

Abstract
The advent of single cell transcriptome analysis has permitted the discovery of cell-to-cell variation in transcriptome expression of even presumptively identical cells. We hypothesize that this variability reflects a many-to-one relationship between transcriptome states and a cell’s phenotype. In this relationship the molecular ratios of the subsets of RNA are determined by the cell systems’ stoichiometric constraints, which underdetermine the transcriptome state. Further the variability is in part induced by the tissue context and is important for system-level function. This theory is analogous to theories of literary deconstruction where multiple “signifiers” work in opposition to one another to create meaning. By analogy, transcriptome phenotypes should be defined as subsets of RNAs comprising selected RNA systems where the system associated RNAs are balanced with each other to produce the associated cellular function. This idea provides a framework for understanding cellular heterogeneity in phenotypic responses to variant conditions such as disease challenge.
Keywords: transcriptome, phenotype, biological constraints
Deconstruction and Variation
The literary concept of deconstruction developed largely by Jacques Derrida derives, in part, from the idea that meaning derives from the contrasts between opposing words[1]. The word “black” has meaning in the context of being a contrasting state to “white”, “blue”, “red”, etc. The function of words is to bring meaning but if that meaning is contextual then this context must be understood through the opposing contrasts (oppositions) that give rise to that meaning. This deconstructionist view is not alien to biology. For example, the meaning of the term “gene” has become ambiguous with growing complexity of the genome and proposals have been made to define the term within the variational context of all genomes as contrasting states [2]. In cells, the correlate of the deconstructionist idea is that the function of a protein or mRNA manifests in the variational context of other molecules. Use of phrases like “necessary and sufficient” to describe the function of a molecule hides the shared implicit assumption of the contrasting context of all other factors. While biology requires the function of particular molecules, their presence alone is insufficient for function; processes such as intermediary metabolism are emergent properties resulting from the complex interplay of the contrasting functions of other molecules. Contextual functions of the RNA constituents of the transcriptome or the proteins made from these RNAs define the pleiotropic relation of the molecular components to biological function.
Cells also display contrasting context in tissues and organs. Rather than comprising identical units, fully differentiated cells display heterogeneity in function, responses, and dysfunction. For example, both normal and disease-related cognitive decline involve select brain subsystems and differential dysfunction at the cellular level [3–7]. Increasing ability to discriminate molecular states, especially levels of gene expression, has revealed molecular heterogeneity underlying seemingly homogeneous phenotypes at all scales down to the level of single cells [8–17]. The first papers presenting single cell transcriptome data [18, 19] have been followed by a multitude of papers that have carried out single cell measurements, especially for the transcriptome [18–31]. All of these papers show significant variation between individual cells for both mRNAs and more recently non-coding RNA [32]. Many models and hypotheses have been generated to explain cellular heterogeneity [13, 15, 33]. Here we first examine several possibilities concerning the nature of single cell variability. Then we present our main hypothesis that the relationship between transcriptome states and the cell’s phenotype should naturally be many-to-one. We propose that the heterogeneity of cells is a natural relaxed state reflecting both the many-to-one relationship and the ecological interaction of the cells. We then present a conceptual model that suggests that changes in the transcriptome to phenotype relationship in relation to changes in the cell’s environmental context explain heterogeneous cellular responses to external forces such as stress, aging, therapeutics, etc.
Perhaps single cell variation is due to technical problems
Many factors affect the accurate recovery of transcriptome measurements in single cell experiments [34, 35] with one of the most significant steps being first strand cDNA synthesis required for amplification. For single cell transcriptome variation, the unit of measurement cannot be replicated without techniques for non-destructive whole genome assays, making it difficult to assess technical variability. Nevertheless, a multitude of papers have demonstrated biological signals from single cell whole-transcriptome measurements; for example, using split-pool experiments to identify biological versus technical variance factors [34, 36], as well as spike-in and single molecule FISH validations [37]. One important control experiment involves using in vitro dilutions of well-defined bulk RNA or other control RNA such as the External RNA Reference Consortium (ERCC) RNA [38], which has been used to model technical noise of single cell transcriptome while demonstrating biological variation beyond technical noise [31, 39]. Indeed, using both commercial bulk RNA (UHRR, Agilent; Human Brain Reference, Life Tech) as well as ERCC RNA, dilution experiments suggest a quantitative detection of 6–12 input molecules (unpublished data). As seen in other studies, single cell RNA from five different mouse cell types (cardiomyocytes, brown adipose tissue cells, pyramidal neurons from hippocampus and the cortex, and serotonergic neurons) [31] revealed that for every cell type such transcriptomes show a high degree of cell-to-cell variability. For certain cell types, there is more within cell type transcriptome variance than the difference between the average transcriptome of two different cell types. However, in a subset of genes that have previously been annotated as marker genes for respective cell types, the resulting subset of transcriptomes cluster by input cell types with a high degree of significance (Fig 1). This recapitulates many published works that show single cell whole transcriptome measurements contain biologically relevant quantitative information. As reviewed in [40], the idea that only technical noise is captured in single cell measurements can be easily rejected. Further, while the focus of this article is on cellular variation of the transcriptome, there are other types of single cell variation that are detected using techniques that are less molecularly cumbersome and hence are independent of RNA amplification and sequencing technical error such as high content imaging of cellular morphology [41] or live cell biological processes [42]. These examples serve to illustrate that biological “cell-to-cell” variation exists regardless of the contributions of technical noise.
Figure 1.
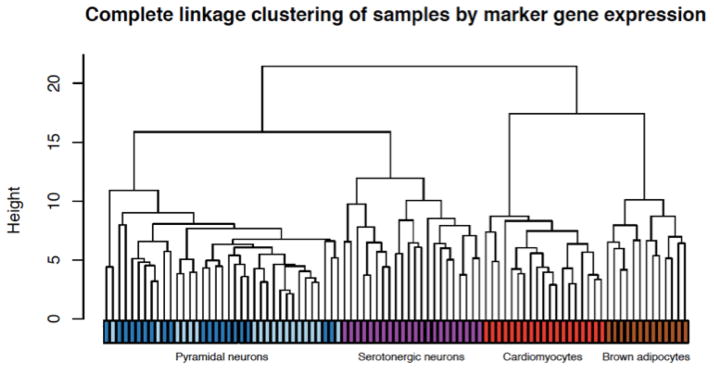
Individual cells of different mouse cell types clustered using cell type marker genes subset from whole single cell transcriptomes measured using in vitro transcription amplification and RNAseq. Reprinted with permission from [31].
Perhaps all cells are different
It is estimated that the human body has ~100 trillion cells, every one of them originating from a single zygote. The development of a complex multi-cellular body requires cell lineage differentiation and diversification, but even cells of the same lineage may differ in their size, shape, epigenetic states, etc. By some estimates, every cell contains at least one somatic mutation and thus all cells may differ in their genome as well. Some recent studies even suggest that each cell might express differing alleles of a locus in a random fashion with the implication that every cell might have a nearly unique allelic expression combination [27, 43]. It could be trivially argued that all cells are different from each other in some subset of molecules. Therefore, the question is not whether individual cells are different from each other but whether there are substantial differences that previously were thought to only exist between functionally and phenotypically distinct sets of cells. Single cell genome-wide gene expression datasets suggest the existence of such large-scale non-trivial differences between phenotypically identical cells. For example, single cell data from rat hippocampal neurons and astrocytes were analyzed [44] and the divergence of median gene expression within the neurons was 166.4 (log expression units) and for the astrocytes it was 128.3; while between the two cell types it was 202.4. Thus, neurons of the same phenotype displayed ~82% of the divergence between neurons and astrocytes. Similar diversity of single cell transcriptomes, including distinct multimodality of expression (suggesting at least for some subset of genes, there is an ON-OFF switching between different cells) has been seen in other studies [23, 31, 36]. Many single cell studies have uncovered a rich set of hidden subtypes, which manifest as distinct clusters in transcriptome space despite the cell-to-cell variation [20, 21, 45, 46] often resulting in discovery of new rare molecular factors governing distinct subtypes [28, 47]. In sum, while every cell may be different at some molecular level, current studies suggest significant non-trivial differences in the transcriptome of phenotypically identically characterized cells that at a larger-scale form distinct hierarchical clusters of transcriptome states. These patterns suggest that transcriptome variation is significantly structured at all levels of cellular organization and reject the idea that cells are trivially different from each other.
Perhaps transcription is noisy
Recent data suggests that gene transcription is subject to intrinsic and extrinsic noise [15, 48]. At the single molecule-level, transcription and other posttranscriptional processes are subject to molecular noise due to their finite numbers; and, such noise may be amplified through gene regulatory networks [49]. The term “noise” is somewhat of an ambiguous term, and has been used to describe any variation. Here, we differentiate between the stochastic phenomena that arise from statistical mechanical interactions of a small finite number of molecules versus variations that occur between different cells due to other factors. The number of active molecules of a given type is often surprisingly small. For example, careful measurements suggest that there are 100,000 to 1 million mRNA molecules per mammalian cell [50] suggesting the mRNA of most genes are on average 10 to 100 molecules per cell (assuming 10,000 expressed genes). Therefore, transcription of any mRNA species is likely to have stochastic waiting times; and local spatial environment of the DNA or the many-body interactions of the relevant proteins may cause the waiting times to be clustered, or “bursty” [51]. In [52, 53], a statistical model of single cell variation was used to suggest that variation patterns follow that expected from accumulation of quantal molecular noise. Nevertheless, mRNA measures taken with standard RNAseq techniques do not measure real-time transcription but the accumulated mRNA over time (i.e., integration of the transcription over time). The estimated median half-life of mammalian mRNA is on the order of nine hours [54] and therefore, unless the stochastic waiting time arising from statistical mechanical interaction of molecules is on the same time scale, this source of stochastic fluctuations is likely to only affect a small percentage of cell-to-cell variation. Furthermore, if the variation were due to the bursty waiting times of transcription, it would be difficult to generate multi-modal patterns [23] or cell-specific multivariate dependent directions of covariation (other than expression level dependent directions) [26, 31, 55]. Stochastic molecular interactions of a small number of molecules undoubtedly play a role in cell-to-cell variation. However, disparate time-scales, distinct patterns such as multi-modality, and multivariate dependent directions of variation, all suggest that molecular noise likely explains only a small percentage of the observed single cell transcriptome variation.
Perhaps transcriptome variability results from dynamical changes in cellular/molecular physiology
In many cells, there is an intrinsic set of molecular dynamics such as cell cycles [56], circadian rhythms [57], and metabolic switches [58]. Different cells may be at different phases of these dynamical trajectories or even coupled dynamics [59], which would manifest as single cell variability. The effect of such internal dynamics will always play a role in single cell variation. Indeed, in the mouse cell data set shown in Fig 1, the cardiomyocytes were the only proliferating cells measured and the most variable genes in this cell type belonged to the functional category of cell cycles [31]. Even for post-mitotic cells we cannot rule out that different cells are in different phases of other physiological dynamics. One possible hint of the role of physiological dynamics in single cell variation might come from a dimensional analysis. A dynamical system, at least that of a single smooth system, is inherently one-dimensional. If a large number of variable dimensions are found in the data, it may be indicative of factors other than intrinsic molecular physiology dynamics. However, because the one-dimensional temporal trajectory can be folded in complicated ways creating a high-dimensional embedding, it is difficult to tease out temporal dynamics. Fully accounting for such variation will require an experimental design with very large-scale sampling. Some recent studies have begun to tease out the cell physiological dynamics as a component of single cell variation [25, 60]. In particular, cell-cycle contribution to single cell heterogeneity has been explicitly analyzed in [61] and only a small number of genes (n = 27) showed cell-cycle dynamics in a non-proliferating cell. Another single cell study suggested that the cell-cycle contribution to variation ranges between 5–17%, again suggesting that phases of some intrinsic cell cycle dynamics do not contribute greatly to variation [62]. We recall that the average number of mRNA molecules in a typical mammalian cells is between 10–100 molecules and by our measures the relative frequency of the median rank gene in a mammalian cell is ~10−5. Thus, shallow sequencing (e.g., 1 million reads per cell) will not recover full dynamic characteristics of individual cells. In the meanwhile, statistical analysis of dynamic processes requires a large number of samples distributed evenly across the phases of the dynamics, thus costs become an important barrier. We believe as costs come down enabling large-scale measurements with sufficient read depth, dissecting single cell dynamics will become much more feasible. The idea that single cells reveal the phases of intrinsic dynamics also suggests that network and regulatory system models (focus of systems biology) should be estimated with single cell data.
In sum, we cannot rule out the possibility that some of the observed cell-to-cell variation is due to catching cells at different phases of their natural physiological dynamics. However, some direct estimates suggest that the contribution of dynamical phases to overall cell-to-cell variation may be relatively minor. More detailed dynamic analyses from single cell measurements would be highly desirable as the currently available data is limited in either read depth or sample size. We note cell-to-cell variation need not be a single factor concept. All cells may be undergoing, say, circadian rhythm dynamics, while also displaying variation in other dimensions. This also suggests that changes in the dynamics (e.g., by disease or external stimulation) may alter the variability in characteristic ways yielding clues to the cellular responses to the challenge.
Perhaps single cell variation may be a necessary for tissue or organ function
A tissue or an organ’s organismal function is derived from the collective functions of individual cells. A tissue is never homogeneous in its cell composition and even for cells of putative identical phenotype, it may be that plastic variation is necessary to carry out tissue/organ-level function [13, 63]. The idea of “bet hedging” in single cell organisms as an evolutionary strategy has been shown in many examples [64–66]. In multicellular organisms, a kind of bet hedging may be involved in tissue/organ level function; not only in clear examples such as adaptive immunity but also in selective differentiation of sub-functions as in the formation of memory cells [67]. In [68], an intriguing model is proposed for the slow turnover and maintenance of mammalian tissue sizes by low-level stochastic switching of cell states. Development and lineage fate choice may involve initial heterogeneity of cell states that narrow into differentiated substates [33, 69, 70]. Even within a differentiated post-mitotic tissue, cells may be signaling both for heterogeneity and spatial differentiation [29, 30, 60, 71]. Recently, a new method for isolating single cell mRNA using a photo-activated poly-A capture reagent called TIVA was developed [72]. Using this method, the expression range of mouse neurons isolated from primary cell cultures versus those isolated from tissue slices were examined and were found to have a greater range of expression for the latter. All of this data suggests that a cell within a tissue may adapt to its micro-environment, driving cell to cell variability. More intriguingly, tissue level function may require diversified states of individual cells; as much as an ecological community requires diversified roles of constituent individuals. This idea is suggested in part by a recent study [73] showing that mouse embryonic fibroblast expression of components of the NF-kB signaling system are timed to induce cellular heterogeneity in the oscillatory expression of these proteins. This was interpreted as showing that cellular variation is evolutionarily advantageous. Thus, some tissues may induce more heterogeneous cell states while other tissues may involve more homogeneous cells. We also suggest the possibility that the maintenance of a single cell’s phenotypic state may require underlying heterogeneous molecular states of surrounding cells, each providing paracrine signals to support the cell’s phenotype. Finally, while the natural equilibrium state of cells within an organism is heterogeneous, some transient external stimuli may induce a common transcriptional response causing more homogeneous states in the activated part of the gene regulatory network where upon expression variability for the genes in the network might decrease. Consequently, there is considerable evidence that individual cells in a tissue adopt a heterogeneous state either through relaxation of their physiological dynamics or by active signaling and maintenance of an aggregate field. The idea of variation as function is exciting; however, direct tests in native cells and complex cellular systems will be difficult until regulatory factors are identified.
Perhaps multiple transcriptome states support identical cell phenotype
A simple view of a cell’s phenotype is that there is a one-to-one relationship between its transcriptome state and its phenotype state. Therefore, changes in the average expression (averaged over natural dynamics like cell cycles) are expected to lead to changes in the phenotype. Here, we make the argument that the set of transcriptome states that support a phenotype, what we previously called the “equi-phenotypic support” [74], should be a broad combination of gene expression. That is, a cell has a many-to-one relationship between its transcriptome and phenotype.
First, with some exceptions, a vast array of post-transcriptional regulatory mechanisms play a strong role in determining the relationship between transcript levels and protein levels (and types) as well as levels of functional non-coding RNA [75–80]. Even in single cell organisms, such as, Kinetoplastids, no evidence of RNA regulation is shown at the level of transcription [81]. Thus, the number of molecules of a given mRNA is unlikely to simply map to the number of functional mRNA molecules. More importantly, the number of molecules of many molecular species may be dictated by the need to meet the stoichiometric relationship of the chemical reactions that comprise the molecular physiology. If so, the levels of particular mRNAs may need only to meet some minimal bound rather than an exact number—for example, we might require “at least twice the number of A compared to B” rather than “exactly two molecules of A for every molecule of B” (Fig 2). If functional demand only requires meeting inequality bounds (e.g., “at least as many as”) rather than exact constraints, then there will be large domains of transcriptional states that are functionally equivalent (Fig 2).
Figure 2.
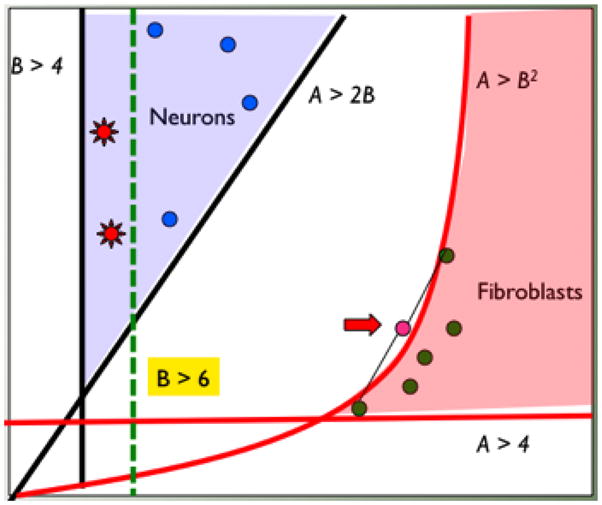
A schematic diagram representing our conceptual model for the relationship between transcriptome and phenotype. Cells of a given external phenotype (e.g., neurons and fibroblasts; blue dots and green dots) are seen to map to broad domains of molecular state space (many-to-one relationship). The domains are determined by stoichiometric constraints of active pathways (schematically represented as inequalities). Some constraints may be nonlinear due to multi-component interactions (A>B2). Non-linear constraints may lead to population averages of cells being outside the state of any cell (red arrow). The constraints may change (e.g., from B>4 to B>6) due to physiological changes (e.g., aging) or external stimuli (e.g., chemotherapy drug) and the response of the cells may be heterogeneous due to original variation (red stars within blue dots representing neurons) explaining phenomena such as drug resistance or heterogeneous degeneration.
Second, if satisfying stoichiometric relationships is the functional rationale for particular levels of mRNA, the question is how many different structural or chemical stoichiometric relationships are relevant for a given cell at any given time. Stoichiometric relationships determine a set of “constraints” on the functionally relevant numbers of RNA molecules, perhaps as a bound rather than equality (e.g., A > 2B). Given the entire transcriptome, the number of such constraints that need to be satisfied to maintain phenotypic function is likely to be considerably under-determined. For example, ~12,000 different genes were observed to be expressed in a single mouse pyramidal neuron [31]. The question is whether there are 12,000 different stoichiometric relationships required by various chemical and structural constraints or whether there are only, say, 11,000. If it is the latter, there will be 1,000 degrees of freedom; that is, 1,000 dimensions in which variation is functionally neutral. We note that this argument is different from the “robustness” argument made above for the long lasting stochasticity of molecular noise upon transcriptional bursting. We propose that there are multiple combinations of transcriptome states that are functionally equivalent because the physiological reactions are agnostic to their variations.
A synthetic view of single cell transcriptome variability
There are likely to be many factors that induce cell-to-cell heterogeneity in the transcriptome state, as well as other molecular and morphological states. These factors are not exclusive of each other and a given cell’s state is likely to be determined by all of the above combinations, especially with some factors governing the variability of certain genes or gene combinations while other factors govern other gene combinations. We suspect that the factors with the largest amplitude in variability is likely to be (i) dynamics of molecular physiology; (ii) micro-environment adaptation and tissue level function; and, (iii) “degrees of freedom” from functional equivalence. The dynamics of molecular physiology may dominate cell heterogeneity in certain cases such as the cell cycle in proliferating cells while other dynamics such as circadian rhythms may have smaller effects. Regardless, such dynamics may manifest in certain gene directions independent of variation in other gene directions. An important implication is that if single cell variability is due to phase differences from dynamics of regulated cell physiology then pooled cell measurements will average out this dynamical information. Thus, system biology models should require measurement at the level of single cells.
A population of cells within an organ is likely to experience a significantly different micro-environment that requires the cell to have different transcriptional states. As mentioned above, organ function may also require differentiated cell function, not only from different types of cells (e.g., glial cells and neurons) but also from different individual cells of the same type. This is certainly true for the neural network function of neurons in the CNS but it is also likely to be true for other cells with regards to their respective function in an organ. Even in single cell micro-organisms, populations of clonal cells differentiate into active cells and quiescent cells by phase regulation [81]. Recapitulation of cell diversity has been observed in clonally selected and propagated lymphocytes [82]. These data suggest that the equilibrium state of cells might be naturally variable. Cells may synchronize and homogenize only at distinct points, e.g., application of drugs, specific stages of development, etc., and then relax back to heterogeneous states. If stimulus affects only certain pathways, such pathways might be identified by changes in single cell variation, which would suggest a new approach towards unbiased screening of the transcriptome for key genes.
We propose above that there is likely to be large degrees of freedom for variation in transcriptome states that have no functional consequences. We previously called this the equi-phenotypic support set [74]; i.e., all the possible combinations of mRNA that satisfy the molecular balances required to support that phenotype. The possible constraints are likely to be comprised of both exact relationships, e.g., A = 2B, and inequality bounds, e.g., A >2B. Given typical reaction kinetics, the constraints are likely to also involve non-linear bounds such as A2 >B (e.g., dimer steady states). Furthermore, the equi-phenotypic support set of a phenotype may be altered by the context of the cell such as during development, aging, and drug treatment. Fig 2 shows a picture of this model in a two-dimensional conceptual representation of the transcriptome. Here, two broad domains of two hypothetical RNA A and RNA B are shown to support the phenotype of neurons (blue) and fibroblasts (red). The two domains are specified by a set of functional molecular balance constraints (e.g, B> 4, A > B2, etc.). There are many possible transcriptome states for each cell type, which would result in single cell variation. The picture shows that averages of different cells of the same type may not result in the same transcriptome as any single cell transcriptome (e.g., the red arrow where the average transcriptome of two fibroblasts do not correspond to any fibroblast transcriptome). In higher dimensions this phenomenon may happen easily and the pooled transcriptome of single cells may correspond neither to the tissue level transcriptome nor to any possible single cell transcriptome. More importantly, such domains of equi-phenoptypic support sets may shift with changing conditions. For example, aging may change one functional constraint from B > 4 to B > 6 as shown in the picture. In this scenario, some of the existing cells may remain functional (blue dots) while others may degenerate (red stars). This model potentially explains the heterogeneous responses of cells to therapeutics, aging, and degenerative diseases. An equally intriguing possibility is that this model might underlie differential responses to drugs such as chemotherapy reagents, where inherent heterogeneity in transcriptional or other epigenetic states form the basis for resistant subpopulations, rather than a somatic mutation and clonal selection mechanism.
Concluding remarks
The idea that biological variation is functionally important derives from a rich history of observation perhaps most notably that of evolutionary diversification between strains or species and the establishment of ecological communities. In fact, the ecologist H. Clements suggested that ecological communities might be considered “super-organisms” [83]; in a complementary view, cells in tissues might be considered ecological communities.
Single cell deconstruction takes the idea of biological variation to the individual cell where there is interplay of different RNAs giving rise to acute transient functional states and more lasting phenotypic states. The process of RNA transcription is regulated through the combinatorial activity of various transcription components such as polymerases, chromosome states, regulatory proteins, etc. As described earlier it is likely that transcription occurs in short bursts of RNA polymerase activity that gives rise to a bolus of RNA that is then processed and transported from the nucleus to the cytoplasm of the cell. Given that it is difficult to quantitatively control the amount of RNA that is transcribed it seems unlikely that this level of control is so tightly regulated that it can account for the balancing of various intracellular systems. Rather this suggests that the system level function of the cells (i.e., tissue/organ) involves broad permissible states of individual molecular components—permissible states that arise both by physical-chemical constraints and evolutionary selection for the system-level function. It is at this level that cellular deconstruction manifests as the functioning of multiple cellular pathways including those apparent from the transcriptome that require contextualization from both the micro-environment and local interactions.
Understanding single cell biology, which will be accomplished as genomics moves more rapidly into single cell and even higher resolution functional measurements, should permit synthesis of an over-arching theory of physiology and organismal system level function (see Outstanding questions). “Deconstructing” the variability of individual components of a physiological system in relation to their “oppositions” will provide for quantifiable patterns of molecular characteristics of organismal function and allow for synthesis of concepts that would have been obscured by non-contextual transcriptome observation and quantitation (Fig 3). We note again that the original idea of literary deconstructionism is opposite of the idea of reductive view of nature; that is, it proposes that understanding of the meaning of words requires deconstruction of the context in which they are written, not a strict definition but rather a relational meaning that portends understanding. We argue that the functional pleiotropy afforded by the equi-phenotypic subsets of RNAs requires a similar deconstruction of cellular language that provides context for the cell’s phenotype state.
Figure 3.

Comparison of Literary and Biological Deconstruction. The use of descriptors including words in literature and RNA for a cell represents an amalgam of other descriptors that through their opposing meanings or function elaborate the concepts of language and cellular phenotype.
Outstanding Questions Box.
How important is the transcriptome in defining cellular phenotype? Many cellular features are used to “define cell type”, is transcriptome one of the most or least important of these features?
What aspect of cellular phenotype requires transcriptome variability? If variability is important to individual cell function or higher system-level function then it should be possible to define the cellular feature(s) that require transcriptome variability.
Does single cell transcriptome variability change in a disease specific manner? Can variability across multiple cells be a biomarker of disease or disease-state?
Does variability in transcriptome change in concert with other highly variable aspects of cell phenotype such as the metabolome?
How is the degree of cell-to-cell variability regulated, cell-autonomously or through communication with other cells or its microenvironment?
What is the role of transcriptome and epigenome variability in differential responses of cells to therapeutic reagents (e.g., chemotherapy) or physiological changes (e.g., aging)?
Trends Box.
Comparisons of transcriptomes between seemingly identical cells show that the transcriptomes have significant biological variation.
Cells exhibit “many to one” sets of equally functional transcriptional states that are defined in part by pathway constraints that are underspecified in comparison to number of genes.
Cellular contextualization occurs from the influence of the microenvironment and local interactions suggesting that variation itself may be important for system level function.
Acknowledgments
The authors thank Jennifer Singh for sequencing of the single mouse cells and Hannah Dueck for providing preliminary analysis of mouse single cell datasets. This work has been supported in part by Ellison Medical Foundation Aging Award, NIH Single Cell Analysis Program grant 5U01MH098953, and Health Research Formula Funds from the Commonwealth of Pennsylvania. The funding bodies had no role in study design, in collection, analysis, interpretation of data, or in the writing of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Derrida J. Writing and difference. London: Routledge & K. Paul; 1978. p. xx.p. 342. [Google Scholar]
- 2.Kim J, Kim M. The mathematical structure of characters and modularity. In: Wagner GP, editor. The character concept in evolutionary biology. Academic Press; San Diego: 2001. pp. 215–236. [Google Scholar]
- 3.Woodruff-Pak DS, et al. Differential effects and rates of normal aging in cerebellum and hippocampus. Proc Natl Acad Sci U S A. 2010;107(4):1624–9. doi: 10.1073/pnas.0914207107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430(7000):631–9. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morrison JH, Hof PR. Life and death of neurons in the aging cerebral cortex. Int Rev Neurobiol. 2007;81:41–57. doi: 10.1016/S0074-7742(06)81004-4. [DOI] [PubMed] [Google Scholar]
- 6.Ginsberg SD, et al. Expression profile of transcripts in Alzheimer’s disease tangle-bearing CA1 neurons. Ann Neurol. 2000;48(1):77–87. [PubMed] [Google Scholar]
- 7.Buckner RL. Memory and executive function in aging and AD: multiple factors that cause decline and reserve factors that compensate. Neuron. 2004;44(1):195–208. doi: 10.1016/j.neuron.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Schubert C. Single-cell analysis: The deepest differences. Nature. 2011;480(7375):133–7. doi: 10.1038/480133a. [DOI] [PubMed] [Google Scholar]
- 9.Elowitz MB, et al. Stochastic gene expression in a single cell. Science. 2002;297(5584):1183–6. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- 10.Levsky JM, et al. Single-cell gene expression profiling. Science. 2002;297(5582):836–40. doi: 10.1126/science.1072241. [DOI] [PubMed] [Google Scholar]
- 11.Alizadeh AA, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503–11. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 12.Kamme F, et al. Single-cell microarray analysis in hippocampus CA1: demonstration and validation of cellular heterogeneity. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003;23(9):3607–15. doi: 10.1523/JNEUROSCI.23-09-03607.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaern M, et al. Stochasticity in gene expression: from theories to phenotypes. Nature reviews. Genetics. 2005;6(6):451–64. doi: 10.1038/nrg1615. [DOI] [PubMed] [Google Scholar]
- 14.Trimarchi JM, et al. Molecular heterogeneity of developing retinal ganglion and amacrine cells revealed through single cell gene expression profiling. The Journal of comparative neurology. 2007;502(6):1047–65. doi: 10.1002/cne.21368. [DOI] [PubMed] [Google Scholar]
- 15.Raj A, van Oudenaarden A. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell. 2008;135(2):216–26. doi: 10.1016/j.cell.2008.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J, et al. Normalization, testing, and false discovery rate estimation for RNA-sequencing data. Biostatistics. 2011 doi: 10.1093/biostatistics/kxr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bahar R, et al. Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature. 2006;441(7096):1011–4. doi: 10.1038/nature04844. [DOI] [PubMed] [Google Scholar]
- 18.Eberwine J, et al. Analysis of gene expression in single live neurons. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(7):3010–4. doi: 10.1073/pnas.89.7.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miyashiro K, Dichter M, Eberwine J. On the nature and differential distribution of mRNAs in hippocampal neurites: implications for neuronal functioning. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(23):10800–4. doi: 10.1073/pnas.91.23.10800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiu IM, et al. Transcriptional profiling at whole population and single cell levels reveals somatosensory neuron molecular diversity. Elife. 2014:3. doi: 10.7554/eLife.04660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaitin DA, et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 2014;343(6172):776–9. doi: 10.1126/science.1247651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sasagawa Y, et al. Quartz-Seq: a highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity. Genome Biol. 2013;14(4):R31. doi: 10.1186/gb-2013-14-4-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shalek AK, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498(7453):236–40. doi: 10.1038/nature12172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xue Z, et al. Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature. 2013;500(7464):593–7. doi: 10.1038/nature12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moignard V, et al. Decoding the regulatory network of early blood development from single-cell gene expression measurements. Nat Biotechnol. 2015;33(3):269–76. doi: 10.1038/nbt.3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen KH, et al. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science. 2015;348(6233):aaa6090. doi: 10.1126/science.aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng Q, et al. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science. 2014;343(6167):193–6. doi: 10.1126/science.1245316. [DOI] [PubMed] [Google Scholar]
- 28.Patel AP, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shalek AK, et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature. 2014;510(7505):363–9. doi: 10.1038/nature13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar RM, et al. Deconstructing transcriptional heterogeneity in pluripotent stem cells. Nature. 2014;516(7529):56–61. doi: 10.1038/nature13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dueck H, et al. Deep sequencing reveals cell-type-specific patterns of single-cell transcriptome variation. Genome Biol. 2015;16(1):122. doi: 10.1186/s13059-015-0683-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cabili MN, et al. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol. 2015;16(1):20. doi: 10.1186/s13059-015-0586-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raj A, et al. Variability in gene expression underlies incomplete penetrance. Nature. 2010;463(7283):913–8. doi: 10.1038/nature08781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu AR, et al. Quantitative assessment of single-cell RNA-sequencing methods. Nat Methods. 2014;11(1):41–6. doi: 10.1038/nmeth.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eberwine J, et al. Quantitative biology of single neurons. J R Soc Interface. 2012;9(77):3165–83. doi: 10.1098/rsif.2012.0417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marinov GK, et al. From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing. Genome Res. 2014;24(3):496–510. doi: 10.1101/gr.161034.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grun D, Kester L, van Oudenaarden A. Validation of noise models for single-cell transcriptomics. Nat Methods. 2014;11(6):637–40. doi: 10.1038/nmeth.2930. [DOI] [PubMed] [Google Scholar]
- 38.External, R.N.A.C.C. Proposed methods for testing and selecting the ERCC external RNA controls. BMC Genomics. 2005;6:150. doi: 10.1186/1471-2164-6-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ding B, et al. Normalization and noise reduction for single cell RNA-seq experiments. Bioinformatics. 2015 doi: 10.1093/bioinformatics/btv122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Junker JP, van Oudenaarden A. Every cell is special: genome-wide studies add a new dimension to single-cell biology. Cell. 2014;157(1):8–11. doi: 10.1016/j.cell.2014.02.010. [DOI] [PubMed] [Google Scholar]
- 41.Sailem HZ, Sero JE, Bakal C. Visualizing cellular imaging data using PhenoPlot. Nat Commun. 2015;6:5825. doi: 10.1038/ncomms6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Campbell PD, et al. Dynamic visualization of transcription and RNA subcellular localization in zebrafish. Development. 2015;142(7):1368–74. doi: 10.1242/dev.118968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eckersley-Maslin MA, et al. Random monoallelic gene expression increases upon embryonic stem cell differentiation. Dev Cell. 2014;28(4):351–65. doi: 10.1016/j.devcel.2014.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sul JY, et al. Transcriptome transfer produces a predictable cellular phenotype. Proc Natl Acad Sci U S A. 2009;106(18):7624–9. doi: 10.1073/pnas.0902161106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Usoskin D, et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci. 2015;18(1):145–53. doi: 10.1038/nn.3881. [DOI] [PubMed] [Google Scholar]
- 46.Treutlein B, et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014;509(7500):371–5. doi: 10.1038/nature13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poulin JF, et al. Defining midbrain dopaminergic neuron diversity by single-cell gene expression profiling. Cell Rep. 2014;9(3):930–43. doi: 10.1016/j.celrep.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li GW, Xie XS. Central dogma at the single-molecule level in living cells. Nature. 2011;475(7356):308–15. doi: 10.1038/nature10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paulsson J. Summing up the noise in gene networks. Nature. 2004;427(6973):415–8. doi: 10.1038/nature02257. [DOI] [PubMed] [Google Scholar]
- 50.Islam S, et al. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods. 2014;11(2):163–6. doi: 10.1038/nmeth.2772. [DOI] [PubMed] [Google Scholar]
- 51.Chong S, et al. Mechanism of transcriptional bursting in bacteria. Cell. 2014;158(2):314–26. doi: 10.1016/j.cell.2014.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Piras V, Selvarajoo K. The reduction of gene expression variability from single cells to populations follows simple statistical laws. Genomics. 2015;105(3):137–44. doi: 10.1016/j.ygeno.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 53.Piras V, Tomita M, Selvarajoo K. Transcriptome-wide variability in single embryonic development cells. Sci Rep. 2014;4:7137. doi: 10.1038/srep07137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schwanhausser B, et al. Global quantification of mammalian gene expression control. Nature. 2011;473(7347):337–42. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 55.Park J, et al. Identifying functional gene regulatory network phenotypes underlying single cell transcriptional variability. Prog Biophys Mol Biol. 2015;117(1):87–98. doi: 10.1016/j.pbiomolbio.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rifkin SA, Kim J. Geometry of gene expression dynamics. Bioinformatics. 2002;18(9):1176–83. doi: 10.1093/bioinformatics/18.9.1176. [DOI] [PubMed] [Google Scholar]
- 57.Baggs JE, Hogenesch JB. Genomics and systems approaches in the mammalian circadian clock. Current opinion in genetics & development. 2010;20(6):581–7. doi: 10.1016/j.gde.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tu BP, McKnight SL. Metabolic cycles as an underlying basis of biological oscillations. Nature reviews. Molecular cell biology. 2006;7(9):696–701. doi: 10.1038/nrm1980. [DOI] [PubMed] [Google Scholar]
- 59.Bieler J, et al. Robust synchronization of coupled circadian and cell cycle oscillators in single mammalian cells. Mol Syst Biol. 2014;10:739. doi: 10.15252/msb.20145218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Durruthy-Durruthy R, et al. Reconstruction of the mouse otocyst and early neuroblast lineage at single-cell resolution. Cell. 2014;157(4):964–78. doi: 10.1016/j.cell.2014.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Buettner F, et al. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat Biotechnol. 2015;33(2):155–60. doi: 10.1038/nbt.3102. [DOI] [PubMed] [Google Scholar]
- 62.McDavid A, et al. Modeling bi-modality improves characterization of cell cycle on gene expression in single cells. PLoS Comput Biol. 2014;10(7):e1003696. doi: 10.1371/journal.pcbi.1003696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Raser JM, O’Shea EK. Control of stochasticity in eukaryotic gene expression. Science. 2004;304(5678):1811–4. doi: 10.1126/science.1098641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Acar M, Mettetal JT, van Oudenaarden A. Stochastic switching as a survival strategy in fluctuating environments. Nat Genet. 2008;40(4):471–5. doi: 10.1038/ng.110. [DOI] [PubMed] [Google Scholar]
- 65.Martins BM, Locke JC. Microbial individuality: how single-cell heterogeneity enables population level strategies. Curr Opin Microbiol. 2015;24:104–112. doi: 10.1016/j.mib.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 66.Veening JW, et al. Bet-hedging and epigenetic inheritance in bacterial cell development. Proc Natl Acad Sci U S A. 2008;105(11):4393–8. doi: 10.1073/pnas.0700463105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. 2012;12(11):749–61. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ahrends R, et al. Controlling low rates of cell differentiation through noise and ultrahigh feedback. Science. 2014;344(6190):1384–9. doi: 10.1126/science.1252079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ohnishi Y, et al. Cell-to-cell expression variability followed by signal reinforcement progressively segregates early mouse lineages. Nat Cell Biol. 2014;16(1):27–37. doi: 10.1038/ncb2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Buganim Y, et al. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell. 2012;150(6):1209–22. doi: 10.1016/j.cell.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Park J, et al. Inputs drive cell phenotype variability. Genome Res. 2014;24(6):930–41. doi: 10.1101/gr.161802.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lovatt D, et al. Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue. Nature methods. 2014;11(2):190–6. doi: 10.1038/nmeth.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Paszek P, et al. Population robustness arising from cellular heterogeneity. Proc Natl Acad Sci U S A. 2010;107(25):11644–9. doi: 10.1073/pnas.0913798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim J, Eberwine J. RNA: state memory and mediator of cellular phenotype. Trends Cell Biol. 2010;20(6):311–8. doi: 10.1016/j.tcb.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Keene JD. RNA regulons: coordination of post-transcriptional events. Nature reviews. Genetics. 2007;8(7):533–43. doi: 10.1038/nrg2111. [DOI] [PubMed] [Google Scholar]
- 76.Winter J, et al. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nature cell biology. 2009;11(3):228–34. doi: 10.1038/ncb0309-228. [DOI] [PubMed] [Google Scholar]
- 77.Eulalio A, Behm-Ansmant I, Izaurralde E. P bodies: at the crossroads of post-transcriptional pathways. Nature reviews. Molecular cell biology. 2007;8(1):9–22. doi: 10.1038/nrm2080. [DOI] [PubMed] [Google Scholar]
- 78.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nature reviews. Genetics. 2008;9(2):102–14. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 79.Glanzer J, et al. RNA splicing capability of live neuronal dendrites. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(46):16859–64. doi: 10.1073/pnas.0503783102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Martin KC, Ephrussi A. mRNA localization: gene expression in the spatial dimension. Cell. 2009;136(4):719–30. doi: 10.1016/j.cell.2009.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Clayton C, Shapira M. Post-transcriptional regulation of gene expression in trypanosomes and leishmanias. Molecular and biochemical parasitology. 2007;156(2):93–101. doi: 10.1016/j.molbiopara.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 82.Germain RN. Open questions: a rose is a rose is a rose--or not? BMC Biol. 2014;12:2. doi: 10.1186/1741-7007-12-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Clements FE. Plant Succession. Washington DC: Carnegie Institution of Washington; 1916. [Google Scholar]