

Abstract
Studies of historical isolates inform on the evolution and emergence of important pathogens and phenotypes, including antimicrobial resistance. Crucial to studying antimicrobial resistance are isolates that predate the widespread clinical use of antimicrobials. The Murray collection of several hundred bacterial strains of pre-antibiotic era Enterobacteriaceae is an invaluable resource of historical strains from important pathogen groups. Studies performed on the Collection to date merely exemplify its potential, which will only be realised through the continued effort of many scientific groups. To enable that aim, we announce the public availability of the Murray collection through the National Collection of Type Cultures, and present associated metadata with whole genome sequence data for over half of the strains. Using this information we verify the metadata for the collection with regard to subgroup designations, equivalence groupings and plasmid content. We also present genomic analyses of population structure and determinants of mobilisable antimicrobial resistance to aid strain selection in future studies. This represents an invaluable public resource for the study of these important pathogen groups and the emergence and evolution of antimicrobial resistance.
Electronic supplementary material
The online version of this article (doi:10.1186/s13073-015-0222-7) contains supplementary material, which is available to authorized users.
Background
Antimicrobial resistance (AMR) in bacteria represents a global public health crisis, and AMR in Enterobacteriaceae is a particularly recognised threat [1, 2]. This bacterial family includes pathogenic genera (e.g. Salmonella, Escherichia/Shigella, Klebsiella) that are responsible for a significant proportion of the global diarrhoeal disease burden [3] as well as systemic and nosocomial infections, often associated with heightened virulence or AMR [4, 5]. To manage these pathogens, it is critical that we understand the emergence and the evolution of clinically relevant phenotypes. Pivotal to understanding pathogen emergence and evolution is the context in which it occurred, and historical isolates have greatly informed theories regarding the emergence, disappearance and primary reservoir hosts of the pathogens that cause plague, leprosy and tuberculosis [6–8]. More recently, isolates of Vibrio cholerae and Shigella flexneri sampled from before the widespread clinical use (and consequent evolutionary pressure) of antimicrobials, i.e. the ‘pre-antibiotic’ era, were used to examine the evolution of virulence and AMR in these pathogens [9, 10]. To expand these studies in our continued efforts to understand the emergence and persistence of AMR, historical isolates must be studied alongside their contemporary counterparts.
The Murray collection (the ‘Collection’) comprises several hundred bacterial strains (mostly Enterobacteriaceae) collected from diverse geographic locations largely in the pre-antibiotic era (between 1917 and 1954) [11]. The Collection was amassed by the late eminent microbiologist Professor Everitt George Dunne Murray over the course of his career [12], and was stored on Douglas digest agar slopes [13]. On E.G. D. Murray’s death in 1964, the collection was passed on to his son, Robert Everitt George Murray, who was also an eminent microbiologist. In the early 1980s, R.E.G. Murray in collaboration with British microbiologists, lyophilised and transferred subcultures of the Collection from The University of Western Ontario, Canada, to the National Collection of Type Cultures (NCTC) at Public Health England, where they are held today.
Use of the Collection to provide historical context has already yielded important insights regarding the state-of-play of enteric pathogens in the first half of the 20th century, and phenotypic shifts that have occurred since those times. Seminal work by scientists who coordinated the international transfer of the Collection showed that the machinery for the accumulation and plasmid-borne transfer of AMR (e.g. Incompatibility group types) [11, 14], were qualitatively similar to those of modern isolates, and this was also demonstrated for mercury resistance and Salmonella virulence determinants [15–17]. Other studies have demonstrated significant phenotypic shifts, including increased virulence and resistance to antimicrobials and antiseptics in Klebsiella sp. [18], and an increase in the magnitude and incidence of AMR in modern Escherichia isolates [19]. These studies however, merely exemplify the potential of the Collection. For example, its use to inform pathogen evolution through dating analyses remains entirely untapped, and enormous scope exists to further study the emergence and evolution of the pathogens, and their AMR and other traits.
In fact, the scale of the remaining work requires the coordinated expertise and effort of multiple microbiological research groups. Here, to serve that purpose, we announce the public release of the Murray collection isolates through formal accession of the 683 strains into the NCTC and provide the associated metadata. In addition to facilitating access to the physical strains, we verify the metadata by bacterial subtyping and analysis of whole genome sequencing data (also released here) generated for 370 of the strains. Finally, we present preliminary phylogenetic and gene content analyses that will aid strain selection for future scientific studies.
Collection composition and associated metadata
The Murray collection (as held by the NCTC) comprises 683 bacterial strains belonging to 447 equivalence groups (Table 1). Equivalence groups (Additional file 1: Table S2) included strains that were related in one of the following three ways: duplicate strains in the original collection with the same name and original date; colony variants detected during subculture in Canada before transfer to the UK; or derivatives (colony variants detected during receipt of strains at NCTC). The isolates were primarily Salmonella, Escherichia and Shigella (which are combined here), Klebsiella and Proteus (Table 1), and fell into variably diverse subgroups e.g. subspecies, serotypes beyond those designations (see Additional file 1: Table S2; Additional file 2: Figure S1; Additional file 3: Figure S2; Additional file 4: Figure S3 and Additional file 5: Figure S4). Bacteria outside of these four main genera (see Other, Table 1) were originally poorly designated e.g. coliform, Enterobacteriaceae, and were subsequently determined (see ‘Confirming the collection’ below) to belong to the main genera, or the following: Morganella, Rauotella, Aeromonas and Enterobacter (Table 2, Additional file 1: Table S2).
Table 1.
Summary of the collection contents by genus and time
| Collection | Sequenced | |||||
|---|---|---|---|---|---|---|
| Total strains | Unique inc. equivalence groups | Years of isolation | Total strains | Unique inc. equivalence groups | Years of isolation | |
| Salmonella | 361 | 222 | 1917 - 1952 | 174 | 127 | 1917 - 1946 |
| E. coli/Shigella | 256 | 174 | 1917 - 1954 | 140 | 121 | 1917 - 1954 |
| Klebsiella | 42 | 30 | 1920 - 1949 | 35 | 26 | 1920 - 1949 |
| Proteus | 18 | 16 | 1919 - 1940 | 14 | 12 | 1919 - 1940 |
| Other sp. | 6 | 6 | 1920 - 1940 | 7 | 6 | 1935 - 1940 |
| Total | 683 | 447a | 370 | 291a | ||
aThese totals affected by an equivalence group containing both Klebsiella (M45) and Escherchia/Shigella (M162). inc. - including
Table 2.
Assembly characteristics of the sequenced Murray collection isolates
| Assembly characteristics [mean (range)] | |||||
|---|---|---|---|---|---|
| Genus | No. | GC content | Total length (bp) | Contigs | N50 (bp) |
| Salmonella | 174 | 52 | 4739744 | 44 | 316870 |
| (51–52) | (4450735–5136048) | (15–126) | (70209–992086) | ||
| Escherichia/Shigella | 140 | 50 | 4679816 | 258 | 64933 |
| (50–51) | (3820214–5434207) | (63–567) | (14204–369379) | ||
| Klebsiella | 35 | 56 | 5287110 | 172 | 117718 |
| (55–57) | (4980231–5582843) | (24–286) | (58784–465957) | ||
| Proteus | 14 | 39 | 3935672 | 35 | 313856 |
| (38–39) | (3823752–3991064) | (18–58) | (201904–763476) | ||
| Morganella | 3 | 51 | 3842744 | 23 | 557210 |
| (NA) | (3744830–3948322) | (19–29) | (403231–664661) | ||
| Enterobacter | 2 | 54 | 5364204 | 58 | 341570 |
| (NA) | (5291805–5436603 ) | (52–64) | (341563–341 576) | ||
| Aeromonas | 1 | 59 | 4494408 | 39 | 166907 |
| Raoultella | 1 | 55 | 5488300 | 33 | 336936 |
The demographic features (e.g. place, person, time) and clinical details of pathogen infection are often crucial in the interpretation of genotypic and phenotypic analyses on the isolated pathogen. Although many of these details are available for the Collection strains, this metadata is incomplete and somewhat imperfect. The diverse geographical origins of the collection “including Europe, Malta, the Middle East, northern Russia, India and North America” has been reported [11], but were not available for individual strains. Metadata held at the NCTC showed the strains originated from diverse clinical specimens, e.g. stool, urine, blood, antral washes, cerebrospinal fluid, but the clinical syndrome, e.g. meningitis, pneumonia, hepatitis and cholecystitis, or patient/supplier name were also alternatively recorded (Additional file 1: Table S2). This ‘Origin’ information was only available for approximately one quarter (n = 150) of the strains. Contrastingly however, the large majority (92 %, 628 of 683) of strains had a date or year noted on the original vial (Additional file 1: Table S2). When these dates were stratified by genus, a unique time signature emerged, perhaps reflecting E.G.D. Murray’s changing research interests over time (Fig. 1a). Notably, these dates were presumed to be the date of isolation for the strains, but could also represent date of strain receipt, or some other event. Overall however, the novel analyses presented in this study largely support the original metadata demonstrating that it is, if imperfect, robust.
Fig. 1.
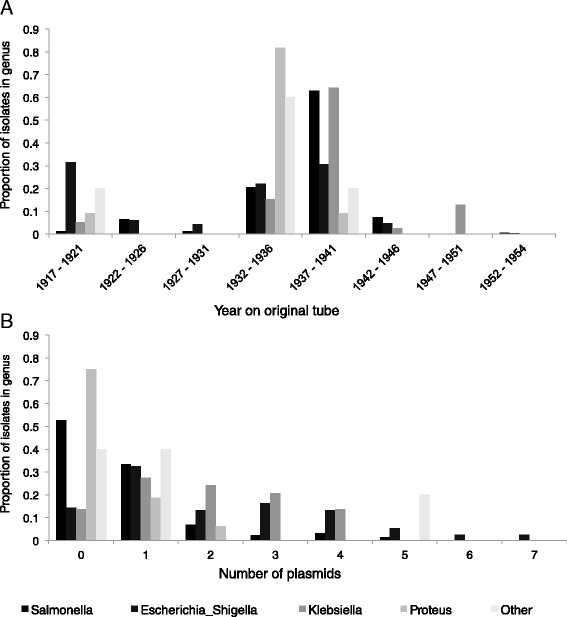
Metadata available for the Collection strains by genus, including year on original vial (a) and number of plasmids (b)
In addition to the published studies on conjugative plasmids that highlighted the importance of the collection for studying mobilisable-AMR [11, 14], efforts to comprehensively determine the full plasmid content of the collection were made in the late 1980s [20]. Using traditional plasmid preparation and gel electrophoresis techniques, this work determined the number and approximate sizes of plasmids contained in each of 489 Collection strain subcultures (from [14]). The findings showed that the strains contained between zero and seven plasmids each, and that certain genera contained more plasmids than others (Fig. 1b, full results reproduced in Additional file 1: Table S2). Plasmids ranged in estimated molecular weight from 1 to 500 Md (though estimates ≥ 150Md were noted as likely to be inaccurate). Attempts to verify this plasmid content metadata among 271 strains that were also whole genome sequenced were made (see Additional file 6: Supplementary Material).
Confirming the collection
In order to confirm the genus designations in the Collection, modern laboratory and in silico tools were applied to a subset of strains. The subset included all ACPD Hazard Group 2 (HG2) organisms and excluded most known HG3 organisms (23 HG3 organisms were included), thereby excluding known Shigella dysenteriae and Salmonella enterica where the serovar was unknown (see Additional file 1: Table S2). Of the total 683 isolates, 359 underwent MALDI-TOF analysis (of which 354 also underwent characterisation by 16 s rRNA sequencing). Outside of the ‘Other’ genera discussed above (and see Table 1), the MALDI-TOF results were generally concordant, with the exception of three isolates (M108, M162, M144) originally designated as Klebsiella that were determined to be Escherichia/Shigella sp., and the misidentification of a Salmonella isolate (M179) as an Escherichia by 16 s rRNA sequencing (Additional file 1: Table S2). Of the isolates that underwent MALDI-TOF analysis, 334 progressed to whole genome sequencing, alongside an additional 36 isolates not characterised by MALDI-TOF. Those revived isolates originally designated to be shigellae also underwent serotyping, and were largely confirmed (for 66 of 72 strains) to be either S. flexneri or S. sonnei as originally designated (Additional file 1: Table S2). Genus identification and in silico multi-locus sequence typing on whole genome sequencing data (Additional file 1: Tables S2 and Additional file 7: Table S3) confirmed the MALDI-TOF designation, or the original genus designation in all cases.
Genomic analysis of the Murray collection
To verify the robustness of the Collection, as well as add value, provide further metadata, and facilitate the development of selection criteria for ongoing studies, 370 strains (representing 291 equivalence groups), mostly representative of the collection (Tables 1, 2, Additional file 1: Table S2 and Additional file 7: Table S3) were whole genome sequenced. Some analyses of these genomes are briefly reported here, and more detail is given in the Additional file 6: Supplementary Material.
Fig. 2.
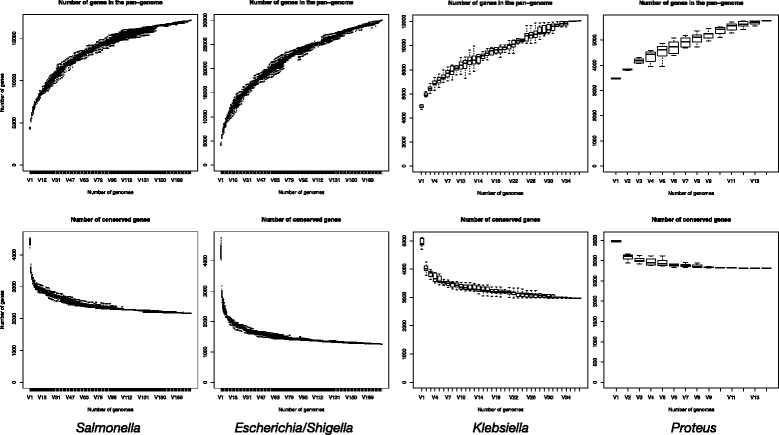
Rarefaction curves for pan- (above) and core- (below) genome sizes by genus
De novo assemblies created to facilitate core genome identification exemplified the unique genomic characteristics of each bacterial genus (Table 2, see Additional file 7: Table S3 for full results), which were similarly reflected in features of the core genomes including the discovery rate and final number and size of the core genome (Table 3, Fig. 2). For example, the Proteus had a lower GC content than the other genera (Table 2) and Salmonella strains had a larger core genome (Table 3) than Escherichia/Shigella, which had a larger accessory genome (Fig. 2).
Table 3.
Core genome size for the main genera in the Collection
| Salmonella | Escherichia/Shigella | Klebsiella | Proteus | |
|---|---|---|---|---|
| Total isolates (inc. refs) | 185 | 185 | 37 | 14 |
| Core genes (≥95 % isolates) | 3002 | 1983 | 3296 | 2870 |
| Core genes (100 % isolates) | 2159 | 1255 | 2966 | 2813 |
| Core genome (CG) length (bp) | 2195115 | 1381269 | 2881098 | 2775840 |
| CG variant sites (bp) | 136888 | 114723 | 64138 | 47079 |
To provide enhanced subgrouping information, core genome phylogenies were constructed from the variant sites in core genes for the main genera (Additional file 2: Figure S1; Additional file 3: Figure S2; Additional file 4: Figure S3 and Additional file 5: Figure S4). In addition to providing context for future strain selection, core genome phylogenies were used to verify the designation of equivalence groups within the Collection.
Antimicrobial resistance
Although no phenotypic studies of AMR were done here, AMR has been reported in the pre-antibiotic era Murray Collection strains, including tetracycline resistance in Proteus sp., ampicillin resistance in the Klebsiella and both ampicillin and kanamycin resistance in Escherichia sp. [11, 18, 19]. To aid the future selection of isolates based on the potential presence and absence of AMR, the presence of antimicrobial resistance genes among the strains was determined (Additional file 8: Table S1). This revealed many resistance genes (often known to be chromosomally encoded) that were present across all members of a genus, particularly across Salmonella, Escherichia/Shigella and Klebsiella whose profiles differed greatly, though unsurprisingly, from the more phylogenetically remote Proteus. Some genes however were differentially present among the genera with differing degrees of correlation to population structure (Additional file 8: Table S1, Fig. 3). For example, the tetC gene was present in nearly all Klebsiella isolates, but only a fraction of Escherichia/Shigella and Salmonella isolates, highlighting the potential of the Collection for studying the early horizontal transmission of AMR among Enterobacteriaceae.
Fig. 3.
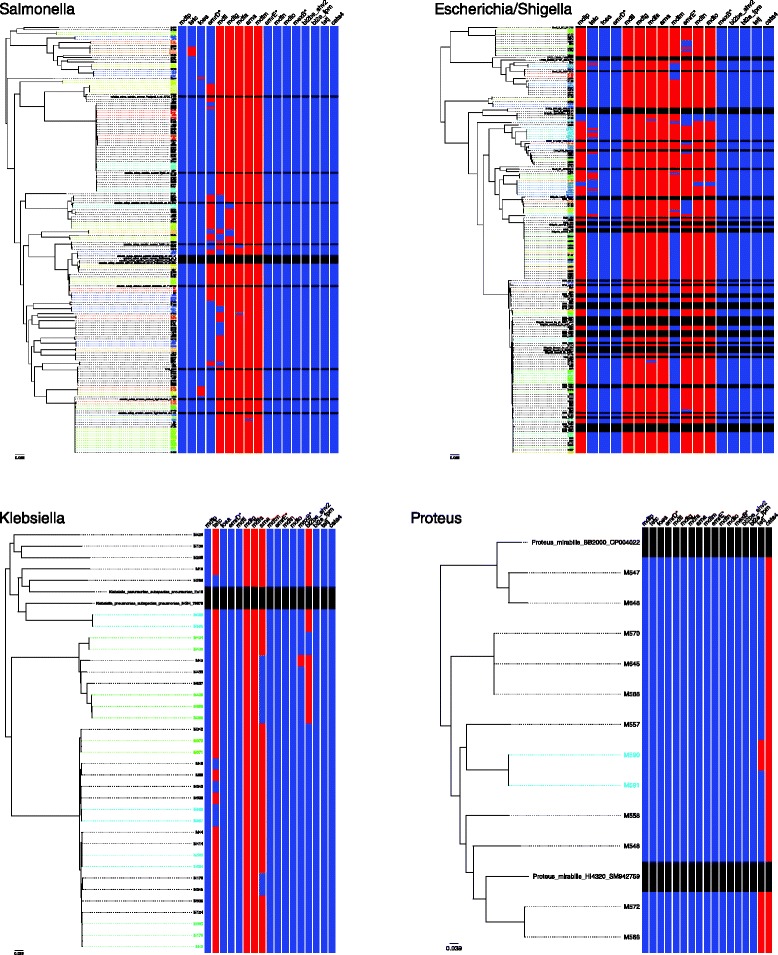
Presence (red) and absence (blue) of variably present antimicrobial resistance genes among the Collections strains overlaid adjacent to core genome phylogenies for each genus. The presence of genes in reference isolates was not determined (black)
Summary
This study comprehensively describes a large collection of diverse bacteria (primarily Enterobacteriaceae) from the pre-antibiotic era, now publicly available from the NCTC, and thus represents an invaluable resource for studying the evolution and emergence of AMR and Enterobacteriaceae. We also created a significant genomic resource for the scientific community in the form of freely available whole genome sequencing data for over half of the strains in the Collection. Using this data, we verified much of the metadata of the Collection including species identification, plasmid content and the existence of equivalence groups among the strains. Finally, we presented additional analyses to guide future scientific studies; defining the phylogenetic subgroups and genetic determinants of mobilisable AMR present in the Collection. The availability of these live isolates, associated sequencing data and preliminary analysis to the scientific community will surely spark a spate of studies into the evolution and epidemiology of these pathogens and their antimicrobial resistances.
Availability of supporting data
The strains in the collection are available at the NCTC under the Murray Collection Identifiers, and accession numbers shown in Additional file 8: Table S1. The whole genome sequencing data is available at the European Nucleotide Archive at (http://www.ebi.ac.uk/ena/data/view/PRJEB3255), according to the strain-specific accession numbers shown in Additional file 1: Table S2.
Additional files
Original Collection metadata and laboratory determination of plasmid content and species. (XLSX 112 kb)
Core genome phylogenetic tree for Salmonella sp. The tree is mid-point rooted. Strains noted to be in equivalence groups are similarly coloured. (PDF 39 kb)
Core genome phylogenetic tree for Escherichia/Shigella sp. The tree is mid-point rooted. Reference genomes representing previously published phylogroups are indicated. Strains noted to be in equivalence groups are similarly coloured. (PDF 40 kb)
Core genome phylogenetic tree for Klebsiella sp. The tree is mid-point rooted. Strains noted to be in equivalence groups are similarly coloured. (PDF 24 kb)
Core genome phylogenetic tree for Proteus sp. The tree is mid-point rooted. Strains noted to be in equivalence groups are similarly coloured. (PDF 23 kb)
Table S4. Selected references for each genus and species. Figure S5. Number of plasmids detected in Collection strains by laboratory and in silico approaches. Marker size is scaled by the number of strains and the trendline represents the overall correlation. (ZIP 175 kb)
Sequencing, assembly and gene content analyses for strains sequenced for this study. (XLSX 146 kb)
Antimicrobial resistance genes in sequenced strains by genus. (DOCX 66 kb)
Acknowledgements
The authors thank David Harris and the WTSI sequencing teams for coordination of sample sequencing, and Philippa Bracegirdle and Steven Brimble for assistance in designing initial work flows at PHE. WTSI authors were funded by grant number 980561. CB and AC are supported by MRC grant G1100100/1. KSB is in receipt of a Wellcome Trust Postdoctoral Training Fellowship for Clinicians (106690/Z/14/Z). The authors are also grateful to Vicki Hughes, Naomi Datta, Tegid Matthews, Peter Sneath, Laurence Rowland Hill, Rita Legros, and R.E.G. Murray who coordinated the transfer of the strains to the NCTC and performed the laboratory analysis of plasmid content.
Abbreviations
- AMR
Antimicrobial resistant/resistance
- CG
Core Genome
- GC
Guanine-Cytosine
- MALDI-TOF
Matrix Assisted Laser Desorption Ionisation - Time of Flight
- MLST
Multi-Locus Sequence Typing
- NCTC
National Collection of Type Cultures
- rRNA
ribosomal Ribose Nucleic Acid
- UK
United Kingdom
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KSB collated metadata, performed the whole genome sequence data analysis and drafted manuscript. EB and HMcG recovered strains, performed identification tests and prepared lysates for sequencing. ADG recovered and prepared lysates of HG3 isolates. CB and GL selected references and provided helpful discussions on the manuscript. AW and AC performed in silico plasmid analysis. NRT provided helpful discussions on the manuscript. JP and JER conceived study and facilitated and guided work performed at the WTSI and NCTC respectively. All co-authors contributed to manuscript writing and read and approved the final manuscript.
References
- 1.Organisation WH. WHO Global strategy for containment of antimicrobial resistance. Switzerland: World Health Organisation; 2001. [Google Scholar]
- 2.CfDCa P. Antibiotic resistance threats in the United States, 2013. Atlanta, USA: US Department of Health and Human Services; 2013. [Google Scholar]
- 3.Mortality GBD, Causes of Death C Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385:117–171. doi: 10.1016/S0140-6736(14)61682-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicolas-Chanoine MH, Blanco J, Leflon-Guibout V, Demarty R, Alonso MP, Canica MM, Park YJ, Lavigne JP, Pitout J, Johnson JR. Intercontinental emergence of Escherichia coli clone O25:H4-ST131 producing CTX-M-15. J Antimicrob Chemother. 2008;61:273–281. doi: 10.1093/jac/dkm464. [DOI] [PubMed] [Google Scholar]
- 5.Walsh TR. Emerging carbapenemases: a global perspective. Int J Antimicrob Agents. 2010;36:S8–S14. doi: 10.1016/S0924-8579(10)70004-2. [DOI] [PubMed] [Google Scholar]
- 6.Bos KI, Harkins KM, Herbig A, Coscolla M, Weber N, Comas I, Forrest SA, Bryant JM, Harris SR, Schuenemann VJ, et al. Pre-Columbian mycobacterial genomes reveal seals as a source of New World human tuberculosis. Nature. 2014;514:494–497. doi: 10.1038/nature13591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wagner DM, Klunk J, Harbeck M, Devault A, Waglechner N, Sahl JW, Enk J, Birdsell DN, Kuch M, Lumibao C, et al. Yersinia pestis and the plague of Justinian 541–543 AD: a genomic analysis. Lancet Infect Dis. 2014;14:319–326. doi: 10.1016/S1473-3099(13)70323-2. [DOI] [PubMed] [Google Scholar]
- 8.Schuenemann VJ, Singh P, Mendum TA, Krause-Kyora B, Jager G, Bos KI, Herbig A, Economou C, Benjak A, Busso P, et al. Genome-wide comparison of medieval and modern Mycobacterium leprae. Science. 2013;341:179–183. doi: 10.1126/science.1238286. [DOI] [PubMed] [Google Scholar]
- 9.Baker KS, Mather AE, McGregor H, Coupland P, Langridge GC, Day M, Deheer-Graham A, Parkhill J, Russell JE, Thomson NR. The extant World War 1 dysentery bacillus NCTC1: a genomic analysis. Lancet. 2014;384:1691–1697. doi: 10.1016/S0140-6736(14)61789-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Devault AM, Golding GB, Waglechner N, Enk JM, Kuch M, Tien JH, Shi M, Fisman DN, Dhody AN, Forrest S, et al. Second-pandemic strain of Vibrio cholerae from the Philadelphia cholera outbreak of 1849. N Engl J Med. 2014;370:334–340. doi: 10.1056/NEJMoa1308663. [DOI] [PubMed] [Google Scholar]
- 11.Hughes VM, Datta N. Conjugative plasmids in bacteria of the ‘pre-antibiotic’ era. Nature. 1983;302:725–726. doi: 10.1038/302725a0. [DOI] [PubMed] [Google Scholar]
- 12.Collip JB. Professor E. G. D. Murray: An Appreciation. Journal of the Canadian Medical Association. 1965;92:95–96. [PMC free article] [PubMed] [Google Scholar]
- 13.Murray RGE. More on bacterial longevity: The Murray Collection. American Society for Microbiology News. 1985;51:261–262. [Google Scholar]
- 14.Datta N, Hughes VM. Plasmids of the same Inc groups in Enterobacteria before and after the medical use of antibiotics. Nature. 1983;306:616–617. doi: 10.1038/306616a0. [DOI] [PubMed] [Google Scholar]
- 15.Essa AM, Julian DJ, Kidd SP, Brown NL, Hobman JL. Mercury resistance determinants related to Tn21, Tn1696, and Tn5053 in enterobacteria from the preantibiotic era. Antimicrob Agents Chemother. 2003;47:1115–1119. doi: 10.1128/AAC.47.3.1115-1119.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones CS, Osborne DJ. Identification of contemporary plasmid virulence genes in ancestral isolates of Salmonella enteritidis and Salmonella typhimurium. FEMS Microbiol Lett. 1991;64:7–11. doi: 10.1111/j.1574-6968.1991.tb04627.x. [DOI] [PubMed] [Google Scholar]
- 17.Jones C, Stanley J. Salmonella plasmids of the pre-antibiotic era. J Gen Microbiol. 1992;138:189–197. doi: 10.1099/00221287-138-1-189. [DOI] [PubMed] [Google Scholar]
- 18.Wand ME, Baker KS, Benthall G, McGregor H, McCowen JW, Deheer-Graham A, Sutton JM. Characterisation of pre-antibiotic era Klebsiella pneumoniae isolates with respect to antibiotic/disinfectant susceptibility and virulence in Galleria mellonella. Antimicrob Agents Chemother. 2015;59:3966–3972. doi: 10.1128/AAC.05009-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Houndt T, Ochman H. Long-term shifts in patterns of antibiotic resistance in enteric bacteria. Appl Environ Microbiol. 2000;66:5406–5409. doi: 10.1128/AEM.66.12.5406-5409.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matthews T. Plasmid Populations of pre-antibiotic era Enterobacteriaceae. Bristol Polytechnic, Plasmid section of the National Collection of Type Cultures; Porton Down, UK. 1987.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Original Collection metadata and laboratory determination of plasmid content and species. (XLSX 112 kb)
Core genome phylogenetic tree for Salmonella sp. The tree is mid-point rooted. Strains noted to be in equivalence groups are similarly coloured. (PDF 39 kb)
Core genome phylogenetic tree for Escherichia/Shigella sp. The tree is mid-point rooted. Reference genomes representing previously published phylogroups are indicated. Strains noted to be in equivalence groups are similarly coloured. (PDF 40 kb)
Core genome phylogenetic tree for Klebsiella sp. The tree is mid-point rooted. Strains noted to be in equivalence groups are similarly coloured. (PDF 24 kb)
Core genome phylogenetic tree for Proteus sp. The tree is mid-point rooted. Strains noted to be in equivalence groups are similarly coloured. (PDF 23 kb)
Table S4. Selected references for each genus and species. Figure S5. Number of plasmids detected in Collection strains by laboratory and in silico approaches. Marker size is scaled by the number of strains and the trendline represents the overall correlation. (ZIP 175 kb)
Sequencing, assembly and gene content analyses for strains sequenced for this study. (XLSX 146 kb)
Antimicrobial resistance genes in sequenced strains by genus. (DOCX 66 kb)