

Abstract
Obesity is linked to type 2 diabetes (T2D) and cardiovascular diseases; however, the underlying molecular mechanisms remain unclear. We aimed to identify obesity-associated molecular features that may contribute to obesity-related diseases. Using circulating monocytes from 1,264 Multi-Ethnic Study of Atherosclerosis (MESA) participants, we quantified the transcriptome and epigenome. We discovered that alterations in a network of coexpressed cholesterol metabolism genes are a signature feature of obesity and inflammatory stress. This network included 11 BMI-associated genes related to sterol uptake (↑LDLR, ↓MYLIP), synthesis (↑SCD, FADS1, HMGCS1, FDFT1, SQLE, CYP51A1, SC4MOL), and efflux (↓ABCA1, ABCG1), producing a molecular profile expected to increase intracellular cholesterol. Importantly, these alterations were associated with T2D and coronary artery calcium (CAC), independent from cardiometabolic factors, including serum lipid profiles. This network mediated the associations between obesity and T2D/CAC. Several genes in the network harbored C-phosphorus-G dinucleotides (e.g., ABCG1/cg06500161), which overlapped Encyclopedia of DNA Elements (ENCODE)-annotated regulatory regions and had methylation profiles that mediated the associations between BMI/inflammation and expression of their cognate genes. Taken together with several lines of previous experimental evidence, these data suggest that alterations of the cholesterol metabolism gene network represent a molecular link between obesity/inflammation and T2D/CAC.
Introduction
Obesity is a major risk factor for type 2 diabetes (T2D) and cardiovascular disease. Traditionally, obesity-related insulin resistance and atherosclerosis have been viewed as lipid accumulation disorders, with fatty acid accumulation in insulin target tissues, including liver, muscles, and adipose tissue, and cholesterol accumulation in atheromatous plaques (1). In the past decade, experimental studies have demonstrated an essential role for immune cells, especially monocytes/macrophages, and chronic inflammation in the progression of insulin resistance and atherosclerosis (2–6). However, the precise molecular mechanisms accounting for these relationships remain uncertain. Genome-wide analyses of gene expression modifications associating with obesity could help define the cellular features of obesity and suggest potential molecular mechanisms that contribute to obesity-associated diseases (7). Our goal is to better understand the biological mechanisms underlying the obesity-related risk for common diseases by identifying T2D- and/or subclinical cardiovascular disease–associated molecular characteristics that are common in the blood monocytes of obese individuals. Accordingly, using purified peripheral monocytes from 1,264 Multi-Ethnic Study of Atherosclerosis (MESA) participants, we identified transcriptomic features associated with BMI and determined which of these features were also associated with T2D and coronary artery calcium (CAC).
Research Design and Methods
Study Population
The MESA study was designed to investigate the progression of subclinical cardiovascular disease in a community-based cohort of 6,814 participants. Since its inception in 2000, extensive clinical, sociodemographic, lifestyle and behavior, and laboratory data were collected at five clinic visits (examinations) (8). The present analysis is primarily based on analyses of purified monocyte samples from the April 2010–February 2012 examination (exam 5) of 1,264 randomly selected MESA participants from four MESA field centers (Baltimore, MD; Forsyth County, NC; New York, NY; and St. Paul, MN). At exam 5, these participants also underwent an assessment of BMI, T2D, and CAC. The study protocol was approved by the institutional review board at each site. All participants signed informed consent.
Purification of Monocytes
Monocytes were isolated with anti-CD14 monoclonal antibody–coated magnetic beads using autoMACs automated magnetic separation unit (Miltenyi Biotec, Bergisch Gladbach, Germany) (see the Supplementary Data for details).
DNA/RNA Extraction
DNA and RNA were isolated from samples simultaneously using the AllPrep DNA/RNA Mini Kit (Qiagen, Inc., Hilden, Germany) (see the Supplementary Data for details).
Global Expression Quantification
The Illumina HumanHT-12 v4 Expression BeadChip and Illumina BeadArray Reader were used to perform the genome-wide expression analysis, following the Illumina expression protocol (see the Supplementary Data for details). These data have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (GEO) and are accessible through GEO Series accession number (GSE56045).
Epigenome-Wide Methylation Quantification
The Illumina Infinium HumanMethylation450 BeadChip and HiScan Reader were used to perform the epigenome-wide methylation analysis (see the Supplementary Data for details). These methylation data have been deposited in GEO and are accessible through GEO Series accession number (GSE56046).
Quality Control and Preprocessing of Microarray Data
Data preprocessing and quality control (QC) analyses were performed in R software (http://www.r-project.org/) using the Bioconductor (http://www.bioconductor.org/) packages (see the Supplementary Data for details). The Illumina HumanHT-12 v4 Expression BeadChip included 48,000 transcripts. Statistical analyses were limited to probes retained after applying the following QC elimination: nondetectable expression in ≥90% of MESA samples using a detection P value cutoff of 0.0001, overlap with a repetitive element or region, low variance across the samples (<10th percentile), or putative and/or not well-characterized genes (i.e., gene names starting with KIAA, FLJ, HS, Cxorf, MGC, or LOC). We included 14,619 gene transcripts (10,898 unique genes) for analysis.
The Illumina Infinium HumanMethylation450 BeadChip included probes for 485,000 C-phosphorus-G (CpG) sites. Of these 485,000 CpG sites, 448,588 passed the QC elimination criteria that included “detected” methylation levels <90% of MESA samples using a detection P value cutoff of 0.05, existence of any single nucleotide polymorphisms within 10 base pairs of the targeted CpG site, or overlap with a repetitive element or region.
These preprocessing pipelines effectively removed large effects of sample position on chips. However, probe-specific position effects existed for some CpG sites. Therefore, sample position on chips was adjusted in the analysis of methylation data.
Measurement of BMI, Inflammatory Stress, T2D, and CAC
Weight was measured with a Detecto platform balance scale to the nearest 0.5 kg. Height was measured with a stadiometer (Accu-Hite with level bubble) to the nearest 0.1 cm. BMI was defined as weight in kilograms divided by square of height in meters. Plasma interleukin-6 (IL-6) was measured by ultrasensitive ELISA (Human IL-6 Quantikine HS; R&D Systems, Minneapolis, MN). Plasma C-reactive protein (CRP) was measured using the BN II nephelometer (N High Sensitivity CRP; Dade Behring Inc., Deerfield, IL).
T2D was defined as fasting glucose ≥7.0 mmol/L (≥126 mg/dL) or use of hypoglycemic medication, and impaired fasting glucose was defined as fasting glucose 5.6–6.9 mmol/L (100–125 mg/dL). Fasting serum glucose at each examination was measured by rate reflectance spectrophotometry using thin-film adaptation of the glucose oxidase method on the VITROS analyzer (Johnson & Johnson Ortho-Clinical Diagnostics, Rochester, NY). To achieve consistency of the serum glucose assay over examinations, 200 samples from each examination were reanalyzed. The original observations were then recalibrated. The CT Reading Center for cardiac scans in the MESA is at the Los Angeles Biomedical Research Institute at Harbor-UCLA Medical Center. CAC is scored using the Agatston method, which accounts for lesion area and calcium density using Hounsfield brightness. The reread agreement for the Agatston score (intraclass correlation coefficient, 0.99) was excellent.
Association Analysis
Association analyses were performed using the linear model (lm) function and the stepAIC function of the MASS package in R. To identify gene transcripts or methylation sites associated with BMI, we fit separate linear regression models with BMI as a predictor of each transcript expression or the M-value for each CpG site. Covariates were age, sex, race/ethnicity, study site, technical covariates (expression/methylation chip, sample position on chips for methylation analyses), and residual sample contamination with nontargeted cells (e.g., nonmonocytes, see below). To identify methylation sites associated with cis-gene expression, we fit separate linear regression models with the M-value for each CpG site (adjusted for methylation chip and position effects) as a predictor of transcript expression for any autosomal gene within 1 Mb of the CpG site in question. Covariates were age, sex, race/ethnicity, study site, expression chip, and residual sample contamination with nontargeted cells. Analyses stratified by sex and ethnicity were performed as an internal validation and check of generalizability. P values were adjusted for multiple testing using the q-value false discovery rate (FDR) method (9). To minimize false-positive results, we used a commonly used FDR threshold of 0.05. All FDR control was performed at the genome- and epigenome-wide scales for the tested gene transcripts and methylation probes, respectively.
Stability Analysis–Enhanced Weighted Gene Co-expression Network Identification
We applied the weighted correlation network analysis as implemented in the R package Weighted Gene Co-expression Network Analysis (WGCNA) (10) to construct network modules of highly correlated transcripts. We used the less stringent threshold of FDR ≤0.10 to preselect a subset of 2,807 BMI-associated genes to satisfy the scale-free topology criterion. We first obtained a weighted network based on the pairwise correlations among all transcripts considered, with an adjustment to produce a scale-free topology. Then, a topological overlap measure to estimate the network interconnectedness was used to hierarchically cluster the transcript. With respect to the default parameters of WGCNA, we changed only the correlation type from Pearson to biweight midcorrelation (which is more robust to outliers) and set the minimum size for module detection to 10 (see the Supplementary Data for a detailed assessment of the stability of the identified networks).
Using this novel approach of stability analysis–enhanced WGCNA, we then computed, for each consensus module, the eigengene defined as the first right-singular vector in the singular value decomposition of the standardized pm × N expression matrix (or the first eigenvector of the N × N correlation matrix, n = 1,264) based on the pm genes within module m. Association analyses of the eigengenes of individual subnetworks (modules) with T2D and CAC were performed using logistic and linear regression, respectively. CAC was log-transformed (log [Agatston score + 1]). We fitted separate models with the first eigenvector for each network as a predictor of T2D or CAC. Covariates were age, sex, race/ethnicity, study site, expression/methylation chip, methylation position (for CpG methylation analyses only), and residual sample contamination with nonmonocytes.
Functional Annotation Analysis
Database for Annotation, Visualization and Integrated Discovery (DAVID) Bioinformatics Resources 6.7 was used to examine enrichment of gene ontology pathway genes among BMI-associated genes (FDR <0.05) (11). The Encyclopedia of DNA Elements (ENCODE) (12) project data were accessed through the University of California, Santa Cruz, Genome Browser (13) (http://genome.ucsc.edu/) to evaluate potential functional regions surrounding BMI-associated methylation sites. Downloaded data sets included histone modifications detected in a CD14+ sample (ChIP-seq: H3K4m1/H3K4me3) from the Broad Institute Bernstein Laboratory, as well as DNaseI hypersensitivity data in a CD14+ sample by Digital DNaseI from the University of Washington ENCODE group. These data were released by the ENCODE Consortium/Data Coordination Center to be freely downloaded, analyzed, and published.
Analyses of Risk Gene Dosage
Within the identified cholesterol (cyan) module of the coexpression network, we converted the continuous gene expression values to a dichotomized score coded 0 vs. 1 (−1) for an expression profile with positive (negative) association with BMI, with 1 (−1) representing expression values greater than the median value of this profile in the study population. We then summed the score for the 11 genes as the “risk score,” defined as the number of risk genes for each participant. Association analyses for risk gene dosage with T2D and CAC were performed using the logistic and linear model, respectively. CAC was log-transformed (log [Agatston score + 1]). We fit separate models with risk gene dosage as a predictor of T2D or CAC. Covariates were age, sex, race/ethnicity, and study site. BMI, HDL cholesterol, triglycerides, systolic blood pressure, cigarette smoking, and physical activity were further adjusted in the models.
Mediation Analyses
Methylation of DNA, which occurs predominately at CpG dinucleotides, has been viewed as an important potential regulator of gene expression (14). To investigate DNA methylation as a potential molecular link between BMI and gene expression, we performed mediation analyses under an assumed causal model in which BMI leads to a change in the methylation level of a CpG site, which at least partially mediates the effects of BMI on the expression of a nearby gene. The mediation analyses accounted for the biological and technical covariates and were performed by robust structural equation modeling (SEM) as implemented in the R package lavaan (15). SEM analysis in general, and as implemented in lavaan, is based on maximum likelihood and the normal distribution but provides several approaches to effectively deal with nonnormal data. A first approach consists of computing robust SEs by sandwich-type covariance matrices and scaled test statistics, in particular the Satorra-Bentler statistic, whose amount of rescaling reflects the degree of kurtosis, whereas the second approach uses specific bootstrapping methods to obtain SE and test statistics (15). The results we report are based on bootstrapping, which we found to be somewhat more conservative than the use of robust SE and the Satorra-Bentler statistic. Lastly, mediation analyses for the eigengene of the cholesterol metabolism network (cyan) module were conducted in the same way and under the assumed causal model that BMI leads to a change in T2D or CAC, which is at least partially mediated by a change in gene expression in the cyan module.
RNA Sequencing
A subset of 374 samples was randomly selected from the 1,264 MESA monocyte samples for RNA sequencing of mRNA (see the Supplementary Data for details).
Replication Study
Gene expression profiles from monocytes enriched from the whole blood of 1,285 German men and women were used to replicate main findings (see the Supplementary Data for details).
Bisulfite Treatment of Genomic DNA and Pyrosequencing
A subset of 176 samples was randomly selected from the 1,264 MESA monocyte samples for bisulfite treatment of genomic DNA and pyrosequencing (see the Supplementary Data for details).
Weight-Loss Intervention Study
We quantified the transcriptome in the pre- and postintervention monocytes of 16 sedentary obese men and women, aged 65–80 years, by leveraging an ongoing 5-month weight-loss intervention trial. The weight-loss intervention involved provision of a hypocaloric (−250 to 400 kcal/day) diet, behavioral counseling with a registered dietitian, and 4 days/week of supervised moderate-intensity aerobic exercise training (16). An estimate of insulin sensitivity by the HOMA score (17) was calculated with the formula: [fasting plasma insulin (μU/mL) × glucose (mmol/L)]/22.5. We used the same protocol for monocyte purification and mRNA quantification as in MESA. The paired t test was used to compare cyan module gene expression before and after the intervention. The Pearson correlation coefficient was used to assess the relationships between changes in the cyan module gene expression and changes in HOMA measures.
Results
Transcriptome-Wide Analysis of BMI
The study population consisted of 1,264 MESA participants; of whom, 47% were Caucasian, 32% Hispanic, 21% African American, and 51% female and were aged 55–94 years (Supplementary Table 1). To determine if an obesity-associated transcriptional signature exists in circulating monocytes, we examined the relationships between BMI and the individual gene expression level of 10,898 genes detected in the monocytes. Using an FDR threshold of 0.05, we identified 1,741 BMI-associated genes (Fig. 1 and Supplementary Fig. 1), among which the top 10 most significantly BMI-associated gene transcripts are listed in Supplementary Table 2a. Similar associations were observed for waist circumference (Supplementary Table 2b). The top BMI-associated gene was guanine nucleotide binding protein, gamma 10 (GNG10, P = 3.44 × 10−23; Supplementary Table 2a). GNG10 encodes a G-protein-γ subunit that facilitates G-protein activation of phospholipase A2 (PLA2) and the subsequent hydrolysis of phospholipids to form fatty acid and lysophospholipid products. Gene set enrichment analysis using DAVID (11) revealed that overall, lipid biosynthesis genes were significantly overrepresented among all of the BMI-associated transcripts. Other biological processes significantly enriched among the BMI-associated genes included mitochondrial transport, apoptosis, inflammation, and glucose metabolism (Supplementary Table 3).
Figure 1.

Schematic summary of main findings in MESA.
Coexpression Network Analysis of BMI: Identification of the Cholesterol Metabolism Gene Network
Next, we performed a weighted gene coexpression network analysis using the R package WGCNA (10) and a network stabilization procedure to identify robust networks of genes with coordinated gene expression patterns associated with BMI. The identified consensus modules are labeled by different colors (with gray denoting genes not assigned to modules); hence, we will refer to specific modules by their color (e.g., cyan). We identified 15 consensus modules associated with BMI (P = 1.60 × 10−18 to 7.39 × 10−6) (Fig. 2 and Supplementary Table 4a and b). Gene ontology or Kyoto Encyclopedia of Genes and Genomes terms were significantly enriched (FDR <0.05) in 5 of the 15 modules identified, including sterol metabolism (cyan), antigen processing and presentation (magenta), noncoding RNA metabolism (brown), and ribosome machinery/translation (black and light cyan) (Fig. 2 and Supplementary Table 5).
Figure 2.
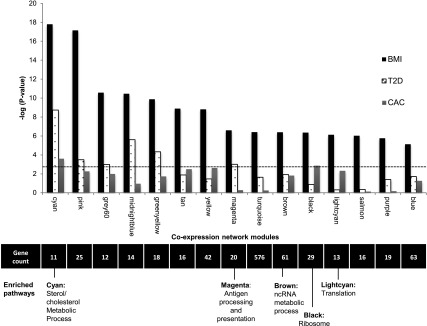
Gene coexpression network modules identified associating with BMI. Significance (−log10 P value on the y-axis) resulting from the association of each consensus network module of coexpressed genes (eigengene) with BMI, T2D, and CAC in 1,264 monocyte samples; dashed line represents a Bonferroni correction for 15 modules (α = 0.05). The number of genes and the biological pathways significantly enriched within each module (from DAVID, FDR <0.05) are also shown below, corresponding to each coexpression network module (x-axis). See also Supplementary Tables 4 and 5. ncRNA, noncoding RNA.
The coexpressed network module most significantly associated with BMI was the cyan module (P = 1.6 × 10−18), which contained 11 functionally coupled genes related to sterol metabolism (Fig. 3 and Supplementary Table 5), including 8 genes that were upregulated with increasing BMI (LDLR, HMGCS1, FDFT1, SQLE, CYP51A1, SC4MOL, SCD, and FADS1), and 3 that were downregulated with increasing BMI (MYLIP, ABCG1, and ABCA1). These genes are known to be key contributors to cholesterol uptake (↑LDLR, ↓MYLIP) (18,19), fatty acid and cholesterol synthesis (↑SCD, FADS1, HMGCS1, FDFT1, SQLE, CYP51A1, SC4MOL) (20–22), and cholesterol efflux (↓ABCA1, ABCG1) (23). Collectively, the expression profiles of the cyan module genes suggest that obesity is associated with cholesterol and lipid accumulation in circulating monocytes.
Figure 3.

BMI-associated expression patterns of the 11 intercorrelated genes in the cyan module reveal altered cholesterol regulation. Absolute correlation coefficients between expression of cyan module genes in 1,264 monocyte samples range from 0.3 to 0.7, indicated by line thickness. Positive correlations between genes are indicated by red lines (negative = blue), positive associations between gene expression and BMI are indicated by red nodes (negative = blue), and the strength of the correlation between genes and the cyan module eigengene is indicated by node size. The anticipated effects of BMI-associated gene expression on intracellular cholesterol levels and fatty acid synthesis are indicated by brackets/arrows. Cyan module genes include 3-hydroxy-3-methylglutaryl-CoA synthase 1 (HMGCS1); farnesyl-diphosphate farnesyltransferase 1 (FDFT1); squalene epoxidase (SQLE); methylsterol monooxygenase 1 (SC4MOL); cytochrome P450, family 51, subfamily A, polypeptide 1 (CYP51A1); stearoyl-CoA desaturase (SCD); fatty acid desaturase 1 (FADS1); low density lipoprotein receptor (LDLR); myosin regulatory light chain interacting protein (MYLIP); ATP-binding cassette, subfamily G, member 1 (ABCG1); and ATP-binding cassette, subfamily A, member 1 (ABCA1).
To determine whether the relationship between cyan module gene expression and BMI was driven by the serum lipid profiles, we examined the association between the eigengene for the cyan module and BMI after adjustment for plasma HDL, LDL, and triglycerides measured at the same time as the blood draw for monocyte separation. In this analysis, the association with BMI was attenuated but remained significant (P = 8.7 × 10−8). We observed that LDL, HDL, and triglycerides levels were negatively associated with cholesterol synthesis/uptake gene expression (P < 2.2 × 10−16) and positively associated with cholesterol efflux gene expression (P < 2 × 10−16). To further evaluate the internal validity of the cyan module, we examined the association of individual cyan module genes with BMI across sex and race subgroups. In general, the associations were qualitatively consistent across subgroups, and all P values for interactions between sex or race and gene expression levels were nonsignificant (P > 0.05) (Supplementary Table 6). In addition, we validated the gene expression signals using RNA sequencing data in a subset of 374 samples (Supplementary Table 7).
We then performed similar analyses of the cyan module genes in 1,285 German men and women from the Gutenberg Heart Study (GHS) using monocytes enriched from whole blood with the RosetteSep monocyte enrichment cocktail (24). Seven of the 11 cyan module genes (ABCG1, MYLIP, SQLE, ABCA1, SC4MOL, CYP51A1, and FDFT1) were replicated in the GHS cohort (genome-wide FDR <0.05) (Supplementary Table 8). However, the cyan module was not detected in the GHS, which could be due to the expected low purity of monocytes in the GHS given the different methods we had for monocyte collection (see the Supplementary Data).
Weight Loss and the Cholesterol Metabolism Gene Network
We further investigated the effects of a 5-month weight-loss intervention on cyan module gene expression in 16 obese older adults. Mean weight loss was –6.7 ± 1.1%, and the HOMA measure of insulin resistance decreased an average of 33 ± 8%. The intervention significantly downregulated SQLE (P = 0.04), whereas its association with other members of the cyan modules did not reach statistical significance (P = 0.06–0.83) (Supplementary Table 9), although the effect directions were all consistent with the observed BMI associations in MESA. Furthermore, changes in the HOMA measure of insulin resistance were inversely associated with changes in ABCA1 and MYLIP (P = 0.05), whereas the association of HOMA changes with other members of the cyan modules did not reach statistical significance (P = 0.08–0.95) (Supplementary Table 9), although the effect directions were all consistent with the observed T2D associations in MESA.
Associations of the Cholesterol Metabolism Gene Network With T2D and CAC
To determine whether the cyan network (or any of the other BMI-associated networks) was related to T2D or cardiovascular disease, we examined the association between the eigengene of each BMI-associated network and prevalent T2D (excluding individuals with impaired fasting glucose) or CAC in the MESA participants using a Bonferroni-adjusted significance threshold of P < 0.003 (0.05 of 15 BMI-associated networks). This analysis showed the cyan module was the most significantly associated with T2D and CAC (Fig. 2 and Supplementary Tables 4a and b). These associations did not differ by the status of stain use (Supplementary Table 10) and were attenuated but significant after adjustment for statin use (T2D: odds ratio per SD increment, 1.47 [P = 2.6 × 10−5]; CAC: fold change per SD increment, 1.13 [P = 0.046]). To alleviate the concern that the detection of the cyan module may be influenced by statin use or preselection of BMI-associated genes, we performed an unsupervised WGCNA using the full set of genes after adjusting the expression values for the statin effect and confirmed that the cyan module remained present.
We also created a cyan module risk score, defined as the number of risk genes for each participant. There was an increase in the odds of having T2D (odds ratio 5.4; 95% CI 3.0–9.4; P = 5.9 × 10−9; excluding individuals with impaired fasting glucose) associated with a risk score of 9–11 versus a score of 0–2 genes, and the test for a linear trend across the full range of the risk score (0–11) was also highly significant (Ptrend = 5.07 × 10−10) (Fig. 4). Likewise, the amount of CAC was significantly increased in subjects with a score of 9–11 vs. 0–2 (fold change 2.1; 95% CI 1.4–3.2; P = 0.001), and the corresponding Ptrend was 2.02 × 10−3. These associations were attenuated but remained significant after additional adjustment for smoking, BMI, HDL cholesterol, triglycerides, systolic blood pressure, and physical activity. BMIs were positively associated with T2D (P = 3.43 × 10−21) and CAC (P = 6.05 × 10−4). Mediation analyses using robust SEM showed significant indirect effects of BMI on T2D and CAC that were mediated through the eigengene of the cyan network (P = 9.27 × 10−4 and P = 0.015, respectively). However, this type of mediation analysis is not sufficient to derive a causal relationship because we cannot rule out other possible competing causal models.
Figure 4.

Odds of T2D and amount of CAC increase as intracellular cholesterol accumulation risk score increases. Odds ratio of T2D (individuals with impaired fasting glucose were excluded) (A) and fold change of CAC (B) (with 95% CI), by risk gene dosage group. Dosage groups defined by the number of “risk genes” (expression levels predicted to promote intracellular cholesterol accumulation) from the cyan network: reference group (ref.) = 0–2, low = 3–5, medium = 6–8, high = 9–11. Model 1 was adjusted for age, sex, race, and study sites; model 2 was adjusted for all variables in model 1 plus smoking, HDL cholesterol, triglycerides, systolic blood pressure, and physical activity; and model 3 was adjusted for all variables in model 2 plus BMI.
Associations of the Cholesterol Metabolism Gene Network With Inflammatory Mediators
To investigate systemic inflammation in relation to the cholesterol metabolism gene network variation, we also tested systemic inflammatory markers, plasma IL-6 and CRP, measured at MESA exam 1, as correlates for the gene expression network profiles measured 9 years later in monocytes (MESA exam 5). In these analyses, the cyan module eigengene significantly associated with logIL-6 (β = 0.007, P = 2.0 × 10−7) and logCRP measurements (β = 0.004, P = 1.7 × 10−7) (Supplementary Table 4b).
Methylation Associations With the Cholesterol Metabolism Gene Network
To investigate epigenetic modification of DNA as a potential regulator of the cholesterol metabolism gene expression, we integrated genome-wide DNA methylation data from our methylomics of gene expression study (25), which was measured in the same 1,264 monocyte samples, and tested for associations between expression of the cholesterol metabolism network genes and DNA methylation of CpG sites located near the network genes.
Using an FDR threshold of 0.001 (a more stringent FDR threshold used to retain fewer false-positives), we identified 30 CpG sites (of 6,253 CpG sites located within 1 MB of any cyan network gene) that were cis-gene expression–associated methylation sites (eMS), whose degree of methylation was linked to the expression of 9 of the cyan network genes (ABCA1, ABCG1, CYP51A1, FADS1, FDFT1, LDLR, SC4MOL, SCD, and SQLE) (Supplementary Table 11). Among these 30 eMS, only 4 CpG sites had methylation profiles also associated with BMI (cg06500161, cg05323251, cg10192877, and cg05119988, FDR <0.001) (Table 1). Mediation analyses, using SEM and accounting for population and technical covariates, provided statistical evidence that the relationship between BMI and the expression of three cyan module genes (ABCG1, SC4MOL, and LDLR) was at least partially mediated through methylation (see “Indirect” effects in Table 1). ABCG1 harbored two eMS, including the eMS most strongly correlated with BMI (cg06500161, P = 2.8 × 10−7, FDRepigenome-wide = 5.4 × 10−4) (Supplementary Fig. 2 and Table 1). Methylation profiles of these two ABCG1-eMS were independently associated with decreasing expression of ABCG1 (from multiple regression analysis, P < 0.05). ABCG1-eMS are located within the ABCG1 gene body and occupy ENCODE-annotated regulatory sites (12,13), characterized by features indicative of active cis-regulatory sequences (DNaseI hypersensitive hotspots and enhancer histone methylation marks, H3K4m1/H3K4m3). In a subset of 176 samples, pyrosequencing-based validation of the ABCG1-eMS corroborated the association between BMI and methylation of cg06500161 (P = 3.35 × 10−4 for pyrosequencing methylation vs. BMI).
Table 1.
eMS that potentially mediate the relationship between BMI and cholesterol network gene expression
| eMS | BMI-methylation |
BMI-gene expression |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Indirect |
Direct |
|||||||||
| CpG ID | CpG location | β | SE | P value | FDR | Gene | β | P value | β | P value |
| cg06500161 | Body (ABCG1) | 0.005 | 0.001 | 2.8E-07 | 5.4E-04 | ABCG1 | −0.005 | 2.6E-06 | −0.025 | 3.7E-12 |
| cg05323251 | Body (ICAM3) | 0.003 | 0.001 | 3.0E-06 | 2.9E-03 | LDLR | 0.001 | 2.4E-03 | 0.012 | 1.5E-07 |
| cg10192877 | Body (ABCG1) | 0.004 | 0.001 | 5.3E-05 | 0.02 | ABCG1 | −0.002 | 1.1E-03 | −0.025 | 3.6E-14 |
| cg05119988 | 5′UTR (SC4MOL) | −0.006 | 0.001 | 1.0E-04 | 0.03 | SC4MOL | 0.001 | 1.2E-03 | 0.009 | 1.2E-06 |
Effects of BMI on methylation of cyan module gene eMS (β, SE, P value, and genome-wide FDR) and the indirect (mediated) and direct effects (β, P value) of BMI on gene expression. Indirect effects were estimated using SEM for methylation mediating the effect of BMI on gene expression (log2 fold change of expression per 1-unit increase in BMI [kg/m2]). Some eMS were located within gene bodies (gene containing CpG in parentheses) or in the 5′ untranslated regions (UTR) of genes. Analyses included 1,264 CD14+ monocyte samples and were adjusted for age, sex, race, study site, and residual sample contamination with nonmonocytes.
Discussion
In summary, we discovered a large number of genes whose expression is strongly associated with BMI. Notable among these genes is a well-defined network of coexpressed cholesterol metabolism genes (the cyan network) whose coordinated action would be expected to increase intracellular cholesterol content. Furthermore, we discovered that expression of the coexpressed cholesterol metabolism genes at exam 5 was also correlated with IL-6 and CRP measured at exam 1, although our data cannot infer their temporal relationships. These findings are intriguing because cellular cholesterol homeostasis is expected to be tightly regulated by a cholesterol-mediated feedback system that depends on two transcription factors, sterol response element–binding proteins and liver X receptor-α. When plasma LDL levels are high, more LDL uptake by cells through the LDL receptor potentially leads to a compensatory downregulation of cellular cholesterol synthesis and uptake genes, which may explain the observed negative relationship between the plasma LDL levels and the expression of cholesterol synthesis and uptake genes. However, this negative feedback regulation of cholesterol metabolism can be overridden by inflammatory stress. In vitro and in vivo studies confirm that inflammatory stress results in altered expression of the network genes and can increase intracellular cholesterol content in several cell types, including hepatocytes, vascular smooth muscle cells, and macrophages (26–30). In addition, human experimental data from us (monocytes) and others (adipose tissue) (31–34) show that weight loss rebalanced the expression of members of the cholesterol metabolism gene network. These data suggest that obesity and/or inflammatory stress may predispose intracellular cholesterol accumulation.
Importantly, we discovered that the same lipid metabolic coexpression network is also strongly associated with both CAC and T2D. Cholesterol loading of macrophages (foam cell formation) is typically viewed as a critical cellular component of the atherosclerotic lesion (35), whereas the pathophysiology of obesity-related T2D involves fatty acids and triglycerides rather than cholesterol as in atherosclerosis. However, several lines of recent experimental evidence (36–43) have shown that disruption of cellular cholesterol homeostasis in several cell types (e.g., macrophage, β-cell, adipocyte) can lead to pathological processes preceding T2D. Our data for the first time made the link between potential disruption of cellular cholesterol homeostasis and CAC or T2D using human monocytes in the population setting. The longitudinal relationships between changes in members of the cyan module gene expression and changes in the HOMA measure observed in the small weight-loss intervention study indirectly support the cross-sectional findings in MESA. Taken together with the aggregation of previously reported evidence, our data suggest an emerging hypothesis that the cholesterol metabolism gene network represents a molecular link between obesity/inflammation and its most important complications—T2D and cardiovascular disease. If confirmed, it will provide a rationale for developing novel therapeutics targeting intracellular cholesterol by systematically modulating this cholesterol metabolism gene network, rather than individual genes, for optimizing the prevention and treatment of obesity-related diseases.
We also report a convergence of DNA methylation and gene expression data. Our findings of BMI-associated increases in methylation that are linked to expression alterations of several members of the cholesterol metabolism gene network, coupled with the mediation analyses, suggest a hypothesis that obesity leads to changes in the molecular architecture of monocytes, in part through alteration of DNA methylation. ABCG1 plays a critical role in mediating cholesterol efflux and preventing cellular lipid accumulation (44); however, effects of genetic polymorphisms on ABCG1 expression have not been reported from expression quantitative trait loci studies or observed in our study. The predicted regulatory regions of ABCG1 that harbor expression-associated methylation sites (such as the genomic loci surrounding cg06500161) merit special attention as potential targets for novel therapies designed to attenuate the adverse metabolic and clinical effects of obesity. Additional functional evaluation of the relationship between methylation of this genomic loci and gene expression is warranted.
As a cross-sectional study, the associations observed in MESA do not support causation (in either direction). Although our weight-loss intervention data support that obesity precedes the cholesterol metabolism gene changes in monocytes, inferences should be made with caution due to the small sample size and lack of a nonweight-loss and nonexercise control group. We also cannot discern whether obesity itself or other unmeasured obesity-related factors are directly related to the expression of the cholesterol metabolism genes. Furthermore, the weight-loss intervention trial cannot reveal the temporal relationships between the cholesterol metabolism gene network and cardiometabolic traits. More research is needed to further clarify the causes as well as the functional and clinical consequences of obesity-related alterations in expression of this complex network of genes. BMI is a commonly used indicator of general obesity but is suboptimal for estimation of visceral adiposity, which may be the best predictor for obesity-related metabolic and cardiovascular risks (45). However, our data showed that the BMI associations were similar to the associations with waist circumference, a surrogate of visceral adiposity (46).
A distinction of our study is the use of purified circulating monocytes, as opposed to mixed human cell types (e.g., whole blood or adipose tissue) (47–49), which may produce type I and type II errors (50,51). We choose to study monocytes because they are key cells of innate immunity and major contributors to the pathogenesis of inflammatory diseases, including T2D and cardiovascular disease (52). The involvement of circulating monocytes has been further indicated by other lines of evidence. For example, high-fat meals can promote lipid loading in circulating monocytes even before their migration into tissues and differentiation to macrophages (53). Another plausible hypothesis is that the associations with the cholesterol metabolism network genes that we have observed in monocyte samples may also reflect changes in other tissues or cells that are not accessible in the population setting (e.g., hepatic cells, β-cells). Supporting this notion, inflammatory stimuli produced gene expression changes in several cell types, including hepatocytes and vascular smooth muscle cells (27,30), which are consistent with the correlations we observed between IL-6 and the network genes in human monocytes samples. We and others have also highlighted the importance of β-cell cholesterol homeostasis in T2D (41,43,). Collectively, our data suggest that circulating monocytes can serve as a useful in vivo model to better understand complex molecular mechanisms underlying obesity-related diseases.
Supplementary Material
Article Information
Acknowledgments. The authors thank the other investigators, the staff, and the participants of the MESA study for their valuable contributions and thank Carole Grenier (Duke University, Durham, NC) for provision of technical support. A full list of participating MESA investigators and institutions can be found at http://www.mesa-nhlbi.org.
Funding. The MESA Epigenomics and Transcriptomics Study was funded by National Institutes of Health grant R01-HL-101250 to Wake Forest University Health Sciences. The weight-loss intervention study was supported by the National Institutes of Health grant R01-HL-093713 and the Wake Forest University Claude D. Pepper Older Americans Independence Center (P30-AG21332). This research was supported by National Heart, Lung, and Blood Institute contracts N01-HC-95159 through N01-HC-95165 and N01-HC-95169 and by National Center for Research Resources grants UL1-RR-024156 and UL1-RR-025005.
Duality of Interest. A provisional patent application relevant to this work has been filed. No other potential conflicts of interest relevant to this article were reported.
Author Contributions. For MESA investigators: J.D. performed experiments, researched the data, and wrote the manuscript. L.M.R., R.P.T., I.H., D.H., and C.E.M. performed experiments, researched the data, and helped write the manuscript. K.L., B.J.N., S.B.K., Z.H., A.d.l.F., N.S., R.E.S., C.-C.C., T.H., J.S.P., S.M., D.R.J., and W.P. performed experiments and researched the data. N.X., M.O.G., Y.-D.I.C., J.I.R., and D.S.S. researched the data. Y.L. oversaw the study, carried out experiments, researched the data, and wrote the manuscript. For GHS investigators: T.Z. and P.S.W. acquired data. T.Z. and C.M. analyzed and interpreted data. T.Z., C.M., and S.B. reviewed and edited the manuscript. P.S.W. and S.B. obtained funding. T.Z. and S.B. supervised the study. Y.L. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db14-1314/-/DC1.
References
- 1.Rocha VZ, Libby P. Obesity, inflammation, and atherosclerosis. Nat Rev Cardiol 2009;6:399–409 [DOI] [PubMed] [Google Scholar]
- 2.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003;112:1796–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 2003;112:1821–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arkan MC, Hevener AL, Greten FR, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med 2005;11:191–198 [DOI] [PubMed] [Google Scholar]
- 5.Hotamisligil GS. Inflammation and metabolic disorders. Nature 2006;444:860–867 [DOI] [PubMed] [Google Scholar]
- 6.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011;145:341–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature 2010;465:721–727 [DOI] [PubMed] [Google Scholar]
- 8.Bild DE, Bluemke DA, Burke GL, et al. Multi-ethnic study of atherosclerosis: objectives and design. Am J Epidemiol 2002;156:871–881 [DOI] [PubMed] [Google Scholar]
- 9.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 2003;100:9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dennis G Jr, Sherman BT, Hosack DA, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 2003;4:3. [PubMed] [Google Scholar]
- 12.Dunham I, Kundaje A, Aldred SF, et al.; ENCODE Project Consortium . An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489:57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenbloom KR, Sloan CA, Malladi VS, et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res 2013;41:D56–D63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 2012;13:484–492 [DOI] [PubMed] [Google Scholar]
- 15.Rosseel Y. lavaan: an R package for structural equation modeling. J Stat Softw 2012;48:1–36 [Google Scholar]
- 16.Nicklas BJ, Gaukstern JE, Legault C, Leng I, Rejeski WJ. Intervening on spontaneous physical activity to prevent weight regain in older adults: design of a randomized, clinical trial. Contemp Clin Trials 2012;33:450–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985;28:412–419 [DOI] [PubMed] [Google Scholar]
- 18.Brown MS, Goldstein JL. Receptor-mediated endocytosis: insights from the lipoprotein receptor system. Proc Natl Acad Sci U S A 1979;76:3330–3337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zelcer N, Hong C, Boyadjian R, Tontonoz P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science 2009;325:100–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flowers MT, Ntambi JM. Role of stearoyl-coenzyme A desaturase in regulating lipid metabolism. Curr Opin Lipidol 2008;19:248–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glaser C, Heinrich J, Koletzko B. Role of FADS1 and FADS2 polymorphisms in polyunsaturated fatty acid metabolism. Metabolism 2010;59:993–999 [DOI] [PubMed] [Google Scholar]
- 22.Seth G, McIvor RS, Hu WS. 17Beta-hydroxysteroid dehydrogenase type 7 (Hsd17b7) reverts cholesterol auxotrophy in NS0 cells. J Biotechnol 2006;121:241–252 [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Collins HL, Ranalletta M, et al. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J Clin Invest 2007;117:2216–2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeller T, Wild P, Szymczak S, et al. Genetics and beyond—the transcriptome of human monocytes and disease susceptibility. PLoS One 2010;5:e10693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, Ding J, Reynolds LM, et al. Methylomics of gene expression in human monocytes. Hum Mol Genet 2013;22:5065–5074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao L, Chen Y, Tang R, et al. Inflammatory stress exacerbates hepatic cholesterol accumulation via increasing cholesterol uptake and de novo synthesis. J Gastroenterol Hepatol 2011;26:875–883 [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, Moser AH, Shigenaga JK, Grunfeld C, Feingold KR. Downregulation of liver X receptor-alpha in mouse kidney and HK-2 proximal tubular cells by LPS and cytokines. J Lipid Res 2005;46:2377–2387 [DOI] [PubMed] [Google Scholar]
- 28.Khovidhunkit W, Kim MS, Memon RA, et al. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res 2004;45:1169–1196 [DOI] [PubMed] [Google Scholar]
- 29.Ruan XZ, Moorhead JF, Fernando R, Wheeler DC, Powis SH, Varghese Z. Regulation of lipoprotein trafficking in the kidney: role of inflammatory mediators and transcription factors. Biochem Soc Trans 2004;32:88–91 [DOI] [PubMed] [Google Scholar]
- 30.Chen Y, Ku H, Zhao L, et al. Inflammatory stress induces statin resistance by disrupting 3-hydroxy-3-methylglutaryl-CoA reductase feedback regulation. Arterioscler Thromb Vasc Biol 2014;34:365–376 [DOI] [PubMed] [Google Scholar]
- 31.Mutch DM, Pers TH, Temanni MR, et al.; DiOGenes Project . A distinct adipose tissue gene expression response to caloric restriction predicts 6-mo weight maintenance in obese subjects. Am J Clin Nutr 2011;94:1399–1409 [DOI] [PubMed] [Google Scholar]
- 32.Dahlman I, Linder K, Arvidsson Nordström E, et al. Changes in adipose tissue gene expression with energy-restricted diets in obese women. Am J Clin Nutr 2005;81:1275–1285 [DOI] [PubMed] [Google Scholar]
- 33.Johansson LE, Danielsson AP, Parikh H, et al. Differential gene expression in adipose tissue from obese human subjects during weight loss and weight maintenance. Am J Clin Nutr 2012;96:196–207 [DOI] [PubMed] [Google Scholar]
- 34.Shea J, French CR, Bishop J, et al. Changes in the transcriptome of abdominal subcutaneous adipose tissue in response to short-term overfeeding in lean and obese men. Am J Clin Nutr 2009;89:407–415 [DOI] [PubMed] [Google Scholar]
- 35.Tabas I. Consequences of cellular cholesterol accumulation: basic concepts and physiological implications. J Clin Invest 2002;110:905–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bie J, Zhao B, Song J, Ghosh S. Improved insulin sensitivity in high fat- and high cholesterol-fed Ldlr-/- mice with macrophage-specific transgenic expression of cholesteryl ester hydrolase: role of macrophage inflammation and infiltration into adipose tissue. J Biol Chem 2010;285:13630–13637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bie J, Zhao B, Marqueen KE, Wang J, Szomju B, Ghosh S. Macrophage-specific transgenic expression of cholesteryl ester hydrolase attenuates hepatic lipid accumulation and also improves glucose tolerance in ob/ob mice. Am J Physiol Endocrinol Metab 2012;302:E1283–E1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Le Lay S, Ferré P, Dugail I. Adipocyte cholesterol balance in obesity. Biochem Soc Trans 2004;32:103–106 [DOI] [PubMed] [Google Scholar]
- 39.Yu BL, Zhao SP, Hu JR. Cholesterol imbalance in adipocytes: a possible mechanism of adipocytes dysfunction in obesity. Obes Rev 2010;11:560–567 [DOI] [PubMed] [Google Scholar]
- 40.de Haan W, Bhattacharjee A, Ruddle P, Kang MH, Hayden MR; de . ABCA1 in adipocytes regulates adipose tissue lipid content, glucose tolerance, and insulin sensitivity. J Lipid Res 2014;55:516–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fryirs M, Barter PJ, Rye KA. Cholesterol metabolism and pancreatic beta-cell function. Curr Opin Lipidol 2009;20:159–164 [DOI] [PubMed] [Google Scholar]
- 42.Maxfield FR, Tabas I. Role of cholesterol and lipid organization in disease. Nature 2005;438:612–621 [DOI] [PubMed] [Google Scholar]
- 43.Brunham LR, Kruit JK, Pape TD, et al. Beta-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat Med 2007;13:340–347 [DOI] [PubMed] [Google Scholar]
- 44.Kennedy MA, Barrera GC, Nakamura K, et al. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab 2005;1:121–131 [DOI] [PubMed] [Google Scholar]
- 45.Britton KA, Massaro JM, Murabito JM, Kreger BE, Hoffmann U, Fox CS. Body fat distribution, incident cardiovascular disease, cancer, and all-cause mortality. J Am Coll Cardiol 2013;62:921–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Després JP, Lemieux I. Abdominal obesity and metabolic syndrome. Nature 2006;444:881–887 [DOI] [PubMed] [Google Scholar]
- 47.Emilsson V, Thorleifsson G, Zhang B, et al. Genetics of gene expression and its effect on disease. Nature 2008;452:423–428 [DOI] [PubMed] [Google Scholar]
- 48.Hulsmans M, Geeraert B, De Keyzer D, et al. Interleukin-1 receptor-associated kinase-3 is a key inhibitor of inflammation in obesity and metabolic syndrome. PLoS One 2012;7:e30414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feinberg AP, Irizarry RA, Fradin D, et al. Personalized epigenomic signatures that are stable over time and covary with body mass index. Sci Transl Med 2010;2:49ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhong H, Yang X, Kaplan LM, Molony C, Schadt EE. Integrating pathway analysis and genetics of gene expression for genome-wide association studies. Am J Hum Genet 2010;86:581–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dimas AS, Deutsch S, Stranger BE, et al. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science 2009;325:1246–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol 2011;11:762–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Varela LM, Ortega A, Bermudez B, et al. A high-fat meal promotes lipid-load and apolipoprotein B-48 receptor transcriptional activity in circulating monocytes. Am J Clin Nutr 2011;93:918–925 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.