T helper type 17 (Th17) cell clones: (a) FACS-sorted CD4+ CD45RA− CD27+ memory T cells were plated in the presence or absence of the Th17 cytokine cocktail, with and without BIRB796. After 5 days the cells were harvested and stimulated with PMA/ionomycin for 4 hr in the presence of brefeldin and then stained with specific surface markers. The expression of IL-17 was assessed by flow cytometry. Representative plots are shown. (b) Bar graph shows the frequency of CD4+ IL-17+ memory cells after 5 days. Error bar represents the SE from the mean of three independent experiments. (c) Graph represents the frequency of IL-17+ cells gated on alive CD4+ memory cells after polarization into Th17 cells in the presence of different dilutions of BIRB796. A representative experiment is shown. (d) FACS-sorted Th17 cell clones were stimulated with PMA/ionomycin for 4 hr in the presence of brefeldin, with and without BIRB796. Representative plots from one experiment out of 11 are shown. (e) Graph represents the frequency of IL-17+ cells gated on live CD4+ cell clones. ***P < 0·001, *P < 0·01.
The MNK/eIF-4E pathway regulates human Th17 cell commitment
Map-kinase signal-integrating kinases can be activated by the p38 cascade and are involved in cytokine regulation.31 Recent evidence indicates that MNKs selectively contribute to IL-17 protein synthesis in Th17 cells in mice.7 We therefore investigated whether the impact of p38 signalling pathway on Th17 commitment in humans shares the same mechanism as in mice. First, we tested whether p38 activation actually leads to eIF-4E phosphorylation in human isolated CD4 naive T cells. Lymphocytes were sorted based on CD4, CD45RA and CD27 expression and then stimulated for 20 min with PMA and ionomycin. Expression of phospho-p38 and phospho-eIF-4E was tested by Western blot, which revealed that p38 phosphorylation results in the MNK/eIF-4E pathway activation (Fig.5a). We therefore performed a differentiation assay in the presence of the MNK inhibitor and then measured the frequency of IL-17-releasing cells after 5 days in culture. The fraction of Th17 differentiated cells was significantly reduced in the presence of the inhibitor and this reduction appeared to be dose-dependent (Fig.5b lower panel, c), without affecting cell viability (Fig.5b, upper panel).
Figure 5.

eIF-4E activation is required for T helper type 17 (Th17) generation and interleukin-17 (IL-17) release in human CD4+ naive cells: (a) FACS-sorted naive CD4+ T cells were stimulated for 20 min with PMA/ionomycin and then checked for p38 phosphorylation and eIF-4E expression by Western blot. One experiment out of three is shown. Tubulin was used as loading control. (b) FACS-sorted naive T cells were polarized into Th17 in the presence or absence of the MAPK interacting protein kinases (MNKs) inhibitor. After 5 days the frequency of vital cells (upper panels) and of IL-17-releasing cells (lower panels) was assessed by flow cytometry. Plots from a representative experiment out of three are shown. Graphs represent the frequency of live cells (upper graph) and IL-17+ cells (lower graph) gated on CD4+ lymphocytes. Error bar represents the SE from the mean of three distinct experiments. (c) CD4+ naive T cells were polarized into Th17 lymphocytes in the presence of different dilutions of the MNK inhibitor. (d) Graph represents the levels of IL-17 in the supernatants with different concentrations of iMNK. *P < 0·05.
We finally evaluated by ELISA the amount of IL-17 released after polarization with the Th17 cocktail and again the MNK inhibitor drastically diminished the quantity of IL-17 secreted in comparison with the control cultures (Fig.5d).
RR-MS patients show an altered responsiveness of the p38 cascade upon stimulation
As the p38 signalling pathway is involved in Th17 commitment in healthy donors, we explored the impact of p38 activation on the polarization process of CD4+ naive T cells isolated from five patients with RR-MS. When tested in the differentiation assay in vitro, BIRB796 was able to dramatically decrease the percentage of committed Th17 lymphocytes after 5 days in culture (Fig.6a). The release of IL-17 evaluated by ELISA was also significantly reduced in CD4+ T cells isolated by patients with RR-MS, showing that the role played by p38 in the modulation of Th17 commitment was consistent in patients and in healthy donors (Fig.6b).
Figure 6.
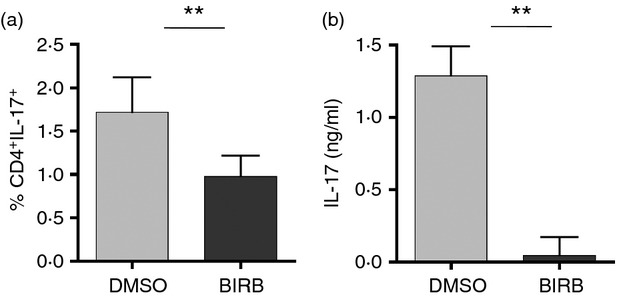
p38 activation is required for interleukin-17 (IL-17) release in patients with relapsing–remitting multiple sclerosis (RR-MS): FACS-sorted CD4+ CD45RA+ CD27+ naive T cells isolated from patients with RR-MS were polarized into T helper type 17 (Th17) in the presence or absence of BIRB796. The expression of IL-17 was assessed by flow cytometry (a) and by ELISA (b). Error bars represent the SE from the mean of three distinct experiments. **P < 0·01.
The MAPK family is considered crucial in the pathogenesis of another autoimmune disorder: rheumatoid arthritis.32–34 The c-Jun N-terminal kinases (JNKs) and extracellular-signal-regulated kinases (ERKs) pathways have been already studied in patients affected by RR-MS, but little is known of the p38 cascade.35 We therefore investigated the expression of the p38 MAPK in CD4+ cells isolated from the peripheral blood of patients with RR-MS and healthy donors, to evaluate if there is an alteration of this pathway during disease. We selected 18 patients affected by RR-MS and 18 healthy donors. As MS is a widely heterogeneous disorder, we chose a group of individuals that were comparable for gender, age and disease progression (see Supplementary material, Table S1). We first measured by flow cytometry the level of expression of the p38 kinase ex vivo, and found that there was no difference in the basal level of p38 total protein and phosphorylated protein between donors and patients (Fig.7a). However, when the levels of p38 phosphorylation were measured in isolated CD4-positive T cells upon stimulation with PMA and ionomycin, we uncovered a completely different situation: CD4+ lymphocytes from RR-MS individuals in fact showed a significantly higher propensity to phosphorylation than their counterparts isolated from healthy donors (Fig.7b,c). After stimulation the fold increase of p38 MFI in CD4 cells was 33% higher in patients than in healthy donors. To validate the results obtained by flow cytometry, we performed the same experiment in Western blot, isolating CD4+ T cells from patients and donors and measuring p38 phosphorylation levels upon stimulation. Lymphocytes isolated from patients with RR-MS showed a higher level of p38 activation in comparison with cells isolated from healthy donors, confirming the cytometric results (Fig.7d).
Figure 7.
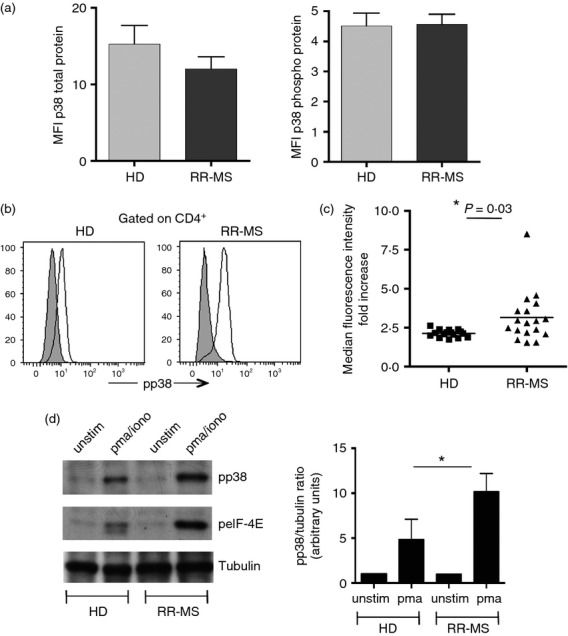
The p38 cascade is up-regulated in patients with relapsing–remitting multiple sclerosis (RR-MS): (a) Graph represents the median fluorescence intensity (MFI) of p38 total protein (left) and phosphorylated protein (right) in peripheral blood mononuclear cells (PBMCs) from healthy donors and patients with MS ex vivo. Error bars represent the SE from the mean of 10 individuals from each group. (b) PBMCs isolated from one donor and one MS patient, were stimulated and tested for p38 phosphorylation. Representative plots from one donor and one patient are shown. Histograms show a representative phospho-staining for p38 before (grey) and after (white) stimulation. (c) PBMCs isolated from 18 healthy donors and 18 patients were stimulated for 20 min with PMA/ionomycin and then stained for phospho-p38. Graph represents the increase of median fluorescence intensity (MFI) after stimulation. (d) FACS-sorted CD4+ T cells were isolated from one donor and one MS patient, stimulated and checked for p38 and eIF-4E phosphorylation by Western blot. Tubulin was used as loading control. One representative experiment out of five is shown. *P < 0·05.
Discussion
In the present work we studied the role played by p38 MAPK in lymphocyte commitment in humans and we investigated the level of p38 activation in CD4+ cells isolated from patients with MS. We show that p38 activation is required for IL-17 generation from human CD4+ T cells. p38 regulates inflammatory responses following its phosphorylation downstream of T-cell receptor activation, promoting the production of pro-inflammatory and T-cell-polarizing cytokines like IL-1β, IL-12, IL-6 and IL-23. In particular, a number of studies have revealed a role for p38 in the production of IL-17 by Th17 cells both in vitro and in vivo.14,15,36 Th17 generation from CD4+ naive T cells has been extensively studied and described, because of the essential role played by IL-17-releasing lymphocytes in physiological and pathological immune responses. Interestingly, it has been previously shown that IL-17 production in the mouse model is regulated at the translational level through the activation of the eukaryotic translational initiation factor 4E/MAPK-interacting kinase pathway.7 Nevertheless, previous studies showed that important differences do exist between humans and mice in the stimuli and the cues that lead to Th17 development, suggesting that the signalling cascade downstream of these stimuli may be different between the two species.9 Several cytokines, known to be regulated by p38-MAPK, such as IL-1β and IL-6, are higher in MS patients than in healthy individuals. Importantly, these cytokines are required to elicit EAE.
Pharmacological inhibition of p38 MAPK in mice, by means of several different small-molecule inhibitors have been revealed to be efficacious in autoimmune diseases in pre-clinical studies37,38 and able to ameliorate EAE, when continually administered after induction of disease.6,15 In this context it is noteworthy that p38 signalling is required for onset and progression of EAE39 in accordance with its aptitude to promote a Th17-mediated response and it is therefore crucial to investigate the impact of p38 activation in Th17 differentiation in humans. The IL-23–Th17 axis has been shown to be essential in the inflammatory response in various autoimmune diseases and the discovery of new mechanisms that regulate Th17 generation and functionality is therefore crucial in the attempt to control the autoimmune response in humans. The capacity of p38 to modulate human Th17 differentiation consequently appears to be particularly attractive. T-cell expression of a dominant negative p-38 MAPK transgene in mice inhibited EAE and IL-17 production.7 As p38 inhibitors have shown great promise in pre-clinical models of rheumatoid arthritis and Crohn’s disease, diseases that share some molecular mechanisms with MS, we believe that the use of p38 inhibitors in MS is worth investigating and may have a strong clinical impact in this setting. Importantly, here we show that p38 inhibitors interfere with the generation of IL-17-producing cells from naive CD4 cells isolated from healthy donors as well as from patients affected by RR-MS. Noteably, p38 activation is required for the transcription of RORγt mRNA, suggesting that, contrary to the mouse model, in humans the p38 cascade is actually implicated in the differentiation process of CD4+ naive T cells into Th17 and not only in the modulation of IL-17 secretion. In addition to this it is remarkable that the p38 signalling pathway is also crucial for IL-17 release from already committed central memory lymphocytes. As patients with MS show an increased frequency of central memory Th17 cells in the peripheral blood,17,18 the present findings appear to be even more prominent. Indeed, our results suggest that p38 inhibition may regulate both Th17 cell generation from naive cells and IL-17 release from differentiated lymphocytes, so possibly resulting in an ultimately stronger effect in vivo.
Interestingly, when we tested the impact of p38 inhibition on Th17 polarization, we found for the first time that IL-21 release from naive CD4+ cells exposed to the cocktail was significantly increased in the presence of BIRB796. Interleukin-21 has been shown to counteract T regulatory cell suppression and to induce Th17 generation in mice, and it is considered crucial in many inflammatory diseases.29,40 We strongly believe that this aspect needs to be taken into consideration and further investigated, for example in the aim to enhance the effect of the p38 inhibitors currently under study for the treatment of certain autoimmune pathologies.
Many clinical trials with p38 inhibitors have been recently performed in inflammatory pathologies.41–44 However, little is known on the activation of the p38 cascade in individuals with RR-MS, and the efficacy of p38 inhibitors in the cure of MS has not been tested yet. Here we show for the first time that responsiveness of the p38 signalling pathway upon activation appears to be significantly increased in cells from patients, in comparison to healthy donors. p38 activation has been previously studied in cells from EAE lesions, where it was upregulated in astrocytes and glial cells.34 To our knowledge, this is the first time that the p38 cascade is investigated in CD4+ T cells from patients with MS. The reasons for the resonse being so much higher are not easy to discern. It is possible to speculate that an environment rich in pro-inflammatory cytokines may determine the altered threshold of activation seen in lymphocytes from patients. In addition to this, it has been shown that highly differentiated lymphocytes are defined by an elevated expression of phosphorylated p3845 and an increased frequency of such effector T cells in patients with MS has been previously observed by us (unpublished observation) and others.46 Hence, the combination of high levels of cytokines with increased numbers of differentiated effector cells may account for the elevated responsiveness of the p38 signalling pathway seen in CD4+ from patients with MS. Of course this aspect deserves to be further investigated.
The capacity of the p38 cascade to modulate the biosynthesis of multiple pro-inflammatory cytokines has been largely investigated.47,48 In the light of this, the observed higher activation of the p38 cascade in cells from patients with RR-MS may be, at least to some extent, responsible for the inflammatory process that underlies the disease. If this were to be confirmed, the use of p38 inhibitors in the treatment of MS would appear even more reasonable. One more aspect that is worth considering is that the efficacy of p38 inhibitors on inflammatory syndromes in humans appears to be low and complicated by adverse effects like neurotoxicity or hepatotoxicity.49,50 However, here we confirm previous data from the mouse model, showing that the MNK/eIF-4E pathway downstream of p38 is involved in human Th17 cell commitment. This finding could help to develop more targeted compounds in the same attempt to reduce Th17 cell development and functionality in vivo, so reducing broad adverse effects. Interestingly MNK proteins have been shown to regulate the inflammatory process and they therefore appear to be promising targets for anti-inflammatory therapies.31
In summary, our results indicate that the p38 cascade modulates Th17 cell functionality in both healthy donors and in patients affected by RR-MS, and highlight the higher propensity of lymphocytes isolated from patients to activate the p38 signalling pathway upon stimulation, in comparison to lymphocytes from healthy donors. These data suggest that p38 MAPK may provide a critical contribution to the pathogenesis of multiple sclerosis and open new perspectives for the use of MAPK inhibitors as treatment for autoimmune diseases.
Acknowledgments
We thank David Kipling for providing us with the BIRB796. We also thank G. Ruocco for cell sorting and M. Lanuti for assistance in figure preparation.
D.D.M and M.S. were supported by a research fellowship FISM – Fondazione Italiana Sclerosi Multipla – cod.2010/B/3, cod.2011/B/3. This work was partially supported by the Italian Multiple Sclerosis Society FISM – Fondazione Italiana Sclerosi Multipla cod 2013/R/2 to L.B., cod. 2013/R/23 to E.V. and C.S., by the Italian Ministry of Health (Progetto Finalizzato) to L.B., by the Italian Association for Cancer Research (AIRC) to C.S.
Author contributions
D.D.M. designed and performed research, analysed data and wrote the paper; M.L. performed research; M.D.B. performed cell sorting; M.C. and E.V. contributed to research; D.C. and C.G. were responsible for recruitment and selection of RR-MS patients; C.S. and A.A. contributed to experimental design and paper revision; M.S. and G.B. contributed to experimental design and paper writing; L.B. supervised the study.
Disclosures
Dr Claudio Gasperini received funds for research by TEVA. He has served as consultant for Merck Serono and Biogen. He received speaker honoraria from Novartis, Teva, Merck Serono, Bayer Sheering and Biogen. Prof Diego Centonze is an Advisory Board member of Almirall, Bayer Schering, Biogen, Genzyme, GW Pharmaceuticals, Merck-Serono, Novartis and Teva, and has received honoraria for speaking or consultation fees from Almirall, Bayer Schering, Biogen Idec, Genzyme, GW Pharmaceuticals, Merck Serono, Novartis, Sanofi-Aventis and Teva. He is also an external expert consultant of the European Medicine Agency (EMA), and the principal investigator in clinical trials for Bayer Schering, Biogen Idec, Merck Serono, Mitsubishi, Novartis, Roche, Sanofi-aventis and Teva. His pre-clinical and clinical research was supported by grants from Bayer, Biogen, Merck Serono, Novartis and Teva. Prof. Arne Akbar is a consultant for GSK and OnoPharmaceuticals. Dr Luca Battistini received funds for research from TEVA. He also received speaker honoraria by Genzyme. The other authors have no disclosures to report.
Supporting Information
Table S1. Listed patients with relapsing–remitting multiple sclerosis were all female, were all in the remission phase at the time of the sampling and did not undergo any treatment since at least 3 months before the present study.
References
- Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646–55. doi: 10.1016/j.immuni.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Chen Z, O’Shea JJ. Th17 cells: a new fate for differentiating helper T cells. Immunol Res. 2008;41:87–102. doi: 10.1007/s12026-007-8014-9. [DOI] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- Park H, Li Z, Yang XO, et al. A distinct lineage of Cd4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–41. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupé P, Barillot E, Soumelis V. A critical function for transforming growth factor-β, interleukin 23 and proinflammatory cytokines in driving and modulating human Th17 responses. Nat Immunol. 2008;9:650–7. doi: 10.1038/ni.1613. [DOI] [PubMed] [Google Scholar]
- Lu L, Wang J, Zhang F, et al. Role of Smad and Non-Smad signals in the development of Th17 and regulatory T cells. J Immunol. 2010;184:4295–306. doi: 10.4049/jimmunol.0903418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noubade R, Krementsov DN, Del Rio R, et al. Activation of P38 MAPK in CD4 T cells controls IL-17 production and autoimmune encephalomyelitis. Blood. 2011;118:3290–300. doi: 10.1182/blood-2011-02-336552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Natarajan K, Shanmugam N. High glucose driven expression of pro-inflammatory cytokine and chemokine genes in lymphocytes: molecular mechanism of IL-17 family gene expression. Cell Signal. 2014;26:528–39. doi: 10.1016/j.cellsig.2013.11.031. [DOI] [PubMed] [Google Scholar]
- Annunziato F, Romagnani S. Mouse T helper 17 phenotype: not so different than in man after all. Cytokine. 2011;56:112–5. doi: 10.1016/j.cyto.2011.06.009. [DOI] [PubMed] [Google Scholar]
- Diehl NL, Enslen H, Fortner KA, et al. Activation of the P38 mitogen-activated protein kinase pathway arrests cell cycle progression and differentiation of immature thymocytes in vivo. J Exp Med. 2000;191:321–34. doi: 10.1084/jem.191.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rincon M, Pedraza-Alva G. Jnk and P38 Map kinases in CD4+ and CD8+ T cells. Immunol Rev. 2003;192:131–42. doi: 10.1034/j.1600-065x.2003.00019.x. [DOI] [PubMed] [Google Scholar]
- Cook R, Wu CC, Kang YJ, Han J. The role of the P38 pathway in adaptive immunity. Cell Mol Immunol. 2007;4:253–9. [PubMed] [Google Scholar]
- Salvador JM, Mittelstadt PR, Guszczynski T, Copeland TD, Yamaguchi H, Appella E, Fornace AJ, Ashwell JD. Alternative P38 activation pathway mediated by T cell receptor-proximal tyrosine kinases. Nat Immunol. 2005;6:390–5. doi: 10.1038/ni1177. [DOI] [PubMed] [Google Scholar]
- Jirmanova L, Giardino Torchia ML, Sarma ND, Mittelstadt PR, Ashwell JD. Lack of the T cell-specific alternative P38 activation pathway reduces autoimmunity and inflammation. Blood. 2011;118:3280–9. doi: 10.1182/blood-2011-01-333039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namiki K, Matsunaga H, Yoshioka K, et al. Mechanism for p38α-mediated experimental autoimmune encephalomyelitis. J Biol Chem. 2012;287:24228–38. doi: 10.1074/jbc.M111.338541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–67. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, et al. Il-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zepp J, Wu L, Li X. Il-17 receptor signaling and T helper 17-mediated autoimmune demyelinating disease. Trends Immunol. 2011;32:232–9. doi: 10.1016/j.it.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172:146–55. doi: 10.2353/ajpath.2008.070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. Il-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–73. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343:938–52. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- Goverman J, Woods A, Larson L, Weiner LP, Hood L, Zaller DM. Transgenic mice that express a myelin basic protein-specific T cell receptor develop spontaneous autoimmunity. Cell. 1993;72:551–60. doi: 10.1016/0092-8674(93)90074-z. [DOI] [PubMed] [Google Scholar]
- Matusevicius D, Kivisakk P, He B, Kostulas N, Ozenci V, Fredrikson S, Link H. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler. 1999;5:101–4. doi: 10.1177/135245859900500206. [DOI] [PubMed] [Google Scholar]
- Durelli L, Conti L, Clerico M, et al. T-helper 17 cells expand in multiple sclerosis and are inhibited by interferon-β. Ann Neurol. 2009;65:499–509. doi: 10.1002/ana.21652. [DOI] [PubMed] [Google Scholar]
- Lock C, Hermans G, Pedotti R, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–8. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- Hautecoeur P, Forzy G, Gallois P, Demirbilek V, Feugas O. Variations of Il2, Il6, Tnf α plasmatic levels in relapsing remitting multiple sclerosis. Acta Neurol Belg. 1997;97:240–3. [PubMed] [Google Scholar]
- Vladic A, Horvat G, Vukadin S, Sucic Z, Simaga S. Cerebrospinal fluid and serum protein levels of tumour necrosis factor-α (TNF-α) interleukin-6 (Il-6) and soluble interleukin-6 receptor (Sil-6r Gp80) in multiple sclerosis patients. Cytokine. 2002;20:86–9. doi: 10.1006/cyto.2002.1984. [DOI] [PubMed] [Google Scholar]
- Li C, Beavis P, Palfreeman AC, Amjadi P, Kennedy A, Brennan FM. Activation of P38 mitogen-activated protein kinase is critical step for acquisition of effector function in cytokine-activated T cells, but acts as a negative regulator in T cells activated through the T-cell receptor. Immunology. 2011;132:104–10. doi: 10.1111/j.1365-2567.2010.03345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peluso I, Fantini MC, Fina D, Caruso R, Boirivant M, MacDonald TT, Pallone F, Monteleone G. IL-21 counteracts the regulatory T cell-mediated suppression of human CD4+ T lymphocytes. J Immunol. 2007;178:732–9. doi: 10.4049/jimmunol.178.2.732. [DOI] [PubMed] [Google Scholar]
- Huber S, Schrader J, Fritz G, et al. P38 map kinase signaling is required for the conversion of CD4+CD25− T cells into iTreg. PLoS ONE. 2008;3:e3302. doi: 10.1371/journal.pone.0003302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxade M, Parra JL, Rousseau S, et al. The Mnks are novel components in the control of TNF-α biosynthesis and phosphorylate and regulate hnRNP A1. Immunity. 2005;23:177–89. doi: 10.1016/j.immuni.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Lopez-Santalla M, Salvador-Bernaldez M, Gonzalez-Alvaro I, et al. Tyr(3)(2)(3)-dependent P38 activation is associated with rheumatoid arthritis and correlates with disease activity. Arthritis Rheum. 2011;63:1833–42. doi: 10.1002/art.30375. [DOI] [PubMed] [Google Scholar]
- Thalhamer T, McGrath MA, Harnett MM. MAPKs and their relevance to arthritis and inflammation. Rheumatology (Oxford) 2008;47:409–14. doi: 10.1093/rheumatology/kem297. [DOI] [PubMed] [Google Scholar]
- de Launay D, van de Sande MG, de Hair MJ, et al. Selective involvement of ERK and JNK mitogen-activated protein kinases in early rheumatoid arthritis: a prospective study aimed at identification of diagnostic and prognostic biomarkers as well as therapeutic targets. Ann Rheum Dis. 2012;71:415–23. doi: 10.1136/ard.2010.143529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin T, Ahn M, Jung K, Heo S, Kim D, Jee Y, Lim YK, Yeo EJ. Activation of mitogen-activated protein kinases in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2003;140:118–25. doi: 10.1016/s0165-5728(03)00174-7. [DOI] [PubMed] [Google Scholar]
- Commodaro AG, Bombardieri CR, Peron JP, et al. p38α MAPKinase controls IL17 synthesis in Vogt–Koyanagi–Harada syndrome and experimental autoimmune uveitis. Invest Ophthalmol Vis Sci. 2010;51:3567–74. doi: 10.1167/iovs.09-4393. [DOI] [PubMed] [Google Scholar]
- Badger AM, Bradbeer JN, Votta B, Lee JC, Adams JL, Griswold DE. Pharmacological profile of Sb 203580, a selective inhibitor of cytokine suppressive binding protein/P38 kinase, in animal models of arthritis, bone resorption, endotoxin shock and immune function. J Pharmacol Exp Ther. 1996;279:1453–61. [PubMed] [Google Scholar]
- Lee JC, Laydon JT, McDonnell PC, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–46. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- Huang G, Wang Y, Vogel P, Kanneganti TD, Otsu K, Chi H. Signaling via the kinase p38α programs dendritic cells to drive Th17 differentiation and autoimmune inflammation. Nat Immunol. 2012;13:152–61. doi: 10.1038/ni.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn T, Bettelli E. Bettelli E. Gao W. Awasthi A. Jäger A. Strom TB. Oukka M. Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory TH17 cells. Nature. 2007;448:484–7. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Fleischmann R. Kinase inhibitors: a new approach to rheumatoid arthritis treatment. Curr Opin Rheumatol. 2010;22:330–5. doi: 10.1097/BOR.0b013e3283378e6f. [DOI] [PubMed] [Google Scholar]
- Kumar S, Boehm J, Lee JC. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov. 2003;2:717–26. doi: 10.1038/nrd1177. [DOI] [PubMed] [Google Scholar]
- Bonilla-Hernan MG, Miranda-Carus ME, Martin-Mola E. New drugs beyond biologics in rheumatoid arthritis: the kinase inhibitors. Rheumatology (Oxford) 2011;50:1542–50. doi: 10.1093/rheumatology/ker192. [DOI] [PubMed] [Google Scholar]
- Goldstein DM, Kuglstatter A, Lou Y, Soth MJ. Selective p38α inhibitors clinically evaluated for the treatment of chronic inflammatory disorders. J Med Chem. 2010;53:2345–53. doi: 10.1021/jm9012906. [DOI] [PubMed] [Google Scholar]
- Di Mitri D, Azevedo RI, Henson SM, et al. Reversible senescence in human CD4+CD45RA+CD27− memory T cells. J Immunol. 2011;187:2093–100. doi: 10.4049/jimmunol.1100978. [DOI] [PubMed] [Google Scholar]
- Thewissen M, Linsen L, Somers V, Geusens P, Raus J, Stinissen P. Premature immunosenescence in rheumatoid arthritis and multiple sclerosis patients. Ann N Y Acad Sci. 2005;1051:255–62. doi: 10.1196/annals.1361.066. [DOI] [PubMed] [Google Scholar]
- Yang Y, Kim SC, Yu T, Yi YS, Rhee MH, Sung GH, Yoo BC, Cho JY. Functional roles of p38 mitogen-activated protein kinase in macrophage-mediated inflammatory responses. Mediators Inflamm. 2014;2014:352371. doi: 10.1155/2014/352371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulop T, Le Page A, Fortin C, Witkowski JM, Dupuis G, Larbi A. Cellular signalling in the aging immune system. Curr Opin Immunol. 2014;29:105–11. doi: 10.1016/j.coi.2014.05.007. [DOI] [PubMed] [Google Scholar]
- Dambach DM. Potential adverse effects associated with inhibition of P38α/β map kinases. Curr Top Med Chem. 2005;5:929–39. doi: 10.2174/1568026054985911. [DOI] [PubMed] [Google Scholar]
- Hammaker D, Firestein GS. Go upstream, young man: lessons learned from the P38 saga. Ann Rheum Dis. 2010;69:i77–82. doi: 10.1136/ard.2009.119479. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Listed patients with relapsing–remitting multiple sclerosis were all female, were all in the remission phase at the time of the sampling and did not undergo any treatment since at least 3 months before the present study.