

Abstract
In the nitrogenase molybdenum-iron (MoFe) protein, we have identified five potential substrate access pathways from the protein surface to the FeMo-cofactor (the active site) or the P-cluster using experimental structures of Xe pressurized into MoFe protein crystals from Azotobacter vinelandii and Clostridium pasteurianum. Additionally, all published structures of the MoFe protein, including those from Klebsiella pneumoniae, were analyzed for the presence of nonwater, small molecules bound to the protein interior. Each pathway is based on identification of plausible routes from buried small molecule binding sites to both the protein surface and a metallocluster. Of these five pathways, two have been previously suggested as substrate access pathways. While the small molecule binding sites are not conserved among the three species of MoFe protein, residues lining the pathways are generally conserved, indicating that the proposed pathways may be accessible in all three species. These observations imply that there is unlikely a unique pathway utilized for substrate access from the protein surface to the active site; however, there may be preferred pathways such as those described here.
Nitrogen fixation is the process by which atmospheric dinitrogen (N2) is reduced to a biologically active form of nitrogen, ammonia (NH3). This reaction is achieved on the industrial scale by the Haber–Bosch process, producing enough ammonia for nitrogen fertilizers to sustain 27–40% of the world’s population.1 Because of the dependence of the Haber–Bosch process on molecular hydrogen obtained from natural gas, this process accounts for more than 1.5% of the global energy consumption each year.2 Industrial nitrogen fixation uses heterogeneous iron catalysts, pressures near 250 atm, and temperatures between 400 and 600 °C to reduce dinitrogen.2,3 In contrast, the biological catalyst, nitrogenase, reduces N2 to NH3 at ambient temperature and atmospheric pressure. Understanding the process by which nitrogenase functions may facilitate the development of environmentally cleaner alternatives to the Haber–Bosch process, making nitrogenase an attractive enzyme to study for biotechnological NH3 production. Although the enzyme has been studied for many decades, the detailed mechanism of N2 reduction remains poorly understood. For example, certain stoichiometric aspects of the standard model of biological nitrogen fixation (Scheme 1) are still under discussion, including the ATP/e– ratio and the obligatory nature of H2 evolution.4 Furthermore, N2 reduction requires at least six protons, but the specific form of ammonia evolved (NH3 versus NH4+) and the possibility of H2 evolution may require up to 10 or more protons for N2 reduction.
Scheme 1. Standard Model for Biological Nitrogen Fixation.
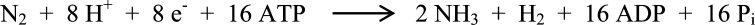
Nitrogenase consists of two proteins: the hetero-tetrameric molybdenum-iron (MoFe) protein and the homodimeric iron (Fe) protein. The Fe protein houses two ATP binding sites and the [4Fe:4S] cubane cluster. The MoFe protein consists of two αβ dimers and contains three types of metal centers: (1) two [8Fe:7S] “P-clusters” at the α- and β-subunit interfaces, (2) two active sites, a [7Fe:9S:Mo:C:R-homocitrate] cluster called the FeMo-cofactor in the α-subunits, and (3) two mononuclear iron sites, named Fe16, between the β- and β′-subunits.5 During substrate turnover, electrons flow from the [4Fe:4S] cluster to the P-cluster to the FeMo-cofactor, at which most, if not all, substrate reduction occurs upon sufficient buildup of protons and electrons.6 Considering only the inorganic components, the FeMo-cofactor adopts near C3v symmetry, with a central, trigonal prismatic core composed of three faces and three edges parallel to the C3 axis that are made from six Fe atoms, numbered Fe2, 3, 4, 5, 6, and 7. A particular face may be identified by listing the four Fe atoms composing that face, such as Fe2,3,6,7. Similarly, an edge is identified by listing the two Fe atoms composing that edge. Examined in isolation, the edges and faces would be indistinguishable from each other, but variation in neighboring protein residues creates nonequivalent environments around the FeMo-cofactor inside the protein. The edges and faces may therefore each have different mechanistic roles. For example, CO has been shown to bind in a bridging fashion to Fe2,6.7
In this work, five possible pathways for substrate access from the protein surface to the FeMo-cofactor or P-cluster are experimentally identified. To accomplish this, Azotobacter vinelandii (Av) and Clostridium pasteurianum (Cp) MoFe protein crystals were pressurized with xenon (Xe) gas. Additionally, all published structures of the MoFe protein, including those from Klebsiella pneumoniae (Kp), were analyzed for the presence of nonwater, small molecules bound to the protein interior. The native Av, Cp, and Kp MoFe proteins are called Av1, Cp1, and Kp1, respectively, and the corresponding Xe-pressurized proteins are referred to as Av1-Xe and Cp1-Xe. Kp1 and Av1 are structurally similar (73% sequence identity), while their comparison to Cp1 shows an insertion and a deletion, each ∼50 residues, as well as primary structure differences (36% sequence identity between Cp1 and Av1).
Although Xe is monatomic, unlike nitrogenase substrates, several advantages exist for using Xe as a model for nitrogenase substrates such as N2: (1) Xe and N2 are neutral, polarizable, water-soluble, and unable to form hydrogen bonds; (2) the atomic radius of Xe (1.08 Å) is comparable to the NN bond distance (1.10 Å), so sterically, N2 may travel similar pathways as those penetrable by Xe; and (3) Xe is easily detected by X-ray crystallography due to its high electron density and strong anomalous scattering. Furthermore, Xe pressurization is well-established as a tool for probing gaseous substrate pathways in several biological molecules, including myoglobin,8−11 copper amine oxidase,12,13 laccase,14 methane monooxygenase hydroxylase,15,16 cytochrome c oxidase,17 cytochrome ba3 oxidase,18,19 acetyl-CoA synthase/carbon monoxide dehydrogenase,20 and antibodies that oxidize water.21 Finally, several molecular dynamic simulations support the use of Xe binding sites as markers for substrate pathways.22−26
X-ray crystallography, 129Xe-NMR, and computational work on previous Xe studies indicate that Xe typically induces little distortion in the protein structure and occupies existing cavities in a protein either by displacing water molecules or filling otherwise empty pockets.8−10,12−15,17,20,21 Reflecting the inertness of Xe, it tends to bind to the protein using mostly noncovalent, weak van der Waals forces with limited polarization interactions.22 The Xe binding sites are usually hydrophobic, and the closest contacts (3.5–6.0 Å) are typically aliphatic and aromatic side-chains but can be polar groups.8−10,12−15,17,20,21 These studies also show that pathways tend to travel parallel to secondary structure elements rather than through them.22
Previous studies on nitrogenase have identified four possible pathways to the active site. First, a water channel extending from the protein surface to the FeMo-cofactor, called the interstitial channel, was identified from structural analysis of Av1, Cp1, and Kp1, and has been expected to facilitate access to the active site for protons and possibly larger substrates.27−31 Use of the water-filled interstitial channel as an access pathway to the active site does not preclude the existence of other pathways since nonpolar substrates may prefer a less polar route. Second, Seefeldt and co-workers used the program CAVENV from the CCP4 suite with a probe radius of 2.5 Å to identify a hydrophobic substrate pathway, herein called the IS pathway (for authors Igarashi and Seefeldt).32 The third previously proposed pathway, called the NH3 egress pathway by its authors, was identified by a computational cavity analysis of Av1 and Kp1; it extends through the protein scaffold from the β-subunit surface to the FeMo-cofactor.30 Fourth, molecular dynamic calculations yielded a possible substrate pathway that traces the shortest path from the protein surface to the FeMo-cofactor.26 Like the NH3 egress and IS pathways, it does not utilize any water channels but rather tunnels through the protein scaffold. Since nitrogenase has a relatively leisurely turnover rate of about 1 N2/sec per active site, migration through the protein scaffold in the absence of permanent pathways should not be rate limiting, by analogy to O2 binding to the buried heme of myoglobin and hemoglobin.33,34 Compared to the four previously proposed pathways, only the interstitial water channel and the IS pathway coincide with pathways identified in the present work, suggesting that there are multiple potential pathways connecting the surface to the active site of nitrogenase.
Experimental Procedures
Cell Growth and Protein Purification
Av1 and Cp1 protein were obtained using cell growth and protein purification procedures previously described.35,36
Crystallization
Crystals were grown in 24-well plates using the sitting-drop method at room temperature in an anaerobic chamber with an atmosphere of ∼95% argon and ∼5% hydrogen. All crystallization solutions were purged with argon prior to use. Av1 crystals were obtained as described previously.35 For Cp1, the reservoir and crystallization solutions consisted of double-distilled water, 23% polyethylene glycol (MW 3350 g/mol, Hampton Research), 0.2 M lithium citrate (Aldrich), and 5 mM sodium dithionite (J.T. Baker). Several crystals of hexagonal and block morphology formed after 2 days, but only the block crystals diffracted well.
Xenon Pressurization
Outside the anaerobic environment, crystals were slowly lifted through a cryoprotectant layer (Fomblin Y 16/6 mineral oil, Sigma-Aldrich) and then very quickly moved to the pressurization chamber of a homemade gas pressurization device.37 While lightly venting Xe gas (Matheson) through the device, the pressurization chamber was closed. The crystals were pressurized at 14 atm for 10–15 min, after which they were quickly transferred to liquid nitrogen for storage.
Data Collection and Refinement
Diffraction data for Cp1-Xe was collected remotely from the Stanford Synchrotron Radiation Light source (SSRL) on beamline 12-2 with a DECTRIS Pilatus 6 M detector. Reference sets of 1440 diffraction images were collected at 12999.97 eV with an oscillation angle of 0.25° over 360° rotation. To confirm the identity of the Xe sites, diffraction data were also collected at 6690.11 eV using the same strategy. Although well above the L-edge, Xe exhibits significant anomalous scattering at this energy with Δf″ ≈ 10 electrons. Diffraction data for Av1-Xe was collected in-house on a Rigaku MicroMax 007-HF X-ray generator with a Rigaku RAXIS-IV++ detector. All data sets were integrated with the XDS program package.38 Scaling was carried out with the CCP4 suite,39 and phasing was determined by molecular replacement against Av1 (PDB ID 3U7Q) and Cp1 (4WES).36,40 Initial refinement was carried out with CNS,41 and alternative conformations and isotropic B-factors were refined with REFMAC5.42,43 Simulated annealing was performed using PHENIX.44
Determination of Small Molecule Binding Sites
The presence and occupancy of each Xe site were evaluated by examination of electron density maps, anomalous difference Fourier peaks, and the B-factor of Xe and the surrounding residues (Table 2). Electron density and anomalous difference maps are shown for each Xe binding site in Figure S1, Supporting Information.
Table 2. Properties of Small Molecule Binding Sites in Av1 and Cp1 Xe-Pressurized Crystals.
| distancea (Å) to | |||||||
|---|---|---|---|---|---|---|---|
| crystal | site | displaced species in native protein | B-factor (Å2) | occupancy (%) | peak heights in anomalous Fourier map (σ) | FeMo-cofactor | surface |
| Av1-Xe (4WNA) | Xe1 | fills empty pocket | 27.30 | 77 | 18.51 | 11.2 | 10.9 |
| 28.16 | 81 | 20.23 | |||||
| Xe2 | HOH | 31.29 | 49 | 8.48 | 15.1 | 0 | |
| 31.40 | 46 | 7.12 | |||||
| Xe3 | imidazole | 31.48 | 62 | 9.62 | 33.1 | 0 | |
| 29.51 | 54 | 10.84 | |||||
| Cp1-Xe (4WN9) | Xe1 | HOH | 41.89 | 28 | 5.20 | 28.1 | 0 |
| 39.52 | 34 | 5.78 | |||||
| Xe2 | MPD | 64.15 | 46 | 5.93 | 23.8 | 0 | |
| 44.89 | 50 | 7.64 | |||||
| Xe3 | HOH | 29.20 | 22 | 4.18 | 13.6 | 7.4 | |
| 33.79 | 19 | 5.53 | |||||
| PRL | HOH | 20.65 | 100 | N/A | 24.5 | 7.7 | |
| 23.76 | 100 | N/A | |||||
Distances were measured from Xe or PRL to the closest nonsolvent atom in the FeMo-cofactor and at the protein surface.
Nonprotein electron density was evident in the Cp1-Xe structure that was modeled as a proline ligand (PRL, Figure S2). We propose PRL for use in the model because it nicely fits the observed electron density; however, the actual identity and origin of this species are not conclusively known. It is unlikely that the electron density represents ill-defined water molecules because neighboring water molecules are well-defined, and 6–7 water molecules would be needed to accurately model the observed electron and difference density, which is more than the space can accommodate. Furthermore, the ring of PRL could favorably interact with the five neighboring aromatic side chains in this binding pocket.45,46 Finally, the acid group could interact with β-Lys424 and β-Glu323 via hydrogen bonding. While we will refer to the species throughout the manuscript as PRL (to distinguish it from proline in the peptide chain), we cannot unambiguously identify the species at this site. As it is clearly a nonwater ligand, however, we include it in the present analysis.
Pathway Calculations and Display
Pathways were calculated using the software CAVER.47 Coordinates of the small molecules were provided as the starting point for pathway calculations. CAVER calculates pathways from the grid point closest to the provided coordinates, so some starting points are slightly offset from the Xe atoms. The probe radius, shell radius, and shell depth were set to 0.5, 4, and 5 Å, respectively. For each small molecule, two pathways were selected: one from the protein surface to the small molecule binding site and the other from the binding site to a cofactor. For any given starting point, many pathways exist; however, the most probable pathways are those with the shortest length and largest width, and are prioritized by CAVER. Pathways are displayed throughout the manuscript as surfaces generated in PyMOL.48
Results and Discussion
Xe sites were determined from the X-ray crystal structures of one Av1 and two Cp1 Xe-pressurized crystals (Figure 1 and Table 1). Similar to previous Xe binding studies,8−10,12−17,20,21 the Xe atoms in the MoFe proteins displace water or other small molecules or fill empty pockets, rather than displacing residue side chains (Table 2). The three Xe sites in Av1-Xe are conserved in both crystallographically independent αβ dimers of the protein, as are the three Xe binding sites in Cp1-Xe. The root-mean-square deviations (RMSD) between the Xe-pressurized protein structures compared to their native structures is ∼0.20 Å. The RMSD of only the Xe binding pockets is between 0.15 and 0.23 Å, indicating little protein distortion from Xe incorporation (Table S1).
Figure 1.
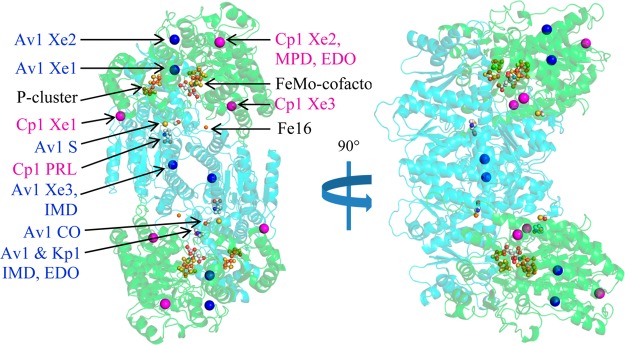
Ribbon representation of Cp1 with the locations indicated for all Xe, PRL, and other small molecule binding sites. The α- and β-subunits are colored in green and cyan, respectively. The FeMo-cofactor, P-cluster, Fe16, and small molecules are displayed as small spheres colored by element. Av1 and Cp1 Xe sites are shown in large blue and magenta spheres, respectively. Binding sites observed in Av1 and Kp1 structures are superposed onto the Cp1 structure. (Cp1-Xe PDB ID: 4WN9; Av1-Xe PDB ID: 4WNA).
Table 1. X-ray Crystallographic Data Collection and Refinement Statistics for Av1 and Cp1.
| Av1-Xe (4WNA) | Cp1-Xe (4WN9) | |||
|---|---|---|---|---|
| Data Collection Statistics | ||||
| resolution range (Å) | 39.62–2.00 (2.00–2.11) | 39.75–1.90 (1.90–1.93) | ||
| wavelength (Å) | 1.5418 | 0.9537 | ||
| space group | P21 | P1 | ||
| unit cell constants | a = 77.12 Å | α = 90° | a = 67.31 Å | α = 73.47° |
| b = 129.8 Å | β = 108.9° | b = 73.45 Å | β = 87.56° | |
| c = 107.5 Å | γ = 90° | c = 108.7 Å | γ = 83.98° | |
| unique reflections | 133045 (6500)a | 137866 (6709)a | ||
| completeness (%) | 98.6 (98.6)a | 96.7 (86.1)a | ||
| redundancy | 3.5 (3.3)a | 3.9 (3.7)a | ||
| I/σ(I) | 9.2 (3.2)a | 16.0 (2.0)a | ||
| Rmerge | 0.079 (0.301)a | 0.055 (0.660)a | ||
| Refinement Statistics | ||||
| protein residues | 1998/2054 | 1951/1984 | ||
| mean B value (Å2) | 23.0 | 31.0 | ||
| Rwork | 0.177 | 0.192 | ||
| Rfree | 0.225 | 0.246 | ||
| Ramachandran outliers | 10 (0.51%) | 14 (0.73%) | ||
| RMSD bond lengths (Å) | 0.009 | 0.008 | ||
| RMSD bond angles (deg) | 1.29 | 1.21 | ||
Numbers in parentheses represent data in the highest resolution shell.
Xe binding sites in Av1-Xe (PDB ID 4WNA) and Cp1-Xe (PDB ID 4WN9), as well as the PRL site in the Cp1-Xe structure, were analyzed for potential access routes to the FeMo-cofactor. Access to the P-cluster was also explored. In addition, imidazole (IMD), ethylene glycol (EDO), carbon monoxide (CO), and sulfur (S) binding sites from previously reported Av1, Cp1, and Kp1 structures were examined (Table S2).7,36,40,49 For all these small molecule binding sites (with the exception of the S site that may be derived from a cofactor sulfur7), we can conclude with certainty that a route from the protein surface to the binding site exists; however, routes from the binding sites to the cofactors are inherently less certain, and indeed, may not exist. Potential pathways were generated using the program CAVER. For reference, Figure 2 and the movie provided in the Supporting Information summarize all of the known water channels and proposed substrate pathways from this study and previously published studies. Residues involved in all pathways are provided in Table S3, and close contacts for each small molecule binding site are provided in Tables S6–S13.
Figure 2.
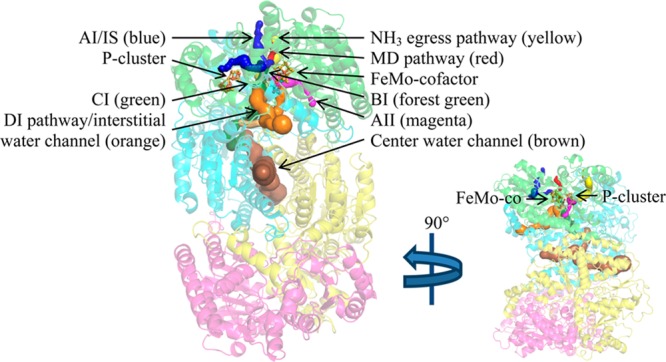
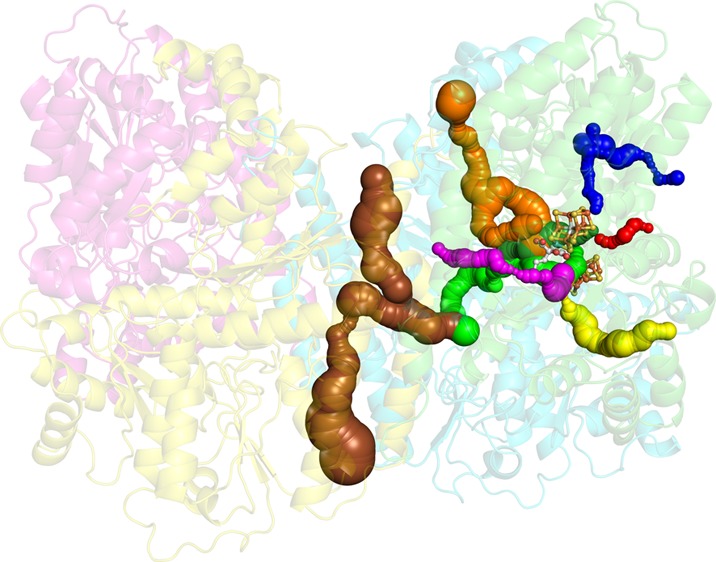
Ribbon representation of Av1 illustrating the channels and pathways discussed in this study, as viewed from two perpendicular orientations, one of which is scaled down. The α-subunits are colored green and magenta, and the β-subunits are colored cyan and yellow. The cofactors are shown in ball-and-stick representation colored by element. Pathways were calculated using CAVER and are displayed as surfaces within the protein structure.47 This figure was created in PyMOL.48
AI/IS and AII Pathways: FeMo-Cofactor Access Based on Xe Binding Sites
Xe binding sites in Cp1 and Av1 were examined to identify potential substrate pathways to the active site. We focused on buried Xe, as these sites have already penetrated into the protein interior. We further focused on Xe sites found in the α-subunit, since these are closer to the FeMo-cofactor compared to the two Xe residing in the β-subunit: Xe in the α-subunit (Av1-Xe1, Av1-Xe2, Cp1-Xe2) are 15, 13, and 23 Å away from the closest Fe atom in the FeMo-cofactor, respectively, while Xe in the β-subunit (Cp1-Xe1 and Av1-Xe3) are 28 and 33 Å away, respectively. These constraints narrow the relevant Xe sites to Av1-Xe1 and Cp1-Xe3.
We propose two substrate pathways based on Av1-Xe1 and Cp1-Xe3, called AI and AII, respectively (Figure 3). In both pathways, two routes were calculated: one from the protein surface to the binding site and another from the binding site to the FeMo-cofactor. The AI pathway, as calculated by CAVER, may include the surface site Av1-Xe2 as the point of substrate penetration through the protein surface; it is in close proximity (11 Å) to Av1-Xe1. A portion of the AI pathway is conserved in the previously published pathway by Seefeldt and co-workers (herein called the IS pathway) using the program CAVENV.32 The IS pathway differs from the AI pathway at the point that it breaches the protein surface. The program utilized in this paper, CAVER, also identified the breaching point of the IS pathway as more favorable than the breaching point of the AI pathway: the average bottleneck radius of the IS and AI pathways from the Av1-Xe1 atom to the protein surface are 0.83 and 0.48 Å, respectively, and the lengths of the pathways are 18.5 and 21.2 Å, respectively. In fact, the IS pathway is the most favored pathway as calculated by CAVER. However, binding of the Av1-Xe2 suggests that there may be multiple entry routes for the AI/IS pathway. Therefore, we present both entry/exit points as possible substrate pathways. Notably, the AI/IS pathway is predominantly hydrophobic, with the exception of residues at the protein surface and a couple around the water surrounding the FeMo-cofactor. These features support Seefeldt’s postulation that this pathway is likely used by nonpolar substrates and/or reduction products.
Figure 3.
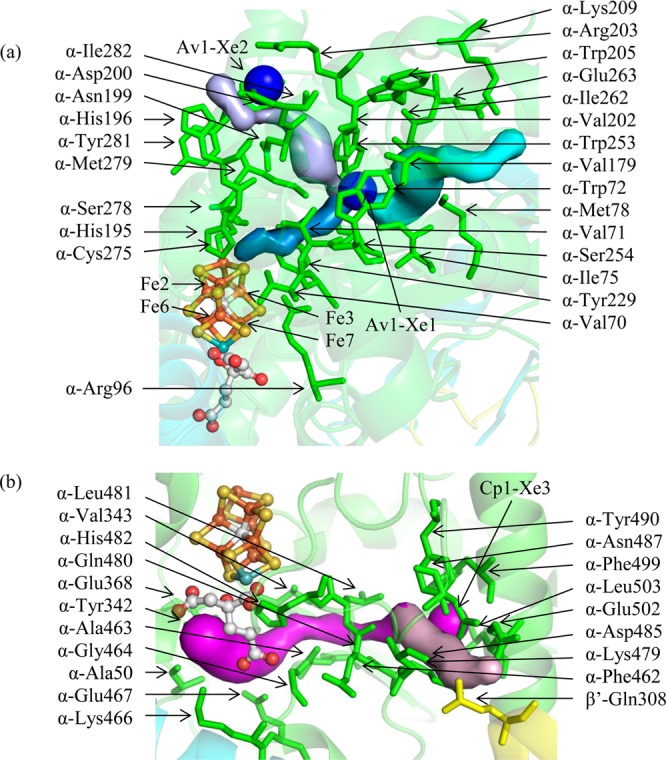
(a) The AI/IS pathway. From the protein surface to the Xe binding site, the AI and IS pathways follow the light purple and cyan pathways, respectively. From the Xe binding site to the FeMo-cofactor, the pathways (slate blue) are the same. Substrates may penetrate the protein surface at the Av1-Xe2 binding site following the light purple pathway or the cyan pathway. Upon reaching the Av1-Xe1 binding site, substrates may continue toward the FeMo-cofactor following the slate blue pathway. The Fe atoms accessed on the FeMo-cofactor by this pathway are labeled. Av1-Xe1 and Av1-Xe2 are displayed as large blue spheres. (b) The AII pathway (magenta and light pink surfaces). Substrates may reach the Cp1-Xe3 binding site following the light pink pathway and then continue toward the FeMo-cofactor following the magenta pathway. Cp1-Xe3 is displayed as a large magenta sphere. In both figures, residues lining the pathways are labeled. The α, β, and β′ subunits are shown in green, cyan, and yellow, respectively.
The AI/IS and AII pathways provide access to two of the three faces of the FeMo-cofactor, namely, the Fe2,3,6,7 and Fe3,4,5,7 faces. Since Xe is nonpolar and the interior Xe sites do not overlap with any polar species from other MoFe crystal structures, the AI and AII pathways may be primarily used by nonpolar substrates and/or reaction products.
BI Pathway: FeMo-Cofactor Access Based on PRL Binding Sites
The nearest neighbors of PRL in Cp1 are five aromatic residues, together with β-Glu323 and β-Lys424 (Figure S2 and Table S12). PRL resides in an arm of the center channel that curves toward the cofactors (Figure 4). The arm terminates before reaching the FeMo-cofactor; thus, substrates would need to continue through the protein scaffold to reach the active site. With CAVER, we deduced a possible substrate pathway from the PRL binding site to the FeMo-cofactor (pathway BI), which accesses the FeMo-cofactor at the Fe2,3,6,7 face.
Figure 4.
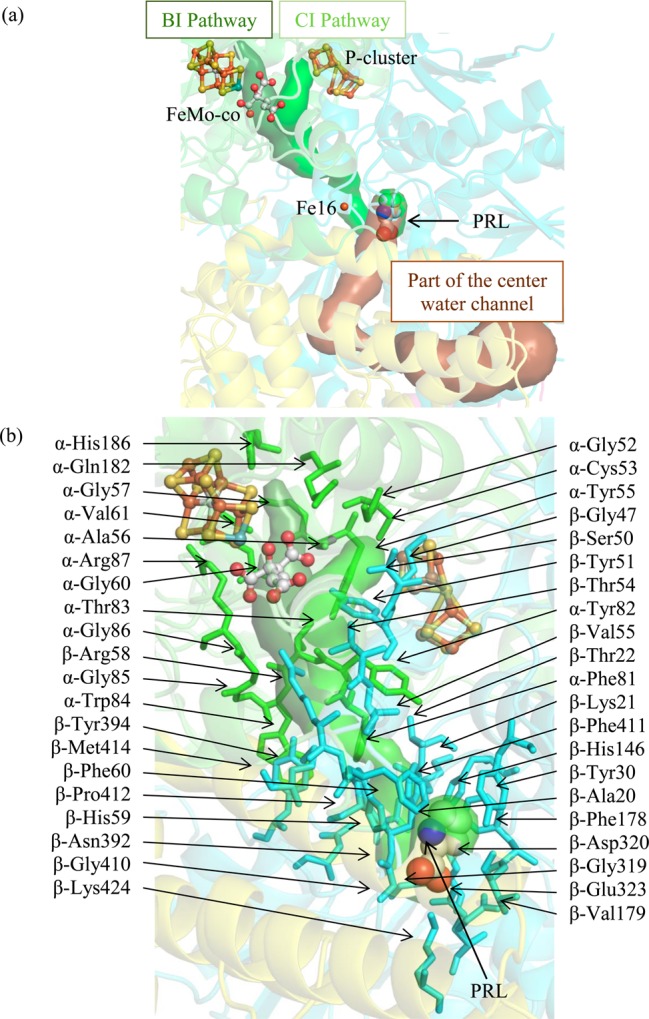
(a) PRL binds in an arm of the center channel (brown surface) that reaches toward the cofactors. The arm terminates before reaching the FeMo-cofactor, so substrates must continue to the cofactors within the protein scaffold. The most likely pathway (in terms of size) is between the α- and β-subunits. Branching from this pathway, substrates may either head toward the FeMo-cofactor (forest green surface, pathway BI) or toward the P-cluster (green surface, pathway CI). (b) A close-up view of the BI and CI pathways leading to the FeMo-cofactor and P-cluster, respectively. In both figures, the α, β, and β′ subunits are shown in green, cyan, and yellow, respectively. Residues lining the pathway are shown in sticks and labeled. The cofactors and PRL are displayed as spheres colored by element.
CI Pathway: P-Cluster Access Based on PRL Binding Sites
Although generally considered as functioning in electron transfer between the Fe protein and FeMo-cofactor, ligand access to and from the P-cluster may be necessary since there is likely elimination of a sulfur atom during P-cluster biosynthesis.50 Also, the redox properties of the P-cluster indicate that it could potentially reduce protons and perhaps other substrates.4,51 A possible substrate pathway, CI, from the PRL site to the P-cluster (Figure 4) was calculated by CAVER. The PRL is 16 Å away from the P-cluster and 24 Å away from the FeMo-cofactor; however, the closest metal center to the PRL binding site is Fe16, at a distance of 14 Å. Although the identity of this third metal site has been confirmed, its function is not currently known.5
Because the volume of the center channel is in excess of 1500 Å3, the walls of the center channel are essentially an extension of the protein surface. As such, the MoFe protein resembles an oblong donut, in which the center channel is the donut hole.52 Water molecules, nonpolar atoms (Xe), and polar molecules (IMD, EDO, MPD) are all found on the protein surface so it is not unexpected that these species have binding sites in the center channel as well. Hence, the BI and CI pathways may facilitate access to the FeMo-cofactor for all species.
DI Pathway/Interstitial Water Channel: FeMo-Cofactor Access Based on IMD, EDO, CO, and S Binding Sites
All published structures of native Av1, Cp1, and Kp1 were investigated for additional nonwater, small molecule binding sites. Those containing small molecules are listed in Table S2. These guest molecules come from crystallization solutions, cryoprotectants, or pressurized gas and include imidazole (IMD), 2-methyl-2,4-pentanediol (MPD), Mg2+, 1,2-ethanediol (EDO), and carbon monoxide (CO); the sulfur (S) may be derived from the FeMo-cofactor.7,36,40,49
We focused on small molecules bound to the protein interior, of which there are five from previously reported MoFe protein structures (Table S2). Several of these bind in or near the interstitial channel (Figure 5), which has been previously proposed to function as a substrate access pathway, as deduced from the conserved water network in Av1, Cp1, and Kp1. The exogenous small molecules observed to bind within this channel include IMD in Av1 (3U7Q) and EDO in Kp1 (1QGU) (Table S4).40,49 The CO and S in Av1 (4TKV) bind in a protrusion from the interstitial channel.7 This protrusion (purple surface in Figure 5) extends through the β-subunit to the protein surface; however, it is narrower and longer than the DI pathway. Therefore, only the short protrusion from the DI pathway containing the CO and S atoms is shown. The IMD and EDO sites directly overlap and are 4.8 and 7.7 Å from the CO and S, respectively. The polarity of IMD, EDO, and CO indicates that this channel may be utilized as an access pathway for polar substrates in addition to water or protons. The DI pathway accesses both the Fe2,3,6,7 and Fe3,4,5,7 faces, which are also accessed by the AI and AII pathways.
Figure 5.
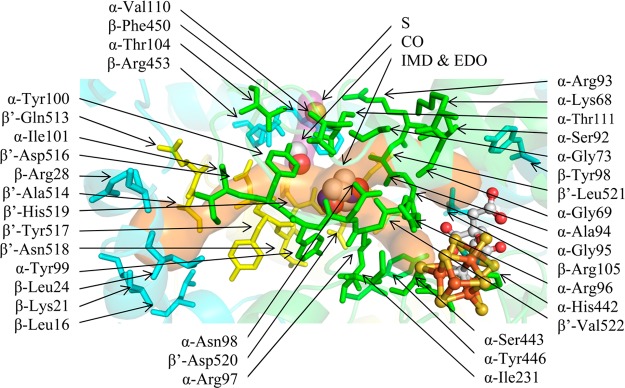
DI pathway (orange surface). The EDO and IMD molecules bind in the DI pathway/interstitial channel that connects the protein surface to the Fe2,3,6,7 and Fe3,4,5,7 FeMo-cofactor faces (orange surface). The CO and S bind in a channel that extends from the DI pathway (purple surface). The α, β, and β′ subunits are shown in green, cyan, and yellow, respectively. Residues lining the pathway are shown in sticks and labeled. The substrates and FeMo-cofactor are displayed as spheres colored by element. The IMD and EDO molecules are superposed in the figure because the binding sites directly overlap.
Pathway Conservation
It is noteworthy that the Av1 and Cp1 Xe binding sites differ, given that Xe is used as an electron dense surrogate for crystallographic analysis of gas binding sites in enzymes. Furthermore, while a diverse set of small molecules have been found to bind to the protein interiors of Cp1, Av1, and Kp1, it is also the case that these binding sites are not identical between the structures. To assess whether the different pathways may be generally relevant to the functioning of nitrogenase or instead primarily reflect the behavior of specific MoFe proteins, the conservations of specific residue and residue type (hydrophobic or hydrophilic) in Av1 and Cp1 were evaluated for (1) all residues in the protein, (2) surface residues, and (3) nonsurface residues (Table S5). This was compared to the conservation of specific residues and residue type for residues lining the substrate binding pockets and proposed pathways (Tables S14–S21). The conservation of specific residue for all nonsurface residues compared to that of residues lining the substrate binding pockets and pathways is 38% and 63% respectively, and the conservation of residue type is 63% and 86%, respectively. This shows higher conservation of specific residue and residue type at the substrate pockets and in the proposed pathways, indicating that the AI, AII, BI, and CI pathways may be accessible in Av1 and Cp1. The differences in Xe and another small molecule binding sites in these structures may reflect details of the surrounding residues that alter the thermodynamics of ligand binding, but not necessarily the dynamic accessibility.
Comparison to Other Forms of the MoFe Protein
The access pathways were compared to two other forms of the MoFe protein. First, examination of the proposed access pathways in complexes of the MoFe and Fe proteins53,54 indicates that the docking of the Fe protein onto the MoFe protein does not block any of the proposed substrate pathways (Figure S3). This suggests that binding of the Fe protein may not sterically interfere with substrate access between the protein interior and exterior. This observation is of interest since the Thorneley–Lowe kinetic model assumes that substrates and products can only bind/leave the free MoFe protein.55 Of course, differences in internal structure or protein dynamics could alter the behavior of the MoFe-protein between free and complexed states. Second, an overlay of the FeMo-cofactor-deficient Av1 protein structure shows that the AII pathway partially overlaps with the channel utilized by the FeMo-cofactor to access its binding pocket (Figure S4).56 This suggests that the funnel between the α-subunit domains mediating the transfer of the FeMo-cofactor into the active site region of the FeMo-cofactor-less protein may have multiple roles.
Conclusion
On the basis of the Xe binding sites, we have identified in Av1 and Cp1, together with small molecule binding sites observed in Av1, Cp1, and Kp1, three new substrate and/or product pathways that can potentially connect the protein surface and the nitrogenase metalloclusters. The AI and AII pathways, deduced from Xe binding sites, are possible pathways for nonpolar substrates. Notably, the AI pathway is mostly conserved in the previously published pathway based on computational analysis by Seefeldt and co-workers. From the PRL binding site, there is a possible pathway to both the FeMo-cofactor (pathway BI) and the P-cluster (CI), the latter of which may provide a pathway for proton access. Both pathways contain part of the center water channel and then extend into the protein scaffold toward the metalloclusters. Given the polarity of the small molecules and the binding pocket environment, the BI and CI pathways may facilitate metallocluster access for both polar and nonpolar substrates/products. IMD, EDO, CO, and S sites in Av1 and Kp1 suggest that the DI pathway/interstitial channel may be used as a polar substrate pathway; it is conserved in all MoFe proteins. All pathways access the Fe2,3,6,7 and/or Fe3,4,5,7 faces of the FeMo-cofactor; however, this does not necessarily indicate that these faces are the primary targets for substrate binding since substrates may be able to move around the FeMo-cofactor. Overall, our studies establish that a variety of small molecules can access the interior of the MoFe-protein through multiple pathways (see the movie in the Supporting Information). This is based on experimental identification of nonwater, small molecule binding sites in the interior of Av1 and Cp1, which are two of the most structurally divergent bacterial MoFe proteins known. While there may be more favored pathways, given the variety of potential routes available, these observations indicate that there is unlikely to be a unique pathway utilized for substrate access from the protein surface to the active site; in effect, this is a molecular-level example of “all roads lead to Rome”.
Acknowledgments
We acknowledge the Gordon and Betty Moore Foundation, the Beckman Institute, and the Sanofi-Aventis Bioengineering Research Program at Caltech for their generous support of the Molecular Observatory at Caltech. We thank the staff at Beamline 12-2, Stanford Synchrotron Radiation Lightsource (SSRL), operated for the DOE and supported by its OBER and by the NIH, NIGMS (P41GM103393), and the NCRR (P41RR001209). We thank Jens Kaiser and James Howard for helpful discussions as well as Thomas Spatzal for discussions and his assistance with the xenon pressurization.
Glossary
Abbreviations:
- Cp
Clostridium pasteurianum
- Av
Azotobacter vinelandii
- Kp
Klebsiella pneumonia
- IMD
imidazole
- EDO
1,2-ethanediol
- MPD
2-methyl-2,4-pentanediol
- PRL
proline (ligand)
Supporting Information Available
(1) RMSD of native and Xe-pressurized proteins; (2) small molecules in previously published MoFe protein structures; (3) residues lining pathways; (4) PRL binding site; (5) IMD, CO, S, and EDO binding sites in Av1 and Kp1; (6) conservation of specific residues and residue type in Av1 and Cp1; (7) close contacts for and conservation of residues around Av1 and Cp1 Xe, PRL, and IMD sites; (8) electron density and anomalous difference maps of Xe binding sites; (9) docking of Fe protein and FeMo-cofactor insertion pathway in relation to substrate pathways; (10) movie of protein illustrating all substrate pathways, xenon sites, P-cluster, and FeMo-cofactor. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The structural model and structure factors have been deposited with the Protein Data Bank. The PDB ID for the Av1-Xe and Cp1-Xe structures are 4WNA and 4WN9, respectively.
The authors declare no competing financial interest.
This material is based upon work supported by the National Science Foundation Graduate Research Fellowship (Grant DGE-1144469 to C.N.M.), the National Institute of Health (NIH Grant GM45162 to D.C.R.), the Howard Hughes Medical Institute (to D.C.R.), the European Research Council (Grant 310656 to O. E.), and Deutsche Forschungsgemeinschaft (Grants Ei-520/7 and IRTG 1478 to O.E.).
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Erisman J. W.; Sutton M. A.; Galloway J.; Klimont Z.; Winiwarter W. (2008) How a century of ammonia synthesis changed the world. Nature Geosci. 1, 636–639. [Google Scholar]
- Kitano M.; Inoue Y.; Yamazaki Y.; Hayashi F.; Kanbara S.; Matsuishi S.; Yokoyama T.; Kim S.-W.; Hara M.; Hosono H. (2012) Ammonia synthesis using a stable electrode as an electron donor and reversible hydrogen store. Nat. Chem. 4, 934–940. [DOI] [PubMed] [Google Scholar]
- Hartwig J. F. (2010) Organotransition Metal Chemistry: From Bonding to Catalysis, p 540, University Science Books, Mill Valley, CA. [Google Scholar]
- Rees D. C.; Howard J. B. (2000) Nitrogenase: Standing at the crossroads. Curr. Opin. Chem. Biol. 4, 559–566. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Kaiser J. T.; Meloni G.; Yang K.-Y.; Spatzal T.; Andrade S. L. A.; Einsle O.; Howard J. B.; Rees D. C. (2013) The sixteenth iron in the nitrogenase MoFe protein. Angew. Chem. 125, 10723–10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duval S.; Danyal K.; Shaw S.; Lytle A. K.; Dean D. R.; Hoffman B. M.; Antony E.; Seefeldt L. C. (2013) Electron transfer precedes ATP hydrolysis during nitrogenase catalysis. Proc. Natl. Acad. Sci. U.S.A. 110, 16414–16419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spatzal T.; Perez K. A.; Einsle O.; Howard J. B.; Rees D. C. (2014) Ligand binding to the FeMo-cofactor: Structures of CO-bound and reactivated nitrogenase. Science 345, 1620–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott E. E.; Gibson Q. H.; Olson J. S. (2001) Mapping the pathways for O2 entry into and exit from myoglobin. J. Biol. Chem. 276, 5177–5188. [DOI] [PubMed] [Google Scholar]
- Teeter M. M. (2004) Myoglobin cavities provide interior ligand pathway. Protein Sci. 13, 313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radding W.; Phillips G. N. Jr. (2004) Kinetic proofreading by the cavity system of myoglobin: Protection from poisoning. BioEssays 26, 422–433. [DOI] [PubMed] [Google Scholar]
- Schoenborn B. P.; Watson H. C.; Kendrew J. C. (1965) Binding of xenon to sperm whale myoglobin. Nature 207, 28–30. [DOI] [PubMed] [Google Scholar]
- Duff A. P.; Trambaiolo D. M.; Cohen A. E.; Ellis P. J.; Juda G. A.; Shepard E. M.; Langley D. B.; Dooley D. M.; Freeman H. C.; Guss J. M. (2004) Using xenon as a probe for dioxygen-binding sites in copper amine oxidases. J. Mol. Biol. 344, 599–607. [DOI] [PubMed] [Google Scholar]
- Johnson B. J.; Cohen J.; Welford R. W.; Pearson A. R.; Schulten K.; Klinman J. P.; Wilmot C. M. (2007) Exploring molecular oxygen pathways in Hansenula polymorpha copper-containing amine oxidase. J. Biol. Chem. 282, 17767–17776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallio J. P.; Rouvinen J.; Kruus K.; Hakulinen N. (2011) Probing the dioxygen route in Melanocarpus albomyces laccase with pressurized xenon gas. Biochemistry 50, 4396–4398. [DOI] [PubMed] [Google Scholar]
- Whittington D. A.; Rosenzweig A. C.; Frederick C. A.; Lippard S. J. (2001) Xenon and halogenated alkanes track putative substrate binding cavities in the soluble methane monooxygenase hydroxylase. Biochemistry 40, 3476–3482. [DOI] [PubMed] [Google Scholar]
- McCormick M. S.; Lippard S. J. (2011) Analysis of substrate access to active sites in bacterial multicomponent monooxygenase hydroxylases: X-ray crystal structure of xenon-pressurized phenol hydroxylase from Pseudomonas sp. OX1. Biochemistry 50, 11058–11069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson-Ek M.; Abramson J.; Larsson G.; Törnroth S.; Brzezinski P.; Iwata S. (2002) The X-ray crystal structures of wild-type and EQ(I-286) mutant cytochrome c oxidases from Rhodobacter sphaeroides. J. Mol. Biol. 321, 329–339. [DOI] [PubMed] [Google Scholar]
- Luna V. M.; Fee J. A.; Deniz A. A.; Stout C. D. (2012) Mobility of Xe atoms within the oxygen diffusion channel of cytochrome ba3 oxidase. Biochemistry 51, 4669–4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna V. M.; Chen Y.; Fee J. A.; Stout C. D. (2008) Crystallographic studies of Xe and Kr binding within the large internal cavity of cytochrome ba3 from Thermus thermophilus: Structural analysis and role of oxygen transport channels in the heme-Cu oxidases. Biochemistry 47, 4657–65. [DOI] [PubMed] [Google Scholar]
- Darnault C.; Volbeda A.; Kim E. J.; Legrand P.; Vernede X.; Lindahl P. A.; Fontecilla-Camps J. C. (2003) Ni-Zn-[Fe4-S4] and Ni-Ni-[Fe4-S4] clusters in closed and open subunits of acetyl-CoA synthase/carbon monoxide dehydrogenase. Nat. Struct. Biol. 10, 271–9. [DOI] [PubMed] [Google Scholar]
- Wentworth P.; Jones L. H.; Wentworth A. D.; Zhu X.; Larsen N. A.; Wilson I. A.; Xu X.; Goddard W. A.; Janda K. D.; Eschenmoser A.; Lerner R. A. (2001) Antibody catalysis of the oxidation of water. Science 293, 1806–1811. [DOI] [PubMed] [Google Scholar]
- Tilton R. F. Jr.; Singh U. C.; Weiner S. J.; Connolly M. L.; Kuntz I. D. Jr.; Kollman P. A.; Max N.; Case D. A. (1986) Computational studies of the interaction of myoglobin and xenon. J. Mol. Biol. 192, 443–456. [DOI] [PubMed] [Google Scholar]
- Tilton R. F. Jr.; Singh U. C.; Kuntz I. D. Jr.; Kollman P. A. (1988) Protein-ligand dynamics: A 96 ps simulation of a myoglobin-xenon complex. J. Mol. Biol. 199, 195–211. [DOI] [PubMed] [Google Scholar]
- Yaël M.; Patricia A.; Anne V.; Xavier V.; Hatchikian E. C.; Martin J. F.; Michel F.; Juan C. F.-C. (1997) Gas access to the active site of Ni-Fe hydrogenases probed by X-ray crystallography and molecular dynamics. Nat. Struct. Mol. Biol. 4, 523–526. [DOI] [PubMed] [Google Scholar]
- Czerminski R.; Elber R. (1991) Computational studies of ligand diffusion in globins: I. Leghemoglobin. Proteins: Struct., Funct., Bioinf. 10, 70–80. [DOI] [PubMed] [Google Scholar]
- Smith D. M. A.; Danyal K.; Raugei S.; Seefeldt L. C. (2014) A substrate channel in nitrogenase revealed by a molecular dynamics approach. Biochemistry 53, 2278–2285. [DOI] [PubMed] [Google Scholar]
- Dance I. (2013) Nitrogenase: A general hydrogenator of small molecules. Chem. Commun. 49, 10893–10907. [DOI] [PubMed] [Google Scholar]
- Durrant M. C. (2001) Controlled protonation of iron-molybdenum cofactor by nitrogenase: A structural and theoretical analysis. Biochem. J. 355, 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barney B. M.; Yurth M. G.; Dos Santos P. C.; Dean D. R.; Seefeldt L. C. (2009) A substrate channel in the nitrogenase MoFe protein. J. Biol. Inorg. Chem. 14, 1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dance I. (2013) A molecular pathway for the egress of ammonia produced by nitrogenase. Sci. Rep. 3, 3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dance I. (2012) The controlled relay of multiple protons required at the active site of nitrogenase. Dalton Trans. 41, 7647–7659. [DOI] [PubMed] [Google Scholar]
- Igarashi R. Y.; Seefeldt L. C. (2003) Nitrogen fixation: The mechanism of the Mo-dependent nitrogenase. Crit. Rev. Biochem. Mol. Biol. 38, 351–84. [DOI] [PubMed] [Google Scholar]
- Rees D. C., Kim J., Georgiadis M. M., Komiya H., Chirino A. J., Woo D., Schlessman J., Chan M. K., Joshua-Tor L., Santillan G., Chakrabarti P., and Hsu B. T. (1993) Crystal structures of the iron protein and molybdenum-iron protein of nitrogenase, In Molybdenum Enzymes, Cofactors, and Model Systems, pp 170–185, American Chemical Society, Washington, D.C. [Google Scholar]
- Ringe D.; Petsko G. A.; Kerr D. E.; Ortiz de Montellano P. R. (1984) Reaction of myoglobin with phenylhydrazine: a molecular doorstop. Biochemistry 23, 2–4. [DOI] [PubMed] [Google Scholar]
- Einsle O.; Tezcan F. A.; Andrade S. L. A.; Schmid B.; Yoshida M.; Howard J. B.; Rees D. C. (2002) Nitrogenase MoFe-protein at 1.16 Å resolution: A central ligand in the FeMo-cofactor. Science 297, 1696–1700. [DOI] [PubMed] [Google Scholar]
- Zhang L.-M., Morrison C. N., Kaiser J. T., Rees D. C.. Nitrogenase MoFe-protein from Clostridium pasteurianum at 1.08 Å resolution: Comparison to the Azotobacter vinelandii MoFe-protein. Acta Crystallogr., Sect. D: Biol. Crystallogr. 71, 274–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowell M. H. B.; Soltis S. M.; Kisker C.; Peters J. W.; Schindelin H.; Rees D. C.; Cascio D.; Beamer L.; Hart P. J.; Wiener M. C.; Whitby F. G. (1996) A simple device for studying macromolecular crystals under moderate gas pressures (0.1–10 MPa). J. Appl. Crystallogr. 29, 608–613. [Google Scholar]
- Kabsch W. (2010) XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Spatzal T.; Aksoyoglu M.; Zhang L.; Andrade S. L.; Schleicher E.; Weber S.; Rees D. C.; Einsle O. (2011) Evidence for interstitial carbon in nitrogenase FeMo-cofactor. Science 334, 940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger A. T.; Adams P. D.; Clore G. M.; DeLano W. L.; Gros P.; Grosse-Kunstleve R. W.; Jiang J.-S.; Kuszewski J.; Nilges M.; Pannu N. S.; Read R. J.; Rice L. M.; Simonson T.; Warren G. L. (1998) Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr., Sect. D: Biol. Crystallogr. 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., D: Biol. Crystallogr. 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubak P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., D: Biol. Crystallogr. 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine P. V.; Grosse-Kunstleve R. W.; Echols N.; Headd J. J.; Moriarty N. W.; Mustyakimov M.; Terwilliger T. C.; Urzhumtsev A.; Zwart P. H.; Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr., Sect. D: Biol. Crystallogr. 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zondlo N. J. (2012) Aromatic–proline interactions: Electronically tunable CH/π interactions. Acc. Chem. Res. 46, 1039–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitgeb B.; Tóth G. (2005) Aromatic–aromatic and proline–aromatic interactions in endomorphin-1 and endomorphin-2. Eur. J. Med. Chem. 40, 674–686. [DOI] [PubMed] [Google Scholar]
- Chovancova E.; Pavelka A.; Benes P.; Strnad O.; Brezovsky J.; Kozlikova B.; Gora A.; Sustr V.; Klvana M.; Medek P.; Biedermannova L.; Sochor J.; Damborsky J. (2012) CAVER 3.0: A tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput. Biol. 8, e1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, Version 1.7.4.2, Schrödinger, LLC.
- Mayer S. M.; Lawson D. M.; Gormal C. A.; Roe S. M.; Smith B. E. (1999) New insights into structure-function relationships in nitrogenase: A 1.6 Å resolution X-ray crystallographic study of Klebsiella pneumoniae MoFe-protein. J. Mol. Biol. 292, 871–891. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Ribbe M. W. (2013) Nitrogenase assembly. Biochim. Biophys. Acta 1827, 1112–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupnik K.; Lee C. C.; Wiig J. A.; Hu Y.; Ribbe M. W.; Hales B. J. (2014) Nonenzymatic synthesis of the P-cluster in the nitrogenase MoFe protein: Evidence of the involvement of all-ferrous [Fe4S4]0 intermediates. Biochemistry 53, 1108–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Woo D.; Rees D. C. (1993) X-ray crystal structure of the nitrogenase molybdenum-iron protein from Clostridium pasteurianum at 3.0-Å resolution. Biochemistry 32, 7104–7115. [DOI] [PubMed] [Google Scholar]
- Schmid B.; Einsle O.; Chiu H.-J.; Willing A.; Yoshida M.; Howard J. B.; Rees D. C. (2002) Biochemical and structural characterization of the cross-linked complex of nitrogenase: Comparison to the ADP-AlF4–-stabilized structure. Biochemistry 41, 15557–15565. [DOI] [PubMed] [Google Scholar]
- Tezcan F. A.; Kaiser J. T.; Mustafi D.; Walton M. Y.; Howard J. B.; Rees D. C. (2005) Nitrogenase complexes: multiple docking sites for a nucleotide switch protein. Science 309, 1377–1380. [DOI] [PubMed] [Google Scholar]
- Thorneley R. N.; Lowe D. J. (1983) Nitrogenase of Klebsiella pneumoniae. Kinetics of the dissociation of oxidized iron protein from molybdenum-iron protein: Identification of the rate-limiting step for substrate reduction. Biochem. J. 215, 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid B.; Ribbe M. W.; Einsle O.; Yoshida M.; Thomas L. M.; Dean D. R.; Rees D. C.; Burgess B. K. (2002) Structure of a cofactor-deficient nitrogenase MoFe protein. Science 296, 352–356. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.