

Abstract
HIV has been reported to induce podocyte injury through down regulation of vitamin D receptor (VDR) and activation of renin angiotensin system (RAS); however, the involved mechanism is not clear. Since HIV has been reported to modulate gene expression via epigenetic phenomena, we asked whether epigenetic factors are contributing to down regulation of VDR. Kidney cells in HIV transgenic mice as well as HIV-infected podocytes (HIV/HPs) displayed enhanced expression of SNAIL, a repressor of VDR. To elucidate the mechanism, we studied the effect of HIV on expression of molecules involved in SNAIL repressor complex formation and demonstrated that HIV enhancesexpression of histone deacetylase (HDAC) 1, DNA methyl transferase (DNMT) 3b and DNMT1. 293T cells, when stably transfected with SNAIL (SNAIL/293T), displayed suppressed transcription and translation of VDR. In SNAIL/293T cells, co-immunoprecipitation studies revealed the association of HDAC1, DNMT3b, DNMT1, and mSin3A with SNAIL. Chromatin immunoprecipitation (ChIP) experiments confirmed the presence of the SNAIL repressor complex at the VDR promoter. Consistent with the enhanced DNA methyl transferase expression in HIV/HPs, there was an increased CpG methylation at the VDR promoter. ChIP assay confirmed occurrence of H3K4 trimethylation on SNAIL promoter. Neither VDR agonist (VDA) nor an HDAC inhibitor (HDACI) nor demethylating agent (DAC) individually could optimally upregulate VDR in HIV milieu. However, VDA and HDACI when combined were successful in de-repressing VDR expression. Our findings demonstrate that SNAIL recruits multiple chromatin enzymes to form a repressor complex in HIV milieu which down regulates VDR expression.
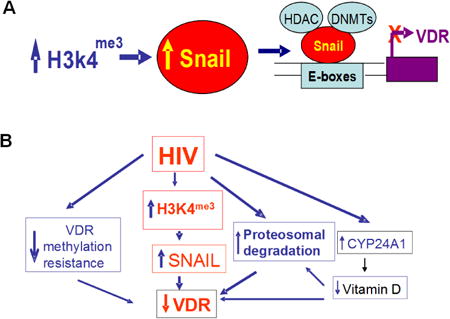
Emerging reports suggest the involvement of epigenetic mechanisms in the development of chronic kidney diseases, specifically in diabetic nephropathy (1, 2). Environmental factors associated with diabetic nephropathy such as hyperhomocysteinemia, dyslipidemia, inflammation, and oxidative stress are known to induce DNA methylation (3-5). Several of these factors are also present in patients with HIV-associated nephropathy (HIVAN) (6, 7); however, the role of epigenetic factors in the development of HIVAN has not been studied so far (8). The renin angiotensin system (RAS) has been shown to play important roles in the development and progression of HIVAN (9-13). For example, infusion of Ang II enhanced progression of HIVAN in an experimental mouse model of the disease (9). Conversely, blockade and inhibition of Ang II production slowed down HIVAN progression both in human and animal models (10-12). Similarly, enhanced renin production accelerated progression of renal lesions, whereas inhibition of renin attenuated progression of renal lesions in Vpr transgenic mice (an HIVAN model) (13). Recent reports indicate that lack of VDR in the genome of T cells is associated with the activation of the RAS (14). HIV has also been reported to down regulate VDR in T cells as well as in kidney cells including both podocytes and tubular cells (15-17). Moreover, HIV induced down regulation of VDR in T cells as well as in kidney cells was associated with the activation of the RAS (7, 15-17). However, the involved mechanism in HIV-induced down regulation of VDR is not clear. We hypothesized that HIV down regulates VDR in podocytes through epigenetic modulation.
VDR, a nuclear receptor, is degraded when not bound with its ligand, active vitamin D3 (1-α 25 [OH)2D3) or a VDR agonist (VDA) (18, 19). Vitamin Ds are fat-soluble secosteroids which participate in an intestinal absorption of calcium, iron, magnesium, phosphate and zinc (20). The most important compounds in this group are vitamin D3 (cholecalciferol) and vitamin D2 (ergocalciferol). Sun exposure to skin and nutrition are important source of cholecalciferol, however, its activation requires enzymatic conversion in the liver and kidney. In the liver, cholecalciferol is converted to calcidiol, (25-hydroxyvitamin D3, abbreviated as 25[OH[D3). In the kidney, calcidiol is converted to active vitamin D3 (1-α 25 [OH]2D3) by CYP27B1 which then degraded by CYP24A1 (21). TO regulate its concentration, active vitamin D3 enhances expression of CYP24A1 and downregulates expression of CYP27A1(22). VDR depending on the presence or absence of its ligand (active vitamin D3) forms either repressor (unliganded VDR) or activator (liganded VDR) complex (23). Both unliganded and liganded VDR serve different function and play a role in the maintenance cellular integrity (23). Liganded VDR has been suggested to be a negative endocrine regulator of transcription of renin (24). On that account, VDAs are currently used as a therapeutic strategy to decrease proteinuria in human and experimental animal models of kidney diseases (25-27).
VDR null mice have been demonstrated to display activated RAS (26), which has been implicated in the development of hypertension and cardiac hypertrophy in this model (24). Similarly, down regulation of VDR has been demonstrated to contribute to the progression of renal lesions in animal experimental models of diabetic nephropathy and obstructive nephropathy (26, 27). In these models, the restoration of VDR and associated down regulation of the RAS through the administration vitamin D, partially ameliorated the progression of renal lesions.
HIV-induced generation of reactive oxygen species (ROS) and associated DNA damage have been attributed to the down regulation of tubular cell VDR expression (17); conversely, restoration of VDR expression provided partial protection against HIV-induced tubular cell DNA damage. Similarly, HIV-induced down regulation of VDR expression was associated with ROS generation, lysosomal cathepsin-L leak and loss of integrity of podocyte actin cytoskeleton (16); whereas, restoration of VDR expression provided protection. These studies indicated that VDR expression is critical for the maintenance of kidney cell phenotype in HIV milieu.
Oxidative stress leads to cellular as well as DNA damage, and has the potential to induce epigenetic changes (28-36). Epigenetic mechanisms including DNA methylation, histone modifications, and non-coding RNAs alter genome function without changing the gene sequences (36). DNA methylation usually occurs in cytosine at consecutive Cytosine phosphate Guanine (CpG) dinucleotides (32-35). Since 5-methylcytosine residues are spontaneously deaminated and are prone to mutate, methylated CpG sites are only found at CpG islands (36). On that account, these epigenetic changes have the potential to turn into permanent DNA methylation patterns. Environmental factors contribute to these epigenetic changes via interactions of DNA methyltransferases (DNMT), such as DNMT1, DNMT3A, and DNMT3B (37). HIV has been shown to down regulate VDR expression in T cells through up regulation of DNMT3B (38). We propose that HIV-induced VDR methylation may also be contributing to down regulation of VDR expression in podocytes.
A human cell sustains DNA damages approximately 10,000 times each day; and although most are repaired, epigenetic alterations persist (34). For example, a double strand break in DNA has potential to embark silencing of a gene either via DNA methylation or by enhancing silencing types of histone modifications (35). These alterations modulate chromatin condensation status, a determinant of genetic transcription. Chromatin needs to be loosened for transcription to occur and tightened to end transcription. Modulation of chromatin condensation can be attained by methylation of histones on specific sites via histone methyl transferases which transfer methyl groups to the lysine or arginine residues of the H3 and H4 histones. Methylated histones carry potential to either repress or activate transcription (39). Usually, H3K4 me2/3 and H3K7/9me3 interaction at the promoter enhance transcriptional activity, whereas methylation of H3K9me2/3, H3K27me2/3, and H4K20me3 lead to repression (39). We hypothesized that HIV could be enhancing podocyte SNAIL expression via trimethylation of histone-3 at 4 lysine residue (H3K4me3). Since SNAIL is a repressor of transcription of VDR (40), HIV-induced up regulation of SNAIL will lead to down regulation of podocyte VDR expression.
In the present study, we evaluated the effect of HIV on DNA methylation and histone modification (methylation and acetylation) in podocytes and renal tissues of HIV transgenic mice. Additionally, we have studied the effects of a VDR agonist (VDA), a demethylating agent (DAC), and an HDAC inhibitor (HDACI) alone or in combination on HIV-modulated podocyte VDR expression.
Results
Kidney cells in HIVAN mice display enhanced expression of SNAIL and mitigated expression of VDR
To determine the kidney cell SNAIL expression in HIVAN, renal cortical sections of 4 week old FVB/N and Tg26 mice (n=4) were immunolabeled and evaluated for either SNAIL or VDR. Representative microphotographs of control and Tg26 mice are shown in Figs. 1A and 1B. Renal cortical section of FVB/N mice displayed a minimal expression of SNAIL by podocytes; on the other hand, podocytes displayed robust expression of SNAIL in the renal cortex of a Tg26 mouse (Fig. 1A). Renal cortical sections of FVB/N displayed moderate expression of VDR in podocytes, whereas, podocytes displayed attenuated expression of VDR in a Tg26 mouse (Fig. 1B).
Fig. 1. Podocytes display enhanced expression of SNAIL but mitigated expression of VDR in HIVAN.
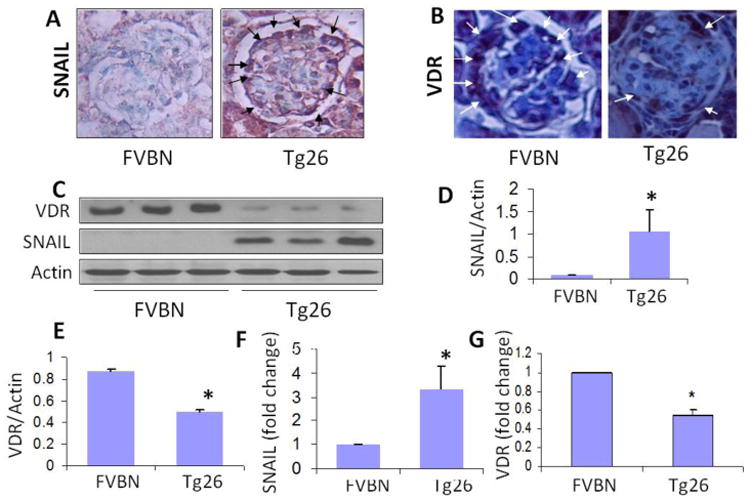
- Representative microphotographs of renal cortical sections displayed minimal expression of SNAIL by podocytes in a FVBN mouse; whereas, podocytes in a glomerulus from a Tg26 mouse displayed moderate expression of SNAIL (indicated by arrows)
- Representative microphotographs of renal cortical section of a FVB/N mouse displayed a robust expression of VDR by podocytes (indicated by arrows); on the other hand, podocytes in a glomerulus from a Tg26 mouse displayed minimal expression of VDR.
- Protein blots from renal tissues of four FVBN and Tg26 mice were probed for SNAIL and the same blots were reprobed for VDR and actin (n=4). Gels from the tissue lysates from three mice are displayed.
- Cumulative densitometric analysis of SNAIL/Actin (n=4) of the protocol C, are shown in bar graphs. *P<0.05 vs. FVBN.
- Cumulative densitometric analysis of VDR/Actin (n=3) of the protocol C, are shown in bar graphs. *P<0.05 vs. FVBN.
- RNAs were extracted from renal tissues of FVBN and Tg26 mice (n=3). cDNA were amplified with primer for Snail.*P<0.05
- RNA were extracted from renal tissues of FVBN and Tg26 mice (n=3). cDNA were amplified with primer for Vdr. Cumulative data are shown in bar graphs. *P<0.05 vs. FVBN.
To quantify kidney cell SNAIL expression in HIVAN, proteins were extracted from renal tissues of 4 week old FVB/N and Tg26 mice (n=4) and protein blots were probed for SNAIL and VDR. Gels of renal tissues of three Tg26 mice are displayed (Fig. 1C). Tg26 mice displayed enhanced expression of SNAIL when compared to respective renal tissues of FVB/N mice. Cumulative density analysis (n=4) are displayed in bar graphs (Fig. 1D). Tg26 mice displayed sixfold increase (P<0.05 vs. FVBN) in SNAIL expression. On the other hand, renal tissues of Tg26 mice displayed down regulation of VDR (Fig. 1C). Cumulative densitometric data are shown as bar graphs (Fig.1E).
To determine mRNA expression of Snail in FVBN and Tg26, RNAs were extracted and cDNAs were amplified with specific primer for Snail. Renal tissues of Tg26 mice showed 3-fold increase in Snail mRNA expression (Fig 1F). To determine whether HIV-induced SNAIL upregulation modulates renal Vdr transcription, RNA was extracted from renal tissues of FVBN and Tg26 mice (n=3). cDNA was amplified with specific primer for Vdr. As shown in Fig.1G, renal tissues of Tg26 mice displayed attenuated mRNA expression of Vdr.
HIV enhances SNAIL expression but attenuates VDR expression in podocytes (in vitro studies)
To determine the dose response effect of HIV on podocyte SNAIL and VDR expressions, podocytes were transduced with variable units of HIV (0, 50, 100, 150, and 200 GEU). Protein blots were probed for SNAIL and after stripping, reprobed for VDR and actin. HIV enhanced podocyte SNAIL expression but down regulated podocyte VDR expression in a dose dependent manner (Supplementary data, Fig S1).
To confirm the effect of HIV on podocyte SNAIL and VDR expression, protein blots of EV/HPs and HIV/HPs (n=3, different lysates) were probed for SNAIL and the same blots were reprobed for VDR and actin. Gels are displayed in Fig. 2A. HIV/HPs displayed enhanced expression of SNAIL when compared to EV/HPs. However, HIV/HPs displayed attenuated expression of VDR when compared to EV/HPs. Cumulative densitometric data on SNAIL/actin and VDR/Actin ratios are shown in Fig.2B and Fig. 2C respectively.
Fig. 2. HIV enhances SNAIL expression but attenuates VDR expression in podocytes (in vitro studies).
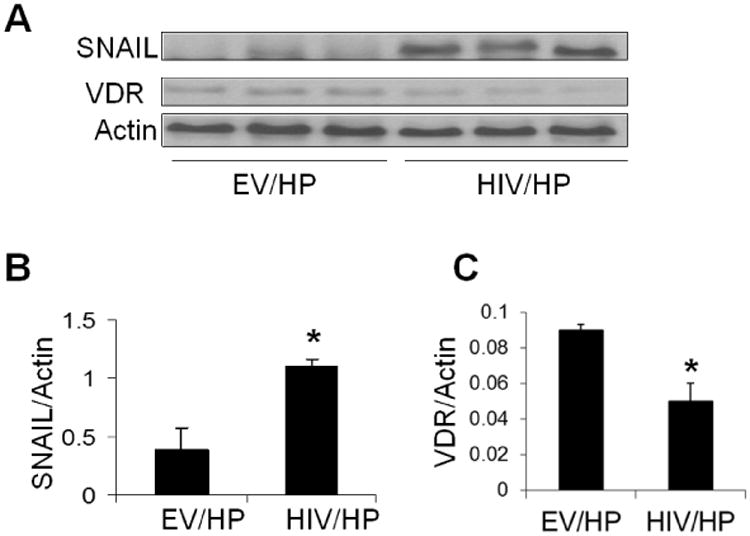
- Protein blots of EV/HPs and HIV/HPs (n=3), were probed for SNAIL and the same blots were reprobed for VDR and actin. Gels from three different lysates are displayed.
- Cumulative densitometric data of gels displayed in B (SNAIL/Actin ratio) are shown in a bar diagram. *P<0.01 vs. EV/HP.
- Cumulative densitometric data of gels displayed in B (VDR/Actin ratio) are shown in bar graphs. *P<0.05 vs. EV/HP.
Modulation of SNAIL expression is associated with altered podocyte VDR expression
EV/HPs were transfected either with siRNA-SNAIL or scrambled siRNA (SCR). Protein blots were probed for SNAIL. The same blots were reprobed for VDR and actin (Supplementary data, Fig. S2).
To determine whether SNAIL expression modulates VDR expression, protein blots of EVHPs, HIV/HPs, siRNA/EV/HPs and siRNA/HIV/HPs were probed for SNAIL and reprobed for VDR and actin (n=3). Representative gels are shown in Fig. 3A. Cumulative data (n=3) are shown as bar graphs in Fig. 3B. HIV enhanced SNAIL expression (Fig. 3A). HIV/HPs displayed down regulation of VDR when compared HIV/HPs silenced for SNAIL (P<0.05).
Fig. 3. SNAIL is critical for repression of VDR expression.
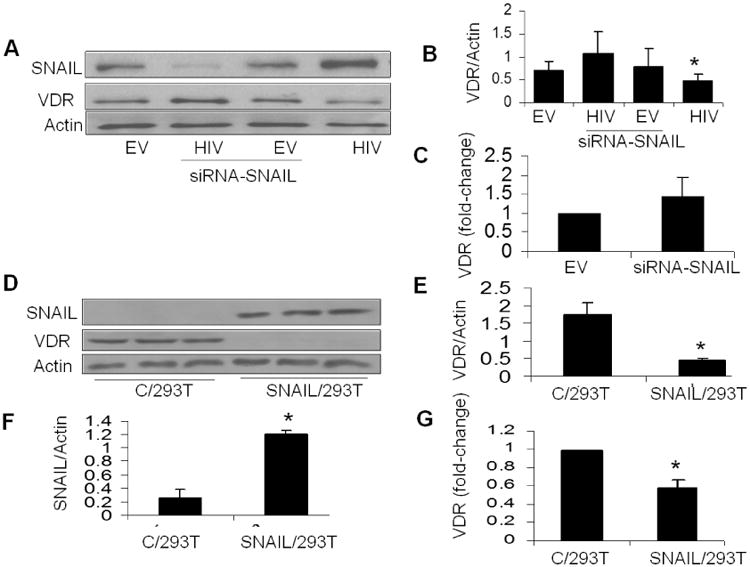
- Protein blots of EV/HPs, siRNA-SNAILEV/HPs, HIV/HPs and siRNA-SNAIL-HIV/HPs were probed for SNAIL and the same blots were reprobed for VDR and actin (n=3). Representative gels are displayed.
- Cumulative densitometry data of the gels displayed in B are shown. *P<0.05 compared to siRNA-SNAIL/HIV/HP
- RNAs were extracted from EV/HPs and siRNASNAIL/EV/HPs. cDNA were amplified with VDR specific primer and mRNA was quantified by real time PCR. Cumulative data (n=3) are displayed in bar graphs.
- Protein blots of C/293T and SNAIL/293T cells were probed for SNAIL and the same blots were reprobed for VDR and actin (n=3). Gels from three different samples are displayed.
- Cumulative densitometric data on VDR/actin ratios of gels shown in E. *P<0.01 vs C/293T.
- Cumulative densitometric data on SNAIL/actin ratios of gels shown in E are displayed as a bar diagram (n=3). *P<0.01 vs C/293T.
- RNA was extracted from control and SNAIL/293T cells and cDNAs were amplified with VDR specific primer and mRNA was quantified by real time PCR. Cumulative data (n=3) are displayed in bar graphs. *P<0.05
To determine the effect of silencing of SNAIL on VDR transcription, mRNA expression of VDR was determined in EV/HP and EV/HPs silenced for SNAIL. EV/HPs silenced for SNAIL did not show any alteration in VDR transcription (Fig. 3C). These findings indicate that only overexpressed SNAIL down regulates VDR expression. Silencing of overexpression of SNAIL in podocytes also induces reversal of repressed VDR.
C/293Ts were transfected with either SNAIL plasmid or pcDNA (control). Protein blots of c/293T, pcDNA/293T and SNAIL/293T were probed for SNAIL. Representative gels are shown (Supplementary data, Fig. S3).
Protein blots of control and SNAIL/293T cells were probed for SNAIL and the same blots were reprobed for VDR and actin (n=3). As shown in Fig. 3D, C/293T cell displayed VDR expression only (no SNAIL expression). SNAIL/293T cells displayed enhanced expression of SNAIL but decreased expression of VDR. Cumulative data on VDR/actin and SNAIL/actin ratios in control and SNAIL expressing 293T cells are shown in Figs. 3E and 3F, respectively. These findings confirm that SNAIL expression induces down regulation of VDR.
To confirm whether SNAIL represses the transcription of VDR, RNA was extracted from control and SNAIL/293T cells and cDNAs were amplified with VDR specific primers. As shown in Fig. 3G, SNAIL/293T displayed attenuated mRNA expression of VDR when compared to C/293T (control) cells.
HIV enhances trimethylation of histone H3 lysine 4 (H3K4me3) of kidney cells both in vitro and in vivo
Since trimethylation of histone H3 lysine 4 has been reported to enhance SNAIL expression in cancer cells (40), we asked whether HIV enhances trimethylation of histone H3 lysine 4 in podocytes. To determine the trimethylation status of H3 lysine 4 in EV/HPs and HIV/HP (n=3 different lysates), protein blots of EV and HIV/HPs were probed for H3K4me3 and re probed for actin. HIV/HPs displayed enhanced expression of H3K4me3 when compared to EV/HPs (Fig. 4A). Cumulative data are shown in bar graphs (Fig. 4B). Thus, it appears that enhanced trimethylation of histone H3 lysine 4 contributes to enhanced podocyte expression of SNAIL in HIV milieu.
Fig. 4. HIV enhances trimethylation of histone H3 lysine 4 (H3K4me3) of kidney cells at SNAIL promoter site.
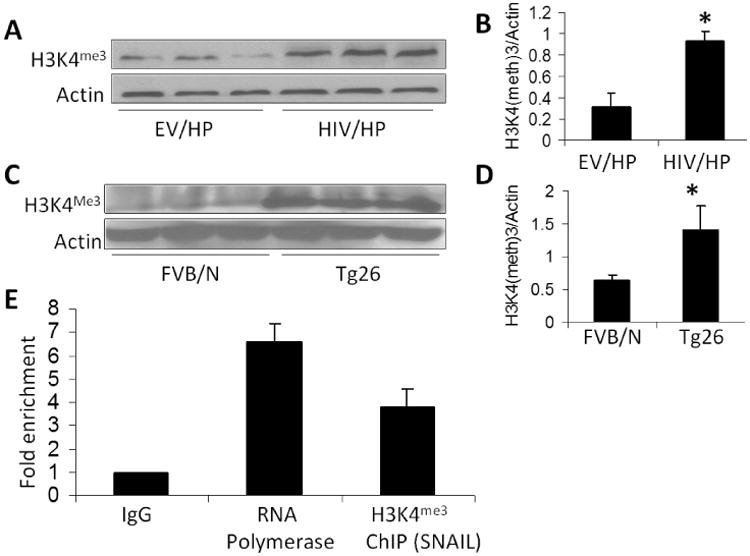
- Protein blots of EV and HIV/HPs (n=3) were probed for H3K4me3 and reprobed for actin. Gels from three different lysates are displayed.
- Cumulative densitometric analysis of gels displayed in A. *P<0.05 vs. EV/HP.
- Protein blots from renal tissues of FVBN and Tg26 mice (n=3) were probed for H3K4me3 and reprobed for actin. Gels from each mouse are displayed.
- Cumulative densitometric analysis of gels displayed in C. *P<0.05 vs. EV/HP.
- Chromatin from lysates of HIV/HPs was immunoprecipitated with either rabbit IgG (as a negative control), anti-RNA polymerase II antibody (as a positive control), or anti-H3K4me3. RNA was extracted and real time PCR was carried out for SNAIL-specific primer. ChIP data expressed as fold enrichment are shown in bar graphs.
To confirm whether HIV-induced trimethylation of histone H3 lysine 4 was also contributing to HIVAN phenotype in Tg26 mice, proteins were extracted from renal cortices of 4 week old FVB/N (n=3) and Tg26 (n=3) mice. Protein blots were probed for H3K4me3 and reprobed for actin. Renal tissues of Tg26 mice displayed enhanced expression of H3K4me3 when compared to FVB/N mice (Fig. 4C). Cumulative data are shown in bar graphs (Fig. 4D).
Since trimethylation of histone H3 lysine 4 is known to activate gene transcription (58, 59), it appears that HIV enhanced SNAIL expression via epigenetic mechanisms in kidney cells.
H3K4 trimethylation of SNAIL
Palmer et al previously described that histone methylation and acetylation was associated with SNAIL activation (40). To determine induction of H3K4 trimethylation of SNAIL, chromatin from lysates of HIV/HPs were immunoprecipitated with either IgG (as a negative control), anti-RNA polymerase II antibody (as a positive control), or anti-H3K4 trimethylation antibody. DNA was extracted and real time PCR was carried out for SNAIL promoter. Data of ChIP assay are shown in Fig 4E. HIV/HPs displayed H3K4 trimethylation of SNAIL.
HIV enhanced podocyte DNA methyl transferase production and also increases VDR DNA methylation
To evaluate whether HIV also enhanced podocyte DNMT3b protein expression, cellular lysates of EV/HPS (control) and HIV/HP (HIV) were subjected to electrophoresis and protein blots were probed for DNMT3b. Expression of DNMT3b and actin in two different cellular lysates of EV/HPs and HIV/HPs are shown in Fig. 5A. Cumulative densitometric data are also shown below. HIV/HPs displayed more than 2.0-fold increase (P<0.01) in DNA methylation specific enzyme.
Fig. 5. HIV enhances podocyte DNA methyl transferase production and increases VDR methylation.
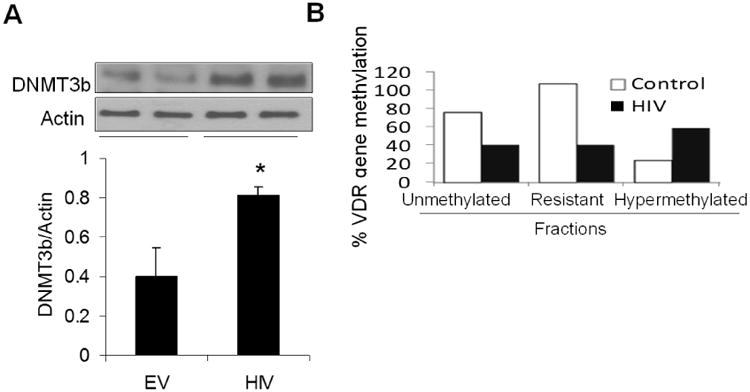
- Protein blots of EV/HPS (control) and HIV/HP (HIV) were probed for DNMT3b and reprobed for actin (n=7). Gels from two different lysates are displayed. Cumulative data (n=7) are shown in bar graphs.
- DNA was extracted from EV/HPs and HIV/HPs and CpG DNA methylation qPCR and methylation qPCR Assay with VDR specific primers were carried out. Methylated fraction was calculated by measuring unmethylated, hypermethylated and resistance to get methylated fractions. Cumulative data are shown in bar graphs. *P<0.05 vs. EV.
To determine CpG methylation status at the VDR gene, genomic DNA samples from control and experimental cells were prepared. Subsequently, VDR specific primers were used to determine the methylation status of CpG islands in VDR. Methylated fraction was calculated by measuring unmethylated, hypermethylated and resistance to methylation fraction (DNA copies resistant to enzyme digestion). As shown in Fig 5B, unmethylated as well as methylation resistant VDR fractions were lower (two-fold) in HIV/HPs when compared to EV/HPs. However, hypermethylated fraction was two-fold higher in HIV/HPs when compared to EV/HPs. Additionally, bisulfite sequencing of genomic DNA revealed partial methylation of CpG islands of podocytes in HIV milieu (Supplementary data, Fig. S4).
Both demethylating agent (DAC) and HDAC inhibitor (HDACI) augment VDA-induced podocyte VDR expression in HIV milieu
Since VDA was not able to enhance podocyte expression of VDR optimally in HIV milieu, we hypothesized that either DAC or HDACI would optimize the effect of VDA.
To evaluate the effect of empty vector on VDA-, HDACI-, and DAC-induced VDR expression, control HPs and EV/HPs were pretreated with either buffer or DAC (demethylating agent, DAC, 5 μM) for 48 hours at 33°C. In parallel sets of experiments, control HPS and EV/HPs were incubated in media containing either buffer, SAHA (10 μM, an HDACI), or VDA (1 nM) for 48 hours at 37°C. Protein blots were probed for VDR and actin (Supplementary data, Fig. S5), HDACI, VDA, and DAC enhanced expression of VDR both in control and EV/HPs. To determine the effect of VDA in the presence or absence of demethylating agents on HIV-induced podocyte VDR expression, EV/HPs and HIV/HPs were pretreated with either buffer or VDA (1nM) for 4 hours followed by treatment with azacytidine (demethylating agent, DAC, 5 μM) for 48 hours at 33°C. Protein blots were probed for VDR and reprobed for actin. HIV downregulated podocyte VDR expression (Fig. 6A). Although both VDA and DAC enhanced podocyte VDR expression under control conditions but neither VDA nor DAC could enhance podocyte VDR expression more than basal levels in HIV milieu.
Fig.6. Both demethylating agents and deacetylating agents (HDAC inhibitors) augment VDA-induced podocyte VDR upregulation in HIV milieu.
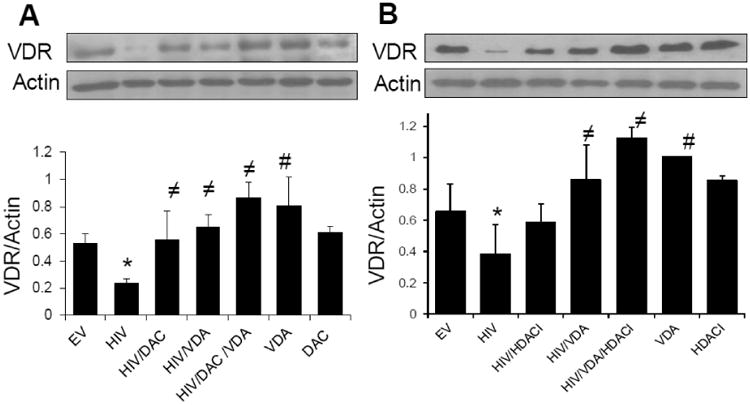
- EV/HPs and HIV/HPs were incubated in media containing either buffer, VDA (1 nM) for 4 h followed by treatment with or without azacytidine (DAC, 5 μM) for 48 hours at 33°C (n=3). Protein blots were probed for VDR and reprobed for actin. Representative gels are displayed. Cumulative data are shown in bar graphs. *P<0.05 compared to all other variables; ≠P<0.05 compared to HIV alone; #P<compared to EV.
- EV/HPs and HIV/HPs were pretreated with either buffer or HDACI (SAHA, 10 μM) for 2h followed incubation in media containing either buffer, VDA (1 nM) in the presence or absence of SAHA for 48 hours (n=3). Protein blots were probed for VDR and reprobed for actin. Representative gels are displayed. Cumulative data are shown in a bar diagram. *P<0.05 compared to EV; ≠P<0.05 compared to HIV alone; #P<0.05 compared to EV.
However, VDA in combination with DAC enhanced podocyte VDR expression in HIV milieu (Fig. 6A). Cumulative data (n=3) are shown in bar graphs. To determine the effect of HDAC inhibitors on VDA-induced podocyte VDR expression in HIV milieu, EV/HPs and HIV/HPs were incubated in media containing either buffer, VDA (EB1089, 1 nM) in the presence or absence of SAHA (HDAC inhibitor, HDACI, 10 μM) for 48 hours. Protein blots were probed for VDR, stripped and reprobed for actin. HIV downregulated podocyte VDR expression but this effect of HIV was partially inhibited both by VDA and SAHA. Only combined therapy displayed optimal podocyte VDR expression in HIV milieu (Fig. 6B). Cumulative data are shown as a bar diagram below (Fig. 6B).
HIV enhances transcription of molecules involved in SNAIL repressor complex
Since SNAIL forms a repressor complex contains mSin3A,HDAC1/2 and DNMTs and binds at VDR promoter site, RNA was extracted from EV/HPs and HIV/HPs and cDNAs were amplified with HDAC1, DNMT1, DNMT3b and SNAIL specific primers. As shown in Fig. 7A, HIV enhanced 3-fold in mRNA expression for HDAC1 and DNMT3b, 5-fold increase in mRNA expression for DNMT1 and 7-fold increase in mRNA expression for SNAIL.
Fig. 7. HIV enhances transcription and binding of molecules involved in SNAIL repressor complex.
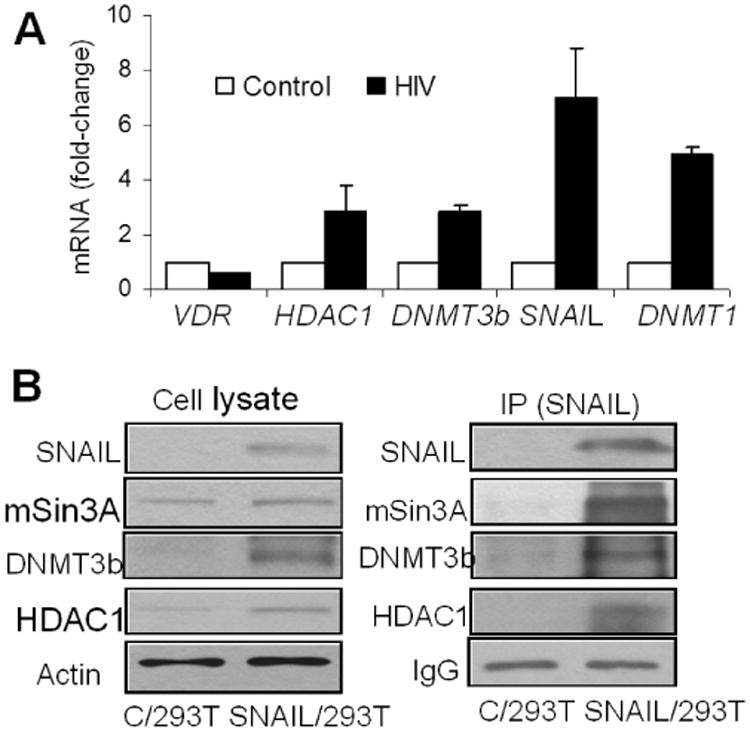
- RNA was extracted from EV/HPs and HIV/HPs and cDNA was amplified with primers for HDAC1, DNMT1, DNMT3b and SNAIL (n=3). Cumulative data are shown in bar graphs.
- 293T cells were transfected with SNAIL plasmid (SNAIL/293T). Cellular lysates of control (C/293T) and SNAIL/ 293T cells were probed for different molecules of SNAIL repressor complex and reprobed for actin. Gels are displayed. Cellular lysates of C/293T and SNAIL/293T cells were immunoprecipitated (IP) with SNAIL antibody. IP fraction was reprobed for SNAIL and other molecular markers. Gels are displayed.
To prepare SNAIL over expressing 293T cells, C/293Ts were transfected with either SNAIL plasmid or pcDNA (control). Protein blots of C/293T, pcDNA/293T and SNAIL/293T were probed for SNAIL (Supplementary data, Fig. S6). To study the presence of the reported SNAIL repressor constituents, protein blots of C/293T and SNAIL/293T cells were probed for SNAIL and reprobed for HDAC1, mSin3A, DNMT3b, DNMT1, and actin. To confirm the interaction of these molecules with SNAIL, lysates of C/293T cells and SNAIL/293T cells were immunoprecipitated (IP) with anti-SNAIL antibody. IP fractions were probed for SNAIL and reprobed for HDAC1, mSin3A, DNMT3b, DNMT1, and IgG. As shown in Fig. 7B, SNAIL repressor complex displayed enhanced expression of SNAIL, HDAC1, and DNMT3b.
To determine the molecules involved in HIV-induced podocyte SNAIL repressor complex, protein blots of EV/HPs and HIV/HPs were probed for SNAIL and reprobed for HDAC1, mSin3A, DNMT3b and actin(Fig. 8A). Cumulative densitometric data are shown as bar graphs (Fig. 8B, n=3).
Fig. 8. Compostion of SNAIL repressor complex in HIV milieu.
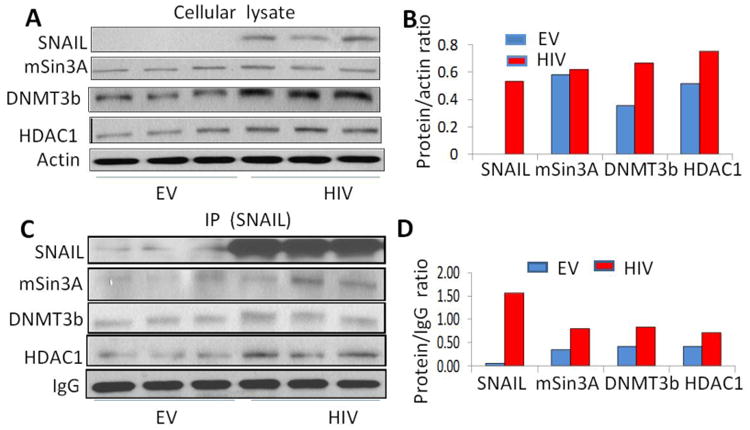
- Protein blots of EV/HPs and HIV/HPs were probed for indicated molecules of SNAIL repressor complex and reprobed for actin (n=3). Gels are displayed.
- Cumulative densitometric data are shown as bar graphs.
- Cellular lysates of EV/HPs and HIV/HPs were immunoprecipitated (IP) with SNAIL antibody. IP fraction was reprobed for SNAIL and other indicated molecules. Gels are displayed.
- Cumulative densitometric data are shown as bar graphs.
To determine the presence of molecule in SNAIL repressor complex, lysates of EV/HPs and HIV/HPs were immunoprecipitated with anti-SNAIL antibody. IP fractions were probed for SNAIL and reprobed for HDAC1, mSin3A, DNMT33b, and IgG. As shown in Fig. 8C, SNAIL repressor complex displayed enhanced expression of SNAIL, HDAC1, mSin3A and DNMT3b. Cumulative densitometric data are shown as bar graphs (Fig. 8D).
SNAIL binds at podocyte VDR promoter region in HIV milieu
The VDR promoter region is shown with the putative E-boxes (Supplementary data, Fig. S7). The ChIP Q-PCR primer recognized the E-boxes upstream of the start site.
To determine the binding of SNAIL at VDR promoter region, 293T cells were transfected with SNAIL plasmid. Chromatin from cellular lysates of SNAIL/293T cells was immunoprecipitated with either IgG (as a negative control), anti-RNA polymerase II antibody (as a positive control), or anti-SNAIL antibody. Subsequently, real time PCR was carried out with primers that recognize the VDR promoter within the first E-Box upstream of the start site. Binding of SNAIL to VDR promoter region is shown in Fig. 9A. Samples were run on agarose gel, input included as a control for loading (Fig 9B).
Fig. 9. SNAIL binds at podocyte VDR promoter region in HIV milieu.
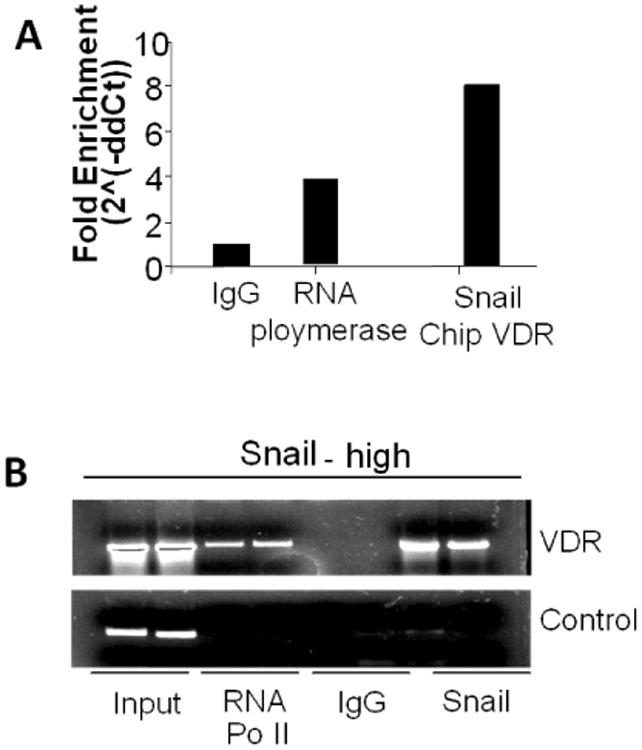
- 293T cells were transfected with SNAIL plasmid. Chromatin from cellular lysates of SNAIL/293T cells was immunoprecipitated (IP) with either rabbit IgG (as a negative control), anti-RNA polymerase II antibody (as a positive control), or anti-SNAIL antibody. Subsequently, real time PCR was carried out with primers that recognize the VDR promoter within the first E-Box upstream of the start site. The fold enrichment of binding to VDR promoter region is displayed.
- Aliquots from the above mentioned samples were run on agarose gel and are displayed. Input was included as control for loading.
Proposed mechanisms of HIV-induced down regulation of VDR are shown in Fig. 10 (A and B). HIV induces down regulation VDR by multiple mechanisms:
Enhancement of podocyte SNAIL expression by trimethylation of H3K4,
Hyper-methylation of VDR, 3. Proteosomal degradation of VDR, and 4. Enhanced podocyte expression of CYP24A1 leading to degradation of vitamin D and thus exposing the unliganded VDR for proteosomal degradation (23). Additionally, HIV-induced enhanced expression of CYP24A1 (23) will enhance degradation of 1-25α(OH)2D3 and decrease concentration of 1-25α(OH)2D3 in the incubation media. Since 1-25α(OH)2D3 also induces transcription of VDR (60), a decreased level of 1-25α(OH)2D3 would be associated with attenuated expression of VDR.
Fig. 10. Proposed mechanisms of HIV-induced down regulation of VDR.
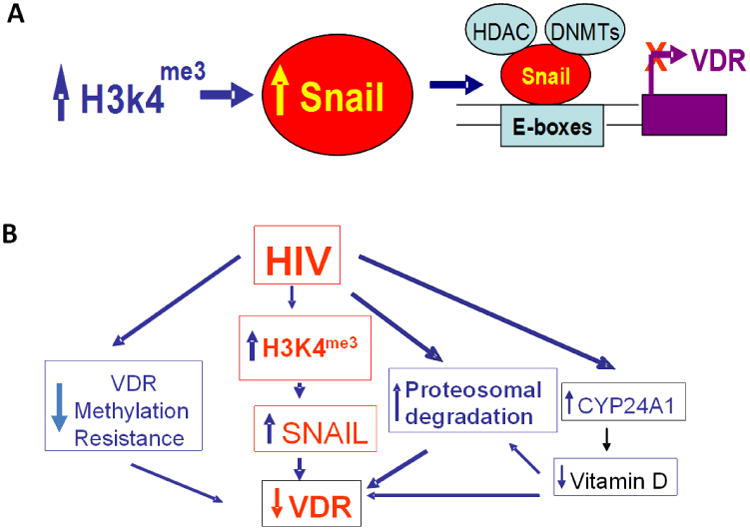
- HIV enhances podocyte SNAIL expression via trimethylation of H3K4 and deacetylation of H3K9/14ac.
- HIV uses multiple strategies for down regulation of podocyte VDR expression : 1. Enhancement of podocyte SNAIL expression by trimethylation of H3K4 and deacetylation of H3K9/14ac, 2. Hyper-methylation of VDR, 3. Proteosomal degradation of VDR, and 4. Enhanced podocyte expression of CYP24A1 leading to degradation vitamin D and thus exposing the unbound VDR for proteosomal degradation. Additionally, HIV-induced enhanced expression of CYP24A1 (23) will enhance degradation of 1-25α (OH)2D3 and decrease concentration of 1-25α(OH)2D3 in the incubation media. Since vitamin D is a ligand for induced VDR transcription (60), its decreased levels would also be associated with decreased transcription of VDR.
Discussion
In the present study, HIV-induced down regulation of kidney cell VDR expression was associated with CpG methylation at the VDR promoter site and upregulation of SNAIL via H3K4 trimethylation. Of note, VDA upregulated VDR optimally only under basal state but could not do so in HIV milieu. However, combined with HDACI/DAC successfully upregulated VDR expression in HIV milieu. Thus, it appears that for modulation of HIV- induced podocyte VDR expression, one should take into account the modification of chromatin structure as a part of the therapeutic strategy.
Pereria et al., demonstrated that vitamin D induces JMJD3 gene coding for a histone demethylase that is involved in epigenetic regulation of SW480-ADH cells (41). JMJD3 mediated the effect of vitamin D on subset of target genes and down regulated the expression of SNAIL (41). However, vitamin D was not able to alter global levels of H3K27me3 in these studies (41). Nonetheless, their observations are consistent with our findings highlighting the effect of VDA on down regulation of podocyte SNAIL expression.
DNA dependent processes including repair, transcription and replication are determined by chromatin condensation status (42, 43). The chromatin template is constantly modified by acetyl and methyl moieties (42, 43). Several lysine methyl transferases have been linked to cancer development and thus suggest an important role of methyl-lysine in homeostasis of cellular growth (44). In colonic cancer cells, up regulation of SNAIL and down regulation of VDR has been reported (40, 45). On that account, VDAs have been used to down regulate the growth of cancer cells (46). Interestingly, in several instances, VDA was not able to reverse cellular growth of cancer cells (45, 47). Yang et al., demonstrated that over expression of SNAIL in SaO2 cells (osteosarcoma cell line) induced vitamin D (1-α 25[OH]2D3) resistance (to its growth inhibitory effects), whereas, silencing of SNAIL reversed this effect (56). In that scenario, 1-α 25[OH]2D3 resistance seems to be related to its inability to up regulate VDR (optimally) in SNAIL- over expressing cells. We speculate that 1-α 25[OH]2D3 resistance may be the consequence of the involvement of epigenetic mechanisms in down regulation of VDR. In 1-α 25[OH]2D3 responsive cells, down regulation of VDR could have occurred predominantly as a consequence of enhanced proteosomal degradation (a consequence of 1-α 25[OH]2D3 deficient state) which could be prevented by binding of VDR with 1-α 25[OH]2D3. Additionally, adverse milieus like HIV also enhance cellular expression of CYP24A1, an enzyme that degrades 1-α 25[OH]2D3 and thus inducing 1-α 25[OH]2D3 deficient state (23). In the present study, VDA by itself could not optimally amplify podocytes VDR expression; however, VDA in combination with either a demethylating agent or an inhibitor of HDAC amplified podocyte VDR expression optimally.
VDR promoter methylation has also been reported to cause truncation of VDR transcripts in breast tumors (48); however, DNA methylation blockers were able to restore the VDR transcription levels (49). On the other hand, a VDA could not enhance VDR expression but addition of a DNA methylation blocker to a VDA, optimally amplified VDR expression. These findings are consistent with the observations in the present study.
In the present study, bisulfite sequencing revealed partial methylation of CpG islands of podocytes in HIV milieu and EepiTect data displayed an increase in hypermethylated VDR fraction and a decrease in the fraction of resistance to get methylated in HIV milieu. Since demethylating agent (DAC) treatment partially reversed HIV-induced down regulated VDR in podocytes, this wouldthat methylation of VDR contributed to down regulation of podocyte VDR in HIV milieu.
HIV is known to subvert cellular machinery of the host. Recently, HIV has been demonstrated to maintain high renin status in T cells to maintain its enhanced replication (15). High T cell renin generation in HIV milieu was associated with down regulation of VDR (50). In these studies, HIV enhanced T cell DNA methylation in general and increased VDR methylation in particular. Since demethylating agents were able to reverse HIV-induced down regulation of VDR, a causal relationship between hypermethylation and down regulation of VDR exists. In our current study too, HIV displayed very low resistance of podocyte CpG islands at VDR promoter to get methylated; moreover, a demethylating agent could inhibit HIV-induced down regulation of VDR. These studies establish a causal relationship between down regulation of podocyte VDR expression and hypermethylated status of VDR in HIV milieu. Since, a demethylating agent was able to partially reverse HIV-induced down regulation of VDR, it appears that some other mechanisms could also be contributing to the podocyte down regulation of VDR such as proteosomal degradation of VDR and upregulation of CYP24A1.
To evaluate putative SNAIL binding sties in colon cancer cells, Palmer et al cloned and analyzed the first 600 nucleotides of human VDR promoter in colonic cells (40). They reported that SNAIL binds at 3 E- boxes within this fragment of the promoter. Cano et al described three consensus E-Boxes for SNAIL binding in 0.5-kb fragment upstream of VDR coding region in osteoblasts (51). ChIP analysis confirmed the binding of SNAIL on E-Boxes II and III in the presence of Tamoxifen in SNAIL-ER transgenic mice (52). We have confirmed SNAIL binding sites at VDR promoter in the HIV milieu (Fig. S7, Supplementary data).
In conclusion, we report for the first time that HIV down regulates kidney cell VDR expression via epigenetic mechanisms. Optimal podocyte VDR expression in HIV milieu can be achieved using the combination of demethylating agents/inhibitors of HDAC and VDA.
Material and methods
Tg26 mice
We used age and sex matched FVB/N (control) and Tg26 (on FVB/N background) mice. Breeding pairs of FVBN mice were obtained from Jackson Laboratories (Bar Harbor, ME). Breeding pairs to develop Tg26 colonies were kindly gifted by Prof. Paul E. Klotman M.D., President and CEO, Baylor College of Medicine, Houston, TX). The Tg26 transgenic animal carries the proviral transgene, pNL4-3: d1443, which encodes all the HIV-1 genes except gag and pol and therefore these mice are noninfectious. We are maintaining colonies of these mice in our animal facility. For genotyping, tail tips were clipped, DNA was isolated and PCR studies were carried out using following primers for Tg26.
HIV-F 5′ ACATGAGCAGTCAGTTCTGCCGCAGAC
HIV-R 3′ CAAGGACTCTGATGCGCAGGTGTG
The Ethics Review Committee for Animal Experimentation of Feistein Institute for Medical Research approved the experimental protocol.
Preparation of podocytes
Human podocytes were obtained from Dr. Moin A. Saleem (Children's renal unit and academic renal unit, University of Bristol, Southmead Hospital, Bristol, UK). Human podocytes were conditionally immortalized by introducing temperature-sensitive SV40- T antigen by transfection (53). The cells have additionally been transfected with a human telomerase construct. These cells proliferate at permissive temperature (33°C), and enter growth arrest after transfer to the non-permissive temperature (37°C). The growth medium contained RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 1× Pen-Strep, 1 mM L-glutamine and 1× ITS (Invitrogen) to promote expression of T antigen.
Production of Pseudotyped Retroviral Supernatant
Replication defective viral supernatants were prepared as published previously (54). In brief, GFP reporter gene (from pEGFP-C1; Clontech, Palo Alto, CA) was substituted for gag/pol genes in HIV-1 proviral construct pNL4–3. This parental construct (pNL4-3:∆G/P-GFP) was used to produce VSV. G pseudotyped viruses to provide pleiotropism and high-titer virus stocks. Infectious viral supernatants were produced by the transient transfection of 293T cells using effectene (Qiagen Inc., Valencia, CA) according to the manufacturer's instructions. The HIV-1 gag/pol and VSV.G envelope genes were provided in trans using pCMV R8.91 and pMD.G plasmids, respectively (gifts of Dr. Didier Trono, Salk Institute, La Jolla, CA). As a negative control, virus was also produced from pHR-CMV-IRES2-GFP-∆B, which contained HIV-1 LTR and GFP empty expression vector. The viral stocks were titrated by infecting HeLa tat cells with tenfold serial dilution as reported previously (54). The reciprocal of the lowest dilution showing expression of green fluorescence protein (GFP) was defined as GFP-expressing units (GEU) per ml. Viral stocks ranging from 105 to 106 GEU/ml were used. Podocytes were transduced with NL4-3: EGFP (a viral construct containing all HIV-1 genes except gag and pol, containing EGFP or empty vector (EV).
SNAIL plasmid and SNAIL siRNA transfection
Podocytes were transfected with 20 nM SNAIL and control (scrambled) siRNA (Santa Cruz Biotechnology, Santa Cruz, CA) with Siport Neofax transfection reagent and incubated in optiMEM media for 48 hrs. Control and transfected cells were used in control and experimental conditions
Immunohistochemical studies
Renal cortical sections from control and HIV-1 transgenic (Tg26) mice (n=3) were de-paraffinised and antigen retrieval was done in a double boiler system. The endogenous peroxidase was blocked with 0.3% hydrogen peroxide in methanol for 30 minutes at room temperature (RT). Sections were washed in PBS 3× and incubated in blocking serum solution for 60 minutes at RT followed by incubation with primary antibodies against either VDR (mouse monoclonal, Santa Cruz Biotechnology, Santa Cruz, CA) or SNAIL (mouse monoclonal, Santa Cruz) for overnight at 4°C in a moist chamber. Each of the sections were washed with PBS (3×) and incubated with appropriate secondary antibody at 1:250 dilutions at RT for 1 hour. After washing with PBS (3×), sections were incubated in ABC reagent (Vector Laboratories, Burlingame, CA) for 1/2 hour. Sections were washed in PBS (3×) and then placed in Nova Red /hydrogen peroxide solution, counterstained with methyl green, dehydrated and mounted with a xylene-based mounting media (Permount, Fisher Scientific Corporation, Fair Lawn, NJ). Appropriate positive and negative controls were used.
Western blotting studies
Control and experimental cells as well as renal tissues were lysed in RIPA buffer containing 50 mM Tris-Cl (pH 7.5), 150 mM NaCl, 1mM EDTA, 1% NP-40, 0.25% Deoxycholate, 0.1% SDS, 1× protease inhibitor cocktail I (Calbiochem, EMD Biosciences, Gibbstan, NJ), 1mM PMSF, and 0.2mM sodium orthovanadate. Protein concentration was determined using Biorad Protein Assay kit (Pierce, Rockford, IL). Protein lysates (20 μg) were separated on 4-15% gradient polyacrylamide gels (PAGE, Bio-Rad, Hercules, CA) and transferred onto a nitrocellulose or PVDF membranes in 10% methanol using Bio-Rad mini blot apparatus. The membranes were then blocked in 5% milk and subjected to immunostaining with primary antibodies against VDR (anti-mouse monoclonal, Santa Cruz), SNAIL (anti-rabbit, Santa Cruz), H3K4Me3 (anti-rabbit, Abcam), H3K9/14 Ac (anti-rabbit, Santa Cruz), DNMT3b (anti-rabbit, Abcam), mSin3A (anti-mouse monoclonal, Santa Cruz), HDAC-1 (anti-mouse monoclonal, Santa Cruz), or DNMT1 (anti-rabbit, Cell Signaling) and subsequently with horseradish peroxidase labeled appropriate secondary antibody. The blots were developed using a chemiluminescence detection kit (PIERCE, Rockford, IL) and exposed to X-ray film (Eastman Kodak Co., Rochester, NY). Equal protein loading was confirmed by stripping and reprobing for actin expression (polyclonal (β-Actin antibody, Santa Cruz).
Immunoprecipitation (IP)
293T cells were transfected with SNAIL plasm id (Addgene) and lysates of SNAIL-over expressing 293T cells were first immunoprecipitated following addition of 5 μg of monoclonal Ab to SNAIL (Santa Cruz Biotechnology). The immune complexes were then collected using 25 μl of Protein-A + G sepharose beads (GE Health Care, Life Science), in RIPA buffer. IP was carried out at 4°C, for 4h, on a rotating platform. Following this, the Protein (A+G)-precipitated proteins were pelleted down by centrifugation at 4,500 rpm for 10 min at 4°C. Next, the protein pellet was washed (3×) each time with 1 ml of cold RIPA lysis buffer followed by centrifugation each time for 10 minutes at 2,500 rpm in a microfuge. After washings, beads were resuspended in 100 μl of lysis buffer to which SDS-PAGE sample buffer (50 μl) was added and samples boiled at 100°C, followed by SDS-PAGE and immunoblotted using respective antibodies as indicated.
Real time PCR Analysis
Control and experimental podocytes were used to quantify mRNA expression of molecules pertaining to SNAIL and VDR, and DNA methyltransferases (Dnmts) as described previously (15). Quantitative PCR was carried out in an ABI Prism 7900HT sequence detection system using the primer sequences as shown in Table 1 (Supplementary data). The data are expressed as relative mRNA expression with reference to control, normalized to quantity of RNA input by performing measurements on an endogenous reference gene, GAPDH.
ChIP assay
To investigate the binding of SNAIL at the VDR and H3K4me3 at SNAIL promoter, standard ChIP assays were performed on SNAIL-over expressing 293T cells, which were transfected with SNAIL plasmid (Addgene). Cells were cross-linked with formaldehyde prior to DNA sonication. Chromatin was sheared to an average length of 0.25 to 1 kb for this analysis. Sonicated fractions were treated with specific antibody for overnight. Protein complex was extracted using agarose beads for 1 hour on a rotator at 4°C and reverse cross linking was carried out at 65°C for overnight. DNA was purified using phenol-chloroform and PCR amplification was performed in aliquots of 25 μl with E-boxes specific primers for VDR promoter. Immunoprecipitation of the cross-linked chromatin was carried out with SNAIL (Santa Cruz) and H3K4 me3 (Abcam) antibody. The sensitivity of PCR amplification was evaluated through total DNA collected after sonication (input fraction). VDR and SNAIL promoters were amplified with the primers given in Table 1 using real-time PCR (ABI). Binding of H3K4 me3 on SNAIL promoter and SNAIL on VDR promoter E-box, was calculated by ‘Fold-Enrichment method’, where ChIP signal was divided by the control signals and representing the ChIP signal or signal relative to background. The amplified DNA was separated on 2% agarose gel and visualized with ethidium bromide
Epitech Methyl procedure
The EpiTect Methyl DNA Restriction Kit (Qiagen, Valencia, CA) was used to prepare the genomic DNA samples control and experimental cells according to the manufacturer's instructions. Briefly, four digests (of genomic DNA samples) were performed to detect different methylated DNA fractions. The product of a mock digest contained all of the input genomic DNA. The product of the methylation-sensitive restriction enzyme (Enzyme A) digest contained hypermethylated DNA sequences, while the product of the methylation-dependent restriction enzyme (Enzyme B) digest contained unmethylated DNA sequences. The fraction, which could not be digested by either enzyme was termed as methylation resistant. The product of a double digest measured the background and the success of both enzymatic digestions. Subsequently, EpiTect Methyl dVDR qPCR Primers (Qiagen #MePH16492-1A) were used to analyze the methylation status of CpG islands in the VDR. Dissociation curve was generated to find the Tm and check the specificity of the VDR.
Bisulfite conversion and pyrosequencing
Bisulfite conversion was carried out using an EZ DNA methylation gold kits (#D5005 and #D5006) according to the manufacturer's instructions (Zymo Research, Irvine, CA). Briefly, 0.5 μg of genomic DNA from either empty vector (EV/HP)- or HIV-transduced podocytes (HIV/HPs) cells in a 20 μl volume was mixed with 130 μl of CT (cytosine to thymidine) conversion reagent and was pre-incubated for 10 min at 98°C followed by incubation for 2.5 hours at 64°C. After bisulfite conversion, the unmethylated cytosine(s) were converted to thymidine, and the methylated cytosine(‘s) were not altered. Therefore, the methylated and unmethylated cytosine could be differentiated according to the sequencing results by comparing with original DNA sequence with/without bisulfite treatment as described previously (55). M-binding buffer (600 μl, Zymo Research) was added to the above reaction and DNA was purified using a Zymo spin column. Sequential washes were performed with 100 μl of M-Wash buffer, 200 l of M-sulphonation buffer, and 200 μl of M-wash buffer before eluting the DNA in 30 μl of M-elution buffer. Purified DNA (2 μl) was used as template for PCR reactions with primers (56). Pyrosequencing was performed at the Genomic Core Facility of Albert Einstein College of Medicine, Bronx, NY
Primer pairs were used to amplify the VDR promoter region (AY342401.1) in between -28 to -512 upstream relative to ATG start site and methylation level of each CpG is defined by the sequencing reads of CpGs (57). % methylated CpG = reads (mCpG)/reads (CpG) ×100 Reads (mCpG) present the number of methylated CpG reads at the CpG dinucleotides and reads (CpG) represent the total number of cover reads comprising unmethylated and methylated reads at the CpG dinucleotides. The following primer sequence was used for the amplification of bisulfite converted samples.
| VDR_Meth | F 5 : ATTTTATTTTAATTTGTGGGATTAGGTTGA:-3 R 5: CCAATCCTCTCTTACCAAAAACTCC:3 |
56 |
Statistical analysis
For comparison of mean values between two groups, the unpaired t test was used. To compare values between multiple groups, analysis of variance (ANOVA) was used to calculate a p-value. Statistical significance was defined as P<0.05. Results are presented as mean ± SD. Sequencing data were aligned to the reference genome through Needleman-Wunsch Global Aligner for nucleotide sequences (http://www.ebi.ac.uk/Tools/psa/emboss_needle/nucleotide.html) and further bisulfite sequencing results were screened through ‘Geospiza's FinchTV’ http://www.geospiza.com/Products/finchtv.shtml).
Methylated CpG and CpG content of the VDR promoter region before and after bisulfite conversion were calculated using the formula (43): %Meth CpG = reads (mCpG)/reads (CpG) ×100 (% Meth CpG reads, mCpG).
Supplementary Material
Highlights.
Kidney cells displayed enhanced SNAIL expression in HIV milieu both in vivo and in vitro.
HIV induced podocyte H3K4 trimethylation at SNAIL promoter site.
HIV also induced SNAIL repressor complex formation, which down regulated podoctye VDR expression.
Enhanced CpG methylation at VDR promoter also contributed to attenuated podocyte VDR expression.
Modulation of epigenetic alterations reversed podocyte VDR expression in HIV milieu.
Acknowledgments
This work was supported by grant RO1DK084910, RO1 DK083931, and 1R01DK098074 (PCS) from National Institutes of Health, Bethesda, MD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Smyth LJ, McKay GJ, Maxwell AP, McKnight AJ. DNA hypermethylation and DNA hypomethylation is present at different loci in chronic kidney disease. Epigenetics. 2014;9:366–376. doi: 10.4161/epi.27161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reddy MA, Natarajan R. Epigenetics in diabetic kidney disease. J Am Soc Nephrol. 2011;22:2182–5. doi: 10.1681/ASN.2011060629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ingrosso D, Perna AF. Epigenetics in hyperhomocysteinemic states. A special focus on uremia. Biochim Biophys Acta. 2009;1790:892–899. doi: 10.1016/j.bbagen.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 4.Ekstrom TJ, Stenvinkel P. The epigenetic conductor: a genomic orchestrator in chronic kidney disease complications? J Nephrol. 2009;22:442–449. [PubMed] [Google Scholar]
- 5.Lund G, Zaina S. Atherosclerosis risk factors can impose aberrant DNA methylation patterns: a tale of traffic and homocysteine. Curr Opin Lipidol. 2009;20:448–449. doi: 10.1097/MOL.0b013e3283309928. [DOI] [PubMed] [Google Scholar]
- 6.Medapalli RK, He JC, Klotman PE. HIV-associated nephropathy: pathogenesis. Curr Opin Nephrol Hypertens. 2011;20:306–311. doi: 10.1097/MNH.0b013e328345359a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salhan D, Pathak S, Husain M, Tandon P, Kumar D, Malhotra A, Meggs LG, Singhal PC. HIV gene expression deactivates redox-sensitive stress response program in mouse tubular cells both in vitro and in vivo. Am J Physiol Renal Physiol. 2012;302:F129–40. doi: 10.1152/ajprenal.00024.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ross MJ. Advances in the pathogenesis of HIV-associated kidney diseases. Kidney Int. 2014;86:266–274. doi: 10.1038/ki.2014.167. [DOI] [PubMed] [Google Scholar]
- 9.Ideura H, Hiromura K, Hiramatsu N, Shigehara T, Takeuchi S, et al. Angiotensin II provokes podocyte injury in murine model of HIV-associated nephropathy. Am J Physiol Renal Physiol. 2007;293:F1214–21. doi: 10.1152/ajprenal.00162.2007. [DOI] [PubMed] [Google Scholar]
- 10.Bird JE, Durham SK, Giancarli MR, Gitliz PH, Pandya DG, et al. Captopril prevents nephropathy in HIV-transgenic mice. J Am Soc Nephrol. 1989;9:1441–1447. doi: 10.1681/ASN.V981441. [DOI] [PubMed] [Google Scholar]
- 11.Burns C, Paul SK, Toth IR, Siva L. Effect of angiotensin-converting enzyme inhibition in HIV-associated nephropathy. J Am Soc Nephrol. 1999;8:1140–1146. doi: 10.1681/ASN.V871140. [DOI] [PubMed] [Google Scholar]
- 12.Kimmel PL, Barisoni L, Kopp JB. Pathogenesis and treatment of HIV-associated renal diseases: lessons from clinical and animal studies, molecular pathologic correlations, and genetic investigations. Ann Intern Med. 2003;139:214–26. [PubMed] [Google Scholar]
- 13.Kumar D, Plagov A, Yadav I, Torri DD, Sayeneni S, et al. Inhibition of Renin Activity Slows down the Progression of HIVAN. Am J Physiol Renal Physiol. 2012;303:F711–20. doi: 10.1152/ajprenal.00643.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chandel N, Sharma B, Salhan D, Husain M, Malhotra A, Buch S, Singhal PC. Vitamin D receptor activation and downregulation of renin-angiotensin system attenuate morphine-induced T cell apoptosis. Am J Physiol Cell Physiol. 2012;303:C607–15. doi: 10.1152/ajpcell.00076.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chandel N, Husain M, Goel H, Salhan D, Lan X. VDR hypermethylation and HIV-induced T cell loss. J Leukoc Biol. 2013;93:623–31. doi: 10.1189/jlb.0812383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chandel N, Sharma B, Husain M, Salhan D, Singh T, et al. HIV compromises integrity of the podocyte actin cytoskeleton through downregulation of the vitamin D receptor. Am J Physiol Renal Physiol. 2013;304:F1347–1357. doi: 10.1152/ajprenal.00717.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salhan D, Husain M, Subrati A, Goyal R, Singh T, et al. HIV-induced kidney cell injury: role of ROS-induced downregulated vitamin D receptor. Am J Physiol Renal Physiol. 2012;303:F503–514. doi: 10.1152/ajprenal.00170.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haussler MR, Whitfield GK, Haussler CA, Hsieh JC, Thompson PD, et al. The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J Bone Miner Res. 1998;13:325–349. doi: 10.1359/jbmr.1998.13.3.325. [DOI] [PubMed] [Google Scholar]
- 19.Aranda A, Pascual A. Nuclear hormone receptors and gene expression. Physiol Rev. 2001;81:1269–1304. doi: 10.1152/physrev.2001.81.3.1269. [DOI] [PubMed] [Google Scholar]
- 20.Holick MF. High prevalence of vitamin D inadequacy and implications for health. Mayo Clin Proc. 2006 Mar;81:353–73. doi: 10.4065/81.3.353. [DOI] [PubMed] [Google Scholar]
- 21.Prosser DE, Jones G. Enzymes involved in the activation and inactivation of vitamin D. Trends Biochem Sci. 2004;29:664–73. doi: 10.1016/j.tibs.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 22.Fetahu Irfete S, Höbaus Julia, Kállay Enikő. Vitamin D and the epigenome. Front Physiol. 2014;5:164. doi: 10.3389/fphys.2014.00164. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singh T, Ayosolla K, Rai P, Chandel N, Haque S, Lederman R, Husain M, Vethantham V, Chawla A, Vashistha H, Saleem MA, Ding G, Chander PN, Malhotra A, Meggs LG, Singhal PC. AT1R Blockade in Adverse Milieus: Role of SMRT and Co-repressor complexes. Am J Physiol Renal Physiol. 2015 Jun 17; doi: 10.1152/ajprenal.00476.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li YC, Kong J, Wei M, Chen ZF, et al. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest. 2002;110:229–38. doi: 10.1172/JCI15219. 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larsen Thomas, Mose Frank H, Bech Jesper N, Pedersen Erling B. Effect of paricalcitol on renin and albuminuria in non-diabetic stage III-IV chronic kidney disease: a randomized placebo-controlled trial. BMC Nephrol. 2013;14:163. doi: 10.1186/1471-2369-14-163. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, Deb DK, Zhang Z, Sun T, et al. Vitamin D receptor signaling in podocytes protects against diabetic nephropathy. J Am Soc Nephrol. 2012;23:1977–86. doi: 10.1681/ASN.2012040383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Kong J, Deb DK, Chang A, et al. Vitamin D receptor attenuates renal fibrosis by suppressing the renin-angiotensin system. J Am Soc Nephrol. 2010;21:966–73. doi: 10.1681/ASN.2009080872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mathew JT, Patni H, Chaudhary AN, Liang W, Gupta A, et al. Aldosterone induces mesangial cell apoptosis both in vivo and in vitro. Am J Physiol Renal Physiol. 2008;29:F73–F81. doi: 10.1152/ajprenal.00435.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhat RS, Bhaskaran M, Mongia N, Hitosugi N, Singhal PC. Morphine-induced macrophage apoptosis: Oxidative stress and strategies for modulation. J Leukocyte Biol. 2004;75:1131–1138. doi: 10.1189/jlb.1203639. [DOI] [PubMed] [Google Scholar]
- 30.Kovalchuk O, Baulch JE. Epigenetic changes and nontargeted radiation effects—is there a link? Environ Mol Mutagen. 2008;49:16–25. doi: 10.1002/em.20361. [DOI] [PubMed] [Google Scholar]
- 31.Ilnytskyy Y, Kovalchuk O. Non-targeted radiation effects-an epigenetic connection. Mutat Res. 2011;714:113–25. doi: 10.1016/j.mrfmmm.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 32.Friedl AA, Mazurek B, Seiler DM. Radiation-induced alterations in histone modification patterns and their potential impact on short-term radiation effects. Front Oncol. 2012;2:117. doi: 10.3389/fonc.2012.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holiday R. DNA Methylation and Epigenetic Inheritance. Philosophical Transactions of the Royal Society of London Series B, Biological Sciences. 1990;326:329–338. doi: 10.1098/rstb.1990.0015. [DOI] [PubMed] [Google Scholar]
- 34.Cuozzo C, Porcellini A, Angrisano T, et al. DNA damage, homology-directed repair, and DNA methylation. PLoS Genet. 2007;3(7):e110. doi: 10.1371/journal.pgen.0030110. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.O'Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. In Lee, Jeannie T PLoS Genet. 2008;4:e1000155. doi: 10.1371/journal.pgen.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malanga M, Althaus FR. The role of poly(ADP-ribose) in the DNA damage signaling network. Biochem Cell Biol. 2005;83:354–364. doi: 10.1139/o05-038. [DOI] [PubMed] [Google Scholar]
- 37.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 38.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–32. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- 39.Reinberg Danny, Zhang Yi. Transcription regulation by histone methylation: Interplay between different covalent modifications of the core histone tails. Genes and Development. 2001;15:2343–2360. doi: 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
- 40.Pálmer HG, Larriba MJ, García JM, Ordóñez-Morán P, Peña C, et al. The transcription factor SNAIL represses vitamin D receptor expression and responsiveness in human colon cancer. Nat Med. 2004;10:917–919. doi: 10.1038/nm1095. [DOI] [PubMed] [Google Scholar]
- 41.Pereira F, Barbáchano A, Singh PK, Campbell MJ, Muñoz A, et al. Vitamin D has wide regulatory effects on histone demethylase genes. Cell Cycle. 2012;11:1081–1089. doi: 10.4161/cc.11.6.19508. [DOI] [PubMed] [Google Scholar]
- 42.Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem. 2006;75:243–269. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- 43.Lee S, Roeder RG, Lee JW. Roles of histone H3-lysine 4 methyltransferase complexes, in NR-mediated gene transcription. Prog Mol Biol Transl Sci. 2009;87:343–382. doi: 10.1016/S1877-1173(09)87010-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Copeland RA. Molecular pathways: protein methyltransferases in cancer. Clin Cancer Res. 2013;19:6344–350. doi: 10.1158/1078-0432.CCR-13-0223. [DOI] [PubMed] [Google Scholar]
- 45.Larriba MJ, Martín-Villar E, García JM, Pereira F, Peña C, et al. Snail2 cooperates with Snail1 in the repression of vitamin D receptor in colon cancer. Carcinogenesis. 2009;30:1459–68. doi: 10.1093/carcin/bgp140. [DOI] [PubMed] [Google Scholar]
- 46.González-Sancho JM, Larriba MJ, Ordóñez-Morán P, Pálmer HG, Muñoz A. Effects of 1alpha,25-dihydroxyvitamin D3 in human colon cancer cells. Anticancer Res. 2006;26:2669–2681. [PubMed] [Google Scholar]
- 47.Larriba MJ, Valle N, Pálmer HG, Ordóñez-Morán P, et al. The inhibition of Wnt/beta-catenin signalling by 1alpha, 25-dihydroxyvitamin D3 is abrogated by Snail1 in human colon cancer cells. Endocr Relat Cancer. 2007;14:141–151. doi: 10.1677/ERC-06-0028. [DOI] [PubMed] [Google Scholar]
- 48.Yang H, Zhang Y, Zhou Z, Jinag X, Shen A. Snail-1 regulates VDR signaling and inhibits 1,25(OH)-D3 action in osteosarcoma. Mol cell Pharmacol. 2011;670:341–346. doi: 10.1016/j.ejphar.2011.09.160. [DOI] [PubMed] [Google Scholar]
- 49.Marik R, Fackler M, Gabrielson E, Zeiger MA, Sukumar S, et al. DNA methylation-related vitamin D receptor insensitivity in breast cancer. Cancer Biol Ther. 2010;10:44–53. doi: 10.4161/cbt.10.1.11994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chandel N, Ayasolla K, Lan X, Rai P, Mikulak J, et al. Renin modulates HIV replication in T cells. J Leukoc Biol. 2014;96:601–609. doi: 10.1189/JLB.2A0414-192R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000:276–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 52.de Frutos CA, Dacquin R, Vega S, Jurdic P, Machuca-Gayet I, Nieto MA. Snail1 controls bone mass by regulating Runx2 and VDR expression during osteoblast differentiation. EMBO J. 2009;28:686–96. doi: 10.1038/emboj.2009.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saleem MA, O'Hare MJ, Reiser J, Coward RJ, Inward CD, et al. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol. 2002;13:630–638. doi: 10.1681/ASN.V133630. [DOI] [PubMed] [Google Scholar]
- 54.Husain M, Meggs LG, Vashistha H, Simoes S, Griffiths KO, et al. Inhibition of p66ShcA longevity gene rescues podocytes from HIV-1-induced oxidative stress and apoptosis. J Biol Chem. 2009;284:16648–1658. doi: 10.1074/jbc.M109.008482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y, Jeltsch A. The Application of Next Generation Sequencing in DNA Methylation Analysis. Genes. 2010;1:85–101. doi: 10.3390/genes1010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marik R, Fackler M, Gabrielson E, Zeiger MA, Sukumar S, Stearns V, Umbricht CB. DNA methylation-related vitamin D receptor insensitivity in breast cancer. Cancer Biol Ther. 2010;10:44–53. doi: 10.4161/cbt.10.1.11994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Su J, Yan H, Wei Y, Liu H, Liu H, Wang F, Lv J, Wu Q, Zhang Y. CpG_MPs: identification of CpG methylation patterns of genomic regions from high-throughput bisulfite sequencing data. Nucleic Acids Res. 2013;41:1–15. doi: 10.1093/nar/gks829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Katoh H, Qin ZS, Liu R, Wang L, Li W, et al. FOXP3 orchestrates H4K16 acetylation and H3K4 trimethylation for activation of multiple genes by recruiting MOF and causing displacement of PLU-1. Mol Cell. 2011;44:770–784. doi: 10.1016/j.molcel.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 60.Zella LA, Kim S, Shevde NK, Pike JW. Enhancers located within two introns of the vitamin D receptor gene mediate transcriptional autoregulation by 1,25-dihydroxyvitamin D3. Mol Endocrinol. 2006;20:1231–47. doi: 10.1210/me.2006-0015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.